

Abstract
Hereditary fructose intolerance, caused by mutations in the ALDOB gene, is an unusual cause of hypoglycemia. ALDOB encodes the enzyme aldolase B, responsible for the hydrolysis of fructose 1-phosphate in the liver. Here, we report the case of a 33-year-old female patient who consulted due to repetitive episodes of weakness, dizziness and headache after food ingestion. An ambulatory 72-h continuous glucose monitoring revealed multiple short hypoglycemic episodes over the day. After biochemical exclusion of other endocrine causes of hypoglycemia, hereditary fructose intolerance seemed a plausible diagnosis. Repeated measurements of urinary fructose revealed pathologic fructosuria, but genetic testing for the three most common mutations in ALDOB resulted negative. We decided to perform complete Sanger sequencing of the ALDOB gene and encountered a variant consisting of a T>A substitution in position 1963 of the ALDOB transcript (c.1693T>A). This position is located within the 3′ untranslated region of exon 9, 515 nucleotides downstream the stop codon. After complete withdrawal of dietary fructose and sucrose, the patient presented no new hypoglycemic episodes.
Keywords: Hereditary fructose intolerance, fructosuria, fructose, hypoglycemia, aldolase B
Introduction
Hereditary fructose intolerance (HFI, OMIM 229600) is a fairly rare condition secondary to mutations in the ALDOB gene, which encodes the enzyme aldolase B. The incidence of HFI in different ethnic groups ranges between 1/10,000 and 1/100,000 newborns.1,2 ALDOB is a 15,221-bp long gene, located in the long arm of chromosome 9 (9q31.1) and containing nine exons.3 Aldolase B is responsible for the reversible conversion of fructose 1-phosphate into the three-carbon sugars glyceraldehyde and dihydroxyacetone phosphate (DHAP). Patients with HFI usually have recurrent hypoglycemic episodes after ingesting foods containing fructose. Hypoglycemia seems to be the result of fructose 1-phosphate accumulation, which hinders hepatic glycogen phosphorylase activity by exhausting cytosolic reserves of inorganic phosphate and inhibiting gluconeogenesis.4 The clinical presentation of HFI may include hypoglycemia, hypophosphatemia, hypomagnesemia, abdominal pain and/or vomiting after ingestion of high-fructose meals. Over time, patients may develop hepatomegaly, metabolic acidosis, renal proximal tubulopathy and failure to thrive.4 Most cases develop the associated symptoms during childhood or adolescence, and only a few cases of HFI symptom onset during adulthood have been reported.5 HFI is a diagnosis of exclusion after more prevalent causes of postprandial hypoglycemia are ruled out.6
Here, we report the case of an adult patient with recurrent episodes of symptomatic but mild hypoglycemia and a previously unreported mutation in the aldolase B gene.
Case presentation
A 33-year-old woman from Bogotá, Colombia, consulted due to repetitive episodes of weakness, dizziness and sometimes headache, symmetric numbness in her face and arms and/or sensation of imminent fainting. The reported episodes resolved spontaneously after a few minutes, tended to occur after the main meals and had increased in frequency over the last 5 years. Following advice from relatives and her own personal research, the patient performed blood sugar measurements during the episodes, which yielded results between 40 and 60 mg/dL. She consulted her general practitioner 2 years before, and he recommended lifestyle changes including a decrease in high glycemic index foods and an increase in dietary intake frequency to six times a day, once every 3 h. Adherence to these recommendations led only to partial resolution of the symptoms. The patient had no memory of discomfort after consumption of fruits or juices during childhood but reported an increasing occurrence of the reported postprandial symptoms starting in adolescence that eventually became very uncomfortable and led her to consult. The patient’s parents were not consanguineous, and she did not display hepatomegaly or altered linear growth.
In April 2013, during a routine laboratory work-up that included a pre- and post-meal plasma glucose measurement, the patient experienced an event of asymptomatic hypoglycemia (glycemia 2 h post-meal: 29 mg/dL). She consulted our department with these results. Past medical history included an episode of pulmonary thromboembolism and upper limb deep venous thrombosis in 2011 secondary to protein C deficiency. She received subcutaneous enoxaparin 40 mg a day since 2011 until July of 2015, when she was started on daily oral aspirin 100 mg. Surgical history included appendectomy and herniorrhaphy (×2). She reported a family history of deep venous thrombosis, throat cancer and diabetes mellitus.
We performed an ambulatory 72-h continuous glucose monitoring (CGM) and a biochemical work-up that ruled out adrenal insufficiency and hypothyroidism (results summarized in Table 1). The CGM revealed 48 events of hypoglycemia below 70 mg/dL, the lowest reaching 42 mg/dL. The patient was hospitalized to perform a 72-h fast test, during which no hypoglycemic episodes occurred, ruling out endogenous hyperinsulinism and factitious hypoglycemia. In order to explore the possibility of HFI, we measured urinary fructose levels in three separate samples employing the hexokinase—phosphoglucoisomerase method, the results were 361.2, 379, and 357.1 μM/L (reference range in non-diabetics is 6.1–10.1 μM/L7). Analysis of single-point variations in the three most common mutations in the ALDOB gene (c.448G>C, c.524C>A and c.1005C>G, performed at Genética Molecular de Colombia, SAS—www.geneticamolecular.com.co) revealed no alteration at these genomic sites. However, because the clinical picture of the patient fitted very well a case of HFI, we decided to perform complete sequencing of the ALDOB gene.
Table 1.
Baseline laboratory values.
| Parameter | Value | Reference value |
|---|---|---|
| Aspartate aminotransferase | 19 U/L | 10–45 U/L |
| Alanine aminotransferase | 19 U/L | 10–45 U/L |
| 25-hydroxyvitamin D | 36.5 ng/mL | 30–80 ng/mL |
| Ionized calcium | 1.29 mmol/L | 1.1–1.35 mmol/L |
| Ferritin | 15.8 ng/mL | 11–306 ng/mL |
| Cobalamin | 329 pg/mL | 180–914 ng/L |
| Thyroid-stimulating hormone | 1.72 mIU/L | 0.4–4.2 mIU/L |
| Free T4 | 0.77 ng/dL | 0.7–1.7 ng/dL |
| 8AM cortisol | 248 nmol/L | 193–772 nmol/L |
| Total cholesterol | 161 mg/dL | 50–200 mg/dL |
| HDL cholesterol | 59.6 mg/dL | 40–120 mg/dL |
| LDL cholesterol | 88.8 mg/dL | 0–130 mg/dL |
| Triglycerides | 63 mg/dL | 40–200 mg/dL |
HDL: high-density lipoprotein; LDL: low-density lipoprotein.
Leukocyte DNA was extracted, and the exons 2–9 of the ALDOB gene were amplified through polymerase chain reaction. Primer sequences were designed according to previous reports8 (Table 2). We sequenced the eight amplicons containing exons 2–9 and neighboring regions using Sanger sequencing in an ABI PRISM 3500 genetic analyzer at Universidad de los Andes, Colombia. The sequence of the amplicons was aligned to the human genome using the Basic Local Alignment Search Tool (BLAST) version 2.7.0. We identified a single homozygous point mutation consisting of a T˃A substitution in position 1693 of the ALDOB transcript (c.1693T˃A against cDNA reference sequence NM_000035.3), within the 3’UTR of exon 9, 515 nucleotides downstream the stop codon (Figure 1). Our diagnostic impression was confirmed by molecular analysis, and the patient was initiated on a fructose and sucrose-free diet. Since complete withdrawal of dietary fructose and with very high adherence by the patient, no new hypoglycemic episodes occurred.
Table 2.
ALDOB exons, sequence of primers and size of each amplicon.
| Exon | Exon size (bp) | Forward primer | Reverse primer | Amplicon size (bp) |
|---|---|---|---|---|
| 2 | 122 | CCACAGAATAGAGAGACAGT | GTTGTTATATGATGAGACTG | 506 |
| 3 | 212 | CTAGCCACCTGAGAGCAACCA | TCTCTGTGGGAAGATGACGA | 540 |
| 4 | 55 | GATGCAAACTGTTAGTTAG | GCCTTCATTTCTAGCTTAC | 190 |
| 5 | 161 | ACTCCTTCCCTTTATTA | GGTCCATTTGTAGTTATAGT | 330 |
| 6 | 84 | CTAGGTTCTGAGGCAGCTAG | TTATATGTTAAGTAACAGCTG | 388 |
| 7 | 174 | CTGCAGTGTAAATGTGCCAA | GCTTGGTATTCTGAAGTG | 412 |
| 8 | 200 | CTCAAGCAGGGTATATAAG | CTCAATCCTCATACTGACCTC | 418 |
| 9 | 1327 | TTCCCATGAGAGGCAGA | GACCTTTACTGTTGAAACCC | 710 |
Figure .1.
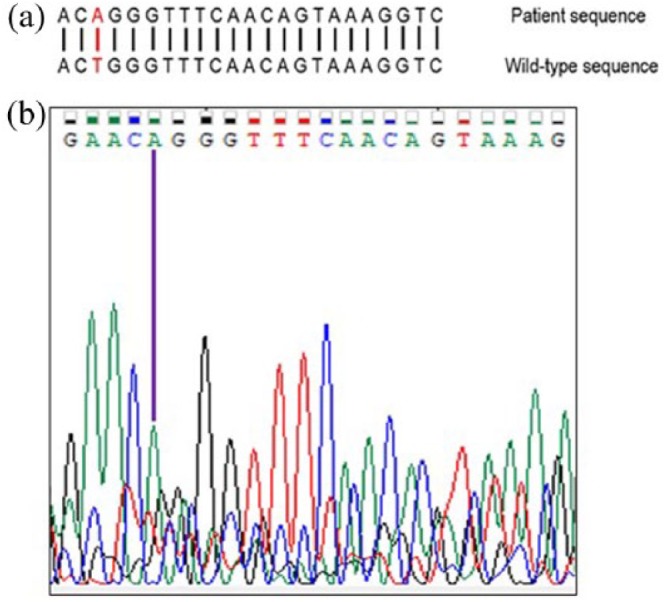
(a) Wild-type sequence of exon 9 of ALDOB around position c.1693 and mutation identified in the patient and (b) electropherogram of the corresponding region.
Discussion
HFI is considered an uncommon cause of hypoglycemia, affected children soon developed an aversion to all foods and protect themselves by self-imposed fructose and sucrose restriction. The strict dietary exclusion leads to normal growth and longevity. Nevertheless, complete elimination of this sugar from the diet is difficult to achieve, especially for undiagnosed adults, without professional advice. These people may suffer symptoms throughout life and represent a diagnostic challenge for attending physicians.9–11
The study of postprandial hypoglycemia in adults involves the exclusion of hyperinsulinemic states and secondary causes of hypoglycemia, followed by tests aimed at less frequent causes of non-hyperinsulinemic hypoglycemia, like urinary fructose. Patients may present fructose elimination in the urine, which can be quantified through enzymatic or chromatographic methods. The diagnosis can be confirmed by the decrease of enzymatic activity of aldolase B in tissue biopsies from liver, kidney or intestinal mucosa or by sequencing the ALDOB gene. Among these options, molecular analysis of ALDOB is the least invasive diagnostic method for the confirmation of the diagnosis. It is particularly important when there is partial response to treatment in order to prevent children who do not have the disease from being submitted to an unnecessarily restrictive diet. Because of these reasons, and also of poor clinical recognition and suspicion of this disease, the diagnosis of HFI in adults represents a huge challenge. The first reported case of HFI diagnosed during adulthood was published in 1978, the patient was a 21-year-old woman with no clinical manifestations during childhood. In this patient, symptoms developed following an intravenous fructose-rich fluid infusion for viral meningitis, and the diagnosis was subsequently confirmed by a fructose tolerance test (a diagnostic procedure no longer in use due to the risk of potentially fatal events).10 Two more cases of HFI diagnosed late in life have been published, one in a 69-year-old man and the other in a 50-year-old woman. In both cases, the diagnosis was suspected based on aversion to sucrose-containing foods. Contrary to our case, both cases manifested symptoms during childhood but had undergone no subsequent clinical or laboratory analyses to study these symptoms. Interestingly, these two patients presented no long-term systemic complications of HFI.9,11 None of these previous reports of adult HFI performed genetic assessment to identify the responsible ALDOB gene mutation.
Molecular and genetic assessment represents the current gold standard to establish a definitive diagnosis of HFI. Up to 60 mutations in ALDOB have been described so far, including 29 missense or nonsense mutations, 9 splicing site mutations, 15 deletions and even 1 regulatory mutation in the promoter.12 However, the most common ALDOB mutations are by far c.448G>C (exon 5), c.524C>A (exon 5) and c.1005C>G (Exon 9).3,12 These common mutations take place in coding regions of the gene and produce harmful modifications in the aldolase B enzyme that result in a truncated polypeptide, major changes in folding, disruption of the substrate-binding site or impaired ability to conform tetramers.13–15 It is interesting to note, however, that HFI has also been reported in a patient heterozygous for the pY204X (c.612T>A) mutation,16 suggesting that HFI may behave as a phenotype with codominance or incomplete penetrance. Exon 9 is the largest of the eight exons of ALDOB, it consists of 1327 base pairs and a 3′ untranslated region (UTR). Exon 9 harbors the c.1005C>G mutation, responsible for up to 8% of all HFI cases.13,15 In our case, amplification and complete sequencing of the ALDOB exons revealed a previously unreported variant located in the 3′ UTR of exon 9. Even though this variant theoretically does not affect the sequence, folding or function of the resulting protein, pathogenic variants of the 3′ UTR have been described in several genetic diseases including hemoglobin H (HbH) disease,17 immunodysregulation polyendocrinopathy enteropathy X-linked (IPEX)18) and aniridia.19 Mutations on the 5′ UTR have been found on hereditary thrombocytopenia,20 breast cancer,21 fragile X syndrome22 and Alzheimer’s disease.23 The mechanisms involve the influence of the 3′ UTR on mRNA stability and translation efficiency.24 Recently, single-point mutations in the p53 mRNA 5′ UTR were associated with dysfunctional activity of p53 and propensity to tumor development.25 Thus, it is biologically plausible that a 3′ UTR variant of ALDOB in our patient may have led to suboptimal aldolase activity levels and the observed phenotype of fructosuria and hypoglycemia.
Conclusion
HFI is one of the least common causes of hypoglycemia, and its detection can be particularly challenging in adult patients. A careful anamnesis and acute clinical suspicion are required to detect HFI in patients with predominantly postprandial, non-hyperinsulinemic hypoglycemia of no apparent cause. This also calls for exclusion of hyperinsulinemic states or exogenous administration of insulin or hypoglycemic drugs. Molecular analysis is the cornerstone of the diagnosis of HFI. More than 60 mutations in ALDOB have been identified to date, but only the three most common mutations in exons 5 and 9 are usually included in the diagnostic study of patients with suspected HFI. In this report, we document a pathogenic variant mutation in the UTR of the ALDOB gene, accompanied by a phenotype of recurrent postprandial hypoglycemia corresponding to that of HFI, albeit with a milder severity. HFI should not be entirely ruled out in the diagnostic algorithm for unexplained hypoglycemia, even in an adult patient or even if the most common mutations of ALDOB are not present.
Acknowledgments
The authors deeply thank the patient whose case is described in this report, for her collaboration, willingness and patience.
Footnotes
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Ethical approval: Our institution does not require ethical approval for reporting individual cases or case series.
Funding: The author(s) received no financial support for the research, authorship and/or publication of this article.
Informed consent: Written informed consent was obtained from the patient(s) for their anonymized information to be published in this article.
ORCID iD: Carlos O Mendivil  https://orcid.org/0000-0001-5546-4206
https://orcid.org/0000-0001-5546-4206
References
- 1. Gruchota J, Pronicka E, Korniszewski L, et al. Aldolase B mutations and prevalence of hereditary fructose intolerance in a Polish population. Mol Genet Metab 2006; 87: 376–378. [DOI] [PubMed] [Google Scholar]
- 2. Santer R, Rischewski J, von Weihe M, et al. The spectrum of aldolase B (ALDOB) mutations and the prevalence of hereditary fructose intolerance in Central Europe. Hum Mutat 2005; 25: 594. [DOI] [PubMed] [Google Scholar]
- 3. Cross NCP, Cox TM, de Franchis R, et al. Molecular analysis of aldolase B genes in hereditary fructose intolerance. Lancet 1990; 335: 306–309. [DOI] [PubMed] [Google Scholar]
- 4. Hommes FA. Inborn errors of fructose metabolism. Am J Clin Nutr 1993; 25: 788S–795S. [DOI] [PubMed] [Google Scholar]
- 5. Douillard C, Mention K, Dobbelaere D, et al. Hypoglycaemia related to inherited metabolic diseases in adults. Orphanet J Rare Dis 2012; 7: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cryer P, Axelrod L, Grossman B, et al. Evaluation and management of adult hypoglycaemic disorders: an Endocrine Society Clinical Practice guideline. J Clin Endocrinol Metab 2009; 94: 709–728. [DOI] [PubMed] [Google Scholar]
- 7. Kawasaki T, Akanuma H, Yamanouchi T. Increased fructose concentrations in blood and urine in patients with diabetes. Diabetes Care 2002; 25: 353–357. [DOI] [PubMed] [Google Scholar]
- 8. Coffee EM, Yerkes L, Ewen EP, et al. Increased prevalence of mutant null alleles that cause hereditary fructose intolerance in the American population. J Inherit Metab Dis 2010; 33: 33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Burmeister LA, Valdivia T, Nuttall FQ. Adult hereditary fructose intolerance. Arch Intern Med 1991; 151: 773–776. [PubMed] [Google Scholar]
- 10. Lameire N, Mussche M, Baele G, et al. Hereditary fructose intolerance: a difficult diagnosis in the adult. Am J Med 1978; 65: 416–423. [DOI] [PubMed] [Google Scholar]
- 11. Yasawy MI, Folsch UR, Schmidt WE, et al. Adult hereditary fructose intolerance. World J Gastroenterol 2009; 15: 2412–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cardiff University Institute of Medical Genetics. The Human Gene Mutation Database, http://www.hgmd.cf.ac.uk/ac/gene.php?gene=ALDOB (accessed 4 September 2018).
- 13. Cross NCP, Stojanov LM, Cox TM. A new aldolase B variant, N334K, is a common cause of hereditary fructose intolerance in Yugoslavia. Nucleic Acids Res 1990; 18: 1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bijarnia-Mahay S, Movva S, Gupta N, et al. Molecular diagnosis of hereditary fructose intolerance: founder mutation in a community from India. JIMD Rep 2015; 19: 85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Davit-Spraul A, Costa C, Zater M, et al. Hereditary fructose intolerance: frequency and spectrum mutations of the aldolase B gene in a large patients cohort from France—identification of eight new mutations. Mol Genet Metab 2008; 94: 443–447. [DOI] [PubMed] [Google Scholar]
- 16. Paolella G, Pisano P, Albano R, et al. Fatty liver disease and hypertransaminasemia hiding the association of clinically silent Duchenne muscular dystrophy and hereditary fructose intolerance. Ital J Pediatr 2012; 38: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Higgs D, Goodbourn S, Lamb J, et al. α-Thalassaemia caused by a polyadenylation signal mutation. Nature 1983; 306: 398–400. [DOI] [PubMed] [Google Scholar]
- 18. Bennett C, Brunkow M, Ramsdell F, et al. A rare polyadenylation signal mutation of the FOXP3 gene (AAUAAA→AAUGAA) leads to the IPEX syndrome. Immunogenetics 2001; 53: 435–439. [DOI] [PubMed] [Google Scholar]
- 19. Chao L, Mishra R, Strong L, et al. Missense mutations in the DNA-binding region and termination codon in PAX6. Hum Mutat 2003; 21: 138–145. [DOI] [PubMed] [Google Scholar]
- 20. Ferrari S, Lombardi A, Putti M, et al. Spectrum of 5’UTR mutations in ANKRD26 gene in patients with inherited thrombocytopenia: c.-140C>G mutation is more frequent than expected. Platelets 2017; 28: 621–624. [DOI] [PubMed] [Google Scholar]
- 21. Wang J, Lu C, Min D, et al. A mutation in the 5’ untranslated region of the BRCA1 gene in sporadic breast cancer causes downregulation of translation efficiency. J Int Med Res 2007; 35: 564–573. [DOI] [PubMed] [Google Scholar]
- 22. Chiang P, Carpenter L, Hagerman P. The 5’ untranslated region of the FMR1 message facilitates translation by internal ribosome entry. J Biol Chem 2001; 276: 37916–37921. [DOI] [PubMed] [Google Scholar]
- 23. Zhou W, Song W. Leaky scanning and reinitiation regulate BACE1 gene expression. Mol Cell Biol 2006; 26: 3353–3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Conne B, Stutz A, Vassalli J. The 3’ untranslated region of messenger RNA: a molecular “hotspot” for pathology? Nat Med 2000; 6: 637–641. [DOI] [PubMed] [Google Scholar]
- 25. Chen J, Kastan MB. 5’-3’-UTR interactions regulate p53 mRNA translation and provide a target for modulating p53 induction after DNA damage. Genes Dev 2010; 24: 2146–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]