

Summary
Osteoarthritis (OA) is the most common joint disease that strongly reduces the quality of life in patients; However, no disease‐modifying therapy is available. For a long time, OA was considered a non‐inflammatory disease that was the result of ‘wear‐and‐tear’ and abnormal mechanics, and therefore many considered the term ‘osteoarthritis’ a misnomer. However, during the last decades the notion arose that inflammation is not only present in the majority of OA patients but, rather, actively involved in the progression of the disease. Influx of immune cells is observed in the synovium and a plethora of inflammatory mediators is present in tissues and fluids from OA patients. These mediators cause the production of degrading enzymes that break down the cartilage matrix, which is the main hallmark of OA. Alarmins, which belong to the group of danger signals, have been implicated in many inflammatory diseases. They are among the first factors to be released upon cell stress due to, for example, infection, damage and inflammation. They attract and activate cells of the immune system and therefore lie at the base of the inflammatory reaction. In this narrative review, an overview of the history of OA, the evolving concept of inflammation as important factor in the OA pathogenesis, and particularly the central role that alarmins play in the initiation and maintenance of the low‐grade inflammatory response in OA, is provided. Moreover, the targeting of alarmins as a promising approach to dampen the inflammation in OA is highlighted.
Keywords: alarmins, inflammation, osteoarthritis, pathology, S100A8/A9
Osteoarthritis
Osteoarthritis (OA) is a highly complex and the most prevalent joint disorder. Worldwide, 9·6% of men and 18·0% of women aged more than 60 years suffer from symptomatic OA, and a total of 242 million people are affected globally 1, 2.
Clinical symptoms include severe pain, joint stiffness and strongly reduced mobility, which together seriously decrease the quality of life 3, 4. Pathologically, OA is characterized by changes in all joint tissues caused by coinciding catabolic and anabolic processes. Cartilage degeneration, ectopic bone formation, subchondral bone sclerosis, damage to ligaments and menisci and fibrosis and inflammation in the synovial membrane that lines the joint cavity are the main disease hallmarks 5.
However, no disease‐modifying osteoarthritis drugs (DMOADs) are available to date. This limits options for therapy to treating symptoms such as pain and often leads to joint‐replacement surgery at end‐stage disease.
The following is a brief narrative overview of the history of OA, and how the view on the disease shifted from being a relatively simple and inevitable ‘wear‐and‐tear’ process towards an active disease of the joint as an organ where inflammation plays an important role in the aetiopathology.
History of osteoarthritis
It is often stated that OA might be the oldest ‘known’ disease, with signs of pathology present in dinosaur skeletons and ancient human skeletons, possibly because bones carrying evidence of OA have withstood the sands of time better than other tissues 6, 7. However, the age of recognition of the disease nowadays known as OA is more debatable, mainly because of ambiguous nomenclature. From the time of Hippocrates until the 18th century all rheumatic complaints were considered to be gout. A first separation came with the description of digitorum nodi by Heberden 8. Although OA of the hip was clinically recognized in the early 1800s, it was put on a par with rheumatoid arthritis (RA), together referred to as arthritis deformans. This term was introduced by Charcot and Trastour and later widely publicized by Virchow in the mid‐19th century, but was used well into the 20th century 9, 10, 11. The term osteo arthritis was most probably introduced by the German orthopaedic surgeon von Volkmann in the 1850s. He was also the first to anatomically and pathologically differentiate OA from RA lesions 9. However, this met with fierce protest, because the term suggests the presence of inflammation, a view that was not endorsed by many at the time.
Osteoarthritis as a mechanical disease
OA has been considered by many a relatively simple ‘wear‐and‐tear’ disease leading to the loss of cartilage. In this view, OA was considered the sole consequence of fragility of the cartilage matrix due to, for example, ageing, which should not be classified as a disease. However, the majority of physicians supported Garrod’s view that processes such as cartilage erosion, osteophyte formation and changes in bone could be the result of a disease process, provided that it was present for a long time 12. As a result, because inflammation was not considered part of the aetiology, many believed OA to be solely driven by mechanical events, which would mean that the disease is caused by or related to physical forces or motion. For this reason the term osteoarthritis, implying inflammation as the major cause, was considered a misnomer. The mechanical origin of OA was thought to be underlined by the evidence that ancient skeletons mainly had signs of OA in the lower back and shoulders, whereas the knees were less affected 13, 14. This is in contrast to that observed in patients nowadays, and could be attributed to differences in physical activity (e.g. due to differences in professions). Furthermore, our current sedentary lifestyle has increased the prevalence of obesity, which strongly associates with knee OA 15, 16. Other clues that abnormal mechanics cause OA came from studies that showed that traumatic event, such as tears of the anterior cruciate ligament and meniscus and varus malalignment, greatly increase the risk of OA development 17, 18, 19, 20, whereas individuals with focal high stress due to femoroacetabular impingement experience excess rates of OA 21, 22.
However, the concept of OA as a mechanical disease implies that cartilage breakdown is accelerated when the cartilage is increasingly loaded. Nevertheless, running, which greatly increases stresses on hip and knee joints, does not aggravate OA incidence and reduced OA pain and hip replacement surgery 23, 24.
Moreover, the notion arose that not all OA could be attributed to mechanical factors. Why is obesity not associated with OA in the hip, whereas this is also a weight‐bearing joint; and why do obese individuals have higher rates of hand OA, whereas they do not experience greater stresses in those joints 25?
The evolving concept of inflammation as driver of pathology in osteoarthritis
Although inflammation has been incidentally mentioned in association with OA since the mid‐19th century, it was not until the last decades that inflammation was increasingly considered to be present and important in the development of OA. In fact, tissue and fluid samples from OA patients have long been used as negative, non‐inflammatory controls in studies investigating rheumatoid arthritis and spondyloarthritis. This concealed the raised levels of proinflammatory factors present in OA tissues and fluids compared to healthy controls and arguably reinforced the idea of OA as a non‐inflammatory disease.
A big step towards the recognition of inflammation in OA was taken in the 1990s, when molecular biology took a leap forwards and showed that soluble mediators, released by various tissues in the joint, could stimulate the production of matrix‐degrading enzymes, such as matrix metalloproteinases (MMPs) and aggrecanases (ADAMTS4/5), which are closely involved in the breakdown of the articular cartilage although, even with these findings, it still took more than 10 years before inflammation was broadly accepted as a critical feature of OA pathogenesis.
Multiple studies have shown clear signs of synovial inflammation that correlate with disease severity, progression and pain sensitization using arthroscopy, ultrasonography or magnetic resonance imaging (MRI) 26, 27, 28, 29, 30. Debate remains about whether synovial inflammation is causative for OA or rather a secondary effect of joint failure and products of matrix degradation. The most broadly accepted view is that synovial inflammation is the result of cartilage fragments, released due to, for example, a traumatic event in the joint, that activate the synovial cells to produce proinflammatory factors and MMPs, thereby further increasing cartilage degeneration 31. A vicious inflammatory circle ensues in this way. An overview of processes that contribute to inflammation in OA is given in Fig. 1.
Figure 1.
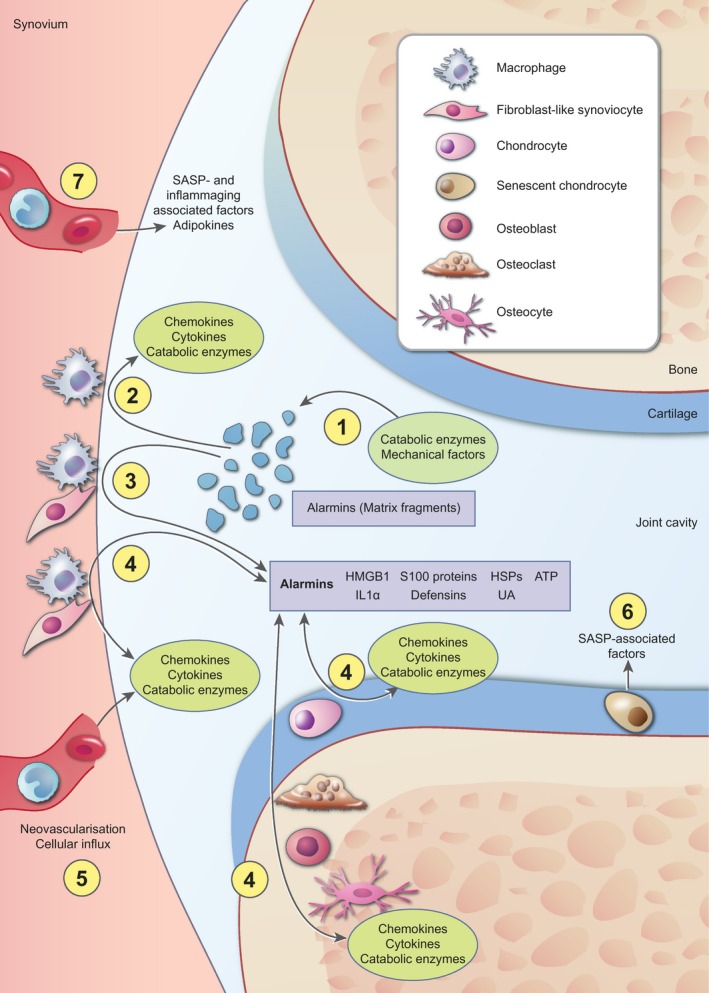
Overview of the processes that contribute to inflammation in osteoarthritis. Nowadays it is well accepted that inflammation is present and actively involved in the pathogenesis of osteoarthritis (OA). The most broadly supported view is that cartilage extracellular matrix fragments, released as the result of an initial trauma or catabolic enzyme activity (a), increase synovial inflammation by stimulation of cells in the synovial lining. Activated synovial cells start to produce proinflammatory factors, such as chemokines, cytokines and catabolic enzymes (b). In addition, these cells secrete high levels of various alarmins, such as high‐mobility group box‐1 (HMGB1) and S100 proteins (c). These alarmins initiate a positive‐feedback loop with reciprocal production of chemokines, cytokines and catabolic enzymes, on one hand, and alarmins on the other hand, not only in synovial cells, but also in cells in the cartilage and periarticular bone (d). Growth factors and chemokines add to the inflammatory process in the joint by stimulating neovascularization and influx of inflammatory cells that also start to produce chemokines, cytokines and catabolic enzymes (e). In addition, cellular senescence is associated with an increased release of factors, such as many cytokines and catabolic enzymes, referred to as senescence‐associated secretory phenotype (SASP), which further stimulates the inflammatory state of the joint (f). Finally, systemic changes are associated with inflammation during OA. Increased systemic levels of inflammatory mediators as the result of SASP or ageing and adipokines as the result of increased fat mass contribute to the inflammatory milieu in the joint (g). Together, these factors are thought to be involved in the pathological processes that take place during OA, such as hypertrophic differentiation of chondrocytes and breakdown of the articular cartilage, fibrosis in the synovial tissue, sclerosis of the subchondral bone and ectopic bone formation (these processes are not depicted in this figure).
OA is mainly linked to activation of innate immunity by binding of damage‐associated molecular patterns (DAMPs) to so‐called pattern recognition receptors (PRRs) 32, 33. Of central importance in the PRR family are the Toll‐like receptors (TLRs). TLR‐2 and TLR‐4 have been shown to bind a multitude of degraded cartilage extracellular matrix fragments. These fragments include low molecular weight hyaluronan, tenascin C, fibronectin, biglycan and aggrecan 34, 35, 36, 37. It is believed that these fragments might cause the initial trigger that starts the inflammatory response in the OA joint. Other local triggers of inflammation include activation of the complement system and stimulation of cells by crystals that are present in the majority of OA patients 38, 39, 40.
Next to local induction of inflammation by danger signals, low‐grade systemic inflammation and ageing‐induced inflammatory responses have been associated with OA development. Low‐grade systemic inflammation is present in many OA patients. Serum levels of various cytokines are increased in OA patients 41. Moreover, OA is strongly linked to obesity and the metabolic syndrome. Increased fat mass has been shown to result in higher systemic inflammatory factors, such as cytokines and various adipose tissue‐produced factors called adipokines that have inflammatory functions 42, 43, 44.
Furthermore, ageing is associated with an altered inflammatory response, also referred to as inflammageing. Cellular senescence claims a central spot in this process. Senescent cells have an increased production of proinflammatory and catabolic mediators 45. Indeed, proteins secreted as the result of this senescence‐associated secretory phenotype (SASP), such as various cytokines, chemokines and MMPs, are abundant in OA tissues and fluids 46.
Together, this shows that inflammation plays a clear role in OA development, although the nature of the inflammatory reaction is different from RA. Whereas RA is characterized by a severe synovial inflammation, a chronic but relatively low‐grade inflammation is found in OA. Activation of the innate immunity in OA results in the production of cytokines, such as interleukin (IL)‐1β, IL‐6, IL‐8 and tumour necrosis factor (TNF)‐α, activation of the complement system and production of matrix‐degrading enzymes such as MMPs and ADAMTS4/5. More in‐depth reviews concerning their involvement in OA can be found elsewhere 47, 48, 49.
Cells of the innate immune system, such as monocytes and macrophages, are mainly associated with the inflammatory response in the OA joint. Whereas OA patients show an altered T cell profile, the clear involvement of these cells in the disease pathogenesis is debatable 50, 51. Depletion of macrophages with intra‐articular injection of clodronate‐laden liposomes strongly reduced the MMP‐mediated breakdown of articular cartilage and the formation of osteophytes in a preclinical model of OA 52, 53. Moreover, depletion of macrophages from a cell suspension made from human OA synovium reduced the cytokine response and activity of matrix‐degrading enzymes 54.
Therefore, whereas OA might not comply with the classical view of inflammation with the presence of rubor, calor, dolor and tumour, and whereas there is no strong evidence of the presence of robust adaptive (auto)immune reactions, there is a clear involvement of both local and systemic inflammation in OA development.
However, results from clinical trials in which inflammatory cytokines, such as IL‐1β and TNF‐α, were targeted have been repeatedly disappointing (elegantly reviewed in 55). Targeting more upstream regulators of acute inflammation that lead to the production and perpetuation of cytokine production might serve as an interesting alternative.
Alarmins
An example of such a group of factors that are released during the first phase of inflammation are the alarmins, a term proposed by Oppenheim et al. Initially defined as molecules that attract and activate antigen‐presenting cells such as dendritic cells, alarmins are nowadays more broadly defined as structurally diverse and evolutionarily unrelated, endogenous molecules that are released upon cell stress, which cause inflammation in vivo 56, 57. However, while osteoarthritis is characterized by an influx and activation of innate immune cells, little is known about the role of dendritic cells in this disease, although no profound adaptive (auto)immune responses are observed.
While defensins and high‐mobility group box‐1 (HMGB1) belong to the first identified, the list of alarmins has grown rapidly since then and now includes, but is not limited to, heat shock proteins (HSPs), uric acid (UA), adenosine triphosphate (ATP), IL‐1α and s100 proteins 57, 58. An in‐depth structural and functional characterization of the various alarmins is beyond the scope of this review and can be found elsewhere 57, 59.
Many alarmins are intracellular proteins that are passively or actively released as the result of stress due to, for example, inflammation and tissue damage. Passive release can be the result of cell injury or death, such as necrosis or netosis. Active release of alarmins is regulated by mechanisms that are independent of the endoplasmatic reticulum and Golgi route. They include degranulation, secretion via the inflammasome and pyroptosis 56, 60, 61, 62.
In the extracellular milieu alarmins bind to a range of receptors, among which the TLRs and receptor for advanced glycosylation end products (RAGE) are the most well‐studied. TLR‐4, in particular, is used by many alarmins 63, 64, 65, 66.
Because of their quick release as the result of cell stress or non‐programmed cell death, alarmins are among the first factors to be secreted and, as such, act as first responders to stimuli. Together with their ability to attract and activate immune cells, this puts them at the base of inflammatory responses.
Alarmins in inflammatory diseases
Alarmins play an important role in a wide range of disorders involving infection‐induced and sterile inflammation, where they are associated with sustaining inflammation and the induction of tissue injury. HMGB1 and S100A8/A9 have been well characterized in the context of sepsis 67, 68, 69. In conditions of sterile inflammation, such as haemorrhagic shock and ischaemic injury in multiple organs, HMGB1 appears to be a crucial factor in promoting inflammation 70, 71, 72, 73.
Moreover, alarmins are key players in tumour immunology, although they have been attributed paradoxical roles. On one hand, factors such as HMGB1 and S100A8/A9 are involved in tumour promotion via their capacities to promote cell proliferation, migration and production of matrix‐degrading enzymes, which together result in tumour growth and metastases 74, 75, 76, 77. Furthermore, S100A8/A9 promotes the production and recruitment of myeloid‐derived suppressor cells that diminish immunity 78. On the other hand, alarmins can promote anti‐tumour immunity by, for example, recruitment and activation of dendritic cells and stimulating T helper type 1 (Th1) immune responses 79, 80, 81.
Finally, it is broadly acknowledged that alarmins play central roles in many chronic inflammatory diseases, including many arthritides, inflammatory bowel disease and atherosclerosis 82, 83, 84, 85, 86. A more detailed overview of the contribution of alarmins to inflammatory diseases is given elsewhere 57, 59.
Alarmins in osteoarthritis
A growing body of evidence shows a central role for alarmins in the initiation and maintenance of the low‐grade inflammatory response that is present during OA. Interestingly, high levels of many alarmins have been described in the synovial fluid of OA patients, including HMGB1, UA, ATP, thymosin β4 and various S100 proteins 40, 87, 88, 89, 90, 91, 92. Other alarmins are described to have increased production and secretion, although the exact extracellular localization remains unknown, such as is the case for HSPs. Release of alarmins has been shown for multiple joint tissues, including the periarticular bone, cartilage and synovium. Their release is stimulated by proinflammatory and catabolic factors, but also by mechanical stress on the tissues via both active release and as the result of cell death in the inflammatory environment. In this section, a selective overview is provided of the alarmins that are present during OA and how they affect the disease pathology.
As described earlier in this narrative, cartilage extracellular matrix fragments such as biglycan, fibronectin, aggrecan and low molecular weight hyaluronan stimulate cell types in the joint via PRRs, leading to the production of inflammatory mediators and the attraction of inflammatory cells, and as such act as alarmins 34, 35, 36, 37, 93. Moreover, they are taken up by synovial macrophages, resulting in the activation of proinflammatory responses in these cells.
IL‐1α is predominantly released by macrophages as the result of stress‐induced activation of the inflammasome 94. Stimulation of chondrocytes with IL‐1α increases nitric oxide production and MMP activity 95. The role of IL‐1 in the development of OA has been well investigated, but debate remains about its importance. Many studies describe the catabolic function of IL‐1 on cartilage, although a preclinical model of OA induced in IL‐1α/β–/– mice showed that both IL‐1α and IL‐1β are not involved in the disease pathology 48, 96.
Other studies have shown increased UA levels in synovial fluid, which correlated with levels of IL‐1β and IL‐18 and with OA severity 40. Furthermore, it was tentatively postulated that monosodium UA crystals, which are key stimulators of inflammation in gout, might promote OA pathology via activation of the inflammasome which leads to the release of IL‐1β and other alarmins 97.
Defensins comprise another group of alarmins that has been linked to a catabolic response in OA. Human β‐defensin 3 expression in chondrocytes is induced by IL‐1β and TNF‐α. Stimulation of chondrocytes with recombinant β‐defensin 3 protein increases the production of the collagenases MMP1 and MMP13, that are among the most potent matrix‐degrading enzymes in OA, whereas it additionally decreases the production of tissue inhibitors of metalloproteinases (TIMP)1 and TIMP2 98. Other studies have shown an increased production of β‐defensins 2 and 4 in OA cartilage and menisci, but further studies are required to elucidate the role played by these alarmins 99, 100.
ATP is released from cells upon cell stress. In turn, ATP has been shown to stimulate both chondrocyte death and the calcification of cartilage 101. Furthermore, a recent study shows the activation of pyroptosis in OA fibroblast‐like synoviocytes by ATP together with TLR‐4 ligands 102, 103. Finally, ATP increases pain sensitivity (hyperalgesia) via binding to the purinergic P2X3 and P2X2/3 receptors 89, 104.
Although these studies suggest the involvement of these alarmins in OA, many aspects about their involvement remain unknown and therefore more (pre‐)clinical studies are needed to further elucidate their importance and mechanism of action in the OA pathogenesis.
HMGB1
HMGB1 levels are increased in synovial fluid of OA patients, which correlates with disease severity. HMGB1 is released from activated synovial cells, chondrocytes and necrotic bone cells under the influence of cytokines such as IL‐1β and TNF‐α 87, 105, 106, 107. Conversely, stimulation of human OA chondrocytes with HMGB1 results in higher secretion of IL‐1β and TNF‐α 106. Interestingly, next to the direct stimulation of immune cells, HMGB1 can form complexes with pathogenic molecules including DNA and RNA but also with proinflammatory cytokines such as IL‐1β, which synergistically activate the immune system 108, 109. In this way, HMGB1 stimulates osteoarthritic synoviocytes to produce the proinflammatory cytokines IL‐6 and IL‐8, and MMP1, MMP3 and MMP13 110, 111.
HSPs
Other alarmins that are involved in OA are HSPs, even though the various family members have been attributed divergent effects on joint homeostasis. Increased levels of HSP60, HSP70 and HSP90 are found within the OA joint 112, 113. HSP90 release results in the progression of cartilage degeneration and activation of the synovium, while increased extracellular levels of HSP60 and HSP70 have a chondroprotective effect and show immunomodulatory activities 114, 115, 116.
S100 proteins
Probably the most well‐studied alarmins in the field of OA are the S100 family members. S100B is expressed by chondrocytes. Once released, S100B is thought to cause proinflammatory and procatabolic effects, mainly via RAGE‐dependent signalling in chondrocytes 117. This results in increased MMP13 expression in chondrocytes 118, 119. S100A4 is expressed in OA cartilage under the influence of, among others, IL‐7, and stimulates chondrocytes to produce MMP13 via RAGE 120, 121. Furthermore, OA has been associated with increased S100A11 production, whose secretion from chondrocytes is stimulated by factors such as TNF‐α and IL‐8 122, 123. In turn, stimulation of chondrocytes with S100A11 stimulates RAGE‐dependent hypertrophic differentiation, a process that is closely associated with the progression of OA 123. S100A12, which is closely related to S100A8 and S100A9, is markedly increased in synovial fluid of OA patients and correlates with disease severity 90, 91. Addition of S100A12 to human chondrocytes increases the expression of MMP13 and vascular endothelial growth factor (VEGF), again via RAGE‐dependent signalling 124.
However, proof for the involvement of these S100 proteins in catabolic effects on chondrocytes that might lead to OA development was mainly obtained with in‐vitro studies, whereas in‐vivo studies using loss‐of‐function and gain‐of‐function experiments have rarely been performed.
S100A8/A9
S100A8 and A9 are by far the best‐studied S100 proteins in the OA context. High levels of S100A8/A9 are present in the synovial fluid and serum of OA patients 92, 125. Among others, basic calcium phosphate (BCP) crystals that are present in the majority of OA patients stimulate the production of S100 proteins in macrophages 126. Interestingly, strongly increased expression of S100A8/A9 has been described with ageing, the dominant risk factor for OA 127. Induction of the experimental collagenase‐induced OA (CiOA) mouse model that involves moderate synovitis in S100a9–/–mice, which additionally lack S100A8 protein in the periphery, results in a strongly decreased synovial inflammation and cartilage degradation, indicating the crucial involvement of S100A8/A9 in this model 125. In contrast, the same study shows that S100A8/A9 is not of importance after induction of the destabilization of the medial meniscus (DMM), in which synovitis is scant. Higher S100A8/A9 serum levels were measured in early symptomatic OA patients that experience progression of joint space width narrowing between baseline measurements and the 2‐year follow‐up measurement compared to non‐progressors 125. S100A8/A9 has been shown to result in increased synovial activation, as determined by a number of inflammatory cell layers 125. A later study confirmed that S100A8/A9 predominantly mobilizes proinflammatory Ly6Chigh monocytes towards the inflamed synovium 128. This phenomenon might be at least partially responsible for the cartilage breakdown via the production of cytokines and matrix‐degrading enzymes. Indeed, stimulation of human OA synovial tissue and macrophages with S100A9 results in increased production of proinflammatory (e.g. IL‐1β, IL‐6, IL‐8, and TNF‐α) and catabolic factors such as MMP1, MMP3 and MMP9, which probably runs via TLR‐4 signalling, as this has been shown to be the dominant S100A8/A9 receptor in myeloid cells 69, 129, 130. However, in addition to possible indirect effects on cartilage degeneration via stimulation of cells in the synovium, direct stimulation of chondrocytes with S100A8 and S100A9 proteins also strongly promotes the production of various proinflammatory cytokines such as IL‐6 and IL‐8, the chemokine monocyte chemoattractant protein 1 and MMP1, MMP3, MMP9 and MMP13, which was shown to run via TLR‐4 as dominant receptor 131. Next to the production of these proinflammatory and catabolic mediators, S100A8/A9 is also involved in nociceptive pain sensation, independent of the degree of synovitis that is associated with different S100 levels (132 and personal unpublished findings).
Interestingly, S100A8/A9 was shown not only to activate the catabolic aspects of OA, but additionally promotes the anabolic process of ectopic bone/osteophytes formation, both in the CiOA experimental model and in early human OA, possibly via MMP‐mediated remodelling of the cartilage matrix that allows osteophytes to increase in size 133. This is underlined by the finding that S100A8/A9 induces Wnt signalling, which has been shown to promote bone formation 134. These findings are in agreement with other studies that address a more immunomodulatory rather than only a proinflammatory effect of S100A8/A9(135, 136 and personal unpublished findings).
Together, these studies make a case for the crucial involvement of alarmins, and particularly S100A8/A9, in the development of disease pathology during OA.
Dampening the alarm as therapy
In this section, a short overview of how these alarmins might serve as therapeutic target for this crippling disease will be given. An overview of possible ways to target alarmins can be found in Fig. 2. A first route to inhibit the function of alarmins is to block their expression, although a clinical application might not be feasible because current techniques, such as siRNA or shRNA, do not allow widespread targeting of the very high expression of many alarmins. Furthermore, care should be taken not to ‘overinhibit’ their intracellular expression because of the pivotal physiological functions that alarmins carry out. This is highlighted by the findings that both S100a8 and Hmgb1‐deficient mice are not viable 137, 138. As a second option, blocking receptors of alarmins using antibodies, blocking peptides or small‐molecule inhibitors might appear to be an attractive therapy, mainly because a multitude of alarmins bind to only a few receptors, such as RAGE, TLR‐2 and TLR‐4. Experimental evidence shows that blocking these receptors can lessen the impact of alarmins in inflammatory processes 131, 139, 140, 141. However, a major drawback of such an approach is that many infectious agents share the same PRRs with alarmins, of which the TLRs are particularly indispensible in host defence. Blocking PRRs will therefore most probably result in undesired adverse effects. Similarly, the use of soluble forms of PRRs, such as soluble TLR‐4 and soluble RAGE, can scavenge alarmins and therefore decrease the cellular signalling, but comprises the same risk of undesired side effects.
Figure 2.

Targeting alarmins as potential therapies to dampen osteoarthritis. Alarmins are thought to play important roles in the inflammatory process during osteoarthritis (OA). This provides a vast amount of opportunities to target these factors. First, the expression of alarmins can be inhibited using RNA interference techniques, such as short inhibitory (si)RNAs or short hairpin (sh)RNAs (a). However, care should be taken with this approach concerning the pivotal (often intracellular) roles that alarmins play under physiological conditions. Another possibility to inhibit the effects of alarmins is to block their pattern recognition receptors (PRRs) (b). This can be achieved using, among others, antibodies, blocking peptides or small molecule inhibitors against the desired receptor. A possible drawback of this approach is that these receptors, such as the family of Toll‐like receptors (TLRs), are crucially involved in the body’s defence against pathogens, meaning that blocking these receptors strongly increases the risk of infections. A third way to inhibit the activity of alarmins is to block their secretion (c). Alarmins are secreted via unconventional ways, involving among others lysosomal, inflammasome‐ or cytoskeleton‐dependent pathways. Interference with these secretion machineries with, for example, colchicine to inhibit S100A8/A9 secretion, ethyl pyruvate or glycyrrhizin to inhibit high‐mobility group box‐1 (HMGB1) secretion or caspase 1 inhibitors to reduce inflammasome‐dependent alarmins therefore prevent alarmins from conducting their proinflammatory functions in the extracellular space. Finally, specific inhibition of alarmins, once secreted into the extracellular environment, could be used to dampen inflammatory responses (d). To this end, specific antibodies, blocking peptides or small‐molecule inhibitors can be used. BoxA has been shown to successfully inhibit HMGB1. Moreover, paquinimod effectively interferes with the binding of S100A9 to TLR‐4. Soluble receptors, such as TLR‐4 and receptor for advanced glycosylation end products (RAGE) can be used to scavenge alarmins and thereby decrease alarmin‐induced cellular signalling.
Two more feasible ways of inhibiting the extracellular function of alarmins on the immune system are to block their secretion or target the alarmins themselves. Because alarmins lack a signal sequence for secretion they are secreted via alternative pathways not involving the endoplasmatic reticulum and Golgi complexes; blocking their release should target these pathways 142, 143, 144. A possible advantage is that this would inhibit the simultaneous release of multiple alarmins without interfering with the secretion of classically released proteins. HMGB1 is released via lysosomes. The natural compound glycyrrhizin has been shown to block the secretion and additionally inhibits the function of HMGB1 145. Furthermore, ethyl pyruvate inhibits the nuclear‐to‐cytoplasmic translation of HMGB1, thereby decreasing its secretion 146, 147. In addition, this compound has been shown to block the NLRP3 inflammasome. The release of several alarmins is thought to follow mechanisms similar to IL‐1, which implies a contribution of the inflammasome. For this reason, compounds such as ethyl pyruvate and specific caspase 1 inhibitors would block the release of these alarmins. S100A8/A9 is secreted by a tubulin‐dependent mechanism 144. Colchicine blocks tubulin polymerization and has been shown to decrease S100A8/A9 release 144, 148; however, the net outcome of these often relatively aspecific methods to block the release of alarmins on the development of OA needs further investigation, given the differential effects of, for example, S100A8/A9 versus HSPs on the cartilage. Moreover, the exact secretion mechanisms for many alarmins remain unknown and the above approaches will only block the active release of alarmins, whereas passive release as the result of cell death is not targeted.
A last approach would consist of selectively blocking the alarmins themselves. In this case it will be important to identify the dominant alarmin in a particular inflammatory disease. Successful attempts to inhibit inflammation have been described using neutralizing antibodies directed against HMGB1 and S100A8/A9 proteins in preclinical studies 149, 150, 151, 152, 153. Of particular interest is the recent identification of the amino acid sequence in the active S100A8/A9 complex that activates TLR‐4 signalling. Specific antibodies directed against this epitope might consequently block the activation of TLR‐4 signalling and activation of the inflammatory response 154. Also, small‐molecule inhibitors, either natural or synthetic, can be used to target alarmins, for example quinoline compounds such as paquinimod and laquinimod for S100A8/A9 155, 156. Interestingly, paquinimod reduced synovial inflammation, osteophyte formation and cartilage damage in a preclinical model for OA 133. However, the efficacy of many of the above approaches to reduce OA pathology remains to be investigated.
Conclusion
Whereas OA might not comply with all the classical signs of inflammation, there is nowadays a wealth of evidence that inflammation is part of the OA pathology and is actively involved in the disease pathogenesis via promotion of catabolic responses, either directly in chondrocytes or by promoting synovial inflammation and its contribution to pain sensation. Of particular interest in this process are the alarmins, which lie at the basis of the inflammatory response. Further understanding of the functioning of alarmins in inflammation brings to light novel and promising targets for the development of innovative DMOADs, in which the S100A8/A9 proteins are expected to claim a central spot based on their broad involvement in the OA disease process.
Disclosure
No specific funding was received from any funding bodies in the public, commercial or not‐for‐profit sectors to carry out the work described in this manuscript. The authors have no competing interests to declare.
References
- 1. World Health Organization . Chronic Rheumatic Conditions. Available at: http://www.who.int/chp/topics/rheumatic/en/ (accessed 5 September 2018). [Google Scholar]
- 2. Global Burden of Disease Study C . Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015; 386:743–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Goldring SR, Goldring MB. Clinical aspects, pathology and pathophysiology of osteoarthritis. J Musculoskelet Neuronal Interact 2006; 6:376–8. [PubMed] [Google Scholar]
- 4. Felson DT. Clinical practice. Osteoarthritis of the knee. N Engl J Med 2006; 354:841–8. [DOI] [PubMed] [Google Scholar]
- 5. Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum 2012; 64:1697–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wells C. The palaeopathology of bone disease. Practitioner 1973; 210:384–91. [PubMed] [Google Scholar]
- 7. Braunstein EM, White SJ, Russell W, Harris JE. Paleoradiologic evaluation of the Egyptian royal mummies. Skeletal Radiol 1988; 17:348–52. [DOI] [PubMed] [Google Scholar]
- 8. Buchanan WW, Kean WF. William Heberden the elder (1710–1801): the compleat physician and sometime rheumatologist. Clin Rheumatol 1987; 6:251–63. [DOI] [PubMed] [Google Scholar]
- 9. Jones RL. Arthritis deformans. Proc R Soc Med 1910; 3 (Balneol Climatol Sect):85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Parish LC. An historical approach to the nomenclature of rheumatoid arthritis. Arthritis Rheum 1963; 6:138–58. [DOI] [PubMed] [Google Scholar]
- 11. Sokoloff L. Some highlights in the emergence of modern concepts of osteoarthritis. Semin Arthritis Rheum 2001; 31:71–107. [DOI] [PubMed] [Google Scholar]
- 12. Garrod A. Discussion on ‘the aetiology and treatment of osteo‐arthritis and rheumatoid arthritis’. Proc R Soc Med 1924; 17:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rogers J, Watt I, Dieppe P. Arthritis in Saxon and mediaeval skeletons. BMJ Clin Res Ed 1981; 283:1668–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jurman RD. Stress and the etiology of osteoarthritis. Am J Phys Anthropol 1977; 46:353–65. [DOI] [PubMed] [Google Scholar]
- 15. Felson DT, Anderson JJ, Naimark A, Walker AM, Meenan RF. Obesity and knee osteoarthritis. The Framingham Study. Ann Intern Med 1988; 109:18–24. [DOI] [PubMed] [Google Scholar]
- 16. Hochberg MC, Lethbridge‐Cejku M, Scott WW Jr, Reichle R, Plato CC, Tobin JD. The association of body weight, body fatness and body fat distribution with osteoarthritis of the knee: data from the Baltimore Longitudinal Study of Aging. J Rheumatol 1995; 22:488–93. [PubMed] [Google Scholar]
- 17. Roos H, Lauren M, Adalberth T, Roos EM, Jonsson K, Lohmander LS. Knee osteoarthritis after meniscectomy: prevalence of radiographic changes after twenty‐one years, compared with matched controls. Arthritis Rheum 1998; 41:687–93. [DOI] [PubMed] [Google Scholar]
- 18. Oiestad BE, Engebretsen L, Storheim K, Risberg MA. Knee osteoarthritis after anterior cruciate ligament injury: a systematic review. Am J Sports Med 2009; 37:1434–43. [DOI] [PubMed] [Google Scholar]
- 19. Sharma L, Chmiel JS, Almagor O et al The role of varus and valgus alignment in the initial development of knee cartilage damage by MRI: the MOST study. Ann Rheum Dis 2013; 72:235–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brouwer GM, van Tol AW, Bergink AP et al Association between valgus and varus alignment and the development and progression of radiographic osteoarthritis of the knee. Arthritis Rheum 2007; 56:1204–11. [DOI] [PubMed] [Google Scholar]
- 21. Nicholls AS, Kiran A, Pollard TC et al The association between hip morphology parameters and nineteen‐year risk of end‐stage osteoarthritis of the hip: a nested case–control study. Arthritis Rheum 2011; 63:3392–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reichenbach S, Juni P, Werlen S et al Prevalence of cam‐type deformity on hip magnetic resonance imaging in young males: a cross‐sectional study. Arthritis Care Res 2010; 62:1319–27. [DOI] [PubMed] [Google Scholar]
- 23. Lo GH, Musa SM, Driban JB et al Running does not increase symptoms or structural progression in people with knee osteoarthritis: data from the osteoarthritis initiative. Clin Rheumatol 2018; 37:2497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Williams PT. Effects of running and walking on osteoarthritis and hip replacement risk. Med Sci Sports Exerc 2013; 45:1292–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yusuf E, Nelissen RG, Ioan‐Facsinay A et al Association between weight or body mass index and hand osteoarthritis: a systematic review. Ann Rheum Dis 2010; 69:761–5. [DOI] [PubMed] [Google Scholar]
- 26. Roemer FW, Guermazi A, Felson DT et al Presence of MRI‐detected joint effusion and synovitis increases the risk of cartilage loss in knees without osteoarthritis at 30‐month follow‐up: the MOST study. Ann Rheum Dis 2011; 70:1804–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ayral X, Pickering EH, Woodworth TG, Mackillop N, Dougados M. Synovitis: a potential predictive factor of structural progression of medial tibiofemoral knee osteoarthritis – results of a 1 year longitudinal arthroscopic study in 422 patients. Osteoarthritis Cartilage 2005; 13:361–7. [DOI] [PubMed] [Google Scholar]
- 28. Kortekaas MC, Kwok WY, Reijnierse M, Stijnen T, Kloppenburg M. Brief report: association of inflammation with development of erosions in patients with hand osteoarthritis: a prospective ultrasonography study. Arthritis Rheumatol 2016; 68:392–7. [DOI] [PubMed] [Google Scholar]
- 29. Petersen KK, Siebuhr AS, Graven‐Nielsen T et al Sensitization and serological biomarkers in knee osteoarthritis patients with different degrees of synovitis. Clin J Pain 2016; 32:841–8. [DOI] [PubMed] [Google Scholar]
- 30. Neogi T, Guermazi A, Roemer F et al Association of joint inflammation with pain sensitization in knee osteoarthritis: the multicenter osteoarthritis study. Arthritis Rheumatol 2016; 68:654–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Homandberg GA, Hui F. Association of proteoglycan degradation with catabolic cytokine and stromelysin release from cartilage cultured with fibronectin fragments. Arch Biochem Biophys 1996; 334:325–31. [DOI] [PubMed] [Google Scholar]
- 32. Gordon S. Pattern recognition receptors: doubling up for the innate immune response. Cell 2002; 111:927–30. [DOI] [PubMed] [Google Scholar]
- 33. Kawai T, Akira S. The role of pattern‐recognition receptors in innate immunity: update on Toll‐like receptors. Nat Immunol 2010; 11:373–84. [DOI] [PubMed] [Google Scholar]
- 34. Hwang HS, Park SJ, Cheon EJ, Lee MH, Kim HA. Fibronectin fragment‐induced expression of matrix metalloproteinases is mediated by MyD88‐dependent TLR‐2 signaling pathway in human chondrocytes. Arthritis Res Ther 2015; 17:320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu‐Bryan R, Terkeltaub R. Chondrocyte innate immune myeloid differentiation factor 88‐dependent signaling drives procatabolic effects of the endogenous Toll‐like receptor 2/Toll‐like receptor 4 ligands low molecular weight hyaluronan and high mobility group box chromosomal protein 1 in mice. Arthritis Rheum 2010; 62:2004–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Barreto G, Soininen A, Ylinen P, et al Soluble biglycan: a potential mediator of cartilage degradation in osteoarthritis. Arthritis Res Ther 2015; 17:379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lees S, Golub SB, Last K, et al Bioactivity in an aggrecan 32‐mer fragment is mediated via Toll‐like receptor 2. Arthritis Rheumatol 2015; 67:1240–9. [DOI] [PubMed] [Google Scholar]
- 38. Wang Q, Rozelle AL, Lepus CM et al Identification of a central role for complement in osteoarthritis. Nat Med 2011; 17:1674–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rosenthal AK. Crystals, inflammation, and osteoarthritis. Curr Opin Rheumatol 2011; 23:170–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Denoble AE, Huffman KM, Stabler TV et al Uric acid is a danger signal of increasing risk for osteoarthritis through inflammasome activation. Proc Natl Acad Sci USA 2011; 108:2088–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fernandez‐Puente P, Mateos J, Fernandez‐Costa C et al Identification of a panel of novel serum osteoarthritis biomarkers. J Proteome Res 2011; 10:5095–101. [DOI] [PubMed] [Google Scholar]
- 42. Messier SP, Mihalko SL, Legault C et al Effects of intensive diet and exercise on knee joint loads, inflammation, and clinical outcomes among overweight and obese adults with knee osteoarthritis: the IDEA randomized clinical trial. JAMA 2013; 310:1263–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Richter M, Trzeciak T, Owecki M, Pucher A, Kaczmarczyk J. The role of adipocytokines in the pathogenesis of knee joint osteoarthritis. Int Orthop 2015; 39:1211–7. [DOI] [PubMed] [Google Scholar]
- 44. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 2003; 112:1796–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence‐associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 2010; 5:99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Greene MA, Loeser RF. Aging‐related inflammation in osteoarthritis. Osteoarthritis Cartilage 2015; 23:1966–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sokolove J, Lepus CM. Role of inflammation in the pathogenesis of osteoarthritis: latest findings and interpretations. Ther Adv Musculoskelet Dis 2013; 5:77–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Goldring MB, Otero M. Inflammation in osteoarthritis. Curr Opin Rheumatol 2011; 23:471–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kapoor M, Martel‐Pelletier J, Lajeunesse D, Pelletier JP, Fahmi H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat Rev Rheumatol 2011; 7:33–42. [DOI] [PubMed] [Google Scholar]
- 50. Rosshirt N, Hagmann S, Tripel E et al A predominant Th1 polarization is present in synovial fluid of end‐stage osteoarthritic knee joints ‐ analysis of peripheral blood, synovial fluid & synovial membrane. Clin Exp Immunol 2018. 10.1111/cei.13230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li YS, Luo W, Zhu SA, Lei GH. T Cells in osteoarthritis: alterations and beyond. Front Immunol 2017; 8:356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Blom AB, van Lent PL, Holthuysen AE et al Synovial lining macrophages mediate osteophyte formation during experimental osteoarthritis. Osteoarthritis Cartilage 2004; 12:627–35. [DOI] [PubMed] [Google Scholar]
- 53. Blom AB, van Lent PL, Libregts S et al Crucial role of macrophages in matrix metalloproteinase‐mediated cartilage destruction during experimental osteoarthritis: involvement of matrix metalloproteinase 3. Arthritis Rheum 2007; 56:147–57. [DOI] [PubMed] [Google Scholar]
- 54. Orlowsky EW, Kraus VB. The role of innate immunity in osteoarthritis: when our first line of defense goes on the offensive. J Rheumatol 2015; 42:363–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chevalier X, Eymard F, Richette P. Biologic agents in osteoarthritis: hopes and disappointments. Nat Rev Rheumatol 2013; 9:400–10. [DOI] [PubMed] [Google Scholar]
- 56. Oppenheim JJ, Yang D. Alarmins: chemotactic activators of immune responses. Curr Opin Immunol 2005; 17:359–65. [DOI] [PubMed] [Google Scholar]
- 57. Yang Han Z, Oppenheim JJ. Alarmins and immunity. Immunol Rev 2017; 280:41–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yang D, Oppenheim JJ. Antimicrobial proteins act as ‘alarmins’ in joint immune defense. Arthritis Rheum 2004; 50:3401–3. [DOI] [PubMed] [Google Scholar]
- 59. Chan JK, Roth J, Oppenheim JJ et al Alarmins: awaiting a clinical response. J Clin Invest 2012; 122:2711–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pruenster M, Vogl T, Roth J, Sperandio M. S100A8/A9: from basic science to clinical application. Pharmacol Ther 2016; 167:120–31. [DOI] [PubMed] [Google Scholar]
- 61. Harris HE, Raucci A. Alarmin(g) news about danger: workshop on innate danger signals and HMGB1. EMBO Rep 2006; 7:774–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Pisetsky DS, Gauley J, Ullal AJ. HMGB1 and microparticles as mediators of the immune response to cell death. Antioxid Redox Signal 2011; 15:2209–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. van Zoelen MA, Yang H, Florquin S et al Role of Toll‐like receptors 2 and 4, and the receptor for advanced glycation end products in high‐mobility group box 1‐induced inflammation in vivo . Shock 2009; 31:280–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ohashi K, Burkart V, Flohe S, Kolb H. Cutting edge: heat shock protein 60 is a putative endogenous ligand of the Toll‐like receptor‐4 complex. J Immunol 2000; 164:558–61. [DOI] [PubMed] [Google Scholar]
- 65. Tewary P, Yang D, de la Rosa G et al Granulysin activates antigen‐presenting cells through TLR4 and acts as an immune alarmin. Blood 2010; 116:3465–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Park JS, Svetkauskaite D, He Q et al Involvement of toll‐like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem 2004; 279:7370–7. [DOI] [PubMed] [Google Scholar]
- 67. Yang H, Ochani M, Li J et al Reversing established sepsis with antagonists of endogenous high‐mobility group box 1. Proc Natl Acad Sci USA 2004; 101:296–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Barnay‐Verdier S, Fattoum L, Borde C, Kaveri S, Gibot S, Marechal V. Emergence of autoantibodies to HMGB1 is associated with survival in patients with septic shock. Intens Care Med 2011; 37:957–62. [DOI] [PubMed] [Google Scholar]
- 69. Vogl T, Tenbrock K, Ludwig S et al Mrp8 and Mrp14 are endogenous activators of Toll‐like receptor 4, promoting lethal, endotoxin‐induced shock. Nat Med 2007; 13:1042–9. [DOI] [PubMed] [Google Scholar]
- 70. Yang R, Harada T, Mollen KP et al Anti‐HMGB1 neutralizing antibody ameliorates gut barrier dysfunction and improves survival after hemorrhagic shock. Mol Med 2006; 12:105–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tsung A, Sahai R, Tanaka H et al The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia‐reperfusion. J Exp Med 2005; 201:1135–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Liu K, Mori S, Takahashi HK et al Anti‐high mobility group box 1 monoclonal antibody ameliorates brain infarction induced by transient ischemia in rats. FASEB J 2007; 21:3904–16. [DOI] [PubMed] [Google Scholar]
- 73. Abraham E, Arcaroli J, Carmody A, Wang H, Tracey KJ. HMG‐1 as a mediator of acute lung inflammation. J Immunol 2000; 165:2950–4. [DOI] [PubMed] [Google Scholar]
- 74. De Ponti A, Wiechert L, Schneller D et al A pro‐tumorigenic function of S100A8/A9 in carcinogen‐induced hepatocellular carcinoma. Cancer Lett 2015; 369:396–404. [DOI] [PubMed] [Google Scholar]
- 75. Ichikawa M, Williams R, Wang L, Vogl T, Srikrishna G. S100A8/A9 activate key genes and pathways in colon tumor progression. Mol Cancer Res 2011; 9:133–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Maeda S, Hikiba Y, Shibata W et al Essential roles of high‐mobility group box 1 in the development of murine colitis and colitis‐associated cancer. Biochem Biophys Res Commun 2007; 360:394–400. [DOI] [PubMed] [Google Scholar]
- 77. Nestl A, Von Stein OD, Zatloukal K et al Gene expression patterns associated with the metastatic phenotype in rodent and human tumors. Cancer Res 2001; 61:1569–77. [PubMed] [Google Scholar]
- 78. Sinha P, Okoro C, Foell D, Freeze HH, Ostrand‐Rosenberg S, Srikrishna G. Proinflammatory S100 proteins regulate the accumulation of myeloid‐derived suppressor cells. J Immunol 2008; 181:4666–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Campana L, Bosurgi L, Rovere‐Querini P. HMGB1: a two‐headed signal regulating tumor progression and immunity. Curr Opin Immunol 2008; 20:518–23. [DOI] [PubMed] [Google Scholar]
- 80. Chen T, Guo J, Han C, Yang M, Cao X. Heat shock protein 70, released from heat‐stressed tumor cells, initiates antitumor immunity by inducing tumor cell chemokine production and activating dendritic cells via TLR4 pathway. J Immunol 2009; 182:1449–59. [DOI] [PubMed] [Google Scholar]
- 81. Zheng H, Dai J, Stoilova D, Li Z. Cell surface targeting of heat shock protein gp96 induces dendritic cell maturation and antitumor immunity. J Immunol 2001; 167:6731–5. [DOI] [PubMed] [Google Scholar]
- 82. Austermann J, Zenker S, Roth J. S100‐alarmins: potential therapeutic targets for arthritis. Expert Opin Ther Targets 2017; 21:739–51. [DOI] [PubMed] [Google Scholar]
- 83. Frosch M, Strey A, Vogl T et al Myeloid‐related proteins 8 and 14 are specifically secreted during interaction of phagocytes and activated endothelium and are useful markers for monitoring disease activity in pauciarticular‐onset juvenile rheumatoid arthritis. Arthritis Rheum 2000; 43:628–37. [DOI] [PubMed] [Google Scholar]
- 84. Vitali R, Stronati L, Negroni A et al Fecal HMGB1 is a novel marker of intestinal mucosal inflammation in pediatric inflammatory bowel disease. Am J Gastroenterol 2011; 106:2029–40. [DOI] [PubMed] [Google Scholar]
- 85. Foell D, Wittkowski H, Ren Z et al Phagocyte‐specific S100 proteins are released from affected mucosa and promote immune responses during inflammatory bowel disease. J Pathol 2008; 216:183–92. [DOI] [PubMed] [Google Scholar]
- 86. Zhou Q, Zhu Z, Hu X, Shu C. HMGB1: a critical mediator for oxidized‐low density lipoproteins induced atherosclerosis. Int J Cardiol 2016; 202:956–7. [DOI] [PubMed] [Google Scholar]
- 87. Ke X, Jin G, Yang Y et al Synovial fluid HMGB‐1 levels are associated with osteoarthritis severity. Clin Lab 2015; 61:809–18. [DOI] [PubMed] [Google Scholar]
- 88. Wei M, Duan D, Liu Y, Wang Z, Li Z. Increased thymosin beta4 levels in the serum and SF of knee osteoarthritis patients correlate with disease severity. Regul Pept 2013; 185:34–6. [DOI] [PubMed] [Google Scholar]
- 89. Kumahashi N, Naitou K, Nishi H et al Correlation of changes in pain intensity with synovial fluid adenosine triphosphate levels after treatment of patients with osteoarthritis of the knee with high‐molecular‐weight hyaluronic acid. Knee 2011; 18:160–4. [DOI] [PubMed] [Google Scholar]
- 90. Wang LC, Zhang HY, Shao L et al S100A12 levels in synovial fluid may reflect clinical severity in patients with primary knee osteoarthritis. Biomarkers 2013; 18:216–20. [DOI] [PubMed] [Google Scholar]
- 91. Han MY, Dai JJ, Zhang Y et al Identification of osteoarthritis biomarkers by proteomic analysis of synovial fluid. J Int Med Res 2012; 40:2243–50. [DOI] [PubMed] [Google Scholar]
- 92. Sunahori K, Yamamura M, Yamana J et al The S100A8/A9 heterodimer amplifies proinflammatory cytokine production by macrophages via activation of nuclear factor kappa B and p38 mitogen‐activated protein kinase in rheumatoid arthritis. Arthritis Res Ther 2006; 8:R69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Midwood K, Sacre S, Piccinini AM et al Tenascin‐C is an endogenous activator of Toll‐like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat Med 2009; 15:774–80. [DOI] [PubMed] [Google Scholar]
- 94. Pelletier JP, Roughley PJ, DiBattista JA, McCollum R, Martel‐Pelletier J. Are cytokines involved in osteoarthritic pathophysiology? Semin Arthritis Rheum 1991; 20 (Suppl. 2):12–25. [DOI] [PubMed] [Google Scholar]
- 95. McNulty AL, Rothfusz NE, Leddy HA, Guilak F. Synovial fluid concentrations and relative potency of interleukin‐1 alpha and beta in cartilage and meniscus degradation. J Orthop Res 2013; 31:1039–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. van Dalen SC, Blom AB, Sloetjes AW et al Interleukin‐1 is not involved in synovial inflammation and cartilage destruction in collagenase‐induced osteoarthritis. Osteoarthritis Cartilage 2017; 25:385–96. [DOI] [PubMed] [Google Scholar]
- 97. Ma CA, Leung YY. Exploring the link between uric acid and osteoarthritis. Front Med (Lausanne) 2017; 4:225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Varoga D, Pufe T, Harder J et al Human beta‐defensin 3 mediates tissue remodeling processes in articular cartilage by increasing levels of metalloproteinases and reducing levels of their endogenous inhibitors. Arthritis Rheum 2005; 52:1736–45. [DOI] [PubMed] [Google Scholar]
- 99. Varoga D, Paulsen FP, Kohrs S et al Expression and regulation of human beta‐defensin‐2 in osteoarthritic cartilage. J Pathol 2006; 209:166–73. [DOI] [PubMed] [Google Scholar]
- 100. Musumeci G, Carnazza ML, Leonardi R, Loreto C. Expression of beta‐defensin‐4 in ‘an in vivo and ex vivo model’ of human osteoarthritic knee meniscus. Knee Surg Sports Traumatol Arthrosc 2012; 20:216–22. [DOI] [PubMed] [Google Scholar]
- 101. Ryan LM, Kurup IV, Derfus BA, Kushnaryov VM. ATP‐induced chondrocalcinosis. Arthritis Rheum 1992; 35:1520–5. [DOI] [PubMed] [Google Scholar]
- 102. Zhao LR, Xing RL, Wang PM et al NLRP1 and NLRP3 inflammasomes mediate LPS/ATPinduced pyroptosis in knee osteoarthritis. Mol Med Rep 2018; 17:5463–9. [DOI] [PubMed] [Google Scholar]
- 103. Shi J, Zhao W, Ying H et al Estradiol inhibits NLRP3 inflammasome in fibroblast‐like synoviocytes activated by lipopolysaccharide and adenosine triphosphate. Int J Rheum Dis 2017. 10.1111/1756-185X.13198. [DOI] [PubMed] [Google Scholar]
- 104. Teixeira JM, Bobinski F, Parada CA, Sluka KA, Tambeli CH. P2X3 and P2X2/3 receptors play a crucial role in articular hyperalgesia development through inflammatory mechanisms in the knee joint experimental synovitis. Mol Neurobiol 2017; 54:6174–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Li ZC, Cheng GQ, Hu KZ et al Correlation of synovial fluid HMGB‐1 levels with radiographic severity of knee osteoarthritis. Clin Invest Med 2011; 34:E298. [DOI] [PubMed] [Google Scholar]
- 106. Terada C, Yoshida A, Nasu Y et al Gene expression and localization of high‐mobility group box chromosomal protein‐1 (HMGB‐1) in human osteoarthritic cartilage. Acta Med Okayama 2011; 65:369–77. [DOI] [PubMed] [Google Scholar]
- 107. Bidwell JP, Yang J, Robling AG. Is HMGB1 an osteocyte alarmin? J Cell Biochem 2008; 103:1671–80. [DOI] [PubMed] [Google Scholar]
- 108. Hreggvidsdottir HS, Ostberg T, Wahamaa H et al The alarmin HMGB1 acts in synergy with endogenous and exogenous danger signals to promote inflammation. J Leukoc Biol 2009; 86:655–62. [DOI] [PubMed] [Google Scholar]
- 109. Sha Y, Zmijewski J, Xu Z, Abraham E. HMGB1 develops enhanced proinflammatory activity by binding to cytokines. J Immunol 2008; 180:2531–7. [DOI] [PubMed] [Google Scholar]
- 110. Garcia‐Arnandis I, Guillen MI, Gomar F, Pelletier JP, Martel‐Pelletier J, Alcaraz MJ. High mobility group box 1 potentiates the pro‐inflammatory effects of interleukin‐1beta in osteoarthritic synoviocytes. Arthritis Res Ther 2010; 12:R165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Qin Y, Chen Y, Wang W et al HMGB1‐LPS complex promotes transformation of osteoarthritis synovial fibroblasts to a rheumatoid arthritis synovial fibroblast‐like phenotype. Cell Death Dis 2014; 5:e1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Kubo T, Towle CA, Mankin HJ, Treadwell BV. Stress‐induced proteins in chondrocytes from patients with osteoarthritis. Arthritis Rheum 1985; 28:1140–5. [DOI] [PubMed] [Google Scholar]
- 113. Takahashi K, Kubo T, Arai Y, Imanishi J, Kawata M, Hirasawa Y. Localization of heat shock protein in osteoarthritic cartilage. Scand J Rheumatol 1997; 26:368–75. [DOI] [PubMed] [Google Scholar]
- 114. Siebelt M, Jahr H, Groen HC et al Hsp90 inhibition protects against biomechanically induced osteoarthritis in rats. Arthritis Rheum 2013; 65:2102–12. [DOI] [PubMed] [Google Scholar]
- 115. Terauchi R, Takahashi KA, Arai Y et al Hsp70 prevents nitric oxide‐induced apoptosis in articular chondrocytes. Arthritis Rheum 2003; 48:1562–8. [DOI] [PubMed] [Google Scholar]
- 116. Broere F, van der Zee R, van Eden W. Heat shock proteins are no DAMPs, rather ‘DAMPERs’. Nat Rev Immunol 2011; 11:565; author reply. [DOI] [PubMed] [Google Scholar]
- 117. Yammani RR. S100 proteins in cartilage: role in arthritis. Biochim Biophys Acta 2012; 1822:600–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Loeser RF, Yammani RR, Carlson CS et al Articular chondrocytes express the receptor for advanced glycation end products: Potential role in osteoarthritis. Arthritis Rheum 2005; 52:2376–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Li H, Wang D, Yuan Y, Min J. New insights on the MMP‐13 regulatory network in the pathogenesis of early osteoarthritis. Arthritis Res Ther 2017; 19:248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Yammani RR, Long D, Loeser RF. Interleukin‐7 stimulates secretion of S100A4 by activating the JAK/STAT signaling pathway in human articular chondrocytes. Arthritis Rheum 2009; 60:792–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Yammani RR, Carlson CS, Bresnick AR, Loeser RF. Increase in production of matrix metalloproteinase 13 by human articular chondrocytes due to stimulation with S100A4: role of the receptor for advanced glycation end products. Arthritis Rheum 2006; 54:2901–11. [DOI] [PubMed] [Google Scholar]
- 122. Amin AR, Islam AB. Genomic analysis and differential expression of HMG and S100A family in human arthritis: upregulated expression of chemokines, IL‐8 and nitric oxide by HMGB1. DNA Cell Biol 2014; 33:550–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Cecil DL, Johnson K, Rediske J, Lotz M, Schmidt AM, Terkeltaub R. Inflammation‐induced chondrocyte hypertrophy is driven by receptor for advanced glycation end products. J Immunol 2005; 175:8296–302. [DOI] [PubMed] [Google Scholar]
- 124. Nakashima M, Sakai T, Hiraiwa H et al Role of S100A12 in the pathogenesis of osteoarthritis. Biochem Biophys Res Commun 2012; 422:508–14. [DOI] [PubMed] [Google Scholar]
- 125. van Lent PL, Blom AB, Schelbergen RF et al Active involvement of alarmins S100A8 and S100A9 in the regulation of synovial activation and joint destruction during mouse and human osteoarthritis. Arthritis Rheum 2012; 64:1466–76. [DOI] [PubMed] [Google Scholar]
- 126. Corr EM, Cunningham CC, Helbert L, McCarthy GM, Dunne A. Osteoarthritis‐associated basic calcium phosphate crystals activate membrane proximal kinases in human innate immune cells. Arthritis Res Ther 2017; 19:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Swindell WR, Johnston A, Xing X et al Robust shifts in S100a9 expression with aging: a novel mechanism for chronic inflammation. Sci Rep 2013; 3:1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Cremers NAJ, van den Bosch MHJ, van Dalen S et al S100A8/A9 increases the mobilization of pro‐inflammatory Ly6C(high) monocytes to the synovium during experimental osteoarthritis. Arthritis Res Ther 2017; 19:217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. van den Bosch MH, Blom AB, Schelbergen RF et al Alarmin S100A9 induces proinflammatory and catabolic effects predominantly in the M1 macrophages of human osteoarthritic synovium. J Rheumatol 2016; 43:1874–84. [DOI] [PubMed] [Google Scholar]
- 130. Fassl SK, Austermann J, Papantonopoulou O et al Transcriptome assessment reveals a dominant role for TLR4 in the activation of human monocytes by the alarmin MRP8. J Immunol 2015; 194:575–83. [DOI] [PubMed] [Google Scholar]
- 131. Schelbergen RF, Blom AB, van den Bosch MH et al Alarmins S100A8 and S100A9 elicit a catabolic effect in human osteoarthritic chondrocytes that is dependent on Toll‐like receptor 4. Arthritis Rheum 2012; 64:1477–87. [DOI] [PubMed] [Google Scholar]
- 132. Miller RE, Belmadani A, Ishihara S et al Damage‐associated molecular patterns generated in osteoarthritis directly excite murine nociceptive neurons through Toll‐like receptor 4. Arthritis Rheumatol 2015; 67:2933–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Schelbergen RF, Geven EJ, van den Bosch MH et al Prophylactic treatment with S100A9 inhibitor paquinimod reduces pathology in experimental collagenase‐induced osteoarthritis. Ann Rheum Dis 2015; 74:2254–8. [DOI] [PubMed] [Google Scholar]
- 134. van den Bosch MH, Blom AB, Schelbergen RF et al Induction of canonical Wnt signaling by the alarmins S100A8/A9 in murine knee joints: implications for osteoarthritis. Arthritis Rheumatol 2016; 68:152–63. [DOI] [PubMed] [Google Scholar]
- 135. Petersen B, Wolf M, Austermann J et al The alarmin Mrp8/14 as regulator of the adaptive immune response during allergic contact dermatitis. EMBO J 2013; 32:100–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Austermann J, Friesenhagen J, Fassl SK et al Alarmins MRP8 and MRP14 induce stress tolerance in phagocytes under sterile inflammatory conditions. Cell Rep 2014; 9:2112–23. [DOI] [PubMed] [Google Scholar]
- 137. Passey RJ, Williams E, Lichanska AM et al A null mutation in the inflammation‐associated S100 protein S100A8 causes early resorption of the mouse embryo. J Immunol 1999; 163:2209–16. [PubMed] [Google Scholar]
- 138. Calogero S, Grassi F, Aguzzi A et al The lack of chromosomal protein Hmg1 does not disrupt cell growth but causes lethal hypoglycaemia in newborn mice. Nat Genet 1999; 22:276–80. [DOI] [PubMed] [Google Scholar]
- 139. Bro S, Flyvbjerg A, Binder CJ et al A neutralizing antibody against receptor for advanced glycation end products (RAGE) reduces atherosclerosis in uremic mice. Atherosclerosis 2008; 201:274–80. [DOI] [PubMed] [Google Scholar]
- 140. Gasparotto J, Ribeiro CT, Bortolin RC et al Anti‐RAGE antibody selectively blocks acute systemic inflammatory responses to LPS in serum, liver, CSF and striatum. Brain Behav Immun 2017; 62:124–36. [DOI] [PubMed] [Google Scholar]
- 141. Gao W, Xiong Y, Li Q, Yang H. Inhibition of Toll‐like receptor signaling as a promising therapy for inflammatory diseases: a journey from molecular to nano therapeutics. Front Physiol 2017; 8:508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Daniels MJ, Brough D. Unconventional pathways of secretion contribute to inflammation. Int J Mol Sci 2017; 18:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Bertheloot D, Latz E. HMGB1, IL‐1alpha, IL‐33 and S100 proteins: dual‐function alarmins. Cell Mol Immunol 2017; 14:43–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Rammes A, Roth J, Goebeler M, Klempt M, Hartmann M, Sorg C. Myeloid‐related protein (MRP) 8 and MRP14, calcium‐binding proteins of the S100 family, are secreted by activated monocytes via a novel, tubulin‐dependent pathway. J Biol Chem 1997; 272:9496–502. [DOI] [PubMed] [Google Scholar]
- 145. Mollica L, De Marchis F, Spitaleri A et al Glycyrrhizin binds to high‐mobility group box 1 protein and inhibits its cytokine activities. Chem Biol 2007; 14:431–41. [DOI] [PubMed] [Google Scholar]
- 146. Li S, Liang F, Kwan K et al Identification of ethyl pyruvate as a NLRP3 inflammasome inhibitor that preserves mitochondrial integrity. Mol Med 2018; 24:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Dave SH, Tilstra JS, Matsuoka K et al Ethyl pyruvate decreases HMGB1 release and ameliorates murine colitis. J Leukoc Biol 2009; 86:633–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Skoufias DA, Wilson L. Mechanism of inhibition of microtubule polymerization by colchicine: inhibitory potencies of unliganded colchicine and tubulin‐colchicine complexes. Biochemistry 1992; 31:738–46. [DOI] [PubMed] [Google Scholar]
- 149. Kokkola R, Li J, Sundberg E et al Successful treatment of collagen‐induced arthritis in mice and rats by targeting extracellular high mobility group box chromosomal protein 1 activity. Arthritis Rheum 2003; 48:2052–8. [DOI] [PubMed] [Google Scholar]
- 150. Hamada T, Torikai M, Kuwazuru A et al Extracellular high mobility group box chromosomal protein 1 is a coupling factor for hypoxia and inflammation in arthritis. Arthritis Rheum 2008; 58:2675–85. [DOI] [PubMed] [Google Scholar]
- 151. Raquil MA, Anceriz N, Rouleau P, Tessier PA. Blockade of antimicrobial proteins S100A8 and S100A9 inhibits phagocyte migration to the alveoli in streptococcal pneumonia. J Immunol 2008; 180:3366–74. [DOI] [PubMed] [Google Scholar]
- 152. Cesaro A, Anceriz N, Plante A, Page N, Tardif MR, Tessier PA. An inflammation loop orchestrated by S100A9 and calprotectin is critical for development of arthritis. PLOS ONE 2012; 7:e45478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Ryckman C, McColl SR, Vandal K et al Role of S100A8 and S100A9 in neutrophil recruitment in response to monosodium urate monohydrate crystals in the air‐pouch model of acute gouty arthritis. Arthritis Rheum 2003; 48:2310–20. [DOI] [PubMed] [Google Scholar]
- 154. Vogl T, Stratis A, Wixler V et al Autoinhibitory regulation of S100A8/S100A9 alarmin activity locally restricts sterile inflammation. J Clin Invest 2018; 128:1852–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Bjork P, Bjork A, Vogl T et al Identification of human S100A9 as a novel target for treatment of autoimmune disease via binding to quinoline‐3‐carboxamides. PLOS Biol 2009; 7:e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Comi G, Jeffery D, Kappos L et al Placebo‐controlled trial of oral laquinimod for multiple sclerosis. N Engl J Med 2012; 366:1000–9. [DOI] [PubMed] [Google Scholar]