

Abstract

Recent research suggests that KCNQ isoforms, particularly the KCNQ4 and KCNQ5 subtypes expressed in smooth muscle cells, are involved in both establishing and maintaining resting membrane potentials and regulating smooth muscle contractility. Retigabine (RTG) is a first-in-class antiepileptic drug that potentiates neuronal KCNQ potassium channels, but poor subtype selectivity limits its further application as a pharmacological tool. In this study, we improved the subtype specificity of retigabine by altering the N-1/3 substituents and discovered several compounds that show better selectivity for KCNQ4 and KCNQ5 channels. Among these compounds, 10g is highly selective for KCNQ4 and KCNQ5 channels without potentiating KCNQ1 and KCNQ2 channels. These results are an advance in the exploration of small molecule modifiers that selectively activate different KCNQ isoforms. The developed compounds could also serve as new pharmacological tools for elucidating the function of KCNQ channels natively expressed in various tissues.
Keywords: KCNQ channel, retigabine, subtype selectivity, agonist
The Kv7 (KCNQ) subfamily of voltage-gated potassium channels consists of five members (KCNQ1–5) and plays important roles in many excitable cells, such as neurons, cardiac myocytes, and vascular smooth muscle cells.1−4 KCNQ1 is the most divergent and is primarily expressed in cardiac tissues. The KCNQ2–5 subtypes are predominantly found in various central and peripheral neurons.5,6 Specifically, the KCNQ2/3 heterotetramers are considered to be the molecular basis for generating M currents that exert inhibitory control over neuronal firing.7 Therefore, the neuronal KCNQ2/3 channels represent interesting targets for the treatment of diseases that involve altered neuronal excitability, such as epilepsy and chronic pain.8−11 The small molecule retigabine (RTG, Figure 1) is an anticonvulsant drug, and it activates KCNQ2–KCNQ5 channels.12,13 However, the poor subtype selectivity of retigabine leads to undesirable side effects, such as urinary retention, which limit its clinical use.14,15
Figure 1.
Structures of RTG and 1.
It has also been suggested that KCNQ isoforms expressed in smooth muscle cells are involved in establishing and maintaining the resting membrane potential as well as in regulating smooth muscle contractility. KCNQ channels have been identified in multiple smooth muscle cells, including vascular smooth muscle such as thoracic artery, carotid artery, femoral artery, and portal vein; and visceral smooth muscle such as gastrointestinal smooth muscle, bladder detrusor, respiratory smooth muscle, and uterine smooth muscle.16−20 Experiments using an array of pharmacological KCNQ channel modulators have supported the crucial role of these channels in regulating smooth muscle contractility.21−23 Therefore, recent reports have refocused attention on the smooth muscle isoforms of KCNQ channels. It is worth noting that most of the visceral tissues that have been tested show high expression of the KCNQ4 and KCNQ5 subtypes, which suggests that KCNQ4 and KCNQ5 channels could be potential targets for visceral smooth muscle-related diseases such as irritable bowel syndrome and overactive bladder syndrome.24 To date, no compounds that selectively activate KCNQ4 and KCNQ5 channels without activating neuronal KCNQ2 and KCNQ3 channels have been developed for clinical use.
By introducing a CF3 group at the 4-position of the benzylamine moiety and a fluorine atom at the 3-position of the aniline ring of retigabine, Tzounopoulos et al. generated SF0034 and RL648_81 as new KCNQ2/3-specific activators, which are more potent and more selective than retigabine.25,26 During our previous studies on KCNQ modulators, P-RTG, an RTG derivative that incorporates a propargyl group at the N-1 position, did not show any changes in potentiation potency, subtype selectivity, or molecular determinants on KCNQ channels compared to RTG.27 In addition, we found that certain chemical modifications at the N-1 and N-3 positions of retigabine gave rise to a KCNQ4 and KCNQ5 channel agonist with improved subtype specificity (compound 1, Figure 1).28 This compound activated the current of the KCNQ4 and KCNQ5 channels at a concentration of 10 μM by 2.12- and 1.49-fold, respectively. Meanwhile, compound 1 inhibited the current of the KCNQ2 channel by 70%. This encouraged us to further explore this class of retigabine derivatives to improve KCNQ4 and KCNQ5 selectivity. Here, we report the synthesis of a series of N-1- and N-3-substituted retigabine derivatives via structural modification of 1 and the evaluation of their activity toward different KCNQ channel subtypes. We found several compounds that selectively activate KCNQ4 and KCNQ5 channels without activating KCNQ2 channels. These results therefore provide hope for the further development of subtype-specific KCNQ channel agonists.
First, compounds 6a–f were synthesized to evaluate the effect of various substituents at the N-1 position of 1. All compounds were synthesized according to Scheme 1. Compound 3 was synthesized from the commercially available compound 2 and 4-fluorobenzaldehyde via a reductive amination reaction. Compound 3 was then catalytically hydrogenated over Pd/C. Intermediately, compound 4 was treated with ethyl chloroformate to give 5. The reaction of compound 5 with the appropriate alkyl bromide reagent produced 6a–f.
Scheme 1. Synthesis of the N-1/3 Retigabine Derivatives 6a–f, 8a–l, 10a–h, and 13.

Reagents and conditions: (a) 4-fluorobenzaldehyde, cat. p-TsOH, toluene, 120°C, 77.2%; (b) NaBH4, 1,4-dioxane:MeOH (4:1), rt, 94%; (c) H2, Pd/C, MeOH, rt, 100%; (d) ethyl chloroformate, DIPEA, 1,4-dioxane, rt, 90%; (e) R1Br, DIPEA, 60°C, DMF; (f) Boc2O, NaHCO3, H2O/THF (1:2), rt, 85%; (g) 3-bromo-1-propyne, DIPEA, 60°C, DMF, 90%; (h) TFA, DCM, 0°C; (i) ClCOOR2, DIPEA, 1,4-dioxane, rt; (j) ethyl chloroformate, DIPEA, 1,4-dioxane, rt, 50%; (k) ClCOOR3, DIPEA, 1,4-dioxane, rt; (l) 3-bromo-1-propyne, DIPEA, 60°C, DMF, 85%; (m) Boc2O, DMAP, THF, 80 °C, 35%; (n) Zn, NH4Cl, MeOH, H2O, reflux; (o) ethyl chloroformate, DIPEA, rt, 60% over two steps.
With these N-1-substituted analogues in hand, their effects on the KCNQ2, 4, and 5 channels were first assessed. Electrophysiology experiments were conducted using the whole-cell patch clamp technique, and compound effects on the amplitude of the outward current (I/I0) were analyzed. I0 is the amplitude of the outward current in the absence of a compound. I is the amplitude of the outward current in the presence of a compound. Compounds resulting in I/I0 > 1 were defined as activators, while compounds that gave I/I0 < 1 were defined as inhibitors. All compounds were tested at a concentration of 10 μM, and the testing voltage was −10 mV unless otherwise stated. As shown in Table 1, the results indicate that N-1 substitution is important for activation and subtype selectivity. Branching on the allylic group (providing increased length and steric hindrance) had a detrimental effect on KCNQ2 inhibition, but the ability to activate KCNQ4 and KCNQ5 channels was retained. Among the newly synthesized derivatives, the N-1 propargyl-substituted compound 6e displayed the best agonist potency. At a concentration of 10 μM, 6e caused 5.68-fold and 3.20-fold increases in the KCNQ4 and KCNQ5 channels, respectively. However, there was no significant change in the outward current when we applied 6e to KCNQ2 channels; for comparison, RTG increased KCNQ2 currents by 1.6-fold (Figure 2C). To determine the specific potency of 6e on the KCNQ2 and KCNQ4 channels, the concentration–response relationship of 6e was established for the KCNQ2 and KCNQ4 currents. As demonstrated in Figure 2B,D, 6e enhanced KCNQ4 currents in a concentration-dependent manner. The EC50 of 6e for KCNQ4 was determined to be 0.89 ± 0.16 μM. In contrast, 6e (1–30 μM) did not affect the current amplitude of homomeric KCNQ2 currents (Figure 2A) at similar concentrations. In fact, 6e at a concentration of 30 μM slightly potentiated KCNQ2 currents by 2 ± 0.9%.
Table 1. Structures of the N-1 Derivatives 6a–f and Their Effects on KCNQ2, 4, and 5 Channels.


The testing concentration was 10 μM. Each compound was tested in more than four cells.
Figure 2.

Effect of 6e on KCNQ2 and KCNQ4 channels. (A,B) Representative current traces of KCNQ2 (A) and KCNQ4 (B) channels activated by 6e using different drug concentrations as indicated are shown. (C) Histogram plotting 6e and RTG effect on KCNQ2 currents measured at −10 mV (n ≥ 4). (D) Concentration–response relationships of 6e on homomeric KCNQ4 currents. The Hill coefficient was 0.33.
To determine the specificity of 6e on other KCNQ subtypes, we individually expressed and then tested KCNQ1 to KCNQ5 in CHO-K1 cells using the whole cell voltage-clamp technique. The depolarized voltage ranged from −90 to +60 mV in 10 mV increments as described in Figure 3A. Except for KCNQ1 and KCNQ2, the outward currents of all KCNQ subtypes were potentiated by extracellular treatment with 10 μM 6e (Figures 3B and 4C). The provided histogram illustrates that the current amplitude produced by 6e decreased in the following order: KCNQ4 > KCNQ5 > KCNQ3.
Figure 3.

Subtype selectivity of 6e for KCNQ channels. (A) In the voltage protocol, cells were held at −80 mV and stepped to a series of voltages ranging from −90 to +60 mV in 10 mV increments for a 1500 ms pulse followed by stepping to −120 mV for 500 ms. (B) Histogram showing the effects of 6e on different subtypes of KCNQ with currents measured at −10 and +60 mV. (C) Whole-cell currents of CHO cells transfected individually with KCNQ1 to KCNQ5 were recorded in the absence (left panels) and presence (right panels) of 10 μM 6e.
Figure 4.
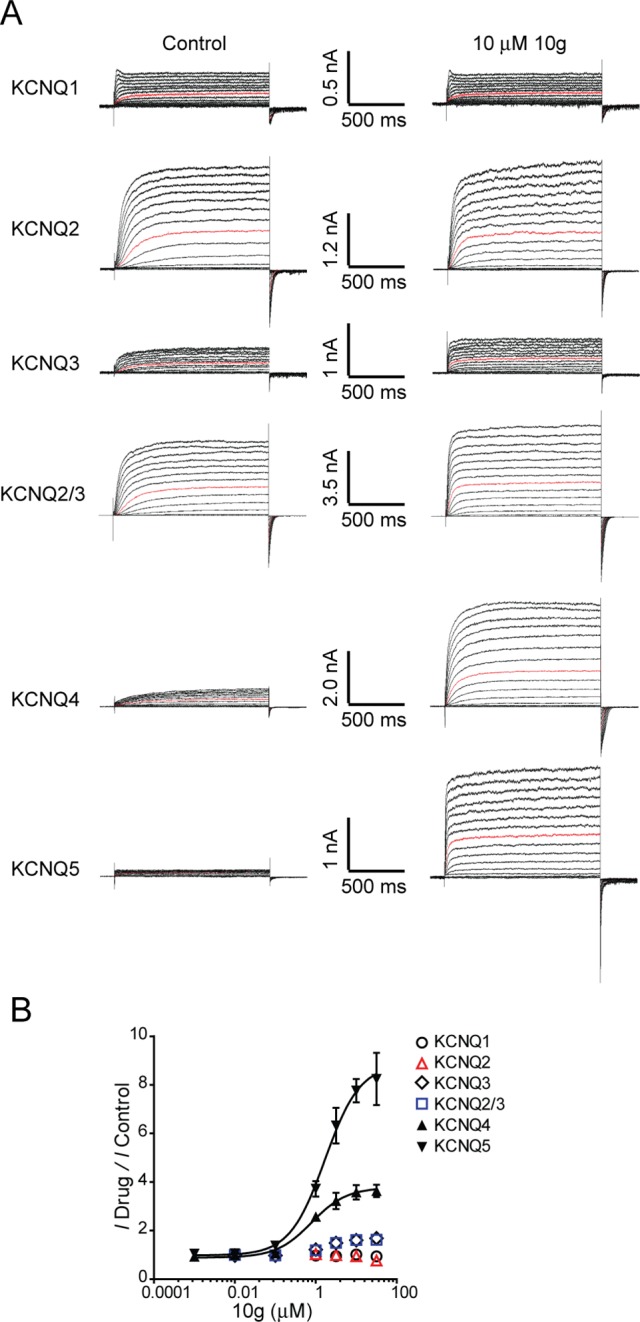
Dose–response curves of 10g on different KCNQ isoforms. (A) Representative traces of homomeric KCNQ1–KCNQ5 channels and heteromeric KCNQ2/3 channels, before and after application of 10 μM 10g. (B) Dose–response relationship of 10g on homomeric KCNQ1–KCNQ5 channels and heteromeric KCNQ2/3 channels.
Next, the N-1 propargyl substituent was maintained, and the N-2 and N-3 substituents were varied. Two new series of N-2- and N-3-substitued analogues (8 and 10) were prepared via a similar synthetic strategy (Scheme 1). Compound 4 was treated with Boc2O to give 7. The reaction of compound 7 with 3-bromo-1-propyne produced 8g. Deprotection of 8g followed by treatment with the appropriate chloroformate gave the final compounds 8a–f and 8h–l. Deprotection of 8g followed by treatment with ethyl chloroformate formed intermediate 9. Compounds 10 were synthesized in the same way from 9.
Accordingly, the two new series of synthesized compounds (8 and 10) were assessed for their effects on the KCNQ2, 4, and 5 channels, and the results are shown in Table 2. When R2 and R3 remained the same, the effects of the substituents were determined from compounds 8a–l. The identities of the functional groups at N-2 and N-3 were very important. For the alkyl substituted compounds 8a–j, a critical difference was observed between the activity of the methyl and tertiary butyl analogues (8a and 8g) and the rest of the analogues; 8a and 8g behaved as weak activators, while the others inhibited the KCNQ2 channel. Changing the length or steric hindrance of the chain caused an ambiguous effect on the activation of KCNQ4 and KCNQ5. In general, alkane-substituted compounds were better agonists than cycloalkane- and arene-substituted compounds. Among these compounds, the bis-tertiary butyl-substituted compound 8g displayed the best agonist potency and selectivity.
Table 2. Structures of N-2/3 Derivatives 8, 10, and 13 and Their Effects on the KCNQ2, 4, and 5 Channels.

|
I/I0a |
|||||
|---|---|---|---|---|---|
| Cpds | R2 | R3 | KCNQ2 | KCNQ4 | KCNQ5 |
| 8a | Me | Me | 1.37 ± 0.11 | 4.77 ± 0.44 | 2.89 ± 0.07 |
| 8b | allyl | allyl | 0.67 ± 0.02 | 2.21 ± 0.11 | 5.26 ± 0.53 |
| 8c | nPr | nPr | 0.78 ± 0.04 | 2.58 ± 0.56 | 2.75 ± 0.84 |
| 8d | iPr | iPr | 0.35 ± 0.04 | 1.33 ± 0.06 | 3.11 ± 0.93 |
| 8e | nBu | nBu | 0.77 ± 0.02 | 2.75 ± 0.40 | 4.13 ± 1.09 |
| 8f | iBu | iBu | 0.83 ± 0.04 | 1.52 ± 0.17 | 1.73 ± 0.10 |
| 8g | tBu | tBu | 1.00 ± 0.09 | 3.05 ± 0.74 | 5.32 ± 0.39 |
| 8h | cyclopropyl | cyclopropyl | 0.64 ± 0.03 | 1.51 ± 0.04 | 2.55 ± 0.38 |
| 8i | cyclobutyl | cyclobutyl | 0.26 ± 0.06 | 1.21 ± 0.09 | 1.33 ± 0.23 |
| 8j | cyclopentyl | cyclopentyl | 0.21 ± 0.05 | 0.88 ± 0.07 | 1.25 ± 0.14 |
| 8k | Ph | Ph | 0.91 ± 0.02 | 1.22 ± 0.05 | 1.16 ± 0.04 |
| 8l | Bn | Bn | 1.25 ± 0.01 | 3.02 ± 0.28 | 3.57 ± 0.31 |
| 10a | Et | Me | 0.61 ± 0.05 | 2.56 ± 0.47 | 3.34 ± 0.54 |
| 10b | Et | Allyl | 0.96 ± 0.04 | 3.68 ± 0.36 | 4.67 ± 0.69 |
| 10c | Et | nPr | 1.07 ± 0.03 | 3.95 ± 0.89 | 5.81 ± 0.62 |
| 10d | Et | iPr | 0.94 ± 0.06 | 5.01 ± 0.83 | 5.71 ± 0.34 |
| 10e | Et | nBu | 1.06 ± 0.03 | 4.16 ± 0.60 | 3.62 ± 0.31 |
| 10f | Et | iBu | 1.04 ± 0.04 | 5.19 ± 0.64 | 4.84 ± 0.32 |
| 10g | Et | tBu | 1.16 ± 0.04 | 6.37 ± 0.84 | 4.58 ± 0.32 |
| 10h | Et | Bn | 0.97 ± 0.15 | 1.27 ± 0.03 | 2.57 ± 0.67 |
| 13 | tBu | Et | 1.07 ± 0.06 | 7.71 ± 0.50 | 4.59 ± 0.56 |
The testing concentration was 10 μM. Each compound was tested in more than four cells.
When R2 was maintained as ethyl and the R3 group was varied, the effects of substituents can be seen by comparing compounds 10a–h. Unlike compounds 8 and with the exception of 10a, which was a weak inhibitor of KCNQ2, compounds 10 had no agonist activity on KCNQ2 and displayed significant activator potency on the KCNQ4 and KCNQ5 channels. Increasing the length or steric hindrance of the alkyl chain had beneficial effects on compound activity (10a vs 10c vs 10e and 10e vs 10f vs 10g). Among the compounds in this series, the N-2 ethyl, N-3 tertiary butyl-substituted compound 10g exhibited the best agonist potency. In addition, an N-2 and N-3 substituent exchanged compound (13) of 10g was designed and synthesized (Scheme 1). Compound 13 displayed agonist activities comparable to that of 10g, and the activity of 13 toward KCNQ4 was even greater than that of 10g.
Compounds 8g, 10g, and 13 were selected for further testing based on their potent activation of the KCNQ4 and KCNQ5 channels. The concentration–response relationships of 8g, 10g, and 13 were then established for the KCNQ4 and KCNQ5 currents. Analysis of the dose–response curves revealed that the EC50 values of the selected compounds ranged from 0.78 to 2.43 μM (Table 3). Compound 10g displayed the best agonist potency against KCNQ4 and KCNQ5 with EC50 values of 0.78 and 1.68 μM, respectively. Then, the evaluation of 10g against all other subtypes was completed (Figure 4). To characterize the activation of KCNQ2/3 heteromeric channels by 10g, we generated a r-KCNQ2/3 tandem construct (see Experimental Section in the Supporting Information). Unfortunately, we observed a maximum increase of approximately 1.50-fold in the outward currents of the KCNQ3 and KCNQ2/3 channels. Notably, at a similar concentration range (1–30 μM), 10g did not affect the current amplitude of homomeric KCNQ2 currents. Compound 10g actually slightly inhibited KCNQ2 currents at higher concentrations (30 μM) (Figure 4), making its effect on the KCNQ channels more specific than that of RTG.
Table 3. Potency (EC50, μM) of RTG, 8g, 10g, and 13 against KCNQ4, KCNQ5, and KCNQ2.
| RTG | 8g | 10g | 13 | |
|---|---|---|---|---|
| KCNQ4 | 5.90 ± 0.18 | 2.03 ± 0.14 | 0.78 ± 0.14 | 1.42 ± 0.04 |
| KCNQ5 | 3.45 ± 0.28 | 2.43 ± 0.17 | 1.68 ± 0.12 | 1.37 ± 0.11 |
| KCNQ2 | 2.17 ± 0.07 | >30 | >30 | >30 |
Investigation of the influence of an activator on V1/2 is very important for fully understanding the effects of an activator on channels. The effects of 6e and 10g on voltage-dependent activation were then analyzed. As described previously,13 the current–voltage G-V curves suggest that one effect of RTG is to produce a 20–30 mV negative shift in the KCNQ current activation curve. However, unlike RTG, 6e and 10g only slightly affected the voltage-dependent activation curves of the KCNQ2 and KCNQ4 channels (data shown in Supplementary Table 1). Particularly at −60 mV or at more negative membrane potentials, the drug 10g does not induce channel opening (data shown in Supplementary Figure 5). We speculated that the RTG-induced G-V shift should be sensitive to structure alterations around N-1, N-2, and N-3 and that the major influence of this class of compounds is on the current amplitude.
The previously reported KCNQ activators have low or no selectivity for KCNQ2–5 channels. Their undesirable side effects are likely due to poor KCNQ2–5 channel selectivity, and nonselective KCNQ modulators may be likely to cause side effects when used clinically. Recently, certain compounds have been reported to be selective for KCNQ4, 5 or KCNQ2 channels. For example, fasudil did not affect KCNQ2 and KCNQ2/3 currents but enhanced KCNQ4 and KCNQ4/5 channels,29 while it originally acted as a potent rho-kinase inhibitor to suppress proliferation/migration and induce apoptosis in urothelial cancer cells.30 AaTXKβ(2–64), a peptide activator isolated from scorpion toxin, increased the maximal currents in homomeric KCNQ4 and heteromeric KCNQ2/3 channels but showed no effect on homomeric KCNQ3 channels.31 In this study, we synthesized a series of selective activators for KCNQ4 and KCNQ5 channels based on the structural core of RTG, an approved antiepileptic drug. Among these newly synthesized derivatives, compound 10g was found to be the most potent activator of KCNQ4 and KCNQ5 and exhibited EC50 values of 0.78 and 1.68 μM, respectively, with no enhancement of the current amplitude for KCNQ2. This result provides a new platform for developing selective KCNQ modulators. However, as a KCNQ4 and KCNQ5-selective probe compound, 10g will be a useful tool for elucidating the mechanism of the interaction between the compounds and channels and for determining the contributions of different KCNQ channel subtypes in various tissues. The slight increase in the outward currents of the KCNQ3 and KCNQ2/3 channels will be an issue in regard to possible side effects; thus, additional experiments are needed to clarify these results. Therefore, experiments are in progress in our lab to further improve the compound specificity.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (No. 81773707) and the Science and Technology Commission of Shanghai Municipality (No. 16431901700 and No. 15431901500).
Glossary
ABBREVIATIONS
- Cpds
compounds
- p-TsOH
p-toluenesulfonic acid
- DIPEA
N,N-diisopropylethylamine
- DMF
N,N-dimethylformamide.
- Boc2O
di-tert-butyl dicarbonate
- DCM
dichloromethane
- rt
room temperature
- DMAP
4-(dimethylamino)pyridine.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00315.
Experimental procedures and characterization data for new compounds (PDF)
Author Contributions
∥ These authors contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Robbins J. KCNQ potassium channels: physiology, pathophysiology, and pharmacology. Pharmacol. Ther. 2001, 90, 1–19. 10.1016/S0163-7258(01)00116-4. [DOI] [PubMed] [Google Scholar]
- Brown D. A.; Passmore G. M. Neural KCNQ (Kv7) channels. Br. J. Pharmacol. 2009, 156, 1185–1195. 10.1111/j.1476-5381.2009.00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood I. A.; Ohya S. New tricks for old dogs: KCNQ expression and role in smooth muscle. Br. J. Pharmacol. 2009, 156, 1196–1203. 10.1111/j.1476-5381.2009.00131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stott J. B.; Jepps T. A.; Greenwood I. A. Kv7 potassium channels: a new therapeutic target in smooth muscle disorders. Drug Discovery Today 2014, 19, 413–424. 10.1016/j.drudis.2013.12.003. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Curran M. E.; Splawski I.; Burn T. C.; Millholland J. M.; VanRaay T. J.; Shen J.; Timothy K. W.; Vincent G. M.; Jager T. de; Schwartz P. J.; Towbin J. A.; Moss A. J.; Atkinson D. L.; Landes G. M.; Connors T. D.; Keating M. T. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat. Genet. 1996, 12, 17–23. 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- Wei A. D.; Butler A. G.; Salkoff L. B. KCNQ-like potassium channels in C. elegans: Conserved properties and modulation. J. Biol. Chem. 2005, 280, 21337–21345. 10.1074/jbc.M502734200. [DOI] [PubMed] [Google Scholar]
- Wang H. S.; Pan Z.; Shi W.; Brown B. S.; Wymore R. S.; Cohen I. S.; Dixon J. E.; McKinnon D. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science 1998, 282, 1890–1893. 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- Xiong Q. J.; Gao Z. B.; Wang W.; Li M. Activation of Kv7 (KCNQ) voltage-gated potassium channels by synthetic compounds. Trends Pharmacol. Sci. 2008, 29, 99–107. 10.1016/j.tips.2007.11.010. [DOI] [PubMed] [Google Scholar]
- Wulff H.; Castle N. A.; Pardo L. A. Voltage-gated potassium channels as therapeutic targets. Nat. Rev. Drug Discovery 2009, 8, 982–1001. 10.1038/nrd2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickenden A. D.; McNaughton-Smith G. Kv7 channels as targets for the treatment of pain. Curr. Pharm. Des. 2009, 15, 1773–1798. 10.2174/138161209788186326. [DOI] [PubMed] [Google Scholar]
- Zheng Y. M.; Xu H. Y.; Zhan L.; Zhou X. D.; Chen X. Q.; Gao Z. B. Activation of peripheral KCNQ channels relieves gout pain. Pain 2015, 156, 1025–1035. 10.1097/j.pain.0000000000000122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Main M. J.; Cryan J. E.; Dupere J. R. B.; Cox B.; Clare J. J.; Burbidge S. A. Modulation of KCNQ2/3 potassium channels by the novel anticonvulsant retigabine. Mol. Pharmacol. 2002, 58, 253–262. 10.1124/mol.58.2.253. [DOI] [PubMed] [Google Scholar]
- Tatulian L.; Delmas P.; Abogadie F. C.; Brown D. A. Activation of expressed KCNQ potassium currents and native neuronal M-type potassium currents by the anti-convulsant drug retigabine. J. Neurosci. 2001, 21, 5535–5545. 10.1523/JNEUROSCI.21-15-05535.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson U. A.; Carson C.; Johnston L.; Joshi S.; Gurney A. M.; McCloskey K. D. Functional expression of KCNQ (Kv7) channels in guinea pig bladder smooth muscle and their contribution to spontaneous activity. Br. J. Pharmacol. 2013, 169, 1290–1304. 10.1111/bph.12210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rode F.; Svalø J.; Sheykhzade M.; Rønn L. C. B. Functional effects of the KCNQ modulators retigabine and XE991 in the rat urinary bladder. Eur. J. Pharmacol. 2010, 638, 121–127. 10.1016/j.ejphar.2010.03.050. [DOI] [PubMed] [Google Scholar]
- Yeung S. Y.; Pucovský V.; Moffatt J. D.; Saldanha L.; Schwake M.; Ohya S.; Greenwood I. A. Molecular expression and pharmacological identification of a role for Kv7 channels in murine vascular reactivity. Br. J. Pharmacol. 2007, 151, 758–770. 10.1038/sj.bjp.0707284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jepps T. A.; Greenwood I. A.; Moffatt J. D.; Sanders K. M.; Ohya S. Molecular and functional characterization of Kv7 K+ channel in murine gastrointestinal smooth muscles. Am. J. Physiol. Gastrointest. Liver. Physiol. 2009, 297, 107–115. 10.1152/ajpgi.00057.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bientinesi R.; Mancuso C.; Martire M.; Bassi P. F.; Sacco E.; Currò D. Kv7 channels in the human detrusor: channel modulator effects and gene and protein expression. Naunyn-Schmiedeberg's Arch. Pharmacol. 2017, 390, 127–137. 10.1007/s00210-016-1312-9. [DOI] [PubMed] [Google Scholar]
- Brueggemann L. I.; Kakad P. P.; Love R. B.; Solway J.; Dowell M. L.; Cribbs L. L.; Byron K. L. Kv7 potassium channels in airway smooth muscle cells: signal transduction intermediates and pharmacological targets for bronchodilator therapy. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2012, 302, 120–132. 10.1152/ajplung.00194.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evseev A. I.; Semenov I.; Archer C. R.; Medina J. L.; Dube P. H.; Shapiro M. S.; Brenner R. Functional effects of KCNQ K+ channels in airway smooth muscle. Front. Physiol. 2013, 4, 277–287. 10.3389/fphys.2013.00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCallum L. A.; Pierce S. L.; England S. K.; Greenwood I. A.; Tribe R. M. The contribution of Kv7 channels to pregnant mouse and human myometrial contractility. J. Cell. Mol. Med. 2011, 15, 577–586. 10.1111/j.1582-4934.2010.01021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung S. Y.; Schwake M.; Pucovský V.; Greenwood I. A. Bimodal effects of the Kv7 channel activator retigabine on vascular K+ currents. Br. J. Pharmacol. 2008, 155, 62–72. 10.1038/bjp.2008.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong X. Z.; Harhun M. I.; Olesen S. P.; Ohya S.; Moffatt J. D.; Cole W. C.; Greenwood I. A. Participation of KCNQ (Kv7) potassium channels in myogenic control of cerebral arterial diameter. J. Physiol. 2010, 588, 3277–3293. 10.1113/jphysiol.2010.192823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haick J. M.; Byron K. L. Novel treatment strategies for smooth muscle disorders: Targeting Kv7 potassium channels. Pharmacol. Ther. 2016, 165, 14–25. 10.1016/j.pharmthera.2016.05.002. [DOI] [PubMed] [Google Scholar]
- Kalappa B. I.; Soh H.; Duignan K. M.; Furuya T.; Edwards S.; Tzingounis A. V.; Tzounopoulos T. Potent KCNQ2/3-specific channel activator suppresses in vivo epileptic activity and prevents the development of tinnitus. J. Neurosci. 2015, 35, 8829–8842. 10.1523/JNEUROSCI.5176-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar M.; Reed N.; Liu R.; Aizenman E.; Wipf P.; Tzounopoulos T. Synthesis and evaluation of potent KCNQ2/3-specific channel activators. Mol. Pharmacol. 2016, 89, 667–677. 10.1124/mol.115.103200. [DOI] [PubMed] [Google Scholar]
- Zhou P. Z.; Zhang Y. M.; Xu H. Y.; Chen F.; Chen X. Q.; Li X. Y.; Pi X. P.; Wang L. P.; Zhan L.; Nan F. J.; Gao Z. B. P-Retigabine: An N-Propargyled Retigabine with Improved Brain Distribution and Enhanced Antiepileptic Activity. Mol. Pharmacol. 2015, 87, 31–38. 10.1124/mol.114.095190. [DOI] [PubMed] [Google Scholar]
- Hu H. N.; Zhou P. Z.; Chen F.; Li M.; Nan F. J.; Gao Z. B. Discovery of a retigabine derivative that inhibits KCNQ2 potassium channels. Acta Pharmacol. Sin. 2013, 34, 1359–1366. 10.1038/aps.2013.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; An H. l.; Li J. W.; Zhang Y. Y.; Liu Y.; Jia Z. F.; Zhang W.; Chu L.; Zhang H. L. Selective activation of vascular Kv7.4/Kv7.5 K+ channels by fasudil contributes to its vasorelaxant effect. Br. J. Pharmacol. 2016, 173, 3480–3491. 10.1111/bph.13639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe H.; Kamai T.; Hayashi K.; Anzai N.; Shirataki H.; Mizuno T.; Yamaguchi Y.; Masuda A.; Yuki H.; Betsunoh H.; Yashi M.; Fukabori Y.; Yoshida K. The Rho-kinase inhibitor HA-1077 suppresses proliferation/migration and induces apoptosis of urothelial cancer cells. BMC Cancer 2014, 14, 412–423. 10.1186/1471-2407-14-412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landoulsi Z.; Miceli F.; Palmese A.; Amoresano A.; Marino G.; Ayeb M. E.; Taglialatela M.; Benkhalifa R. Subtype-selective activation of Kv7 channels by AaTXKβ(2–64), a novel toxin variant from the Androctonus australis Scorpion Venom. Mol. Pharmacol. 2013, 84, 763–773. 10.1124/mol.113.088971. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.