

Abstract

Phosphoinositide 3-kinase (PI3Kγ) is a drug target that has been implicated in the treatment of a range of diseases. We have developed a synthesis of a novel PI3Kγ inhibitor containing a 1,2-dihydro-3H-pyrrolo[3,4-c]pyridin-3-one scaffold. The key step in the synthesis involved a ruthenium-catalyzed [2 + 2 + 2] cyclotrimerization reaction between a diyne and an alkoxycarbonyl isocyanate, a previously unreported coupling partner in such a reaction.
Keywords: cyclotrimerization, PI3Kγ, isocyanate, diyne, medicinal chemistry
The phosphoinositide 3-kinase (PI3K) family of lipid kinases catalyze the phosphorylation of the inositol ring of phosphoinositides. Through this phosphorylation, PI3Ks control a number of important cellular processes such as growth, proliferation, and survival.1,2 PI3Kγ, a member of the most-studied Class I PI3Ks, is activated through G protein-coupled receptors, and its expression is restricted to the hematopoietic system.3,4 Inhibitors of PI3Kγ are expected to have utility in the treatment of autoimmune diseases, such as multiple sclerosis, due to the role of PI3Kγ in lymphocyte chemotaxis and in the production of reactive oxygen species.5−8 The intricate mechanisms involved are still being elucidated.9 There is also evidence linking PI3Kγ inhibition to the treatment of cancer and cardiovascular disease.10,11 A number of selective inhibitors of PI3Kγ have been characterized.5,12−16
Recently, we have described the design and synthesis of isoform selective inhibitors of PI3Kγ based around a 4-aza-isoindolinone core (for example, 1) with the potential for the treatment of multiple sclerosis (Figure 1).5 During the course of a routine structure activity relationship (SAR) study in that program, we required access to analog 2. Because 6-aza-isoindolinones with this substitution pattern had no precedence in the literature at the time,17 we considered the use of ruthenium-catalyzed [2 + 2 + 2] cyclotrimerization methodology described by Yamamoto et al. for the key step of the synthesis.18 The transition metal-catalyzed [2 + 2 + 2] cyclotrimerization reaction between diynes and nitriles or isocyanates has emerged in recent years as a powerful method for the assembly of complex pyridines and pyridones in a single atom-economical and environmentally benign step.19−22
Figure 1.

Reported PI3Kγ inhibitor 1 and target analog 2.
A requirement in our retrosynthetic planning (Figure 2) was that the R group of 4 be labile so that the desired 6-aza-isoindolinone scaffold, 3 could be accessed by a simple deprotection/chlorination/Suzuki sequence on 4. This required that the key cycloaddition step be carried out between amide-diyne 5 and an isocyanate 6, bearing a labile appendage (R). Isocyanates containing easily cleavable pendant groups have not previously been employed, however, in [2 + 2 + 2] cyclotrimerization reactions with diynes of any type. In addition, we needed to confirm that the key cycloaddition step would tolerate the more drug-like, substituted pyrazole group present in coupling partner 5.
Figure 2.
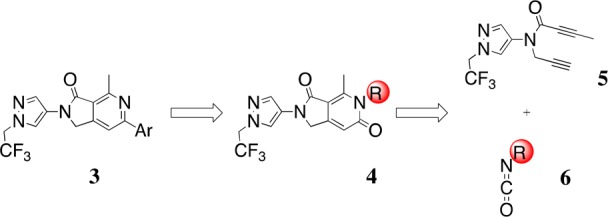
Retrosynthetic analysis of 3. The R group of isocyanate 6 needed to be labile to allow its cleavage from synthetic intermediate 4 en route to the 6-aza-isoindolinone scaffold 3.
In the forward direction (see Scheme 1), readily available 1-(2,2,2-trifluoroethyl)-1H-pyrazol-4-amine23 was alkylated with propargyl bromide to give 7 in 51% yield. The subsequent amide coupling of 7 with but-2-yonoic acid gave amide-diyne 5, the precursor for the key cyclotrimerization step. We were pleased to observe that slow addition of amide-diyne 5 to a mixture of Cp*RuCl(cod) and ethoxycarbonyl isocyanate in tetrahydrofuran at room temperature, followed by heating for 20 min at 65 °C, gave cycloaddition product 8 in an unoptimized yield of 47% as the only detected regioisomer. To the best of our knowledge, this is the first time that an alkoxycarbonyl isocyanate has been employed as a reaction partner in a cyclotrimerization reaction of this type. Acid hydrolysis of the ethoxycarbonyl group of 8 and subsequent chlorination with phosphorus oxychloride gave 9, a substrate suitable for late stage diversification. Suzuki–Miyaura coupling gave dimethoxypyridine 2 in 77% yield.
Scheme 1.

Reagents and conditions: (a) propargyl bromide, K2CO3, DMF, 0 °C to RT, 51%; (b) but-2-ynoic acid, EDCI, DIPEA, DMAP, DCM, 0 °C to RT, 41%; (c) ethoxycarbonyl isocyanate, Cp*RuCl(cod), DCE, 60 °C, 47%; (d) 6 M HCl, THF, 95 °C then POCl3, 95 °C, 68% (2 steps); (e) 2,3-dimethoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine, Pd(PPh3)4, aq. Na2CO3, DMF, 110 °C, 77%.
Considering that this methodology could be a useful way to populate our corporate compound collection with novel drug-like chemical matter, we investigated the scope of the cyclotrimerization reaction with a range of isocyanates, and the results are shown in Table 1. The reaction was very functional-group-tolerant, providing products from the reaction of isocyanates bearing substituents as diverse as ethers (entries 1 and 7), haloalkanes (entries 2 and 4), an acetal (entry 3), an ester (entry 5), a thioether (entry 6), and a sulfone (entry 8). Aryl and heteroaryl isocyanates were moderately competent coupling partners (entries 9, 11, and 12). The reaction also proceeded smoothly with 2-isocyanatospiro[3.3]heptane (entry 10) and a substituted cyclopropyl isocyanate (entry 13). The reason for the moderate to low yields in some of the cyclotrimerization reactions was due to a major byproduct, which was characterized as a dimeric material originating from self-coupling of the diyne component of the reaction. Further experimentation will be necessary to minimize this competing reaction pathway, which appears to dominate as less reactive or very sterically demanding isocyanates are employed (entries 14–16).
Table 1. Scope of the Cyclocotrimerization Reaction.


Compound 2 displayed a high PI3Kγ affinity of 14 nM (Ki) and inhibited the MCP-1 induced chemotaxis of THP-1 cells with an IC50 of 270 nM. The corresponding values for compound 1 are 4 and 47 nM, respectively. We hypothesize that the reason for the loss of potency of 2 in comparison with compounds from the 4-aza-isoindolinone class, such as 1, is due to the energy penalty associated with placement of the heteroaromatic nitrogen of 2 in the hydrophobic environment surrounding Tyr867 (Figure 3), although this potency loss is partially offset by the addition of the C7-methyl group to the core of the scaffold. A detailed report on the SAR at C7 is forthcoming. Compound 2 was less selective than compound 1 for PI3Kγ over the other class Ia isoforms (fold selectivity over α/β/δ for compound 1, 65:31:13; for compound 2, 15:8:6).
Figure 3.
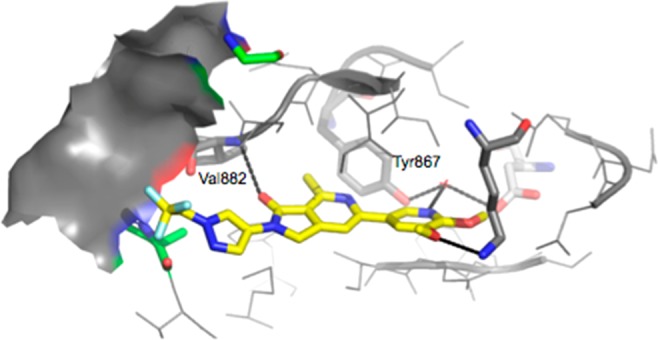
Docking model for 2 bound to PI3Kγ. Receptor used is from the crystal structure of compound 65/PI3Kγ (PDB code: 6C1S). We hypothesize that the loss in affinity is due to placement of a heteroaromatic nitrogen atom in hydrophobic environment surrounding Tyr867.
Much has been reported in the literature regarding the over-reliance on certain classical chemical reaction types in medicinal chemistry, which is leading to narrow population of chemical space.24−27 We believe that addition of more modern synthetic methodologies to the medicinal chemist’s toolbox will be important in serving as a source of novel compounds for the prosecution of the challenging targets and projects of a biopharma industry currently struggling with an innovation crisis.28
In summary, we have developed a practical and efficient synthetic route to a 6-aza-isoindolinone-based inhibitor of PI3Kγ. The key step involved a ruthenium-catalyzed [2 + 2 + 2] cyclotrimerization reaction between a diyne and an alkoxycarbonyl isocyanate, a novel coupling partner in such a reaction. A rationale was proposed for the reduced PI3Kγ potency of compound 2 versus analog 1. Further extension of the scope of the ruthenium-catalyzed [2 + 2 + 2] cyclotrimerization methodology by varying the isocyanate component produced a small library of structurally diverse analogs.
Acknowledgments
We thank Barry Davis for high-resolution mass spectrometry assistance.
Glossary
Abbreviations
- GPCR
G-protein coupled receptor
- PDB
protein data bank
- THF
tetrahydrofuran
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00530.
Description of PI3Kγ inhibition assay, description of cellular assay, synthetic procedures and characterization of compounds, NMR (1H, 19F, 13C) spectra (PDF)
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Vanhaesebroeck B.; Guillermet-Guibert J.; Graupera M.; Bilanges B. The emerging mechanisms of isoform-specific PI3K signaling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. 10.1038/nrm2882. [DOI] [PubMed] [Google Scholar]
- Hawkins P. T.; Anderson K. E.; Davidson K.; Stephens L. R. Signalling through class I PI3Ks in mammalian cells. Biochem. Soc. Trans. 2006, 34, 647–662. 10.1042/BST0340647. [DOI] [PubMed] [Google Scholar]
- Vadas O.; Dbouk H. A.; Shymanets A.; Perisic O.; Burke J. E.; Abi Saab W. F.; Khalil B. D.; Harteneck C.; Bresnick A. R.; Nürnberg B.; Backer J. M.; Williams R. L. Molecular determinants of PI3Kγ-mediated activation downstream of G-protein-coupled receptors (GPCRs). Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 18862–18867. 10.1073/pnas.1304801110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kok K.; Geering B.; Vanhaesebroeck B. Regulation of phosphoinositide 3-kinase expression in health and disease. Trends Biochem. Sci. 2009, 34, 115–127. 10.1016/j.tibs.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Come J. H.; Collier P. N.; Henderson J. A.; Pierce A. C.; Davies R. J.; Le Tiran A.; O’Dowd H.; Bandarage U. K.; Cao J.; Deininger D.; Grey R.; Krueger E. B.; Lowe D. B.; Liang J.; Liao Y.; Messersmith D.; Nanthakumar S.; Sizensky E.; Wang J.; Xu J.; Chin E. Y.; Damagnez V.; Doran J. D.; Dworakowski W.; Jacobs M. D.; Khare-Pandit S.; Mahajan S.; Moody C. S.; Aronov A. M.; Griffith J. P. Design and synthesis of a novel series of series of orally bioavailable, CNS-penetrant, isoform selective phosphoinositide 3-kinaase γ (PI3Kγ) with potential for the treatment of multiple sclerosis. J. Med. Chem. 2018, 61, 5245–5256. 10.1021/acs.jmedchem.8b00085. [DOI] [PubMed] [Google Scholar]
- Lull M. E.; Block M. L. Microglial activation and chronic neurodegeneration. Neurotherapeutics 2010, 7, 354–365. 10.1016/j.nurt.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camps M.; Rückle T.; Ji H.; Ardissone V.; Rintelen F.; Shaw J.; Ferrandi C.; Chabert C.; Gillieron C.; Francon B.; Martin T.; Gretener D.; Perrin D.; Leroy D.; Vitte P.-A.; Hirsch E.; Wymann M. P.; Cirillo R.; Schwarz M. K.; Rommel C. Blockade of PI3Kγ suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat. Med. 2005, 11, 936–943. 10.1038/nm1284. [DOI] [PubMed] [Google Scholar]
- Li D.; Park D.; Abdul-Muneer P. M.; Xu B.; Wang H.; Xing B.; Wu D.; Li S. PI3Kγ Inhibition alleviates symptoms and increases axon number in experimental autoimmune encephalomyelitis mice. Neuroscience 2013, 253, 89–99. 10.1016/j.neuroscience.2013.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazaki Y.; Nishimura Y.; Sabe H. GBF1 bears a novel phosphatidylinositol-phosphate binding module, BP3K, to link PI3Kγ activity with Arf1 activation involved in GPCR-mediated neutrophil chemotaxis and superoxide production. Mol. Biol. Cell 2012, 23, 2457–2467. 10.1091/mbc.e12-01-0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siragusa M.; Katare R.; Meloni M.; Damilano F.; Hirsch E.; Emanueli C.; Madeddu P. Involvement of phosphoinositide 3-kinase γ in angiogenesis and healing of experimental myocardial infarction in mice. Circ. Res. 2010, 106, 757–768. 10.1161/CIRCRESAHA.109.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okkenhaug K.; Graupera M.; Vanhaesebroeck B. Targeting PI3K in cancer: impact on tumor cells, their protective stroma, angiogenesis, and immunotherapy. Cancer Discovery 2016, 6, 1090–1105. 10.1158/2159-8290.CD-16-0716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier P. N.; Martinez-Botella G.; Cornebise M.; Cottrell K. M.; Doran J. D.; Griffith J. P.; Mahajan S.; Maltais F.; Moody C. S.; Huck E. P.; Wang T.; Aronov A. M. Structural basis for isoform selectivity in a class of benzothiazole inhibitors of phosphoinositide 3- kinase γ. J. Med. Chem. 2015, 58, 517–521. 10.1021/jm500362j. [DOI] [PubMed] [Google Scholar]
- Collier P. N.; Messersmith D.; Le Tiran A.; Bandarage U. K.; Boucher C.; Come J.; Cottrell K. M.; Damagnez V.; Doran J. D.; Griffith J. P.; Khare-Pandit S.; Krueger E. B.; Ledeboer M. W.; Ledford B.; Liao Y.; Mahajan S.; Moody C. S.; Roday S.; Wang T.; Xu J.; Aronov A. M. Discovery of highly isoform selective thiazolopiperidine inhibitors of phosphoinositide 3-kinase γ. J. Med. Chem. 2015, 58, 5684–5688. 10.1021/acs.jmedchem.5b00498. [DOI] [PubMed] [Google Scholar]
- Evans C. A.; Liu T.; Lescarbeau A.; Nair S. J.; Grenier L.; Pradeilles J. A.; Glenadel Q.; Tibbitts T.; Rowley A. M.; DiNitto J. P.; Brophy E. E.; O’Hearn E. L.; Ali J. A.; Winkler D. G.; Goldstein S. I.; O’Hearn P.; Martin C. M.; Hoyt J. G.; Soglia J. R.; Cheung C.; Pink M. M.; Proctor J. L.; Palombella V. J.; Tremblay M. R.; Castro A. C. Discovery of a selective phosphoinositide-3-kinase (PI3K)-γ inhibitor (IPI-549) as an immuno-oncology clinical candidate. ACS Med. Chem. Lett. 2016, 7, 862–867. 10.1021/acsmedchemlett.6b00238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pemberton N.; Mogemark M.; Arlbrandt S.; Bold P.; Cox R. J.; Gardelli C.; Holden N. S.; Karabelas K.; Karlsson J.; Lever S.; Li X.; Lindmark H.; Norberg M.; Perry M. W. D.; Petersen J.; Blomqvist S. R.; Thomas M. J.; Tyrchan C.; Eriksson A. W.; Zlatoidsky P.; Oster L. Discovery of highly isoform selective orally bioavailable phosphoinositide 3-kinase (PI3K)-γ inhibitors. J. Med. Chem. 2018, 61, 5435–5441. 10.1021/acs.jmedchem.8b00447. [DOI] [PubMed] [Google Scholar]
- Leahy J. W.; Buhr C. A.; Johnson H. W. B.; Kim B. G.; Baik T.; Cannoy J.; Forsyth T.; Jeong J. W.; Lee M. S.; Ma S.; Noson K.; Wang L.; Williams M.; Nuss J. M.; Brooks E.; Foster P.; Goon L.; Heald N.; Holst C.; Jaeger C.; Lam S.; Lougheed J.; Nguyen L.; Plonowski A.; Song J.; Stout T.; Wu X.; Yakes M. F.; Yu P.; Zhang W.; Lamb P.; Raeber O. Discovery of a novel series of potent and orally bioavailable phosphoinositide 3-kinase γ inhibitors. J. Med. Chem. 2012, 55, 5467–5482. 10.1021/jm300403a. [DOI] [PubMed] [Google Scholar]
- Gadzhili R. A.; Dikusar E. A.; Aliev A. G.; Mamedova G. M.; Potkin V. I.; Alieva S. K. Reactions of ethyl 6-alkyl(phenyl)-2-methyl-4-(chloromethyl)-pyridine-3-carboxylates with some O- and N-nucleophiles. Russ. J. Org. Chem. 2015, 51, 1166–1169. 10.1134/S1070428015080163. [DOI] [Google Scholar]
- Yamamoto Y.; Kinpara K.; Saigoku T.; Takagishi H.; Okuda S.; Nishiyama H.; Itoh K. Cp*RuCl-catalyzed [2 + 2 + 2] cycloadditions of α,ω–diynes with electron-deficient carbon-heteroatom multiple bonds leading to heterocycles. J. Am. Chem. Soc. 2005, 127, 605–613. 10.1021/ja045694g. [DOI] [PubMed] [Google Scholar]
- Broere D. L. J.; Ruijter E. Recent advances in transition-metal-catalyzed [2 + 2+2]-cyclo(co)trimerization reactions. Synthesis 2012, 44, 2639–2672. 10.1055/s-0032-1316757. [DOI] [Google Scholar]
- Alvarez S.; Medina S.; Domínguez G.; Pérez-Castells J. [2 + 2+2] cyclotrimerization of alkynes and isocyanates/isothiocyanates catalyzed by ruthenium-alkylidene complexes. J. Org. Chem. 2013, 78, 9995–10001. 10.1021/jo401538p. [DOI] [PubMed] [Google Scholar]
- Bhatt D.; Patel N.; Chowdhury H.; Bharatam P. V.; Goswami A. Additive-controlled switchable selectivity from cyanobenzenes to 2-alkylpyridines: ruthenium(II)-catalyzed [2 + 2+2] cycloadditions of diynes and alkynylnitriles. Adv. Synth. Catal. 2018, 360, 1876–1882. 10.1002/adsc.201800228. [DOI] [Google Scholar]
- Onodera G.; Suto M.; Takeuchi R. Iridium-catalyzed [2 + 2+2] cycloaddition of α,ω–diynes with isocyanates. J. Org. Chem. 2012, 77, 908–920. 10.1021/jo202083z. [DOI] [PubMed] [Google Scholar]
- Oguro Y.; Miyamoto N.; Takagi T.; Okada K.; Awazu Y.; Miki H.; Hori A.; Kamiyama K.; Imamura S. N-Phenyl-N’-[4-(5H-pyrrolo[3,2-d]pyrimidin-4-yloxy)phenyl]ureas as novel inhibitors of VEGFR and FGFR kinases. Bioorg. Med. Chem. 2010, 18, 7150–7163. 10.1016/j.bmc.2010.08.042. [DOI] [PubMed] [Google Scholar]
- Boström J.; Brown D. G.; Young R. J.; Keserü G. M. Expanding the medicinal chemistry toolbox. Nat. Rev. Drug Discovery 2018, 17, 709–727. 10.1038/nrd.2018.116. [DOI] [PubMed] [Google Scholar]
- Roughley S. D.; Jordan A. M. The medicinal chemist’s toolbox: an analysis of reactions used in the pursuit of drug candidates. J. Med. Chem. 2011, 54, 3451–3479. 10.1021/jm200187y. [DOI] [PubMed] [Google Scholar]
- Cooper T. W. J.; Campbell I. B.; Macdonald S. J. F. Factors determining the selection of organic reactions by medicinal chemists and the use of these reactions in arrays (small focused libraries). Angew. Chem., Int. Ed. 2010, 49, 8082–8091. 10.1002/anie.201002238. [DOI] [PubMed] [Google Scholar]
- Schneider N.; Lowe D. M.; Sayle R. A.; Tarselli M. A.; Landrum G. A. Big data from pharmaceuticals patents: a computational analysis of medicinal chemist’s bread and butter. J. Med. Chem. 2016, 59, 4385–4402. 10.1021/acs.jmedchem.6b00153. [DOI] [PubMed] [Google Scholar]
- Waring M. J.; Arrowsmith J.; Leach A. R.; Leeson P. D.; Mandrell S.; Owen R. M.; Pairaudeau G.; Pennie W. D.; Pickett S. D.; Wang J.; Wallace O.; Weir A. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discovery 2015, 14, 475–486. 10.1038/nrd4609. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.