

Abstract

The further optimization of ER-α degradation efficacy of a series of ER modulators by refining side-chain substitution led to efficacious selective estrogen receptor degraders (SERDs). A fluoromethyl azetidine group was found to be preferred and resulted in the identification of bis-phenol chromene 17ha. In a tamoxifen-resistant breast cancer xenograft model, 17ha (ER-α degradation efficacy = 97%) demonstrated tumor regression, together with robust reduction of intratumoral ER-α levels. However, despite superior oral exposure, 5a (ER-α degradation efficacy = 91%) had inferior activity. This result suggests that optimizing ER-α degradation efficacy leads to compounds with robust effects in a model of tamoxifen-resistant breast cancer. Compound 17ha (GDC-0927) was evaluated in clinical trials in women with metastatic estrogen receptor-positive breast cancer.
Keywords: Estrogen receptor, estrogen receptor degrader, SERD, antagonist, tamoxifen-resistant, breast cancer, chromene, GDC-0927, SRN-927
The estrogen receptor ER-α1 has been an important target in the pharmaceutical industry for many years, with ER antagonists such as tamoxifen (1; Figure 1) and aromatase inhibitors being key therapeutics in the management of ER-positive breast cancer.2 Tamoxifen (1) and 4-hydroxytamoxifen (active metabolite) (2) are selective estrogen receptor modulators (SERMs), meaning they may act as an antagonist or agonist depending on the tissue in which they act.1 Although women with ER-α positive breast cancer typically respond well to tamoxifen, resistance can emerge.3 However, ER-α has been shown to be involved in this resistant state,4 and fulvestrant (3), a selective estrogen receptor degrader (SERD) that antagonizes and degrades ER-α, is clinically beneficial in this patient population.5 However, fulvestrant, which was not designed prospectively as a SERD,5 has poor pharmaceutical properties and must be administered by an intramuscular injection. We sought to identify SERDs where ER-α degradation efficacy was a main driver for structure–activity relationship (SAR) studies and that were orally bioavailable.
Figure 1.

Estrogen receptor ligands.
Much of the previous work on estrogen receptor ligands provides an understanding of the structural motifs required for potent ER-α antagonists, but there has been less disclosure on what is required to maximize ER-α degradation efficacy (i.e., SERD activity). To drive ER-α degradation SAR, we used an in-cell western assay that measured ER-α levels in MCF-7 breast cancer cells. This assay was used to develop the SAR of two chemical series on the program, including the clinical compound GDC-0810/ARN-810 (4),6 a carboxylic acid, and an early lead from an amine-based series (5).7 This Letter describes the further optimization of ER-α degradation efficacy of the amine-based chromene series represented by 5, leading to the clinical compound GDC-0927 (SRN-927). Following the completion of this work, other publications have been released that report SERDs.8,9
The compounds described in this Letter consist of a central chromene core to which an amine-based side chain is appended. The side-chain was installed on the core by two methods: (i) Ullmann coupling between an amino alcohol side-chain and an aryl iodide on the chromene core (General Method 1, Scheme 1) or (ii) alkylation with the desired cyclic amine of a previous installed ethyl mesylate on the chromene core (General Method 2, Scheme 1). See Supporting Information for preparation of the amino alcohol side-chains 10a–c and cyclic amines 6a–g. Intermediate 15 was prepared in an eight-step sequence from 2-(3-methoxyphenyl)acetic acid (12), while mesylate 16 was prepared from 15 in two additional steps. Chromene intermediate 15 was prepared from 2-(3-methoxyphenyl)acetic acid 12.7 Intermediate 16 was prepared from 15 via Ullmann coupling with ethylene glycol, followed by mesylation to afford 16 in 51% over two steps. The final compounds 17h and 17j–k were prepared from 15 using an Ullmann coupling with side-chains 10a–c followed by deprotection of the THP groups using acetic acid. In turn, final compounds 17c–g and 17i were prepared by SN2 displacement of mesylate 16 with the cyclic secondary amines 6a–g, followed by THP deprotection.
Scheme 1. Preparation of Chromene ER Ligands.

Reagents and conditions: (a) SOCl2, DMF, DCM, 0 °C; (b) 1,4-dimethoxybenzene, AlCl3, DCM, 0 °C, 55%; (c) BBr3, DCM, −78 to 0 °C, 57%; (d) DHP, PPTS, DCM, 70%; (e) 4-iodobenzaldehyde, piperidine, DBU, s-butanol, reflux, 75%; (f) MeMgCl, THF, 0 °C to rt, 98%; (g) 80% AcOH/water, 90 °C, 67%; (h) DHP, PPTS, DCM, 55%; (i) ethane-1,2-diol, CuI, 1,10-phenantroline, K2CO3, butyronitrile, 125 °C, 58%; (j) methanesulfonyl chloride, TEA, DCM, 0 °C, 88%; (k) 17h, 17j–k: CuI, K2CO3, butyronitrile, 125–135 °C, 2–4 d, 75–82%; (l) 17c–g, 17i: K2CO3, acetonitrile, 60 °C to reflux, 62–76%; (m) 80% AcOH/water, rt, 27–85%.
The monophenol analogs 20c and 20d (Table 2) were prepared from the corresponding chromene intermediate using an Ullmann coupling with the appropriate amino-alcohol side chains.
Table 2. Fluoromethyl Azetidine Side-Chain Significantly Improves ER-α Degradation Efficacy.

ER-α in-cell western in MCF-7 cells (n ≥ 4).
MCF-7 proliferation assay (n ≥ 3).
Efficacy recorded as percent of efficacy of fulvestrant control.
See ref (7).
Previously we demonstrated that subtle structural changes to the side-chain of the chromene scaffold could lead to large changes in ER-α degradation efficacy.7 For example, installation of an (R)-3-methyl-substituent on the pyrrolidine of 17a, giving 17b, led to a large increase in ER-α degradation efficacy (75% → 89%, Table 1). We continued to optimize ER-α degradation efficacy while maintaining potency by further exploring the substituent at this 3-position on the pyrrolidine, but found that the methoxymethyl (17c) and nitrile (17d) derivatives, although potent ER-α degraders, lost ER-α degradation efficacy. However, when a fluoromethyl-substituted pyrrolidine group was incorporated (17e), a substantial increase in ER-α degradation efficacy over 17b was observed (89% → 97%). It was apparent that the monofluoromethyl at the 3-position of the pyrrolidine was optimal, as 17f and 17g had inferior ER-α degradation efficacy compared to 17e. Addition of the fluoromethyl substituent to either the azetidine (17h) or piperidine (17i) side-chains again led to an increase in ER-α degradation efficacy compared to previously described, nonfluorinated methyl-substituted parent compounds7 (for azetidine 17h, 86% → 97%; for piperidine 17i, 63% → 80%). Previously we found that a (S)-methyl group on the linker ethyl chain led to a boost in ER-α degradation efficacy compared to the unsubstituted system,7 and so we installed a methyl in the linker of azetidine 17h and pyrrolidine 17e, resulting in 17j and 17k, respectively. However, no further increases in ER-α degradation efficacy were obtained, although 17k was found to be another potent, high efficacy ER-α degrader compared to its nonfluorinated parent compound 5 (98% ER-α degradation efficacy versus 91%, respectively). Lead compounds 17e, 17h, and 17k all displayed an MCF-7 proliferation IC50 ≤ 0.2 nM.
Table 1. Importance of a Monofluoromethyl Substituent to ER-α Degradation Efficacy.


ER-α in-cell western in MCF-7 cells (n ≥ 4).
MCF-7 proliferation assay (n ≥ 3).
Efficacy recorded as percent of efficacy of fulvestrant control.
See ref (7).
Of the side-chains shown in Table 1, pyrrolidine 17e was found to have inferior mouse PK compared to azetidine 17h and was deprioritized as a lead side-chain (AUC at 10 mg/kg PO: 0.04 μg·h/mL for 17e versus 0.16 μg·h/mL for 17h). Compounds 17h and 17k and their single active stereoisomers (see below) were profiled further; the fluoromethyl azetidine side-chain of 17h was ultimately selected over the fluoromethyl pyrrolidine of 17k as the lead side-chain due to (i) reduced number of stereocenters (1 versus 3) and (ii) improved performance of the single stereoisomer in a tamoxifen-resistant breast cancer xenograft model (see below).
Previously we showed that replacement of one of the phenols of 5, to give monophenol derivatives such as 20a and 20b, led to a loss of ER-α degradation efficacy (91% → 82–84%) and, in the case of 20a, resulted in loss of antitumor activity in a tamoxifen-resistant breast cancer xenograft model (Table 2).7 Having selected the fluoromethyl azetidine as the preferred side-chain to maximize ER-α degradation on the bis-phenol chromene core, we revisited these monophenol chromenes and prepared 20c and 20d. In contrast to the methyl pyrrolidine monophenols 20a and 20b, fluoromethyl azetidines 20c and 20d have robust levels of ER-α degradation efficacy (98%) equal to that of the bis-phenol chromene 17h. Hence, the fluoromethyl azetidine side-chain not only increases the degradation efficacy of bis-phenol chromenes (compare 5 to 17h), it also maintains that level of ER-α degradation potency and efficacy for monophenol systems (compare 17h to 20c and 20d). As noted for the derivatives in Table 1, 20c and 20d are also potent in the MCF-7 proliferation assay (IC50 < 0.5 nM).
Compounds were synthesized and tested in assays as a 50/50 mixture of stereoisomers at the chromene core. For the lead compounds 17h, 17k, and 20c (and 5) we confirmed that SERD activity was in one stereoisomer by separation by chiral supercritical fluid chromatography (SFC) and then characterized the stereoisomers. As shown in Table 3, the active stereoisomers 17ha, 17ka, and 20ca (from 17h, 17k, and 20c, respectively), all had high potency (IC50 = 0.1 nM) in the ER-α degradation assay compared to the inactive stereoisomers 17hb, 17kb, and 20cb (IC50 > 10 nM). The activity seen in 17hb, 17kb, and 20cb is likely due to residual active stereoisomer (approximately 1% following SFC separation). Analogous results were obtained when these pairs of compounds were tested in an ER-α binding assay and MCF-7 breast cancer cell line antiproliferative assay, indicating that the profiling of enantiomeric or diastereoisomeric mixtures was acceptable during SAR studies.
Table 3. SERD Activity Resides in One Chromene Stereoisomer.

ER-α in-cell western in MCF-7 cells (n ≥ 4).
Efficacy recorded as percent of efficacy of fulvestrant control.
Activity likely due to residual active stereoisomer (approximately 1% following SFC separation).
Previously we described the use of the rat uterine wet weight assay to efficiently triage ER-based pharmacodynamics and to determine tissue-selectivity (breast versus uterine).6,7 Compounds 17ha and 20ca were examined in this assay in agonist mode (no added ethynyl estradiol) and, like 5a, displayed an inverse agonist profile, reducing uterine weights below that of the vehicle treated animals (Figure 2). This is in contrast to tamoxifen (1), which showed partial agonist activity, increasing uterine weights above vehicle in the absence of added estradiol.1
Figure 2.

Compounds 17ha and 20ca are inverse agonists in a rat uterine wet weight assay: compounds administered orally for 3 days (5a, 17ha, and 20ca at 10 mg/kg po; tamoxifen (1) at 60 mg/kg).
As with other ER ligands that also contain two phenol groups, the mouse pharmacokinetics of 5a and 17ha are characterized by high clearance (Cl > 60 mL/min/kg) and low bioavailability (%F < 15). In contrast to 17ha, the monophenol 20ca had improved clearance (Cl = 36 mL/min/kg) and bioavailability (42% F versus 10% F), leading to improved exposure following oral dosing (po AUC = 1.9 μg·h/mL versus 0.16 μg·h/mL). Compounds 17ha and 20ca and the previous lead compound from the chromene series, 5a, were profiled in a MCF-7 based tamoxifen-resistant xenograft model of breast cancer using subcutaneous estradiol pellets to deliver sufficient levels of estradiol to drive tumor growth (Figure 3).10 Importantly, 17ha and 20ca demonstrated superior activity to 5a with 5 of 8 animals in each cohort showing tumor regressions. In contrast, only 1 of 8 animals showed tumor shrinkage in the cohort treated with 5a.
Figure 3.

Compounds 17ha and 20ca demonstrate superior activity to 5a in an MCF-7 tamoxifen-resistant breast cancer xenograft model. MCF-7 tamoxifen-resistant xenograft in mice using subcutaneous 0.72 mg estradiol pellets. Plasma pharmacokinetics measured on day 28 of study during take down.
The inferior profile of 5a compared to 17ha and 20ca is not due to potency against the estrogen receptor (ER-α degradation IC50: 5a = 0.1 nM versus 17ha = 0.1 nM, 20ca = 0.1 nM; MCF-7 proliferation IC50: 5a = 0.1 nM versus 17ha = 0.1 nM, 20ca = 0.2 nM) or pharmacokinetics (day 28 xenograft free AUC = 0.19 μg·h/mL for 5a versus 0.13 and 0.08 μg·h/mL for 17ha and 20ca, respectively). Importantly 5a is a less efficacious ER-α degrader in vitro than 17ha and 20ca (ER-α degradation efficacy: 5a, 91% versus 17ha and 20ca, 97%). As shown in Figure 4, this difference in in vitro ER-α degradation efficacy is reflected in the pharmacodynamic readout of tumor ER-α protein levels on day 28 of the xenograft study. Thus, while compound 5a reduced ER-α levels to those of the vehicle plus estradiol cohort,11 both 17ha and 20ca reduced levels substantially below that of 5a. It is noteworthy that monophenol 20ca had robust activity in this tamoxifen-resistant setting (equal to that of bis-phenol 17ha) since as we previously described, monophenol 20a was inferior to bis-phenol 5 in this same model.7 This result again demonstrates the effectiveness of the optimized fluoromethyl side-chain in maximizing in vitro ER-α degradation efficacy and in vivo xenograft performance.
Figure 4.
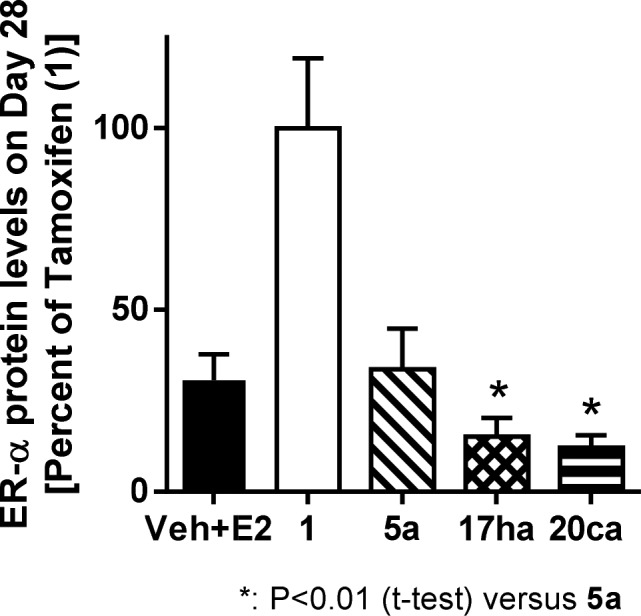
Compared to 5a, ER-α levels are lower in tumors of animals treated with 17ha and 20ca.
The robust performance of 17ha and 20ca in this and other preclinical xenograft models of breast cancer led to consideration of both compounds as potential clinical candidates. Compound 17ha was ultimately selected in part due to results from an assay probing the conformation of ER-α induced by these ligands. In this assay, ligand-induced ER-α conformation was monitored by the interaction of ligand-selective peptide probes with the ligand-bound receptor.12 The peptide probes utilized in this study interact differentially with protein surfaces exposed on ER-α in response to estradiol, 4-hydroxytamoxifen, or fulvestrant binding.13,14 It was found that 17ha and 20ca induce ER-α conformations distinct from fulvestrant and 4-hydroxytamoxifen (see Supporting Information). However, 20ca-bound ER-α interacted with the αIII peptide probe, suggesting that aspects of the 20ca induced ER-α conformation are shared with that induced by the SERM 4-hydroxytamoxifen. In contrast, the ER-α conformations in response to 17ha did not show appreciable interaction with αIII (see Supporting Information). As we were looking to identify a candidate with a profile distinct from tamoxifen, we selected 17ha for development.
Selectivity of 17ha against nuclear hormone receptors was good. In transcriptional reporter assays for the glucocorticoid, mineralocorticoid, progesterone-A, and progesterone-B receptors, 17ha had no significant agonist or antagonist activity. Cytochrome P450 inhibition profiling of 17ha indicated it had little to no competitive inhibitory activity against CYP1A2, CYP2C19, CYP2D6, or CYP3A4 (IC50 > 10 μM) and modest inhibitory effect on CYP2C8 and CYP2C9 (IC50 = 3.0 and 3.2 μM, respectively). In a CEREP panel of radioligand binding assays for 55 targets (protein-free conditions), 17ha at 10 μM displayed >75% binding to six targets. However, in follow-up cell-based functional assays, the interactions with most of these six targets were weak with IC50 values >10 μM. The only interaction with an IC50 < 1 μM was with the dopamine transporter (IC50 = 0.39 μM). When tested for its effect on the hERG channel in a patch clamp assay, 17ha was found to be a moderate inhibitor (IC50 = 4.6 μM). This activity on the dopamine transporter and hERG channel was not considered an issue as selectivity for ER is >1000-fold (MCF-7 proliferation = 0.1 nM; ER-α degradation = 0.1 nM). Compound 17ha was negative in an Ames assay using the TA-98 and TA-100 tester strains.
In summary, we have further maximized ER-α degradation efficacy of a series of ER modulators resulting in highly potent and efficacious chromene SERDs. A fluoromethyl substituent on either a pyrrolidine or azetidine ring at the end of the side-chain was found to be optimal for maximizing ER-α degradation. Fluoromethyl azetidine was determined to be the preferred ring system on the side-chain, leading to the identification of bis-phenol chromene 17ha. In contrast to previous work, when applied to a monophenol chromene core, the fluoromethyl azetidine side-chain gave highly efficacious ER-α degraders such as 20ca. In a tamoxifen-resistant breast cancer xenograft model, 17ha and 20ca (ER-α degradation efficacy = 97%) demonstrated tumor regression, together with robust reduction intratumoral ER-α levels. However, despite higher drug levels, 5a (ER-α degradation efficacy = 91%) had inferior activity. This data suggests optimizing ER-α degradation in vitro maximizes activity in a tamoxifen-resistant breast cancer xenograft derived from the MCF-7 breast cancer cell line. Compound 17ha (GDC-0927 or SRN-927) was selected for development and was evaluated in clinical trials in women with metastatic ER-positive breast cancer.
Glossary
ABBREVIATIONS
- E2
estradiol
- EE
ethynyl estradiol
- ER
estrogen receptor
- ERE
estrogen response element
- SERD
selective estrogen receptor degrader
- SERM
selective estrogen receptor modulator
- UWW
uterine wet weight
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00414.
Experimental information for synthesis of compounds; ligand-induced ER-α conformation; biological assays (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Nilsson S.; Gustafsson J. A. Estrogen receptors: therapies targeted to receptor subtypes. Clin. Pharmacol. Ther. 2011, 89 (1), 44–55. 10.1038/clpt.2010.226. [DOI] [PubMed] [Google Scholar]
- Ariazi E. A.; Ariazi J. L.; Cordera F.; Jordan V. C. Estrogen receptors as therapeutic targets in breast cancer. Curr. Top. Med. Chem. 2006, 6 (3), 181–202. 10.2174/156802606776173483. [DOI] [PubMed] [Google Scholar]
- Riggins R. B.; Schrecengost R. S.; Guerrero M. S.; Bouton A. H. Pathways to tamoxifen resistance. Cancer Lett. 2007, 256 (1), 1–24. 10.1016/j.canlet.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musgrove E. A.; Sutherland R. L. Biological determinants of endocrine resistance in breast cancer. Nat. Rev. Cancer 2009, 9 (9), 631–43. 10.1038/nrc2713. [DOI] [PubMed] [Google Scholar]
- Johnston S. J.; Cheung K. L. Fulvestrant - a novel endocrine therapy for breast cancer. Curr. Med. Chem. 2010, 17 (10), 902–14. 10.2174/092986710790820633. [DOI] [PubMed] [Google Scholar]
- Lai A.; Kahraman M.; Govek S.; Nagasawa J.; Bonnefous C.; Julien J.; Douglas K.; Sensintaffar J.; Lu N.; Lee K.-J.; Aparicio A.; Kaufman J.; Qian J.; Shao G.; Prudente R.; Joseph J. D.; Darimont B.; Brigham D.; Grillot K.; Heyman R.; Rix P.; Hager J.; Smith N. D. Identification of GDC-0810 (ARN-810), an orally bioavailable Selective Estrogen Receptor Degrader (SERD) that demonstrates robust activity in tamoxifen-resistant breast cancer xenografts. J. Med. Chem. 2015, 58 (12), 4888–4904. 10.1021/acs.jmedchem.5b00054. [DOI] [PubMed] [Google Scholar]
- Nagasawa J.; Govek S.; Kahraman M.; Lai A.; Bonnefous C.; Douglas K.; Sensintaffar J.; Lu N.; Lee K.-J.; Aparicio A.; Kaufman J.; Qian J.; Shao G.; Prudente R.; Joseph J. D.; Darimont B.; Brigham D.; Grillot K.; Heyman R.; Rix P.; Hager J.; Smith N. D. Identification of an orally bioavailable chromene-based Selective Estrogen Receptor Degrader (SERD) that demonstrates robust activity in a model of tamoxifen-resistant breast cancer. J. Med. Chem. 2018, 61 (17), 7917–7928. 10.1021/acs.jmedchem.8b00921. [DOI] [PubMed] [Google Scholar]
- De Savi C.; Brdbury R. H.; Rabow A. A.; Norman R. A.; de Almeida C.; Andrews D. M.; Ballard P.; Buttar D.; Callis R. J.; Currie G. S.; Curwen J. O.; Davies C. D.; Donald C. S.; Feron L. J.; Gingell H.; Glossop S. C.; Hayter B. R.; Hussain S.; Karoutchi G.; Lamont S. G.; MacFaul P.; Moss T. A.; Pearson S. E.; Tonge M.; Walker G. E.; Weir H. M.; Wilson Z. Optimization of a novel binding motif to (E)-3-(3,5-difluoro-4-((1R,3R)-2-(2-fluoro-2-methylpropyl)-3-methyl-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indol-1-yl)phenyl)acrylic acid (AZD9496), a potent and orally bioavailable selective estrogen receptor downregulator and antagonist. J. Med. Chem. 2015, 58 (20), 8128–8140. 10.1021/acs.jmedchem.5b00984. [DOI] [PubMed] [Google Scholar]
- Xiong R.; Zhao J.; Gutgesell L. M.; Wang Y.; Lee S.; Karumudi B.; Zhao H.; Lu Y.; Tonetti D. A.; Thatcher G. R. Novel selective estrogen receptor downregulators (SERDs) developed against treatment-resistant breast cancer. J. Med. Chem. 2017, 60 (4), 1325–1342. 10.1021/acs.jmedchem.6b01355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- This E2 is used to drive xenograft tumor growth in vivo and leads to high E2 concentrations (300–400 pg/mL), thus requiring more ER ligand to outcompete the native ligand (E2) and so mediate an inhibitory effect. This is of relevance as plasma E2 levels in the target patient population of postmenopausal women (∼5 pg/mL) are substantially lower. Thus efficacy models run in the context of high E2 plasma concentrations potentially overestimate ER ligand dose and plasma levels necessary to drive efficacy in a postmenopausal setting.
- Of note is that, although an ER agonist, estradiol is also a strong degrader of ER-α. See:; Wijayaratne A. L.; McDonnell D. P. J. Biol. Chem. 2001, 276 (38), 35684–35692. 10.1074/jbc.M101097200. [DOI] [PubMed] [Google Scholar]
- Norris J. D.; Paige L. A.; Christensen D. J.; Chang C. Y.; Huacani M. R.; Fan D.; Hamilton P. T.; Fowlkes D. M.; McDonnell D. P. Peptide antagonists of the human estrogen receptor. Science 1999, 285 (5428), 744–6. 10.1126/science.285.5428.744. [DOI] [PubMed] [Google Scholar]
- Paige L. A.; Christensen D. J.; Gron H.; Norris J. D.; Gottlin E. B.; Padilla K. M.; Chang C. Y.; Ballas L. M.; Hamilton P. T.; McDonnell D. P.; Fowlkes D. M. Estrogen receptor (ER) modulators each induce distinct conformational changes in ER alpha and ER beta. Proc. Natl. Acad. Sci. U. S. A. 1999, 96 (7), 3999–4004. 10.1073/pnas.96.7.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldring N.; Nilsson M.; Buehrer B.; Treuter E.; Gustafsson J. A. Identification of tamoxifen-induced coregulator interaction surfaces within the ligand-binding domain of estrogen receptors. Molecular and cellular biology 2004, 24 (8), 3445–59. 10.1128/MCB.24.8.3445-3459.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.