

Since the discovery of the oncogene in the 1970s with much fanfare including a Nobel Prize to Bishop and Varmus in 1989, the concept of oncogenesis—and the role of the oncogene in this process—has been refined and greatly expanded. While we all thought at that point that we had the etiology of cancer all figured out, subsequent work has resulted in the identification of six “hallmarks” of this disease (1) which include the old concept of sustained proliferative signaling, but also include what in the 1970s would have been unthinkable: metabolic reprogramming (2). This latter phenomenon, which often utilizes the technique of non-targeted metabolomics analysis to identify such pathways, capitalizes on the need of a rapidly growing tumor for an abundance of biochemical materials, and is one of the more recent advances in understanding malignancy as well as a path to new therapies (3).
In the search for new metabolites which associate with cancer, D-2-HG was discovered and later found to be, in most cases, associated with the neomorphic activity of a mutant synthetic enzyme, isocitrate dehydrogenase (IDH). Hence, 2-HG, which was described early in kidney cancer (4) and which lies in the glutamine metabolic pathway, was aptly labeled an “oncometabolite” in parallel nomenclature with oncogene and oncoprotein. The mechanism by which an oncometabolite can transform cells is not as obvious as the other onco-compounds, but probably relates to epigenetic modification (4). Surprisingly, the production of D-2-HG does not always require a mutant IDH enzyme and has also been described in non-malignant, although highly proliferative, renal disease (5). While there has been a fair amount of research reported in glioma, even resulting in the discovery of an inhibitor of mutant IDH now in clinical trials (6), the work on 2-HG in kidney cancer has been more sparse. On this background comes a new paper by Shelar et al. (7) which greatly expands this knowledge base and, more importantly, provides important clinical translation of the data.
The most common form of kidney cancer [also known as renal cell carcinoma (RCC)] is “clear cell” RCC, for which the incidence is rising in developed countries likely due to the obesity epidemic and other factors. Given that survival statistics for metastatic disease, even in the era of immune checkpoint inhibitors, remains dismal, new therapeutic approaches are desperately needed. Moreover, since in most cases there is a large amount of variability in patients in response to all therapies, identifying biomarkers which correlate with aggressivity of RCC would be a major advance. This paper nicely addresses both of these issues.
The study of Shelar et al. has significantly advanced our understanding of the role of L-2-HG in RCC carcinogenesis, by looking at the “flip side” of its metabolism: the degradation enzyme L2HDH. These investigations were made by utilizing different cutting-edge approaches (metabolomics, genomics, transcriptomics and pharmacologic methods) in several in vitro and in vivo models of RCC. Using these approaches the glutamine/α-KG/2-HG pathway (Figure 1) was dissected and the importance of the reduction of L2HGDH catalytic activity, which resulted in elevation of L-2-HG, was investigated in RCC.
Figure 1.
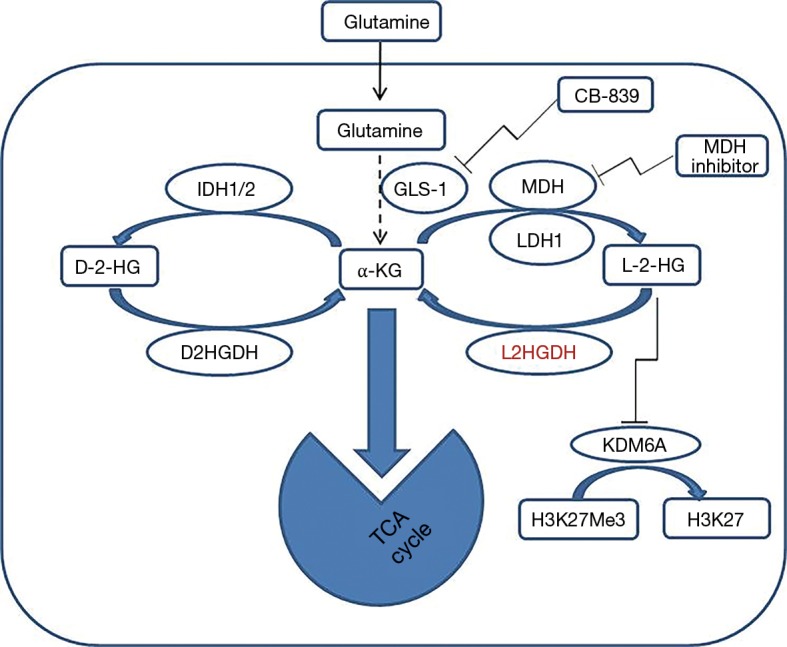
Graphical representation of glutamine/2-HG pathway and therapeutics options in renal cancer based on Shelar et al. Glutamine is the source of 2-HG. Glutamine is metabolized to α-ketoglutarate (α-KG) with glutaminase-1 (GLS-1). IDH1 or 2 (IDH1/2) are involving in metabolizing α-KG to D-2-HG and malate dehydrogenase (MDH) or lactate dehydrogenase 1 (LDH1) will metabolize α-KG to L-2-HG. D-2-HG dehydrogenase (D2HGDH) and L-2HG dehydrogenase (L2HGDH) convert D-2-HG and L-2-HG to α-KG. A decrease in L2HGDH results in accumulation of the oncometabolite L-2-HG. In turn high L-2-HG inhibits activity of histone lysine demethylase 6A (KDM6A) to promote H3K27 trimethylation (H3K27Me3) in RCC cells. The glutaminase-1 (GLS-1) inhibitor CB-839 and MDH inhibitor (derivative of benzylpaullones) can be potential therapeutic targets for RCC. RCC, renal cell carcinoma.
To establish the relationship between L2HGDH expression and RCC tumorigenesis, Shelar et al. started with in vitro studies. Since HK-2 immortalized “normal” renal epithelial cells have relatively high basal L2HGDH expression compared with RCC cells, an shRNA approach (confirmed by mRNA and protein levels) was used to examine the effects of reduced L2HGDH expression in HK-2 cells. Consistent with the role of L2HGDH to regulate the levels of L-2-HG and hence its catalytic activity, L2HGDH knockdown clones demonstrated elevated cellular levels of L-2-HG as determined by gas chromatography-mass spectroscopy (GC-MS). These results show that knockdown of L2HGDH indeed promotes the appearance of in vitro tumor phenotypes in such “normal” renal epithelial cells. Further isotopologue studies using glutamine fully labeled with C13 (U-13C5) demonstrated whether and how many carbons from glutamine were incorporated into L-2-HG. This analysis of carbon sourcing for L-2-HG in RCC cells indicated that glutamine is the predominant carbon source for L-2-HG via the activity of malate dehydrogenase 2 (MDH2), a highly significant finding because it explains, at least in part, the nature of previously reported of glutamine-addiction in RCC (8,9). These data also confirm and highlight the importance of the new potential application of 18F-glutamine-based PET imaging recently proposed for RCC staging and therapeutic stratification (9), and moves these studies closer to the clinic. Real-time imaging of L-2-HG could be achieved by performing high-polarized MRI (HP-MRI) to image metabolic conversion of glutamine to L-2-HG in vivo using 13C-glutamine as described (10). Interestingly, glutaminase inhibition using CB-839 led to a significant reduction of L-2-HG levels in multiple RCC lines in the Shelar et al. study.
Shelar et al. also address the on-target effects of the elevation of the oncometabolite L-2-HG which contribute to RCC carcinogenesis. This was demonstrated by adding exogenous L-2-HG to the immortalized renal epithelial cells (HK-2 cells), which led to elevated cellular levels of L-2-HG and subsequently promoted a migratory phenotype in HK-2 cells mimicking the results of L2HGDH knockdown. To confirm the role of L2HGDH in tumor migration multiple RCC cells were stably transduced with WT L2HGDH cDNA or control vector and L2HGDH found to have inhibitory effect on migratory phenotypes in RCC cells as determined by scratch assay. In addition, the transfected RCC showed that L2HGDH transfection has led to inhibition of in vivo tumor growth dependent on L2HGDH catalytic activity. The role of 2-HG as an oncometabolite has been linked to epigenetic changes that cause inhibition of α-KG-dependent dioxygenases and demethylases, which has been suggested to be a driver for tumorigenesis (11). Similar to D-2-HG, L-2-HG can competitively inhibit dioxygenases due to structural similarity to α-KG. Epigenetic studies performed here have uncovered some mechanisms of L-2-HG to promote RCC growth. Transcriptomic and functional analyses demonstrate that the histone demethylase KDM6A is a target of L-2-HG in RCC (Figure 1).
Finally, in important translational work, the clinical value of L-2-HG/L2HGDH in human RCC in vivo was assessed using the data from the TCGA. In these studies, the biological relevance of the L-2-HG/L2HGDH axis to renal carcinogenesis was examined. The outcomes of RCC patients as a function of L2HGDH showed that patient with tumors in the lower 50% of L2HGDH expression had reduced survival and that lower L2HGDH mRNA expression was associated with higher tumor stage and grade.
In summary, this paper by Shelar et al. adds to our knowledge of RCC oncogenesis with respect to the oncometabolite L-2-HG and provides not only a novel mechanism for manipulating its appearance, but also important data on carbon sourcing of this oncometabolites which has therapeutic implications for this disease. The authors provide biomarker data which have human translational import. In addition to utilizing mutant IDH inhibitors now in clinical trials, work here suggests that alteration of L2HGDH (using MDH inhibitors) is another viable approach as is the previous-proposed use of glutaminase inhibition (Figure 1). Further work in this area will lead to new therapeutic approaches capitalizing on this newly elucidated reprogrammed metabolism of human RCC.
Acknowledgements
Funding: This work was supported by NIH grant 1R01DK082690-01A1, the Medical Service of the US Department of Veterans’ Affairs, and Dialysis Clinics, Inc. (DCI).
Provenance: This is an invited Editorial commissioned by Section Editor Xiao Li (Department of Urology, Jiangsu Cancer Hospital & Jiangsu Institute of Cancer Research & Nanjing Medical University Affiliated Cancer Hospital, Nanjing, China).
Conflicts of interest: The authors have no conflicts of interest to declare.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646-74. 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 2.Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell 2012;21:297-308. 10.1016/j.ccr.2012.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wettersten HI, Aboud OA, Lara PN, Jr, et al. Metabolic reprogramming in clear cell renal cell carcinoma. Nat Rev Nephrol 2017;13:410-9. 10.1038/nrneph.2017.59 [DOI] [PubMed] [Google Scholar]
- 4.Shim EH, Livi CB, Rakheja D, et al. L-2-Hydroxyglutarate: an epigenetic modifier and putative oncometabolite in renal cancer. Cancer Discov 2014;4:1290-8. 10.1158/2159-8290.CD-13-0696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hwang VJ, Kim J, Rand A, et al. The cpk model of recessive PKD shows glutamine dependence associated with the production of the oncometabolite 2-hydroxyglutarate. American Journal of Physiology: Renal Physiology 2015;309:F492-8. 10.1152/ajprenal.00238.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abou Dalle I, DiNardo CD. The role of enasidenib in the treatment of mutant IDH2 acute myeloid leukemia. Ther Adv Hematol 2018;9:163-73. 10.1177/2040620718777467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shelar S, Shim EH, Brinkley GJ, et al. Biochemical and Epigenetic Insights into L-2-Hydroxyglutarate, a Potential Therapeutic Target in Renal Cancer. Clin Cancer Res 2018;24:6433-46. 10.1158/1078-0432.CCR-18-1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shroff EH, Eberlin LS, Dang VM, et al. MYC oncogene overexpression drives renal cell carcinoma in a mouse model through glutamine metabolism. Proc Natl Acad Sci U S A 2015;112:6539-44. 10.1073/pnas.1507228112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abu Aboud O, Habib SL, Trott JF, et al. Glutamine addiction in kidney cancer suppresses oxidative stress and can be exploited for real-time imaging. Cancer Research 2017;77:6746-58. 10.1158/0008-5472.CAN-17-0930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salamanca-Cardona L, Shah H, Poot AJ, et al. In Vivo Imaging of Glutamine Metabolism to the Oncometabolite 2-Hydroxyglutarate in IDH1/2 Mutant Tumors. Cell Metab 2017;26:830-41.e3. 10.1016/j.cmet.2017.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu C, Ward PS, Kapoor GS, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012;483:474-8. 10.1038/nature10860 [DOI] [PMC free article] [PubMed] [Google Scholar]