

Summary
Transcriptional regulation in metazoans occurs through long range genomic contacts between enhancers and promoters, and most genes are transcribed in episodic ‘bursts’ of RNA synthesis. To understand the relationship between these two phenomena and the dynamic regulation of genes in response to upstream signals, we describe the use of live-cell RNA imaging coupled with Hi-C measurements and dissect the endogenous regulation of the estrogen-responsive TFF1 gene. Although TFF1 is highly induced, we observe short active periods and variable inactive periods ranging from minutes to days. The heterogeneity in inactive times gives rise to the widely-observed ‘noise’ in human gene expression and explains the distribution of protein levels in human tissue. We derive a mathematical model of regulation that relates transcription, chromosome structure, and the cell’s ability to sense changes in estrogen and predicts that hypervariability is largely dynamic and does not reflect a stable biological state.
Graphical Abstract
Introduction
There has been tremendous progress in the development of methodologies to interrogate gene expression in single cells, including in situ imaging and single-cell RNA sequencing(Chen et al., 2018). These data demonstrate the vast diversity in gene expression programs present in multicellular organisms. The differences are due both to programmed specialization which arises during differentiation, but also to random processes which result in heterogeneity within a population of cells in a tissue. This latter phenomenon – sometimes called ‘noise’ in gene expression -- is a consequence of the stochastic nature of biochemical reactions. However, to what extent does heterogeneity in gene expression reflect stable sub-populations of cells or simply a transient state? Understanding the dynamic nature of gene expression is essential for interpreting single-cell expression studies and understanding how cells function in a tissue.
Quantitative measurements of RNA and protein in single cells have suggested several underlying principles of non-genetic heterogeneity. First, heterogeneity can be divided into intrinsic and extrinsic noise depending on whether fluctuations are specific to individual genes or experienced across many genes(Elowitz et al., 2002; Xu et al., 2017). Second, the primary contribution to intrinsic noise is transcriptional bursting (period of RNA synthesis activity during which multiple polymerases initiate, separated by inactive periods), which has been observed from bacteria to humans (Lenstra et al., 2016). Third, intrinsic noise can be buffered by transcription from multiple alleles and downstream RNA processing such as splicing, export and decay, which can smooth out fluctuations by time averaging(Battich et al., 2015). Finally, regardless of the type of noise (intrinsic or extrinsic) or the source (transcriptional or post-transcriptional), the resulting heterogeneity can be ergodic or non-ergodic. If variation is ergodic, each cell samples the entire possibility of states.
To interrogate human gene dynamics, we chose the estrogen response in mammary epithelial cells, which has served as a transcriptional paradigm for decades(Masiakowski et al., 1982; May and Westley, 1987). The response is rapid and widespread: within 40 minutes of estradiol (E2) treatment, hundreds of genes are activated or repressed (Hah et al., 2011). Transcriptional activation is regulated through the estrogen receptor (ER), which binds estrogen response elements (EREs) located proximal and distal to putative target genes(Fullwood et al., 2009). However, the role of chromosome structural changes in response to stimulus is unclear. Many enhancer-promoter contacts are pre-formed and become stronger with hormone addition(Hakim et al., 2011; Stavreva et al., 2015), but topological domain boundaries remain largely unchanged(Le Dily et al., 2014). Acute depletion of CCCTC-binding factor (CTCF) and cohesin results in loss of domains but only modest changes in gene expression(Nora et al., 2017; Rao et al., 2017). However, hormone-responsive contacts without CTCF binding may be more relevant for stimulus-dependent regulation(D’Ippolito et al., 2018). Overall, it is unknown how the estrogen response regulates the intrinsic dynamics of endogenous genes, how these dynamics are modulated by genome architecture, and how individual cells harness these dynamics to sense estrogen levels.
We take an integrated approach based on single-molecule imaging, perturbation of cis-and trans-acting regulatory factors, and genome-wide chromosome conformation capture (Hi-C). We used CRISPR/Cas9 to integrate 24 MS2 repeats into multiple endogenous TFF1 loci in human breast cancer cells, thus enabling live-cell (LC) imaging of TFF1 transcription in real time. We find that TFF1 expression variability comes from long stochastic repressive periods for individual alleles that can last > 16 hours, even while other alleles in the same nucleus are active. We identify a cohort of secreted and signal peptide genes which show extreme expression heterogeneity (~ 100-fold) in human and mouse tissue, indicating that long stochastic repressive periods are present in specific gene ontology categories. In competition with this repressive process is a phenomenon we call ‘coupled intrinsic noise’ whereby transcription of one allele makes transcription of another allele more likely, Finally, we derive a mathematical model of TFF1 transcription which integrates dynamics, allele coupling, and genome-wide chromosome conformation.
Results
Live-cell imaging of endogenous TFF1 transcription reveals estrogen-regulated inactive periods and a deep repressive state
To observe regulation of endogenous alleles in single cells, we targeted the estrogen responsive TFF1 gene in MCF7 cells for hCas9 mediated integration of 24 MS2 stem loops into the 3’ UTR (Fig. 1A). As the MS2 loops are transcribed, stably-integrated GFP-MS2 coat protein specifically binds to the RNA loops, enabling LC visualization of RNA at the site of transcription(Janicki et al., 2004). We employed a screening strategy that allowed for the detection of positive clones as correctly integrated alleles yield double band PCR amplicons (Fig. 1B,C). Addition of E2 after hormone depletion of the TFF1-MS2 clonal cell line showed strong induction (Fig. 1D), similar to that observed for the unedited parental cell line. This cell line contains a total of five TFF1 alleles, three of which are labeled with MS2 (Fig. 1E, S1A-C). Individual cells showed multiple TFF1 transcription sites (TS simultaneously in the same cell (Fig. 1F, Movie S1, S2), and individual RNAs could be observed diffusing in the nucleus (Movie S3). In summary, we have labeled three TFF1 alleles at the endogenous loci in human cells, and these alleles show a robust response to E2 and can be imaged with single-molecule sensitivity.
Figure 1.

Live-cell imaging of endogenous TFF1 transcription reveals long and variable inactive periods
(A) Illustration of the 24XMS2 stem loop labeling approach of the TFF1 3’UTR.
(B) Screening methodology for 24XMS2 integrated single-cell clones yields double band PCR amplicons. Genomic PCR primers designed from outside of homology arms are incorporated into the donor plasmid.
(C) Double bands indicate construct integrated at TFF1 in the TFF1-MS2 single-cell clone, and not in the parental unedited cell line.
(D) smFISH quantification of MS2 RNA show the 24X MS2 cell line is inducible by 100nM E2. ls per sample
(E) smFISH shows MS2 labeled RNA is exported into the cytoplasm in the TFF1-MS2 cell line. Bottom panel shows MS2 and TFF1 intron co-localization. Cells are fixed at steady state in saturating E2.
(F) Three alleles can be visualized in the TFF1-MS2 clone upon induction with 100nM E2.
(G) Multiple alleles can be tracked and observed transcribing in the same cell (top). A Hidden Markov Model was used to identify the periods of activity and inactivity (bottom).
(H-I) Distribution of active periods and inactive periods. N=100 alleles from 48 cells.(I) The inset shows an expanded plot of the first 80 bins or ~133 minutes of inactive durations.
(J) Raw intensity traces of 100 alleles plotted as a heatmap. See also Figure S1
LC imaging under saturating estrogen conditions indicates RNA synthesis is both sporadic and discrete (Movie S4). To quantify this active/inactive behavior, we tracked TS and measured the fluorescence intensity over time (Larson et al., 2011)(Fig. 1G, S1D). The duration of active periods is narrowly distributed with a mean of 16.0 ± 0.5 min (Fig. 1H, SEM). However, the duration of inactive periods is broadly distributed with some alleles inactive for more than 12 hours (Figs. 1Iand 1J). The long periods of inactivity constitute a persistent transcriptionally repressive state for single alleles, even when other alleles in the same nucleus are active. Moreover, alleles can be silent for long periods of time and then show intense activity, indicating that long off periods are neither a stable property of the cell nor the allele.
Periods of activity reflect both the time that the promoter is transcriptionally ‘active’ (interval over which nascent transcripts are initiated), and the dwell time of RNA (elongation, cleavage, and release of nascent transcripts)(Coulon et al., 2014). To separate, we use transcription fluctuation analysis(Coulon and Larson, 2016; Ferguson and Larson, 2013; Larson et al., 2011) with Hidden Markov analysis. The transcription bursts are stitched together into one contiguous intensity trace (Fig. S1D,E,F). The analysis indicates that the initiation rate is 0.5 ± 0.02 min, and the dwell time of the nascent RNA is 13.0 ± 0.8 min (Fig. S1G). Therefore, the time that the promoter is ‘active’ is 3.0 ± 1.0 min , and the measured ‘burst’ size (RNA/active period) is 1.5 ± 0.5 transcripts.
Given the highly uniform nature of the bursts for all cells at a given dose, we asked how transcription and cellular mRNA changes with E2 concentration. We measured a steady state dose response by single-molecule FISH (smFISH) (Fig. 2A, B, S2A,B), and obtained an EC50 of 0.02 ± 0.01 nM (Fig. 2C, SEM), agreeing with previous results (0.02 – 0.05nM, (May and Westley, 1987, 1988)). The number of nascent RNA molecules at the TS was 1.67 ± 0.02 (Fig. S2C,D) and did not change with E2 dose (Fig. S2E). We performed LC imaging at doses of 0.05nM, 0.5nM, and typical MCF7 culture conditions, corresponding to the EC50, saturation, and beyond saturation, respectively. Under these conditions, the TFF1-MS2 and the parental cell line have similar mRNA/cell distributions and fraction of cells transcribing (28±1% and 19±2%; Fig. S2F,G). We observed no significant difference between active period durations (Fig. 2D). However, inactive period durations differed between doses (Fig. 2E). Under complete media or 0.5nM, TFF1 bursts every 66 ± 7 and 86 ± 18 min, respectively. In contrast, near EC50, TFF1 bursts every 185 ± 34 min. Thus, E2 regulates the periods of inactivity (frequency of TFF1 activation).
Figure 2.
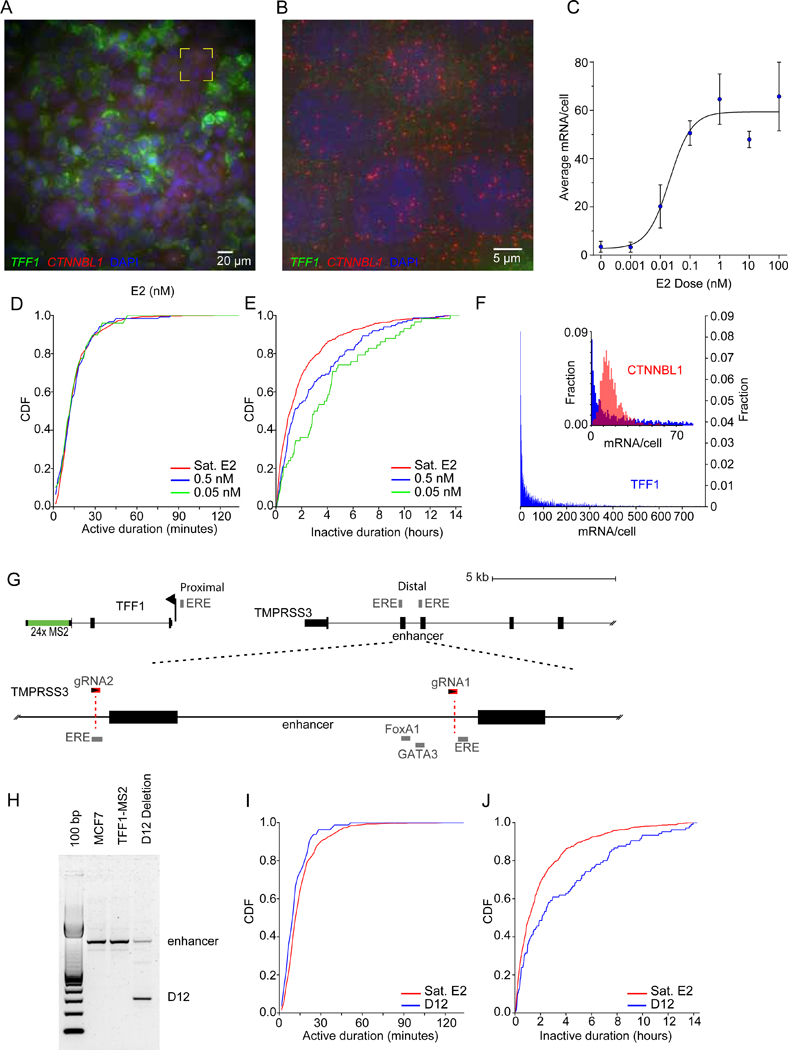
Estrogen receptor regulates the frequency of TFF1 activation
(A) Single molecule FISH imaging of TFF1 (green) and CTNNBL1 RNA (red) in single cells shows broad distribution of TFF1 mRNA in 100nM E2 at steady state. DAPI staining is in blue.
(B) Yellow box in Fig 3a expanded to illustrate cells with low TFF1 and uniform CTNNBL1 mRNA expression.
(C) TFF1 mRNA smFISH average shows strong response to E2 (blue). Sigmoidal dose response fit in black. The EC50 is 0.02 nM, in agreement with previous measures (May and Westley, 1987). Error bars: max and min of 2 biological replicates.
(D-E) Active / inactive cumulative distribution functions (CDF) at different E2 concentrations, respectively (green, 0.05 nM; blue, 0.5 nM; red, complete media).
(F) TFF1 mRNA smFISH illustrates broad TFF1 distribution (blue). The inset shows an expanded plot of the TFF1 distribution in comparison to the housekeeping gene CTNNBL1 (red).
(G) An enhancer of TFF1 is located ~10kb upstream, and contains two EREs (grey). This region is expanded (dotted lines) to show the cofactor binding sites. Two CRISPR guide RNAs (red) were used to delete these cofactor binding sites, and part of an ERE.
(H) PCR amplicons from the parental and deletion TFF1-MS2 cell lines illustrate the deletion amplicon on an agarose gel.
(I-J) Active and inactive CDF of the D12 deletion clone (blue) and parental (red), respectively. Cells were grown and imaged in complete media.
See also Figure S1
Surprisingly, some cells do not contain any mRNA even in the highest E2 concentrations (100 nM) (Fig. 2A,B,F, S2A,B). These cells are true non-expressers for TFF1 because a housekeeping control with similar mean number of mRNA/cell in the same cells showed a normal distribution (CTNNBL1, Fig. 2B,F inset). Although some cells express > 500 mRNA/cell at saturation, the mode is 0 mRNA/cell, and the distribution is monotonically decaying for all E2 concentrations. This result is even more striking considering that for a cell with zero TFF1 mRNA, none of the 5 alleles have transcribed within the half-life of TFF1 message, which is ~43.5 hours(Schueler et al., 2014), consistent with a long or ‘deep’ repressive state.
Activation and re-initiation are coordinately regulated
To understand how different factors contribute to the TFF1 response, we first deleted a region of the proximal enhancer in the TFF1-MS2 cell line with CRISPR Cas9 (Fig. 2G). The TFF1 proximal enhancer is located 10kb upstream and contains binding sites for ER, and the cofactors FoxA1 and GATA3 (Carroll et al., 2005; Hurtado et al., 2011). This deletion removes the cofactor binding sites but leaves a crucial ERE intact and results in diminished transcriptional activity (Pan et al., 2008). We isolated a deletion mutant (Fig. 2H) and imaged the cells under saturation conditions. We observe a slight difference in distribution of the active times (p<0.0003, KS test), and a pronounced difference in the inactive distribution (p<8e-5, KS test) (Fig. 2I,J). The enhancer deletion mutant bursts every 126 ± 27 min in contrast to the parental line which bursts every 66± 7 min. These data indicate that the enhancer primarily modulates how frequently TFF1 is activated.
We next searched for trans-acting factors with a gene-specific role in TFF1 expression and identified the estrogen-specific chromatin binding protein TRIM24(Tsai et al., 2010). Targeting TRIM24 with the chemical inhibitor MD9571, which blocks bromodomain binding to acetylated histones (Palmer et al., 2016; Tsai et al., 2010), we measured a non-monotonic dose-dependent change in TFF1 mRNA levels by smFISH with a ~ 3-fold reduction at 0.5 µM MD9571 (Fig. 3A). CTNNBL1 mRNA levels were marginally reduced. At this dose, the on time decreased to 10.2 ± 0.5 min (Fig. 3B), and the off times were unchanged (Fig. 3C). As validation, we measured the intensity of the TS with smFISH to the TFF1 intron (Fig. 3D,E). TS in treated cells are as abundant but dimmer and rarely show the bright spots indicative of multiple RNA at the locus. Taken together, our data indicate that the TFF1 enhancer determines when the gene fires, but once initiation occurs, binding of the ER co-activator TRIM24 is required for multiple re-initiation events.
Figure 3.
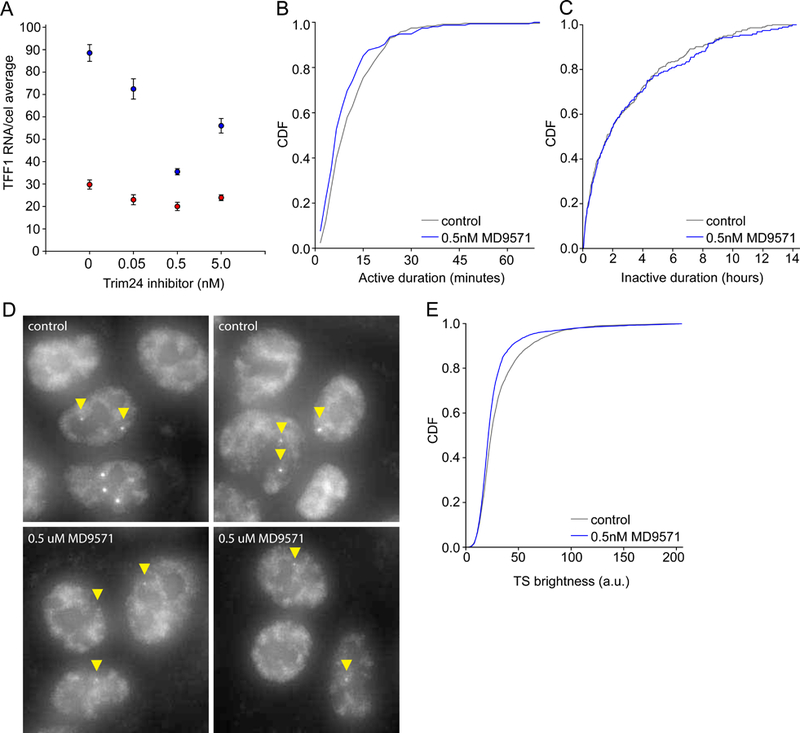
TRIM24 regulates TFF1 initiation rate
(A) TFF1 RNA (blue) is downregulated in response to TRIM24 inhibition by MD9571. Cells were treated with different concentrations of TRIM24 inhibitor. A small decrease in mRNA expression was also observed for CTNNBL1 (red). Error bars represent the SEM, n=3 for TFF1, n=2 for CTNNBL1. An average of over 1000 cells/sample was used.
(B-C) TRIM24 inhibition effects the active time durations and not the periods of inactivity. p=0.035, >0.5 respectively, by Mann-Whitney test.
(D) smFISH validation of TRIM24 effect on TFF1 transcription sites shows less bright transcription sites. Transcription sites were visualized by TFF1 intron probesets.
(E) CDF of transcription site intensity of TFF1 control and TRIM24 inhibitor smFISH data shows a significant decrease in intensity. p<0.001 by Mann-Whitney test.
Alleles in the same nucleus are dynamically coupled
To address whether the variability in transcriptional activity is due to differences between cells, we plotted the summed RNA output of single TFF1 alleles (integrated area under the transcription time series) against the summed output of other TFF1 alleles in the same cell (Fig. 4A). The output of TFF1 alleles in the same cell are correlated (r = 0.65 ± 0.08 SEM,p-value=3.8e-10), while pairs of alleles from different cells are not (Fig. 4B). Likewise, the number of bursts was also correlated (r = 0.48 ± 0.10 SEM,p-value=1.2e-5) (Fig. 4C). Some cells show only a single burst over the entire time course (Fig. 4D, upper), while others are highly active (Fig. 4D middle). Alleles can also show differential behavior, reinforcing the notion that extended off periods are not a consequence of cell state (Fig. 4D, lower).
Figure 4.
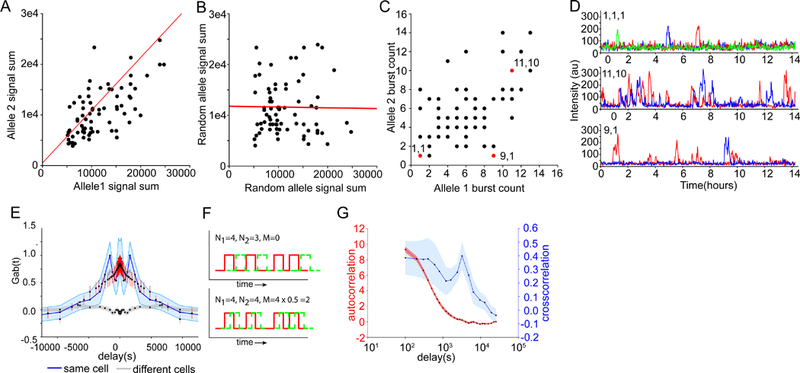
Inter-allelic correlations indicate that intrinsic noise dominates heterogeneity
(A-B) Scatter plot of allele activity in the same and different cells, respectively. Total RNA output is determined by summing the area under the time trace for each allele. Correlation coefficient r =0.65 (p-value = 3.8e-10) and −0.02, slope of 1.04 and −0.015, intercepts 513 and 11783 for intra-cell and inter-cell correlations, respectively. The slopes and intercept were calculated using Reduced Major Axis regression (red line).
(C) Scatter plot of the number of bursts per allele over a 14-hour period in the same cell. Example pairs with highly-correlated (1,1) and (11,10) and non-correlated alleles (9,1) are marked in red.
(D) Traces of example allele pairs in 4C with correlated and non-correlated burst counts.
(E) Gab(t) (blue) denotes the experimental cross-correlation between two alleles as a function of time lag, along with the simulated cross-correlation (red). The random cross correlation (gray) is between alleles in different cells. Error bars are from bootstrap. N=219 alleles, resulting in 159 intra-nuclear correlation traces.
(F) Different scenarios of cross-correlation between transcriptional pulses from two alleles, denoted as red and green for clarity. N1 is the number of bursts from the red allele; N2 is the number of bursts from the green allele. M is the number of co-occurring bursts. Setting burst duration D = 20 min, and total time T=400 min results in G1,2(0) = −1, 1.5 corresponding to 0% and 50% overlap for upper and lower panels, respectively.
(G) Comparison of the autocorrelation (red) and cross-correlation (blue). Autocorrelation: N=219. Cross-correlation: N=159.
See also Figures S3-S5
At first glance, our data suggests a significant amount of extrinsic noise. We tested this by measuring correlations between individual bursts. Inter-allele cross-correlation computed from background-corrected traces (Fig. S3,S4, STAR Methods) showed significant positive correlation between alleles only within the same nuclei (Fig. 4E). When one allele fires, there is a 27 ± 3% greater chance that another allele will fire within the next 2 hours than would be expected by random chance (see STAR Methods). Gab(τ) depends on the number of bursts from each allele and the fraction of bursts which coincide over the observation period. Several simplified scenarios are shown schematically (Fig. 4F). The cross-correlation has a long decay and depends on the spatial separation between alleles, meaning there is often a delayed correlated burst which would not be visible in snapshots of nascent RNA (Fig. 4G, S5). In summary, alleles show spatio-temporal correlations which have not previously been considered in models of gene regulation.
Mathematical model of TFF1 transcription relates transcription dynamics, expression heterogeneity, and protein distribution in human tissue
We next addressed possible kinetic models that could explain the dynamics of TFF1 transcription and steady state levels of TFF1 mRNA. The standard two-state telegraph model (Ko, 1991; Peccoud and Ycart, 1995) does not describe a distribution of off times with a long tail and also does not directly consider RNA synthesis dynamics captured by the on time distribution. To address these limitations, we developed a general and comprehensive model of gene activity that can be compared to both off and on time (mRNA synthesis) dynamics as well as steady state mRNA levels provided by smFISH. The model is fully stochastic and consists of an arbitrary number of “gene states” and “RNA steps “. The standard telegraph model is a subtype of this model with two gene states and no RNA steps. The gene states represent regulatory features underlying transcription before one observes RNA at the TS. Figure 5A shows an example with 3 gene states and 2 RNA steps. One of these gene states is “active” (labeled X), where transcription can be initiated while the rest (labeled Y, Z) are “inactive.” As pre-mRNA is being synthesized it remains attached to chromatin, undergoing a series of transitions such as elongation and release. Allele coupling occurs through a simple mechanism whereby whenever any allele is in the active state (state X), the forward rate to the active state from the penultimate state (state Y) is immediately increased for all the other alleles in the cell by a fixed coupling rate. The influence of E2 concentration is modeled by a change to one of the rates. The number of gene states, RNA steps, E2 effect, and rate parameters are determined by Bayesian model comparison (Fig. S6, see Methods S1).
Figure 5.
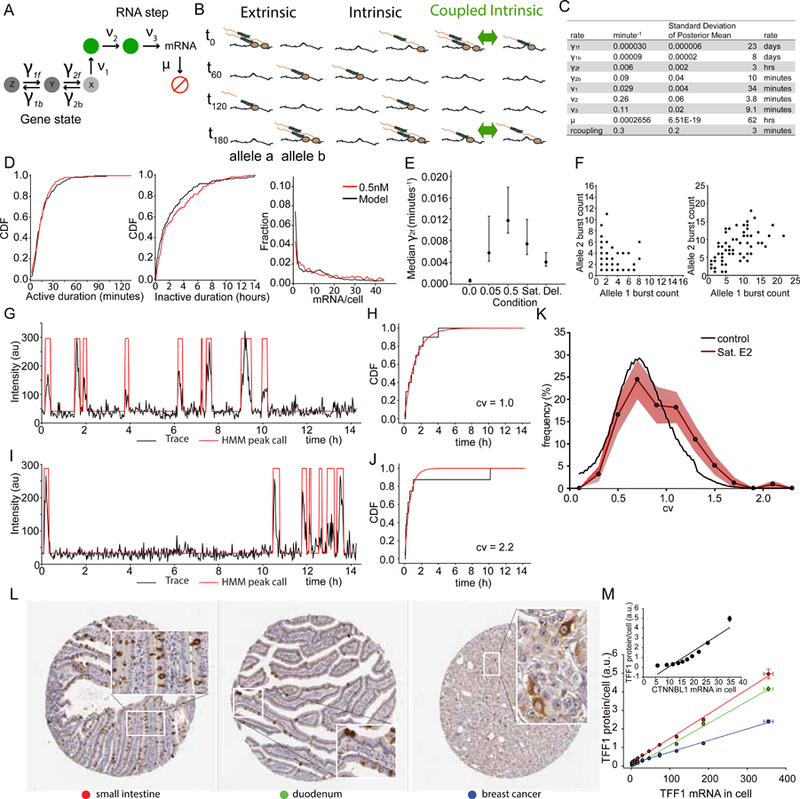
Multi-state model of TFF1 transcription requires interallele coupling
(A) Generalized telegraph model for TFF1 depicts three regulatory gene states and two RNA steps. Only one of the gene states is occupied at any given time while each RNA step can be occupied or unoccupied. Red circle denotes degraded mRNA.
(B) Schematic of the scenarios where extrinsic, intrinsic and coupled intrinsic (green) dominates transcriptional output. Two alleles are shown at 4 time points (t). Polymerase and RNA in beige, MS2-GFP in green. Green arrow denotes an allele coupling factor.
(C) Maximum likelihood rates for TFF1 dynamics in saturating E2 (complete media).
(D) Active, inactive live cell and mRNA smFISH distribution model fits are shown for dose 0.5nM. model in black, data in red.
(E) Median γ2f rates with 95% confidence intervals.
(F) Simulated inter allele total burst correlations for uncoupled (left) and coupled (right) models. The simulated coupled model recapitulates burst output correlations observed in 4C.
(G) Single experimental trace for an allele with Poisson-like distributed inactive times. HMM peak calls in red.
(H) CDF(black) for trace in panel G) and Poisson fit of inactive times(red) show similar agreement and a coefficient of variation (CV) of 1.0.
(I) Single experimental trace (black) from an allele with a very long inactive time. HMM peak calls in red.
(J) CDF (black) for trace (panel I) and Poisson fit of inactive times(red) show disagreement and a coefficient of variation 2.2.
(K) Distribution of CVs for the 100 alleles at saturating estradiol(red) and a simulated Monte Carlo trace (black) with fixed transcription rates drawn from the pool of measured average rates.
(L) TFF1 signal in human tissue images was quantified in three tissues: small intestine(red), duodenum(green) and breast cancer(blue). Image credit to Human Protein Atlas.
(M) Decile to decile plots of TFF1 smFISH mRNA histograms in MCF7 cells and respective tissues from 5L show linear relationship.
See also Figure S6
The model that best fit the data under all conditions while penalizing for model complexity using Bayesian and Akaike information criterion (BIC, AIC) consisted of three gene states and two RNA steps (Fig. 5A, Table S1). The two predicted RNA steps indicate that either zero, one, or two RNA molecules can be present at the TS during a burst, consistent with the direct observation (Fig. 1). Rate parameters for the top model and agreement between the model, LC data, and smFISH are shown in Fig. 5C and 5D. Three features of the model revealed mechanistic insight. First, the model predicts that E2 concentration controls the occupancy probability of gene state X (Fig. 5E): the forward and backward rates between the gene states X and Y had the lowest BIC and AIC values, with the forward rate slightly favored (Fig. 5E). Likewise, the enhancer deletion also perturbs transitions between X and Y. In contrast, TRIM24 inhibition alters initiation rate (ν1, Fig. 5a). Second, the model predicts that the forward rate out of the most inactive state, Z, is extremely long (~ 23 days) (Table S1). This time scale is much longer than the observation time of 14 hours and is an extrapolated time scale based on the shape of the inactive time distribution. Lastly, a single coupling parameter explains the burst correlation between alleles (Fig. 5F), which could not be explained with coupling at other steps. Mechanistically, E2 levels and the cis-acting enhancer only effect Y to X transitions but have no direct effect on the deep repressive Z state, which is the dominant source of heterogeneity in mRNA levels.
As a validation of the model, we independently tested whether each allele visits all states. It is possible that there are stable populations of fast bursters and slow bursters or a continuum of fixed behaviors which appear as distinct states after fitting. To address this, we looked for transitions between dynamical regimes in single trajectories. A single trajectory which obeys a fixed rate constant will obey a Poisson process, with a coefficient of variation (CV = σ/mean) for the off times equal to 1. A trajectory where a hidden transition occurs will show a CV > 1. Experimental examples are shown in Figs. 5G-J. For each measured allele, we computed the CV for off times and binned these data into a histogram (Fig. 5K, red). The null hypothesis -- that every cell is in its own ‘state’ and follows a sing le fixed rate which may be unique to that cell -- is simulated for comparison (Fig. 5K, black). The enrichment in alleles with a CV > 1 indicates that we have captured transitions between states. Thus, multi-state behavior is a property of individual alleles and not a result of static heterogeneity in the population.
We then tested model predictions in gene expression heterogeneity for protein distribution in human tissue. We analyzed immunohistochemical staining of TFF1 in fixed tissue from small intestine, duodenum, and breast cancer and observed the same striking degree of expression heterogeneity in both differentiated and undifferentiated tissues (Fig. 5L, S6). We quantitatively compared the decile distribution of TFF1 protein staining intensity to the decile distribution of mRNA/cell from smFISH (Fig. 5M). There is a linear relationship between TFF1 protein and TFF1 mRNA, but not between TFF1 protein and CTNNBL1 mRNA (Fig. 5M, inset), indicating that the empirical probability distribution of mRNA in MCF7 cells predicts the distribution of protein in multiple human tissues.
Secreted and signal peptide genes are variably expressed
Our data indicate that TFF1 exhibits a deep repressive state which is largely responsible for the observed expression heterogeneity. We determined the generality of the deep repressive state to other genes by identifying genes that were variable in MCF7 single cell RNA-seq (scRNA-seq) data (Rothwell et al., 2014). Since TFF1 has a long RNA half-life, we postulated that genes with both high variability in scRNA-seq expression and long RNA half-lives would be enriched in genes with a deep repressive state. Because the dataset was small, we considered expressed genes (>3 RPKM) with a long RNA half-life (Schueler et al., 2014)(> 1000 minutes), and sorted the list by CV. TFF1 is ranked highly and is also the top variable gene when considering a measure of dispersion for a normal distribution (Fano factor, Table S2). Surprisingly, we observed that the top quintile was enriched in certain gene functional categories: secretion, transmembrane, and signal peptide (Fig. 6A). TFF1 is among these genes as a secreted peptide which is part of the mucosa.
Figure 6.
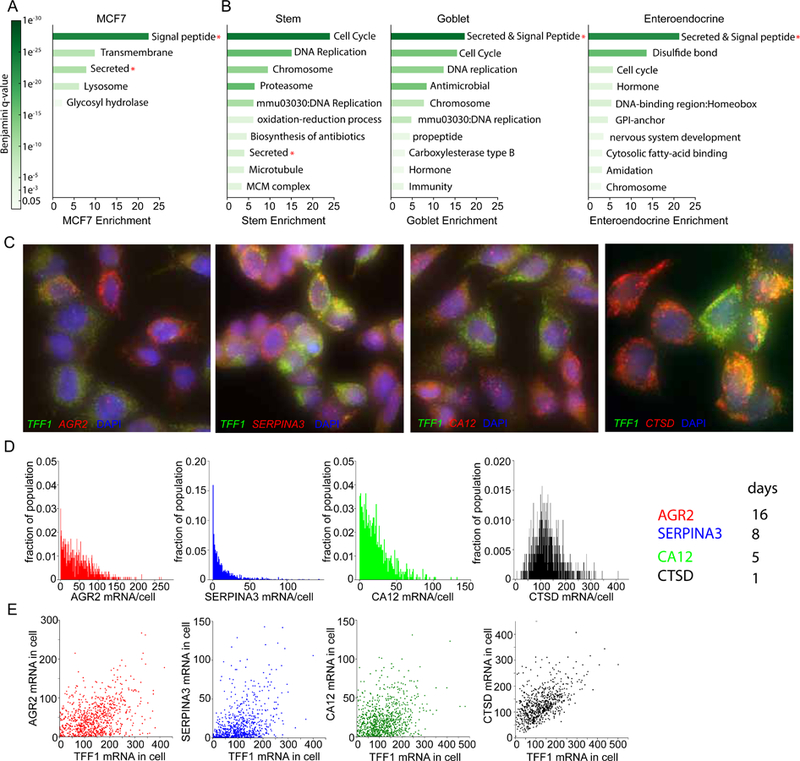
Secreted genes are variably expressed in human and mouse tissue
(A) Secreted and signal peptide genes are variably expressed in MCF7 cells. This analysis uses genes with long RNA half-lives (>1000 minutes) in MCF7 cells. Shown are gene ontology categories and enrichment values. Categories with Benjamini q-values of less than 0.05 were considered.
(B) Several categories are variably expressed within the cell types stem, goblet and enteroendocrine of a small intestine dataset. Secreted and signal peptide genes are variably expressed (Red asterisk). Shown are gene ontology categories and enrichment values. Benjamini pvalues of less than 0.05 were considered.
(C) smFISH validation of secreted and signal peptide candidate genes in MCF7 cells show broad gene expression heterogeneity. Images show co hybridization of TFF1(green) and candidate genes (red). Maximum intensity projections are displayed.
(D) RNA is expressed heterogeneously in single MCF7 cells for 3 of 4 candidates. Plotted are mRNA/cell histograms from the smFISH data (3C). 600–1200 cells used per sample, 3 replicates.
(E) RNA is poorly correlated between estrogen responsive genes in the same cell(r=0.39,0.42,0.26,0.55 respectively). TFF1 mRNA/cell is plotted against candidate gene mRNA/cell in the same cells.
We performed a similar gene ontology analysis on a large published dataset of scRNA-seq from the mouse small intestine (Haber et al., 2017), and identified the most significant variable genes (q-value<0.05). As expected, we observed that variably expressed genes are enriched in the ‘cell cycle’ category in all three subtypes (Fig. 6B; Benjamini p-values <1e-6). We also observed the ‘hormone’ category enriched in goblet and enteroendocrine cells. Importantly, we observed that secreted and signal peptide genes are among top most enriched category in the variable genes (Fig. 6B, asterisks), suggesting a functional role for expression variability in ‘secreted’ and ‘signal peptide’ genes.
We next validated four estrogen-responsive candidate genes (Hah et al., 2011; Massot et al., 1985) from the MCF7 ranked list via smFISH in MCF7s at saturating E2 conditions (Table S3,S4). We observed broad gene expression heterogeneity for 3 of 4 candidates (AGR2, SERPINA3, CA12; Fig. 6 C, D). Surprisingly, co-hybridization with TFF1 resulted in poor RNA correlation, indicating that hypervariable ER target genes show response heterogeneity even in the same cell. We next applied our model to the RNA histograms in conjunction with published RNA half-lives (Schueler et al., 2014) and predicted inactive times for individual alleles of 5 to 16 days (Fig. 6D right). In summary, our analysis identifies a conserved relationship between variable gene expression and secreted and signal peptide genes.
Transcription dynamics and genome architecture can be connected through the concept of entropy
LC imaging, smFISH, and scRNA-seq all point to the existence of multiple off states, one of which is a deep repressive state associated with certain gene ontology categories. Since the hypervariable genes are estrogen-responsive, and since estrogen induces changes in enhancer-promoter contacts (Carroll et al., 2005; Pan et al., 2008), we measured genome-wide chromosome conformation changes in response to E2. We performed 3e Hi-C(Ren et al., 2017) at three different E2 doses (0nM, 0.05nM, Sat. E2, 2 biological replicates) and recapitulated the characteristic contact maps observed for Hi-C (Fig. 7A,B). We observed a monotonic decrease in long range genome-wide contacts in response to increasing estradiol dose (Fig. 7A). Upon closer inspection we observed that topologically associated domains (TADS) appear denser in contacts overall (Fig. 7C), as reported recently(Vian et al., 2018). We also observed increases in off-diagonal contact frequency characteristic of defined enhancer/promoter interactions (Fig. 7D). However, many EREs did not show such obvious enrichments between two specific regions, but rather increasing contact probability with multiple parts of the chromosome and a concomitant loss of contacts with many other regions.
Figure 7.
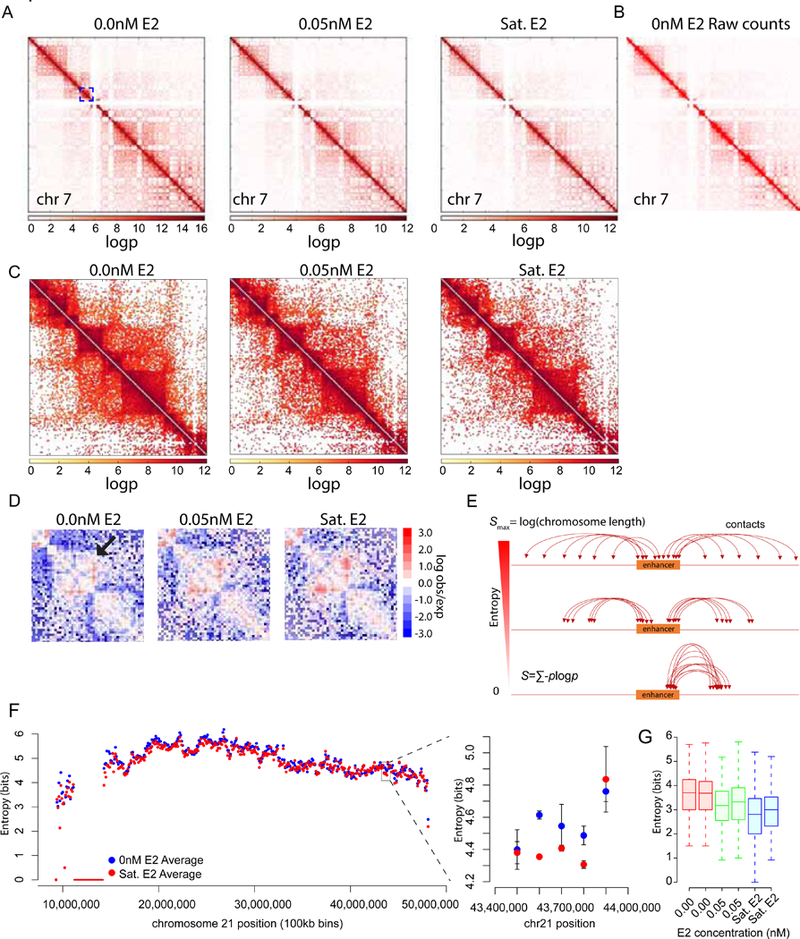
Estrogen regulated enhancers exhibit decreasing entropy and increasing specificity upon induction
(A) Chromosome contacts shift from long range to local contacts upon induction with estradiol. Contact probability map of chromosome 7 is plotted with a scaling multiplier for visualization.
(B) Raw read count contact matrix output from HOMER software plotted in Treeview.
(C) Zoomed-in view of blue box in panel (A). Decrease in long range contacts is observed upon E2 induction.
(D) Example region on chromosome 21(hg19:39,900,000–44,500,000) where contact frequency increases in response to E2. Shown are null model normalized contact map generated by Homer.
(E) Schematic of enhancer entropy. Entropy is defined as the − ∑ P log where p is the probability of an enhancer contacting a region (bin) along the chromosome. An enhancer making equal contacts to all regions of the genome would have maximal entropy. While an enhancer contacting one region would have zero entropy.
(F) Entropy for 100kb bins is calculated across chromosome 21 from two biological replicates (left panel). Entropy for flanking TFF1 region decreases in response to estradiol (right panel).
(G) Entropy was calculated for a list of >700 ER bound enhancers with enhancer transcription using 1kb bins. A decrease in entropy was observed in response to estradiol. Two replicates are plotted. See also Figure S7.
We quantitatively capture this behavior using Shannon entropy as a measure of the contact frequency distributions. Shannon entropy is defined as − ∑ p log p where p is the probability of contact between two regions and is related to the fraction of ligation reads (see STAR Methods), and the sum is over all pairs of regions (Fig. 7E). We applied this measure genome wide to raw count interaction matrices (100kb bins) obtained from HOMER. The raw counts were normalized by converting interaction counts into probability distributions such that the sum of probabilities across a single row of the raw count matrix is equal to 1 (excluding the identity bin). There was a global, but non-uniform, decrease in entropy in response to E2 (Fig. S7). We observed decreases in entropy in the regions immediately flanking TFF1 on chr 21 (Fig. 7F, right panel), but not two bins up or downstream. We then looked only at the entropy of over 700 enhancers which are bound by ER and exhibit enhancer transcription (Hah et al., 2013). Since these enhancers are much smaller (defined as 2kb) than the 100kb bin size used above, we used 1kb bin raw count matrices. The entropy of ER bound enhancers decreases from 3.5 ±0.03 bits to 2.8 ±0.06 bits (Fig. 7G, SEM), indicating that ER enhancers have less entropy or more specific contacts in the presence of E2. Yet, the majority of EREs do not approach the limit of an exclusive, highly specific contact.
To put this quantitative entropy change in a biological context, we compute the mutual information (MI) (Shannon, 1948), which is the difference between the entropy measured from all conditions vs. dose-stratified conditions. MI is the measure of how much information about E2 dose is encoded in chromosomal contacts. The MI, determined from the changes in enhancer entropy with E2, is 2.5 ±0.05 bits (SEM). This value means that each cell is capable of distinguishing 2^2.5 or ~ 5.6 ‘levels’ of E2. Conve rsely, an experimental method for reading out the contact map of all EREs would allow an external observer to distinguish the level of E2 to an error of ~ 1.2 nM. Thus, MCF7 cells contain sufficient information in their estrogen-dependent contacts genome-wide to distinguish gradations in E2 level.
How can this data be reconciled with estrogen-dependent changes in transcription? On one hand, we have the static genome-wide contact probabilities, and on the other hand we have a dynamic model applied to single genes. Shannon entropy can be measured directly in the former case and computed from the kinetic model in the latter case, based on the steady state occupancy of the states (Fig. 5a). The model predicts the existence of at least three gene states, and the transition between state X and state Y is both estrogen-regulated and dependent on the presence of a distal ERE (Fig. 2). Therefore, we hypothesize that the gene states depend in part on changes in chromosome conformation (e.g. enhancers interacting with promoters vs enhancers interacting with other parts of the genome). To account for the fact that EREs make ubiquitous contacts with different parts of the chromosome before E2 addition we consider a Z state that is composed of many energetically degenerate states, either with fast kinetic transitions between them or each connected directly to the Y state with similar kinetics. This modification does not change the kinetic structure (Fig. 5a) or time-resolved predictions for single-molecule data but rather introduces the possibility of ‘hidden’ Z states to better reflect the 3e Hi-C data. The predicted Shannon entropy from the kinetic model can then be written as:
| (1) |
where pi is the model estimated probability of state i and m is the number of equally probable hidden Z states. For the contact entropy of 2.8 bits in the high dose state, this predicts 5 hidden Z states. Thus, even a relatively low number of hidden Z states recapitulates the entropy as measured by 3e Hi-C and predicted from the single-molecule time-lapse data.
Finally, we use MI to quantitatively address how estrogen levels are reflected in single gene transcription, which encompasses not only structural changes visible by 3e Hi-C but also chromatin modifications, transcription factor occupancy, RNA polymerase transitions, etc. Because individual TFF1 bursts are highly similar, the information content is in the time interval between two bursts, and the MI between burst interval and E2 dose is 0.13 bits. To reach 1 bit, which allows an input/output device to distinguish between off and on, requires approximately 8 bursts. Therefore, to determine whether estrogen is present or not from the response of TFF1, the sum of bursts from all TFF1 alleles in the nucleus must be around eight, and to sense gradations in estrogen level requires a greater number of bursts.
Discussion
We have shown how human bursting dynamics are influenced by cis- and trans-acting factors and are physiologically regulated by changes in chromosome structure that occur in response to estrogen stimulation. We find that ‘noise’ in gene expression is driven by long repressive states which are randomly occupied by individual alleles and are not a property of the cell as a whole. Extreme heterogeneity (>500 fold for TFF1 mRNA) is also identified in single-cell RNA seq, present in functionally related classes of genes, and visible at the protein level in both differentiated healthy tissue and undifferentiated human tumors. However, this heterogeneity is a dynamic phenomenon which likely does not reflect a stable biological state or cellular subtype. The heterogeneity which we measure both in transcription dynamics and chromosome conformation places limits on the ability of a cell to decode upstream signals such as estrogen concentration.
The role of intrinsic noise in expression heterogeneity
Stochastic variation in gene expression has typically been attributed to large ‘bursts’ of transcription where many RNA are initiated in rapid succession. However, despite showing tremendous variability in gene expression, the TFF1 burst size is small, resulting in only 1–2 RNAs per burst. Moreover, this small burst size is conserved between yeast (Lenstra et al., 2015), human (this study), and in vitro (Revyakin et al., 2012), leading us to speculate that transcriptional bursts are structurally or mechanistically constrained. One such factor which allows for re-initiation of TFF1 transcription is the tripartite motif containing protein TRIM24, which has a modest effect on mRNA levels (~2–3 fold) because it only changes the burst size but not the frequency of bursting. Previous reports of large burst sizes or burst modulation (Golding et al., 2005; Tantale et al., 2016) may be due to terminology and/or experimental time resolution. For example, if one defines a burst as the time that the gene spends in the X and Y states combined, then the burst size is ~65 RNA.
For the TFF1 gene, the large variation in mRNA levels arises not from large bursts but through two reinforcing processes. First, alleles can transition into a long repressive state which is highly variable and ranges from >14hrs to 23 days. Although E2 increases the burst frequency of TFF1, as has been seen for other enhancer-driven genes (Bartman et al., 2016; Fritzsch et al., 2018; Fukaya et al., 2016; Larson et al., 2013; Skinner et al., 2016), the long repressive state is unresponsive. In fact, alleles spend the majority (> 60%) of the time in this repressive state. Second, once an allele begins making RNA, other alleles in the same nucleus are more likely to become active. Thus, stochastic effects become amplified.
We call this latter process ‘coupled-intrinsic noise’ and distinguish it from previous models of expression variability (Fig. 5B). Historically, stochastic variation has been divided into extrinsic noise and intrinsic noise based on measurements of inter- vs. intra-cell variability (Elowitz et al., 2002). Any variation that affects transcription of all alleles in the cell and is manifested as a distribution along the diagonal of the scatter plot (i.e. Fig. 4A) would be attributed to extrinsic noise. For example, variation in ER expression might result in a lineage of cells that always has higher levels of bound-estrogen and consistently higher activity of TFF1. Variation perpendicular to the diagonal could then be construed as intrinsic noise arising from the stochastic nature of biochemical interactions. Even if both alleles see the same effective concentration of ER and all other activators and repressors, there will still be differences in the firing of the alleles due to Brownian diffusion. In the model of coupled-intrinsic noise, a stochastic event at one allele – post-translational modification of an activator/repressor, generation of a non-coding RNA, etc. – can subseque ntly affect the activity at another allele.Specifically, our data suggest that activation of one allele leads to an increased probability of activation of other alleles in the same nucleus.
Dynamics, architecture, and information transfer in gene regulation
Since EREs are often distally located from the genes they regulate, we carried out 3e Hi-C in MCF7 cells under the same dose and culture conditions we used for imaging. These measurements revealed E2 dose-dependent changes in chromosome contacts such as a decrease in long range contacts and a concomitant increase in density associated with TAD-like structures near the diagonal. We observed clear formation of some de novo loops, but the majority of EREs do not form exclusive contacts with distal parts of the chromosome. In summary, these data agree with previous measurements which show both a strengthening of existing loops (D’Ippolito et al., 2018; Hakim et al., 2011; Stavreva et al., 2015) or an increase in TAD density after transcriptional activation (Vian et al., 2018).
Importantly, the conceptual framework of information theory enables a direct comparison to live-cell single-molecule transcription data. The kinetic model which describes our time-resolved data consists of states with rates of transition between them. At any point in time, the gene exists in a certain gene/RNA state, and the fractional occupancy over time is precisely what is quantified by the Shannon entropy. There is a striking concordance between the entropy changes inferred from our model and the actual entropy changes determined from the contact map. Whilst any model is necessarily a reductionist exercise, and the states in the model could correspond to alternate aspects of transcription, we favor the interpretation that the X and Y states corresponds to different chromosome conformations, with other molecular changes acting downstream of this conformation change. This interpretation is the simplest one which explains our experimental data in its totality, including the dose response, perturbations of cis and trans-acting factors involved in estrogen regulation, correlations between alleles and other genes, and 3e Hi-C data. The corollary to this interpretation is that the Z state reflects the many non-specific contacts which can sequester an enhancer from a particular promoter.
The role – if any – of hypervariability is still th e subject of speculation. We find that single cells are quite capable of accurate sensing of estrogen through the information contained in ERE contacts. However, output of transcription for TFF1 (as represented by RNA synthesis) is much less informative. Due to stochastic variability, information transfer between the upstream signal and downstream output is remarkably inefficient and slow (~ 0.002 bits/min for TFF1), despite a robust dose response. Conversely, information transfer in single-cell eukaryotes can be an order of magnitude faster (Hansen and O’Shea, 2015), indicating there is nothing mechanistically limiting in chromatin remodeling or the transcriptional machinery. One explanation may lie in the observation that this hypervariable expression associated with the deep repressive state occurs for genes functionally associated with secretion. Perhaps in an organismal context, there is a physiological benefit for cells to sense and respond to many inputs as a tissue rather than in a cell-autonomous fashion.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dan Larson (dan.larson@nih.gov).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
The MCF7 cells used in this study are Homo Sapien, female cells with the RRID:CVCL_0031. This cell line has not been authenticated. MCF7 cells were grown in MEM media (Corning) supplemented with 2mM Glutamine (Hyclone SH30034), 1X Penicillin/Strep (100units/mL Penicillin, 100micrograms/mL Streptomycin), and 10% FBS (Sigma). This media is denoted in the text as complete media or Saturated E2. The cell lines TFF1-MS2 and TFF1-MS2-D12 enhancer deletion are derived from this MCF7 parental clone. They were grown in the same conditions.
METHOD DETAILS
Treatment vehicles:
Estradiol (E2) was dissolved in 100% ethanol and maintained at −20C.
MD9571 was dissolved in DMSO.
Gene editing:
Experiments were performed on low passage MCF7 cells from ATCC. The donor construct was initially constructed by the Protein Expression Laboratory (NCI Frederick National Laboratory) and subsequently modified to increase the number of MS2 loops to 24. The genomic integration site was chosen in the middle of the TFF1 3’UTR. The donor construct consisted of left and right homology arms each spanning ~1kb, screening primer sequences, 24 MS2 stem loops, and a PGK-Puromycin-polyA gene flanked by FRT sites. Guide RNAs were designed using the website http://crispr.mit.edu/. A guide RNA targeting the 3’ UTR was cloned into pX330-U6-Chimeric_BB-CBh-hSpCas9(PX330)(Cong et al., 2013) using the standard 1-page protocol. (http://www.addgene.org/crispr/zhang/). All guide RNA sequences used in this study were cloned into PX330 (Table S3). Cells in 6 well plates were transfected with equal amounts of the donor construct and the guide RNA/Cas9 construct using TransfeX (ATCC). The manufacturer’s protocol was followed. Puromycin was added to the cells 7 days post transfection until cell death of control plate completed. Cells were grown until near confluency and plated at low dilution for single cell colony formation on 15 cm dishes. A 96 well plate was also plated for single cell colony formation. Colonies were marked and transferred to 96 well plates with cloning cylinders.
Screening Strategy:
Two primers which amplified the endogenous locus and were located outside of the homology arms were incorporated into the donor construct adjacent to each homology arm (Table S3). Correctly integrated clones will therefore amplify two smaller amplicons, as well as the larger unedited amplicons. 79% (115 of 145) of single cell colonies contained double bands. Clone cA7 contained the correct 24 MS2 stem loop PCR amplicon size (~1.4kb).
PGK-PURO-pA removal:
A guide RNA was designed to the flanking FRT motifs of the donor construct. cA7 cells in 6 well plates were transfected using TransfeX reagent according to the manufacturer’s protocol for MCF7 cells. Cells were plated at low dilution for single cell colony formation on 15cm dishes. Cells were maintained in complete media until visible colonies were observed. Colonies were marked and transferred to 96 well plates using cloning cylinders. Colonies were expanded onto 48 well plates, then replica plated onto 12 mm No. 1.5 glass coverslips. Clones were fixed with 4% paraformaldehyde (PFA) and screened with smFISH (see below) using probes to the puromycin RNA and MS2 repeat RNA. The clone c17 (cA7c17) did not contain puromycin spots.
MS2-GFP:
MS2-GFP was stably integrated into cA7c17 cells in 12 well dishes using lentiviral delivery of MS2-GFP under the control of a ubiquitin promoter as previously described(Larson et al., 2013). Cells were transduced in a 12 well plate by incubating in serum free media for 30 minutes and subsequently incubated with several dilutions of MS2-GFP lentivirus in serum free media supplemented with 2 micrograms/ml polybrene. Plates were tilted every 15–20 minutes for 2 hours in cell culture incubator. Full media was then added to cells and replaced every two days. Lentivirus was prepared by NCI Frederick National Laboratory. This final cell line was subsequently used for live cell experiments (TFF1-MS2).
CRISPR enhancer deletion:
Two guide RNAs were constructed to delete 798bp of the TFF1 upstream enhancer (Table S3). TFF1-MS2 cells in 6 well plates were transfected using Lipofectamine 3000 (Invitrogen) using the standard protocol and equal quantities of the two guide RNA plasmids. 3 days after transfection, cells were plated at low dilution for single cell colony formation. 216 colonies were transferred to 96 well plates, and subsequently replica plated. Genomic DNA was extracted with the Z96 Zymo genomic DNA kit. PCR amplification of the deletion region was performed using Quickload mastermix (NEB). 4 of 192 surviving clones contained deletion bands. The cA5 clone was used for live cell imaging, and designated as clone TFF1-MS2-D12.
smFISH and DNA FISH:
Probesets were designed with “Oligo” software (Ta ble S3) and ordered from Biosearch Stellaris using Quasar 570 and 670 dyes. TFF1 (16 probes) and CTNNBL1 (48 probes) mRNA probe sets were designed to the exons common between all Refseq isoforms. TFF1 intron probesets (79 probes) were designed using the genomic sequence of the first intron. Puromycin screening probesets (22 probes) were designed using the donor construct plasmid sequence. MS2 probsets (2 probes) were designed from the linker regions between the repeats. CA12, SERPINA3, CTSD, AGR2 smFISH probesets were designed using Stellaris Probe Designer (https://www.biosearchtech.com/stellaris-designer ) with the following parameters: masking level at 5, oligo length 20, minimum spacing of 2 nucleotides. 48, 35, 27, and 27 probes were ordered from Biosearch Stellaris in Quasar 670 for CA12, SERPINA3, CTSD, AGR2 respectively. smFISH was performed according to manufacturer’s protocol with minor modifications. Cells grown on 18mm No. 1.5 coverslips in 12 well plates were washed 3 times with HBSS before fixation with 4% PFA in PBS. Fixed samples were washed with 1X PBS, and stored in 70% ethanol at 4C overnight. Hybridized samples were mounted in Prolong Gold with DAPI and allowed to dry overnight.
DNA FISH was done as previously described(Meaburn and Misteli, 2008). Briefly, FISH probes to the TFF1 promoter and the downstream region were generated through nick translation of BAC (CTD-2652C12 and CTD-3095D11, respectively) in the presence of fluorescently labelled UTP (Dy490 and Dy 540 obtained from Dynomics). The probes were ethanol precipitated and resuspended in hybridization buffer (10% [wt/vol] dextran sulfate [Sigma-Aldrich], 50% [vol/vol] formamide [Sigma-Aldrich], 2× SSC, and 1% [wt/vol] Tween 20 [Sigma-Aldrich]) in the presence of 3ug of human COT-1 DNA and 20ug). Cells grown in 384 well plate were fixed with 4%PFA and treated in the following order for FISH: washed with PBS (3X), permeabilized with 5% triton/0.5% Saponin / PBS for 20 min, washed again with PBS(3X), treated with 0.1N HCl for 15min, washed with 2X SSC and incubated with 50% Formamide/ 2XSSC for at least 30 min. Cells were then incubated with the FISH probes at 85°C for 7.5 min and moved to a 37°C water bath overnight. The probed were removed and the cells washed in the following order: 3X 5 min 1XSSC at 42°C 3X 5 min 0.1XSSC at 42°C. The n uclei were stained with DAPI.
Induction and dose response:
Cells for smFISH were plated on glass coverslips and allowed to recover for 2 days. For induction and dose response experiments, cells were subsequently hormone depleted by washing cells twice in Phenol free media supplemented with 10% Charcoal/Dextran Treated FBS (Atlanta Biologicals), 2mM Glutamine and 1X Penstrep (hormone depleted media). Cells were returned to incubator and an hour later media was replaced. This step was performed again. After 4 days, media was replaced with hormone depleted media supplemented with the specific concentration of E2. Cells were allowed to reach steady state and fixed with 4% PFA three days later. MD9571 treated cells for smFISH experiments were treated for 2 days in complete media before fixation.
Cells for live cell imaging were plated onto Labtek Chambered #1.0 dishes (155380). For MEM (saturating E2 conditions), cells were allowed to recover for 2 days. Media was then replaced with Phenol free MEM media supplemented with 10% FBS, 2mM Glutamine and 1X Penstrep. For dose response experiments, cells were washed twice with hormone depleted media, and returned to the incubator. After 4 days, media was replaced with hormone depleted media and supplemented with specific E2 concentration. Media was replaced 3 days later. Imaging started the next day and continued for the following two days. MD9571 treated cells for live cell imaging were immediately imaged for 14 hours upon addition of MD9571.
Microscopy:
Imaging of FISH was performed on two custom built microscopes. Dose response smFISH data was acquired on a custom-built microscope. The microscope consisted of a Zeiss AxioObserver, Zeiss 63x C-Apochromat objective, Hamamatsu ORCA-R2 C10600 camera, and Zeiss Colibri with LEDs for 365nm, 530nM, 630nM for DAPI, Quasar 570 and Quasar 670 excitation respectively. Emission filters specific to these spectra were also used. The microscope was controlled by Micro-Manager(Edelstein et al., 2010). Several fields were imaged with Z-stacks spanning 7 microns at 0.5 micron intervals. The maximum intensity projections were performed and used for analysis.
The image acquisition for the number of active alleles per cell (Fig. S1) and the TFF1-MS2 induction were performed on a custom-built microscope. This microscope consisted of an ASI (www.asiimaging.com) Rapid Automated Modular Microscope System (RAMM) base, Hamamatsu ORCA-Flash4 V2 CMOS camera (www.hamamatsu.com, C11440), Lumencore SpectraX (http://lumencor.com/), ASI High Speed Filter Wheel (FW-1000), ASI MS-2000 Small XY stage. Excitation of DAPI, Quasar 570, and 670 was performed using SpectraX violet, red and green respectively. Emission filters specific to these spectra were used. A quad bandpass filter was used for the TFF1-MS2 induction. The filter wheel with specific channel bandpass filters was used for the TFF1 allele number data acquisition. Image acquisition was performed through Micro-Manager. We obtained multiple z-stacks at 0.5 micron intervals, spanning 10 microns. The maximum intensity projections were performed and used for analysis. The number of cells used per sample used for the analysis ranged from 700–3400 cells. The smFISH dose response data was replicated twice in the laboratory on two separate days. The control data for the number of active TFF1 alleles and the MS2 induction comes from 1 experiment each. MD9571 experiment imaging spanned 7 microns.
Live cell imaging was performed on a Zeiss LSM780 laser scanning confocal microscope using 37C incubation and 5% C02. Imaging was performed using 488 nm excitation, pinhole size of 10, zoom of 0.8 at 1024×1024, pixel size of 0.16 microns, and power of 1 for all data except the enhancer deletion which required power of 1.5. 5 field of views were taken at a time. Z-stacks were acquired spanning 7.5 microns at 0.5micron intervals (16 Z planes). Imaging of Z-stacks occurred every 100 seconds for 512 frames (14.2hrs total). The maximum intensity projections were performed and used for analysis. The number of alleles used for the analysis is as follows: 100 for saturating E2, 33 for 0.5nM, 17 for 0.05nM, and 28 for the deletion mutant at saturating E2. The live cell data for the MEM and enhancer deletion data come from 10 fields of view on 2 different days. The dose response data were each acquired on 1 day each.
Confocal imaging of the diffusing RNA was performed with the same microscope using pixel size of 0.04 microns, pinhole size of 1 and 6% power. One z plane was imaged every 200ms for 256 frames. The movie was bleach corrected using the FIJI and the bleach correction plugin with exponential fit correction.
Three-enzyme Hi-C (3e Hi-C):
Cells were grown in 10cm dishes and treated as above in the dose response smFISH except cells were fixed as previously described for 3e HiC(Ren et al., 2017). Cells were cross-linked with 1% formaldehyde for 10 minutes at 25°C. 2–5 X 106 cells were lysed in 10 ml lysis buffer (10 mM Tris-HCl pH8.0, 10 mM NaCl, 0.2% NP40; 10 µl protease inhibitors (Sigma)) with rotation at 4°C f or 60 minutes. The cells were then treated with 400 µl 1X NEB cutsmart buffer with 0.1% SDS at 65°C for 10 minutes, followed by addition of 44 µl 10% Triton X-100 to quench SDS. Chromatin was subsequently digested with 20 Units CviQ I (NEB), and 20 Units CviA II (NEB) at 25°C fo r 20 minutes, then with 20 Units Bfa I (NEB) at 37°C for 20 minutes. The reaction was stop ped by washing the cells twice with 600 ml wash buffer (10mM NaCl, 1mM EDTA, 0.1% triton-100). The DNA ends were blunted and labeled with biotin by Klenow enzyme in the presence of dCTP, dGTP, dTTP, biotin-14-dATP, followed by ligation using T4 DNA ligase. After reverse crosslinking, the samples were treated with T4 DNA polymerase to remove biotin labels at the DNA ends.
DNA was fragmented to 300–500 bp by sonication with a Bioruptor sonicator (Diagenode UCD-200). Next, The DNA was end-repaired, followed by A-addition as described previously (Lieberman-Aiden et al., 2009). The remaining biotinylated DNA fragments were then captured using Dynabeads MyOne Streptavin C1 Beads (Invitrogen) by incubating for 30 minutes at 25°C with rotation. The DNA on beads was ligated to the Illumina Paired End Adaptors. Following PCR-amplification of the libraries, DNA fragments from 300 to 700 bp were purified from 2% E-gel and sequenced on Hi-Seq 3000.
Two biological replicates were sequenced on a HiSeq 3000, and ~62 million reads/sample were mapped by bowtie 2–2.3.4 using “bowtie2 -p 10 −3 0 –local -x genome fastqfile”. Data was further processed by HOMER 4.8.2 in order to remove small fragment self-ligations and to generate raw-unnormalized and normalized matrices(-norm HOMER option). The HOMER programs and parameters used were the following: “m akeTagDirectory unfiltered_sample samfile_read1,samfile_read2 -tbp 1”,”makeTagDirecto ry filtered_sample -update -genome hg19 -removePEbg -removeSpikes 10000 5”,”analyzeHiC filt ered_sample -res resolution -raw -cpu 16 -pos chr:s-e” Where resolution was 100000 or 1000, and chr=chromosome, s=start, e=end”. Raw count matrices were exported into text files using HOMER and used for the entropy analysis. In addition, 2887 regions were reported as showing increased contacts and 5023 were reported as showing decreased contacts between Sat. E2 and 0nM using interactions found in both replicates with Z-scores of at least 3 and HOMER 4.10. Although there were fewer interactions identified with 4.8.2, the raw count matrices extracted with both HOMER versions on the HOMER 4.8.2 filtered data yielded the same entropy values.
Figures:
Figures were generated in FIJI, R (https://www.r-project.org/), matplotlib (http://matplotlib.org/) and Adobe Illustrator. Figure 2 genome browser view was generated using the genome.ucsc.edu PDF tool and subsequently modified in Adobe Illustrator. Visualization of chromosome contact matrices was performed with HiCPlotter(Akdemir and Chin, 2015) using the probability distributions. We also plotted the HOMER normalized output (-norm option) of a select region on chromosome 21 using Treeview.
QUANTIFICATION AND STATISTICAL ANALYSIS
Shannon Entropy and Mutual Information:
To compute Shannon entropy and mutual information for the 3e Hi-C data, we divided each chromosome into 100kb segments and counted the number of contacts in each segment-segment bin to obtain a contact probability between each segment. The Shannon entropy of a contact given dose d is given by
| (1.1) |
where pi(d) is the probability of a contact “bin” at a given d ose d and the sum is over all bins. The log is always base 2 so the result is in bits. Entropy was also calculated for a list of previously identified ER enhancer locations which span 2kb(Hah et al., 2013). A 1kb bin matrix around each 2kb enhancer was output using HOMER yielding a multi-bin x 2 column matrix. These 2 columns were consolidated by summing the raw counts. The self-bins containing the enhancer were not considered. The mutual information is defined as the difference between the total entropy regardless of dose and conditional entropy (i.e. the average of the dose entropies conditioned on dose):
| (1.2) |
where p(d) is the probability of dose d, which we took to be 1/3.
For the inter-burst mutual information, we computed the OFF-time probability density in closed form from the model (see section on General Telegraph Model) for each dose and computed the corresponding Shannon entropy via for each condition.
Variable gene analysis:
MCF7 single cell data was obtained from GEO accession GSE52716(Rothwell et al., 2014). Genes with a minimum of 3RPKM average over all cells and with a mappable MCF7 RNA half-life from (Schueler et al., 2014) were considered as the expressed background set. From this list, genes with half-lives greater than 1000 minutes (>16hrs, top quarter) were subsequently considered for potential variable candidates. The data was sorted by descending coefficient of variation and the top quintile was used for DAVID GO analysis (https://david.ncifcrf.gov/). Candidates for smFISH were selected by sorting by the Fano factor, and selecting four from the top 20.
Small intestine single cell data from the large cell sort of (Haber et al., 2017) was obtained from GEO accession GSE92332. Data for cells identified as Stem, Goblet, and Enteroendocrine was analyzed as described (Haber et al., 2017) using publicly available R code (https://portals.broadinstitute.org/single_cell/study/small-intestinal-epithelium). Batch effects were analyzed with the same R code and correction was determined unnecessary. Genes with FDR q-values less than 0.05 were used for DAVID GO analysis.
Image analysis:
FISH and live cell analysis was performed with custom IDL software previously described(Coulon et al., 2014). Briefly, FISH spots and live cell transcription sites were identified with the software localize. FISH spots were identified by using the manual bandpass threshold. Cells were segmented into nucleus and cell which includes the cell. These masks were generated using Cellprofiler(Carpenter et al., 2006) (Broad Institute, http://www.cellprofiler.org) and used as input into a modified version of the software FISHAuxiliary for classification of cell spots. This software also outputs the coordinates of the nuclear spots. Colocalization of transcription sites to determine the % of TFF1 intron spots that contained MS2 spots was performed by using these coordinates. Spots with a Euclidean distances less than or equal to 5 pixels were considered localized. All spot finding software is available at www.larsonlab.net.
Sigmoidal dose response analysis was performed with Graphpad Prism 7.01 using the Variable slope, four parameter logistic equation. For the 0 nM concentration, the log value was set to −1. For the live cell transcription site tracking we also used Localize. However, since cells can move considerable during the 14-hour period, we first tracked cells by hand using ImageJ macros to extract mouse coordinates, and then cropped around the cell using these coordinates using custom FIJI macros. These cropped stacks were then used by Localize. Live cell transcription site identification was performed using the option to have the previous frame coordinate used if no spot was found in the current frame. Live cell traces were then generated using the track function in localize. Since multiple alleles could be transcribing in the same cell at different times, traces were manually inspected.
Tissue sections are hematoxylin and eosin plus protein-specific antibody immunohistochemical staining and are publicly available at www.proteinatlas.org. Segmentation and quantification was accomplished through the following steps using IDL. Principal component analysis separates the color channels to enrich for a single channel containing the immunohistochemical stain. The channel containing the greatest enrichment for the antibody stain was used for subsequent analysis. Image thresholding was carried out with a maximum entropy segmentation, followed by morphological closing with a 4×4 kernel. The only user-implemented fitting parameter is the minimum area required of segmented image to be considered a cell. This value varies depending on the magnification of the tissue section. Unique cells were then labeled and identified. The staining intensity of the cell was determined as the sum in the segmented region of the principal component channel.
Other analysis:
Burst timing correlation is computed using standard cross-covariance analysis(Coulon et al., 2014; Coulon and Larson, 2016; Ferguson and Larson, 2013). The theoretical curve for coefficient of variation for single-allele traces (Fig. 5) is computed through Monte Carlo simulations. Each trace can be considered to have a single rate which is measured as number of bursts in 14 hours. This pool of rates can then be sampled to compute in silico trajectories. The CV is computed for each in silico trajectory and then binned into a histogram. For detailed description of analyses see below.
Kolmogorov-Smirnov, correlation significance and reduced major axis linear regression were performed using ks, cor.test, lmodel2, R packages.
Time series analysis
Hidden Markov Model analysis:
The integrated fluorescence of a diffraction-limited transcription site (TS) reflects the number of nascent RNA present at the gene. The time-dependent intensity of the spot – referred to as a ‘trajectory’ – is determined through a Gaussian Mas k fitting routine described in the methods and characterized previously(Thompson et al., 2002). Briefly, the algorithm uses an iterative fitting procedure which is based on a Gaussian distribution to approximate the point-spread function of the microscope. Local background subtraction is performed for each spot in each frame of the time-series, but no other manipulations are carried out to generate the raw data trace (Fig. 1G). For example, the trajectories are not corrected for photobleaching.
These trajectories are then analyzed by a Hidden Markov Model (HMM) algorithm for finding the transcriptional bursts. First, the data is normalized to a peak height of unity. Second, the normalized data are used as input for the IDL program ‘HMM_V5_4_2_text.sav’ described in (Lee, 2009) and available at http://research.chem.psu.edu/txlgroup/HMM/HMM_code.html. The output of this program is a binary trace representing the times when the gene is active and inactive.
Finally, the duration of the inactive and active times are compiled from many trajectories to generate a histogram of OFF and ON times, either as a probability distribution function (Fig. 1H,I) or a cumulative distribution function (Fig. 2D,E). We emphasize that the HMM fitting routine is only used as an automated routine to find bursts and therefore functions as a segmentation routine. Subsequent analyses are carried out on the empirically measured active and inactive times from the segmented trajectory, not on any rate parameters returned from the HMM algorithm directly.
Master trace analysis:
The dynamics evident in a single-molecule fluorescence trajectory are the result of processes relating to regulation (i.e. when a gene is active) and also the molecular mechanisms of elongation, splicing, cleavage, and release(Coulon et al., 2014). To isolate the latter from the former, we artificially ‘removed’ the inactive peri ods to generate a fluorescence master trajectory which only reflects fluorescence signal during active transcription (Fig. S1D,E,F). First, the active periods are identified through the HMM algorithm. Second, the raw trajectory data from the TS during the active periods is concatenated to generate a single fluorescence trajectory (Fig. S1E,F). Although fluctuations are easily visible, the mean of the trace, generated from hundreds of alleles taken from different cell passages and imaged on different days, is strikingly consistent (Fig. S1F).
We then chose a region between frame 1200 and 4200 for autocorrelation analysis, comprising concatenated data from ~ 300 bursts from ~ 50 individual allele traces (Fig. S1F,G). The autocorrelation is generated using a multi-tau algorithm and fit with a model for stem loops in the 3’ UTR, as described previously in depth(Larson et al., 2011). The equation, derived in the supplemental material of ref (Larson et al., 2011), for a 3’ stem loop cassette is:
| (2.1) |
where k is the rate of transition from one stem-loop to the next; χ is the initiation rate for transcription; S is the number of stem loops (24 for the TFF1-MS2 construct); and τ is the autocorrelation delay. From this analysis, we compute a dwell time of a TFF1-MS2 nascent transcript of 13.0 +/− 0.8 min. This dwell time includes the elongation time for the MS2 cassette (~1.4 kb) and any 3’ processing steps such as cleavage and release during which the nascent transcript is still visible at the TS. In addition, the autocorrelation analysis also gives an effective initiation rate of 0.50 +/− 0.02 min−1, the inverse of which is the time between consecutive transcripts. Bursts have an average duration of 16 min from the HMM analysis. So polymerases are capable of initiating over a 3 min window. The experimentally determined burst size is 3 min*0.5 min−1 = 1.5.
We note that these numbers – dwell time and effecti ve initiation rate – are determined here through the autocorrelation analysis of the data using the 3’ UTR stem loop calculation, which is independent from the approach described in the modeling section (part 3, below). This latter approach is based on a global analysis of ON / OFF time histograms and total mRNA at multiple doses and for a deletion mutant. The rates from the autocorrelation analysis are not used to constrain parameters in the global fit. Nevertheless, there is close agreement between values returned from the global fit and the autocorrelation analysis described here, supporting the independent assessments of nascent RNA dynamics independent of gene active / inactive dynamics.
Allele Correlation:
In general, correlations between alleles can be carried out in multiple ways and over many timescales. The cell line has three alleles labeled, but all analysis is carried out as pairwise correlations, and no ternary correlations are considered. Correlations can be computed between alleles in the same nucleus or alleles from different nuclei. Correlations can be computed both from raw trajectories and HMM traces. Correlations can be static measurements which consider only the total output of one allele over a fixed time period compared to the total output of another allele over that same time period. Conversely, correlations can be computed between bursts from different alleles using the full time-dependent trajectories. We have used multiple approaches to study inter-allele correlations, described individually in this section.
Total Output Correlation:
Consider a cell which has multiple bursting alleles visualized over a period T. The total output of that allele is the integrated area under the fluorescence trajectory after background subtraction. This quantity is computed simply by subtracting the median fluorescence intensity from the trajectory (the median is an excellent approximation of the background for sparse bursts) and summing the resulting trace over the observation period T. Thus, each allele is reduced to a single number, which is the integrated output. For a cell where three alleles are active, three points are generated for the scatter plot: allele 1 vs. allele 2, allele 2 vs. allele 3, and allele 1 vs. allele 3.
Likewise, this operation can be carried out on the HMM trace instead of the raw trace. Instead of the integrated area under the HMM trace, we use the number of bursts (Fig. 4). This simplification is motivated by the empirical observation that burst duration is narrowly distributed for the TFF1 gene. However, it is trivial to use the area under the HMM curve as well, and the resulting scatter plot looks very similar (Fig. S3A,B). For each allele, one determines the number of bursts N during the observation period T, and for a three allele time series, three points are generated in the scatter plot, as described above.
This correlation is the equivalent of a ‘rate correlation.’ The rate for each allele is N/T, but since the period of observation is the same for both alleles, we simplify the analysis to the number of bursts for reasons which will become clear later. Importantly, rates of transcription can be highly correlated between alleles without any correlations between bursts. For example, if estrogen receptor (ERα) is the rate determining factor in the nucleus, one expects that alleles might transcribe at similar rates, even though there need be no relationship between the actual times when transcription is observed at each allele, which might be largely random.
Random Controls:
We use multiple random controls to account for possible artifacts in the measurement and cross-correlation analysis. First, we look at correlations between alleles imaged on different days. Second, we look at correlations between alleles in different fields in the same dish and imaged in the same experiment. Third, we consider correlations between alleles in cells located in the same field. None of our analysis has revealed any statistically significant cross-correlations for the above scenarios (see “Experimental Cross-correlatio ns” section below). Thus, the completely general random control simply consists of correlations between alleles in different cells, regardless of the field in which the cell is located or the day on which the data were acquired.
In addition, we also use simulated transcription data as a control for the time-resolved auto- and cross-correlations. This simulated transcription data consists of square pulses resembling the output of the Hidden Markov algorithm. The simulated HMM data consist of pulses or bursts which are identical in width (16 minutes, in agreement with the TFF1-MS2 burst duration) and height. The spacing of the bursts is determined by drawing from a uniform distribution of random numbers.
Regression analysis:
The typical regression method, known as ordinary least squares regression, is sufficient if the x-values are user-controlled parameters known with relatively high precision (i.e., dose, time, etc.). However, if the x-variable is also a random variable with errors which are comparable to the y-variable (i.e. bursts of allele one vs. bursts of allele two), regression of one random variable on to another random variable will result in systematic deviations, a phenomenon known as “regression dilution”(Sokal and Rohlf, 2012). This problem is well known, and a number of alternative regression approaches have been developed. Here, we use reduced major-axis regression, which is an example of Model II regression and is appropriate for data drawn from a bivariate normal distribution(Sokal and Rohlf, 2012).
Cross-Correlation Analysis:
We emphasize that fluctuation analysis occurs in two steps: 1) generation of the correlation function from a fluorescence trajectory and 2) interpretation of the correlation function to infer biological parameters. The former can be carried out in a rigorous mathematical way and essentially amounts to data processing without assumptions. We follow the multi-tau approach which has been developed in the fluorescence correlation spectroscopy literature over several decades (see reference (Wohland et al., 2001) for an example). The latter step (interpretation) depends on the unique biological problem of interest. In this section, we focus largely on the information that is contained in a transcription cross-correlation. A thorough treatment of transcription correlation in general can be found in reference(Coulon and Larson, 2016).
Generating the cross-correlation:
As with the autocorrelation, the cross-correlation is computed using a multi-tau correlation algorithm described earlier(Coulon and Larson, 2016). In these experiments, the alleles are all labeled with identical MS2 stem loops. Alleles can be distinguished within a time series, because the spatial separation and time-resolution of the imaging are sufficient to uniquely identify individual TS. However, although the alleles can be distinguished, it is impossible to establish the unique allelic identity of the TS in the microscope. Thus, there is no natural ‘order’ to a cross-correlation as there would be if the alleles had different colors. Therefore, the cross-correlation is computed between two alleles in both orientations (allele 1 →allele 2; allele 2 →allele 1) and averaged together to produce a symmetric correlation (Fig. S3C-G).
In addition, the background fluorescence, which essentially amounts to the cumulative time that a TS is not visible, strongly effects the interpretation of the cross-correlation, as shown below. In fact, because transcription is rare, background levels make a significant contribution to the correlation. Therefore, correlations functions can be background corrected to obtain certain quantities (such as the fraction of bursts which co-occur from two alleles). The final step in generating the cross-correlation is background correction, described in detail below.
If multiple correlation functions from different alleles are averaged together, which is almost always the case, there are several different methods of averaging to consider. This question is treated in detail in Coulon et al, section 2.3(Coulon and Larson, 2016). The first, and most common, is to simply generate the arithmetic average of the individual correlations. However, this averaging method ignores the fact that some correlations reflect dozens of events, while other may only reflect a few events. Because the correlation function is normalized by mean fluorescence, the normalization may over- or under-weight certain correlation functions in the arithmetic average. Thus, the underlying details of the fluorescence trajectory used to generate the correlation function can introduce an implicit bias. The solution is to use a global mean of all the traces to average the correlation function. In practice, these two methods –local vs. global averaging – diverge when individual trajectories ar e either of vastly different length or contain vastly different transcriptional activity levels. The global averaging method (Eqn. 7 in reference (Coulon and Larson, 2016)) is the preferred method for transcription correlations and is used exclusively in this paper. The error bars are determined from bootstrap estimation(Day et al., 2015).
Interpreting the cross-correlation:
The auto/cross-correlation is defined in the following way:
| (2.2) |
where τ is the autocorrelation delay; Fi(t) is the fluorescence at time t of allele I, and angular brackets denote the average over time; δFi(t) = Fi (t)-<Fi (t)> is the instantaneous fluctuation in fluorescence, defined as the deviation from the average value. Taking the value at zero delay (the y-intercept of the correlation function, also called the “amplitude”) the autocorrelation for a single allele (i=1) becomes:
| (2.3) |
which corresponds to the variance of the fluorescence signal divided by the mean.
In the examples that follow, we consider only this G(0) value for a number of different transcription scenarios. We use a geometric argument for simplicity and to convey the interpretation in terms of the raw trajectories. We assume certain defined features of a transcriptional burst and show later how well these assumptions agree with the actual data.
Single-allele autocorrelation
Consider transcription as occurring in square pulses (or bursts) of equal duration (Δ) and height I over a measurement duration T (Fig. S4A). The relevant values for G(0) can be derived by computing the variance and mean of such a signal using the definition:
| (2.4) |
For square pulses,
| (2.5) |
And the variance becomes:
| (2.6) |
Resulting in:
| (2.7) |
Experimental parameters for TFF1 are T = 854 min, Δ= 16 min, N = 6, resulting in a calculated G(0) of 7.9. Note that this quantity does not depend on the height of the pulse c.
If background is included, which amounts to a non-zero offset to the trajectory, the mean and mean squared fluorescence are:
| (2.8) |
but the variance is unchanged (Eqn. 2.6). Because the variance is unchanged and the mean fluorescence is changed by background, the resulting G(0), which is the ratio of the two quantities becomes:
| (2.9) |
which is the actual G(0) (Eqn. 2.7) reduced by a scaling factor. Thus, background always artificially lowers G(0) beneath the true value. This correction factor takes the familiar form of the well-known correction factor from FCS(Schwille et al., 1999):
| (2.10) |
The sparse nature of transcriptional bursts can make this background correction substantial. Taking the same parameters as above but adding a constant b = 0.1 offset to bursts with a height of c = 1 (i.e. ~10% background) gives a correction factor of 3.6 and artificially lowers the G(0) from 7.9 to 2.2.
Double-allele cross-correlation
In cross-correlation, the variance σ2 becomes a co-variance (σ1,2). Consider then the static co-variance between two alleles (1,2) for a fixed number of bursts N1, N2 in time T. First, consider the case of complete overlap between the bursts of the two alleles (Fig. S4B). In this case, it is clear that G(0) for the cross-correlation is identical to the G(0) for the autocorrelation. This concordance is predicated on the assumption that the alleles are labeled with the same number of stem loops which bind the same number of fluorophores.
Second, consider the case of complete exclusion: the bursts never overlap in time (Fig. S4C).
The co-variance can be written as:
| (2.11) |
Since the first term is 0, the co-variance is the product of the means, and G(0) becomes:
| (2.12) |
Thus, the minimum cross-correlation is −1, independent of the details of the underlying dynamics. However, the peak cross-correlation does depend on dynamics and in the case of perfect autocorrelation where each and every pulse from one allele is accompanied by a pulse from the other allele, takes the value of Eqn. 2.7. Therefore, the limits of the cross-correlation function are asymmetric:
| (2.13) |
If we now include background in the calculation, using the same process to derive Eqn. 2.9, the lower limit of the experimental cross-correlation becomes:
| (2.14) |
Again, a non-zero background always lowers the measured value of G(0) from the true value.
Now consider the occasional overlap scenario, where some pulses from allele 1 show complete overlap with pulses from allele 2 (Fig. S4D):
| (2.15) |
where M is the absolute number of overlap bursts. Without loss of generality since the overlap is linear in M and Δ, one can also consider partial overlap in bursts (Fig. S4E). It is therefore straightforward to show:
| (2.16) |
which leads to:
| (2.17) |
Note that if M=N1=N2, the results is identical to the autocorrelation amplitude defined in Eqn.2.7.
With the expressions for G(0) for both the auto- and cross-correlation in hand, one is now in the position to compute normalized quantities based on the ratio of auto- and cross-correlation:
| (2.18) |
This quantity (M/N2) is the fraction of bursts from allele 2 which co-occur with bursts from allele Note one particular nuance of this result: one divides by the auto-correlation from allele 1 to get the fractional co-occurring bursts of allele 2. Moreover, it is critical to note that because of the constant unit-less offset in both the numerator and denominator, background effects do not normalize out in the ratio. In other words, it is imperative to do the correct background correction before computing the normalized burst fraction.
The full time-dependent behavior of experimental auto- and cross-correlations is shown in Fig. S4G. Eqn. 2.18 could be applied to the zero delay time point in these curves to compute the fraction of bursts which show temporal overlap.
Since we have computed the limits of cross-correlation (Eqn. 2.13) we now calculate random expectations of this metric for two unrelated allele signals. Consider the duty cycle of RNA signal for alleles 1 and 2, which amounts to the probability of seeing RNA signal, P(1), P(2). The random expectation joint probability is the overlap of these two probabilities (P(1|2) = P(1)P(2)):
| (2.19) |
This overlap by random chance can be discretized by Δ and multiplied by the observation duration T to get the absolute number of events, which we defined above as M:
| (2.20) |
Plugging this quantity for M into the cross-correlation amplitude (Eqn. 2.17) gives G1,2(0)=0, which is the expected result.
This same value for M could also be arrived at by setting Eqn. 2.17 to zero and solving for M, yielding a random expectation:
| (2.21) |
Using the experimental averages for TFF1 (N1=N2=6, Δ=16 min, T=854 min) gives an average number of overlap bursts of 0.67, which, when normalized by the average number of bursts from each allele, gives a fractional overlap of ~ 11%. Therefore, for TFF1 the random expectation for a burst of one allele to overlap with another allele is approximately 1 in 10.
Importantly, the fractional overlap depends linearly on the number of bursts from one allele, which is why it is ultimately a flawed estimator for population behavior (Fig. S4F). In other words, for highly active cells (many bursts over the experimental period), one might expect a fraction of co-occurrence for TFF1 alleles nearing 30% from random behavior. In the limit where one allele is always on, the fractional overlap is of course 100%. However, for cells which are minimally active, this random fractional overlap approaches 0%. Two conclusions can be drawn: 1) it is critical to measure the coupling between alleles which arises from extrinsic factors; and 2) interpretation of the co-occurrence of bursts can only be done if the activity of the allele is measured over time in a live-cell assay. In summary, because the fractional overlap random expectation depends on the activity of the alleles, this number cannot be estimated by averaging cells of different activity together.
However, the utility of G1,2(t) is that this activity dependence is removed. Essentially, the number of overlap events are effectively normalized by both N1 and N2. Thus, the full cross-correlation (and hence G1,2(0)) can in fact be averaged between cells in a robust way, but fractional overlap can only be measured on a cell by cell basis. Thus, these measures are related but give slightly different quantities. The benefit of the full cross-correlation is that different cells can be averaged together, but the drawback is that the correlation quantity G1,2(0) is asymmetric and doesn’t have an intuitive interpretation. Whereas for the fractional overlap, the interpretation is very simple but doesn’t allow for averaging across cells.
Because of these advantages which are intrinsic to the full cross-correlation, we can determine a time-integrated measure of the extent to which alleles are coupled:
| (2.22) |
This quantity is a measure of the extent to which alleles show correlated bursts normalized to the random expecation, summed over the decay time scale of the cross correlation. Graphically, it corresponds to the area between the intra-cellular cross correlation (i.e. Fig. 4e, blue curve) and the random control (Fig. 4e, black curve). For TFF1 the quantity computed from the experimental data is 27 ± 3%, which means that due to the coupled intrinsic noise phenomenon, one is ~ 30% more likely to see a burst from allele 2 after a burst from allele 1 within a 2 hour window than would be expected by random chance.
Experimental Cross-correlations:
Cross-correlations were computed from a dataset consisting of 100 allele traces. Of these traces, 73 were from cells which showed transcription from more than one allele, therefore allowing cross-correlation. Cross-correlation was always carried out over the entire time trace (T=854 min).
The cross-correlation amplitude (G1,2(0)) is shown as a function of distance between the two alleles (Fig. S5A,B). As described above, this value is asymmetric with a lower limit of −1 and an upper limit depending on the activity of the alleles (Eqn. 13). The random expectation is zero, independent of allele activity. Ordinary least square regression reveals there is a positive slope to the data (0.27), and the x-intercept is 5.4 μm indicating that alleles which are close together show a bias toward anti-correlation, whereas alleles which are far apart show a bias toward positive correlation. In contrast, the cross-correlation between random alleles shows no distance dependence and is distributed around zero (Fig. S5B). Note that the coordinate system is always based on position within the nucleus, so the RMS distance is arbitrary in the random case. The main point to take away is that the values are symmetrically distributed around 0. This random control is carried out using the same number of pairs as the intra-cell correlations, and we also confirmed by looking at a greater number of random pairs (Fig. S5C).
The distance between alleles is a dynamic parameter which changes over the course of the measurement. In all of our analysis, we reduce this variable to a single number which is the root mean square separation over the entire time course:
| (2.23) |
here (xji, yji) is the spatial position of the jth allele in the ith frame, and here T is the absolute number of frames. An example is shown in Fig. S5D. The changes in this distance over time (Fig. S5E,F) result in variability in the x-axis of the scatter plots (Fig. S5A).
In addition, we computed the fractional overlap bursts (Eqn. 2.18) for each cell as well (Fig. S5G,H). Note that this fractional overlap can be calculated in two ways: the fraction of bursts from allele 1 which overlap with allele 2 and the fraction of bursts from allele 2 which overlap with allele 1, and these measures are only equivalent if the integrated activity from each allele is identical. Again, a distance dependence is observed which is not observed in the random control (Fig. S5I). The fractional overlap should be > 0, so negative values arise from experimental noise. The random expectation determined across the correlations from the entire data set (Fig. S4G) is 9% as computed from Eqn. 2.18, but this random expectation can be different from cell to cell, as described above. However, the best fit line shows an offset of 10% in Fig. S5I, in excellent agreement with the theoretical prediction.
Generalized Telegraph Model
The LC and smFISH data were fit to a fully stochastic generalized telegraph model consisting of arbitrary numbers of gene states and pre-RNA steps. Transitions between gene states were reversible. Each pre-RNA step can be occupied by zero or one molecule. One of the gene states was deemed active; when occupied there was a probability that the first pre-RNA step could become occupied if it is not already occupied. Once the first pre-RNA step is occupied there is a probability of the molecule transitioning to the next pre-RNA step. The alleles were coupled via a mechanism in which the rate from the pre-active gene state to the active gene state for all alleles is increased by a given value if any allele is in the active gene state. The dwell time distributions (on and off LC times) and steady state mRNA distributions (smFISH) were computed directly using direct simulation of the model and also in closed form by solving the chemical Master equation. The free parameters fit were the number of gene states and pre-RNA steps, the rates between all states and steps, and the dose effect of E2, which was assumed to influence one of the rate parameters in the model. The best model was chosen by Bayesian model comparison (BIC and AIC). In depth details of the model are available in the Methods S1.
smFISH Cell Counts:
Cell counts are provided in the Table S4.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Estradiol | Sigma | E8875 |
| TRIM24 inhibitor MD9571 | (Palmer et al., 2016) | MD9571 |
| Deposited Data | ||
| 3e Hi-C | this paper | GSE121443 |
| MCF7 scRNA-seq | GEO | GSE52716 |
| Small Intestine scRNA-seq | GEO | GSE92332 |
| MCF7 Half-lives | GEO | GSE49831 |
| TFF1, small intestine | (Uhlen et al., 2015) | www.proteinatlas.org/images/2170/5769_A_6_2.jpg |
| TFF1, duodenum | (Uhlen et al., 2015) | www.proteinatlas.org/images/2170/5769_A_9_2.jpg |
| TFF1, Breast cancer |
(Uhlen et al., 2015) | www.proteinatlas.org/images/2170/5766_A_5_5.jpg |
| Experimental Models: Cell Lines | ||
| MCF7 | ATCC | RRID: CVCL_0031 |
| TFF1-MS2 | this paper | N/A |
| TFF1-MS2-D12 enhancer deletion | this paper | N/A |
| Oligonucleotides | ||
| PCR screening, see Table S3 | IDT | N/A |
| smFISH probes, see Table S3 | Biosearch Stellaris | N/A |
| Recombinant DNA | ||
| pX330-U6-Chimeric_BB-CBh-hSpCas9 | (Cong et al., 2013) | addgene:42230 |
| M04–24XMS2 Donor plasmid | this paper | M04–24XMS2 |
| Software and Algorithms | ||
| Cellprofiler | (Carpenter et al., 2006) | cellprofiler.org |
| FIJI | (Schindelin et al., 2012) | imagej.net/Fiji |
| Localize | (Larson et al., 2013) | www.larsonlab.net |
| FishAuxiliary | (Larson et al., 2013) | www.larsonlab.net |
| R | R Core Team. 2017 | www.r-project.org |
| small intestine scRNA-seq R code | (Haber et al., 2017) | github.com/adamh-broad/single_cell_intestine |
| DAVID | (Huang da et al., 2009) | david.ncifcrf.gov |
| Bowtie 2–2.3.4 | ( Langmead and Salzberg, 2012) |
bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| HOMER 4.8.2 | (Heinz et al., 2010) | homer.ucsd.edu/homer/interactions |
| Generalized Telegraph Model | this paper | github.com/ccc1685/transcription-model |
HIGHLIGHTS.
Live-cell RNA imaging reveals long repressive states cause expression variability.
Highly variable expression occurs in functional gene ontology categories.
Alleles are not independent.
Information theory explains how estrogen is sensed by the cell.
Time-dependent dynamics of promoter-enhancer associations contribute to transcriptional noise and shape responses to ligand-dependent gene induction.
Acknowledgements
We thank Varun Sood for the DNA FISH images and Michelle Barton for the TRIM24 inhibitor MD9571. We thank Anusha Nagari and Lee Kraus for the list of ER enhancers. PX330 was a gift from Feng Zhang (Addgene plasmid # 42230). This work utilized the computational resources of the NIH HPC Biowulf cluster. (http://hpc.nih.gov). We thank Tatiana Karpova and the NCI Core Fluorescence Imaging Facility for assistance and use of the confocal microscope. We thank Murali Palangat and members of the D.R.L. lab for feedback. This work was supported by the Intramural Research Program of the NIH.
We dedicate this work to the memory of Maxime Dahan.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no competing financial interests.
DATA AND SOFTWARE AVAILABILITY
3e Hi-C data is available via GEO accession GSE121443. Code for the generalized telegraph model is available at https://github.com/ccc1685/transcription-model.
References
- Akdemir KC, and Chin L (2015). HiCPlotter integrates genomic data with interaction matrices. Genome Biol 16, 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartman CR, Hsu SC, Hsiung CC, Raj A, and Blobel GA (2016). Enhancer Regulation of Transcriptional Bursting Parameters Revealed by Forced Chromatin Looping. Mol Cell 62, 237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battich N, Stoeger T, and Pelkmans L (2015). Control of Transcript Variability in Single Mammalian Cells. Cell 163, 1596–1610. [DOI] [PubMed] [Google Scholar]
- Bezanson J, Edelman A, Karpinski S, and Shah VB (2017). Julia: A Fresh Approach to Numerical Computing. Siam Rev 59, 65–98. [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J , et al. (2006). CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 7, R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, Eeckhoute J, Shao W, Hestermann EV, Geistlinger TR , et al. (2005). Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell 122, 33–43. [DOI] [PubMed] [Google Scholar]
- Chen X, Teichmann SA, and Meyer KB (2018). From Tissues to Cell Types and Back: Single-Cell Gene Expression Analysis of Tissue Architecture. Annual Review of Biomedical Data Science 1, 29–51. [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA , et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulon A, Ferguson ML, de Turris V, Palangat M, Chow CC, and Larson DR (2014). Kinetic competition during the transcription cycle results in stochastic RNA processing. Elife 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulon A, and Larson DR (2016). Chapter Seven - Fluctuation Analysis: Dissecting Transcriptional Kinetics with Signal Theory. In Methods in Enzymology, Grigory SF, and Samie RJ, eds. (Academic Press; ), pp. 159–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Ippolito AM, McDowell IC, Barrera A, Hong LK, Leichter SM, Bartelt LC, Vockley CM, Majoros WH, Safi A, Song L , et al. (2018). Pre-established Chromatin Interactions Mediate the Genomic Response to Glucocorticoids. Cell Syst. [DOI] [PubMed] [Google Scholar]
- Day CR, Chen H, Coulon A, Meier JL, and Larson DR (2015). High-throughput single-molecule screen for small-molecule perturbation of splicing and transcription kinetics. Methods (San Diego, Calif) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstein A, Amodaj N, Hoover K, Vale R, and Stuurman N (2010). Computer control of microscopes using microManager. Curr Protoc Mol Biol Chapter 14, Unit14 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elowitz MB, Levine AJ, Siggia ED, and Swain PS (2002). Stochastic gene expression in a single cell. Science 297, 1183–1186. [DOI] [PubMed] [Google Scholar]
- Ferguson ML, and Larson DR (2013). Measuring transcription dynamics in living cells using fluctuation analysis. Methods Mol Biol 1042, 47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritzsch C, Baumgartner S, Kuban M, Steinshorn D, Reid G, and Legewie S (2018). Estrogen-dependent control and cell-to-cell variability of transcriptional bursting. Mol Syst Biol 14, e7678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukaya T, Lim B, and Levine M (2016). Enhancer Control of Transcriptional Bursting. Cell 166, 358–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, Mohamed YB, Orlov YL, Velkov S, Ho A, Mei PH , et al. (2009). An oestrogen-receptor-[agr]-bound human chromatin interactome. Nature 462, 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson MA, and Bruck J (2000). Efficient exact stochastic simulation of chemical systems with many species and many channels. J Phys Chem A 104, 1876–1889. [Google Scholar]
- Golding I, Paulsson J, Zawilski SM, and Cox EC (2005). Real-time kinetics of gene activity in individual bacteria. Cell 123, 1025–1036. [DOI] [PubMed] [Google Scholar]
- Haber AL, Biton M, Rogel N, Herbst RH, Shekhar K, Smillie C, Burgin G, Delorey TM, Howitt MR, Katz Y , et al. (2017). A single-cell survey of the small intestinal epithelium. Nature 551, 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hah N, Danko CG, Core L, Waterfall JJ, Siepel A, Lis JT, and Kraus WL (2011). A rapid, extensive, and transient transcriptional response to estrogen signaling in breast cancer cells. Cell 145, 622–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hah N, Murakami S, Nagari A, Danko CG, and Kraus WL (2013). Enhancer transcripts mark active estrogen receptor binding sites. Genome Res 23, 1210–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakim O, Sung MH, Voss TC, Splinter E, John S, Sabo PJ, Thurman RE, Stamatoyannopoulos JA, de Laat W, and Hager GL (2011). Diverse gene reprogramming events occur in the same spatial clusters of distal regulatory elements. Genome Res 21, 697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen AS, and O’Shea EK (2015). Limits on information transduction through amplitude and frequency regulation of transcription factor activity. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoops S, Sahle S, Gauges R, Lee C, Pahle J, Simus N, Singhal M, Xu L, Mendes P, and Kummer U (2006). COPASI- A COmplex PAthway SImulator. Bioinformatics 22, 3067–3074. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Hurtado A, Holmes KA, Ross-Innes CS, Schmidt D, and Carroll JS (2011). FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat Genet 43, 27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janicki SM, Tsukamoto T, Salghetti SE, Tansey WP, Sachidanandam R, Prasanth KV, Ried T, Shav-Tal Y, Bertrand E, Singer RH , et al. (2004). From silencing to gene expression: real-time analysis in single cells. Cell 116, 683–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko MS (1991). A stochastic model for gene induction. J Theor Biol 153, 181–194. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson DR, Fritzsch C, Sun L, Meng X, Lawrence DS, and Singer RH (2013). Direct observation of frequency modulated transcription in single cells using light activation. Elife 2, e00750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson DR, Zenklusen D, Wu B, Chao JA, and Singer RH (2011). Real-time observation of transcription initiation and elongation on an endogenous yeast gene. Science 332, 475–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Dily F, Bau D, Pohl A, Vicent GP, Serra F, Soronellas D, Castellano G, Wright RH, Ballare C, Filion G , et al. (2014). Distinct structural transitions of chromatin topological domains correlate with coordinated hormone-induced gene regulation. Genes Dev 28, 2151–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T-H (2009). Extracting Kinetics Information from Single-Molecule Fluorescence Resonance Energy Transfer Data Using Hidden Markov Models. The Journal of Physical Chemistry B 113, 11535–11542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenstra T, Coulon A, Chow C, and Larson D (2015). Single-Molecule Imaging Reveals a Switch between Spurious and Functional ncRNA Transcription. Molecular Cell 60, 597–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenstra TL, Rodriguez J, Chen H, and Larson DR (2016). Transcription Dynamics in Living Cells. Annu Rev Biophys 45, 25–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO , et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Zhang J, and Zhou T (2016). Effect of Interaction between Chromatin Loops on Cell-to-Cell Variability in Gene Expression. PLoS Comput Biol 12, e1004917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masiakowski P, Breathnach R, Bloch J, Gannon F, Krust A, and Chambon P (1982). Cloning of cDNA sequences of hormone-regulated genes from the MCF-7 human breast cancer cell line. Nucleic Acids Res 10, 7895–7903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massot O, Baskevitch PP, Capony F, Garcia M, and Rochefort H (1985). Estradiol increases the production of alpha 1-antichymotrypsin in MCF7 and T47D human breast cancer cell lines. Mol Cell Endocrinol 42, 207–214. [DOI] [PubMed] [Google Scholar]
- May FE, and Westley BR (1987). Effects of tamoxifen and 4-hydroxytamoxifen on the pNR-1 and pNR-2 estrogen-regulated RNAs in human breast cancer cells. J Biol Chem 262, 15894–15899. [PubMed] [Google Scholar]
- May FE, and Westley BR (1988). Identification and characterization of estrogen-regulated RNAs in human breast cancer cells. J Biol Chem 263, 12901–12908. [PubMed] [Google Scholar]
- Meaburn KJ, and Misteli T (2008). Locus-specific and activity-independent gene repositioning during early tumorigenesis. J Cell Biol 180, 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora EP, Goloborodko A, Valton AL, Gibcus JH, Uebersohn A, Abdennur N, Dekker J, Mirny LA, and Bruneau BG (2017). Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell 169, 930–944 e922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer WS, Poncet-Montange G, Liu G, Petrocchi A, Reyna N, Subramanian G, Theroff J, Yau A, Kost-Alimova M, Bardenhagen JP , et al. (2016). Structure-Guided Design of IACS-9571, a Selective High-Affinity Dual TRIM24-BRPF1 Bromodomain Inhibitor. J Med Chem 59, 1440–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan YF, Wansa KD, Liu MH, Zhao B, Hong SZ, Tan PY, Lim KS, Bourque G, Liu ET, and Cheung E (2008). Regulation of estrogen receptor-mediated long range transcription via evolutionarily conserved distal response elements. J Biol Chem 283, 32977–32988. [DOI] [PubMed] [Google Scholar]
- Peccoud J, and Ycart B (1995). Markovian Modeling of Gene-Product Synthesis. Theoretical Population Biology 48, 222–234. [Google Scholar]
- Rao SSP, Huang SC, Glenn St Hilaire B, Engreitz JM, Perez EM, Kieffer-Kwon KR, Sanborn AL, Johnstone SE, Bascom GD, Bochkov ID , et al. (2017). Cohesin Loss Eliminates All Loop Domains. Cell 171, 305–320 e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren G, Jin W, Cui K, Rodrigez J, Hu G, Zhang Z, Larson DR, and Zhao K (2017). CTCF-Mediated Enhancer-Promoter Interaction Is a Critical Regulator of Cell-to-Cell Variation of Gene Expression. Mol Cell 67, 1049–1058 e1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revyakin A, Zhang Z, Coleman RA, Li Y, Inouye C, Lucas JK, Park S-R, Chu S, and Tjian R (2012). Transcription initiation by human RNA polymerase II visualized at single-molecule resolution. Genes & Development 26, 1691–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell DG, Li Y, Ayub M, Tate C, Newton G, Hey Y, Carter L, Faulkner S, Moro M, Pepper S , et al. (2014). Evaluation and validation of a robust single cell RNA-amplification protocol through transcriptional profiling of enriched lung cancer initiating cells. BMC Genomics 15, 1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B , et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schueler M, Munschauer M, Gregersen LH, Finzel A, Loewer A, Chen W, Landthaler M, and Dieterich C (2014). Differential protein occupancy profiling of the mRNA transcriptome. Genome Biol 15, R15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwille P, Haupts U, Maiti S, and Webb WW (1999). Molecular Dynamics in Living Cells Observed by Fluorescence Correlation Spectroscopy with One- and Two-Photon Excitation. Biophysical Journal 77, 2251–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon CE (1948). A Mathematical Theory of Communication. At&T Tech J 27, 379–423. [Google Scholar]
- Skinner SO, Xu H, Nagarkar-Jaiswal S, Freire PR, Zwaka TP, and Golding I (2016). Single-cell analysis of transcription kinetics across the cell cycle. Elife 5, e12175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokal RR, and Rohlf FJ (2012). Biometry, 4th edn (New York: W.H. Freeman and Company; ). [Google Scholar]
- Stavreva DA, Coulon A, Baek S, Sung MH, John S, Stixova L, Tesikova M, Hakim O, Miranda T, Hawkins M , et al. (2015). Dynamics of chromatin accessibility and long-range interactions in response to glucocorticoid pulsing. Genome Res 25, 845–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tantale K, Mueller F, Kozulic-Pirher A, Lesne A, Victor JM, Robert MC, Capozi S, Chouaib R, Backer V, Mateos-Langerak J , et al. (2016). A single-molecule view of transcription reveals convoys of RNA polymerases and multi-scale bursting. Nat Commun 7, 12248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RE, Larson DR, and Webb WW (2002). Precise nanometer localization analysis for individual fluorescent probes. Biophysical Journal 82, 2775–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai WW, Wang Z, Yiu TT, Akdemir KC, Xia W, Winter S, Tsai CY, Shi X, Schwarzer D, Plunkett W , et al. (2010). TRIM24 links a non-canonical histone signature to breast cancer. Nature 468, 927–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A , et al. (2015). Proteomics. Tissue-based map of the human proteome. Science 347, 1260419. [DOI] [PubMed] [Google Scholar]
- Vian L, Pekowska A, Rao SSP, Kieffer-Kwon KR, Jung S, Baranello L, Huang SC, El Khattabi L, Dose M, Pruett N , et al. (2018). The Energetics and Physiological Impact of Cohesin Extrusion. Cell 173, 1165–1178 e1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohland T, Rigler R, and Vogel H (2001). The Standard Deviation in Fluorescence Correlation Spectroscopy. Biophys J 80, 2987–2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Carter AC, Gendrel AV, Attia M, Loftus J, Greenleaf WJ, Tibshirani R, Heard E, and Chang HY (2017). Landscape of monoallelic DNA accessibility in mouse embryonic stem cells and neural progenitor cells. Nat Genet 49, 377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.