

Abstract

Dual inhibition of angiotensin-converting enzyme (ACE) and neprilysin (NEP) by drugs such as omapatrilat produces superior antihypertensive efficacy relative to ACE inhibitors but is associated with a higher risk of life-threatening angioedema due to bradykinin elevations. We hypothesized that dual AT1 (angiotensin II type 1 receptor) blockade and NEP inhibition with a single molecule would produce similar antihypertensive efficacy to omapatrilat without the risk of angioedema since ACE (the rate limiting enzyme in bradykinin metabolism) would remain uninhibited. Merging the structures of losartan (an AT1 antagonist) and thiorphan (a NEP inhibitor) led to the discovery of a novel series of orally active, dual AT1 antagonist/NEP inhibitors (ARNIs) exemplified by compound 35 (TD-0212). In models of renin-dependent and -independent hypertension, 35 produced blood pressure reductions similar to omapatrilat and combinations of AT1 receptor antagonists and NEP inhibitors. Upper airway angioedema risk was assessed in a rat tracheal plasma extravasation (TPE) model. Unlike omapatrilat, 35 did not increase TPE at antihypertensive doses. Compound 35 therefore provides the enhanced activity of dual AT1/NEP inhibition with a potentially lower risk of angioedema relative to dual ACE/NEP inhibition.
Keywords: Angiotensin, AT1 antagonist, neprilysin inhibitor, dual pharmacology, heterodimer
Angiotensin II is a potent vasoconstrictive peptide in the renin angiotensin system (RAS) that activates the G-protein coupled AT1 receptor.1 Other primary actions of angiotensin II include stimulation of aldosterone secretion by the kidneys, renal reabsorption of sodium,2 and cardiac stimulation.3 An AT1 receptor antagonist (ARB) blocks the vasoconstrictor and aldosterone-secreting effects of angiotensin II by selectively blocking binding of angiotensin II to the AT1 receptor.4 Numerous ARBs (Figure 1) have been described in the literature, and several have been approved by the FDA for the treatment of conditions such as hypertension and heart failure.5
Figure 1.

Selected FDA-approved ARBs.
Natriuretic peptides [atrial natriuretic peptide (ANP), brain-derived natriuretic peptide (BNP), and C-type natriuretic peptide (CNP)] effect peripheral vasodilation, increased renal sodium excretion, reduction of RAS activity, and promotion of antihypertrophic and antifibrotic effects, which are mediated through activation of guanylate cyclase signaling pathways. NEP is a zinc metalloprotease enzyme,6 inhibition of which potentiates the activity of the cardiac natriuretic peptide system (CNPS) by inhibiting breakdown of the natriuretic peptides, mainly ANP.7,8 To date, only one NEP inhibitor (sacubitril, Figure 2) has been approved by the FDA for clinical use (in a fixed-dose combination with the ARB valsartan).
Figure 2.

Selected NEP and ACE/NEP inhibitors.
Since the RAS and the CNPS are interconnected and both are critical in cardiovascular homeostasis, simultaneous inhibition of both systems has the potential to provide superior reductions in blood pressure, increased responder rates, improved cardiac performance, reduction of target-organ damage, and improvements in long-term outcomes in hypertensives and chronic heart failure (CHF) patients.
Dual inhibition of the RAS and CNPS with a single molecule has been attempted by different companies in a number of R&D programs. Omapatrilat is an agent that inhibits both ACE (RAS) and NEP (CNPS). Clinical hypertension trials established its efficacy in patients with mild to moderate hypertension, and the 25,000 patient OCTAVE study demonstrated broadly superior antihypertensive efficacy of omapatrilat relative to the ACE inhibitor enalapril.9 Heart failure trials showed comparable efficacy between omapatrilat and lisinopril (another ACE inhibitor) in improving exercise tolerance in patients with NYHA class II/III heart failure, with a strong suggestion that omapatrilat improved clinical outcomes in heart failure to a greater extent than lisinopril (IMPRESS trial).10 However, the 3-fold higher incidence of angioedema observed for omapatrilat relative to lisinopril and the incidence of life threatening angioedema in the OCTAVE study led to an unfavorable risk–benefit profile and the program was discontinued in 2002. While ACE is the predominant rate limiting enzyme in the metabolism of bradykinin, NEP, APP, DPP-IV, and other zinc metallopeptidases also play a role in bradykinin breakdown. The prevailing hypothesis is that dual inhibition of ACE and NEP by omapatrilat results in significantly greater elevation of bradykinin levels relative to ACE inhibition alone, which in a subset of patients can lead to excessive vascular leakage, fluid extravasation, and angioedema.11
Our expectation was that inhibition of the RAS with an AT1 antagonist/NEP inhibitor (ARNI) rather than an ACE/NEP inhibitor would result in an efficacy profile similar to that of omapatrilat without the risk for bradykinin elevation since ACE would remain uninhibited. A similar strategy led to the approval in 2015 of Entresto (LCZ696, a fixed dose 1:1 coformulation of the ARB valsartan with the NEP inhibitor prodrug sacubitril) for the treatment of CHF.12 An alternative to coformulation is to design a molecule with potent activity at two different targets.13 Potential benefits of this approach include matched pharmacodynamic (PD) effects on the targets of interest, thereby maximizing the potential synergy between the two mechanisms, and reduced potential for drug–drug interactions compared to drug cocktails. We therefore set out to design an ARNI by chemically merging the structure of an ARB with that of a NEP inhibitor.
The 5-position of the imidazole ring in the ARB losartan has been shown to accommodate a wide range of substituents, including some that provide a second pharmacological activity.14−18 Using a similar strategy, we linked a losartan analog (Intermediate A) to the terminal carboxylic acid of thiorphan, a thiol-based inhibitor of NEP. While thiol-based drugs are associated with complex PK/PD relationships and development risks,19 thiorphan was preferred over other NEP inhibitors due to its low molecular weight and high potency (Scheme 1). Heterodimer 1 maintained potent binding affinity at the AT1 receptor relative to losartan (Table 1) but was significantly less potent than thiorphan in the NEP inhibition assay.
Scheme 1. Synthesis of Losartan–Thiorphan Heterodimer 1.

Reagents and conditions: (a) EDC, HOBt, DIPEA, DMF, rt; (b) NaOH, MeOH, rt.
Table 1. Exploration of S1′ and Biphenyl Substitution.


This data suggested that the AT1 active site was more tolerant than that of NEP with respect to addition of a second pharmacophore. Removing the glycine spacer (compound 2) led to a minor loss of potency at both targets. However, the lower molecular weight and reduced conformational flexibility of 2 was considered more conducive to achieving oral activity, and so 2 became the focal point of our early optimization efforts.
Assuming the thiorphan fragment of 2 binds to NEP in a similar fashion to thiorphan itself, the benzyl substituent (R2) of 2 would occupy the S1′ subsite of the enzyme. Replacement of the benzyl group with another S1′ substituent commonly utilized in NEP inhibitors (rac-isobutyl, 3) had little impact on potency at either target (Table 1). Surprisingly, the first significant improvement in NEP potency was achieved by modification of the ARB region of the molecule. Replacing the tetrazole moiety (R1) on the biphenyl fragment with a carboxylic acid (5 vs 3) afforded a 1.7 log unit gain in NEP pIC50 when the S1′ group was rac-isobutyl. Synthesis of the individual enantiomers of 5 revealed that the AT1 receptor had a clear preference for S-enantiomer 7. An extensive survey of other alkyl and aromatic groups in the S1′ pocket (compounds 8–19) demonstrated that the S-isobutyl group maintained the best balance of potency at the two targets. Replacing the β-thiol zinc-chelator (7) with an α-thiol (21) increased NEP potency to a level similar to that of thiorphan, but simultaneously reduced AT1 potency. Since the α-thiols (low AT1, high NEP activity) provided a distinct profile to the β-thiols (high AT1, low NEP activity), both series were progressed to SAR studies focused on the imidazole headgroup substituents.
Replacement of the imidazole 4-Cl substituent [R3, (Table 2)] with a methyl group (23, 24) reduced the AT1 activity in both the α- and β-thiol series. The bulky i-Bu (28) group was not tolerated by NEP in the α-thiol series. Intermediate-size alkyls such as ethyl (24) and cyclopropyl (26) were relatively well-tolerated by both active sites. At the 2-position of the imidazole (R4), insertion of an oxygen atom into the alkyl chain (30 vs 24) was slightly beneficial in terms of both AT1 and NEP potency. An additional gain in AT1 potency was realized by shortening the alkoxy chain from four atoms to three atoms (32 vs 30). The AT1 potency of this ARNI series was now greater than that of the parent ARB, losartan. Using compounds 32 and 33 as starting points, a limited substitution scan on the biphenyl fragment was performed. Adding a fluorine atom ortho- to the headgroup maintained NEP activity and significantly boosted AT1 potency in the α-thiol series (35 vs 33). Alternative fluorine substitution patterns were neutral at AT1 but deleterious to NEP relative to the parent molecule. Biphenyl substitutions did not have a significant effect in the β-thiol analogs.
Table 2. Exploration of Imidazole Headgroup and Biphenyl Substitution.

| compound | R3 | R4 | R5 | R6 | n | AT1 pKi | NEP pIC50 | % inhibition of ang-II pressor response (3 mg/kg, iv) | urinary cGMP fold increase over vehicle (3 mg/kg, iv) |
|---|---|---|---|---|---|---|---|---|---|
| losartan | 8.3 | <5 | 67 | ||||||
| omapatrilat | <5 | 9.7 | 5.3 | ||||||
| 7 | Cl | n-Bu | H | H | 1 | 7.8 | 7.2 | 11 | 2.2 |
| 21 | Cl | n-Bu | H | H | 0 | 7.2 | 9 | ||
| 22 | Me | n-Bu | H | H | 1 | 7.3 | 7.1 | ||
| 23 | Me | n-Bu | H | H | 0 | 6.7 | 9.2 | ||
| 24 | Et | n-Bu | H | H | 1 | 8.3 | 6.9 | 68 | 2.6 |
| 25 | Et | n-Bu | H | H | 0 | 7.4 | 8.5 | 18 | 3.2 |
| 26 | Cyclopropyl | n-Bu | H | H | 1 | 8.2 | 6.9 | 55 | 1.8 |
| 27 | Cyclopropyl | n-Bu | H | H | 0 | 7.5 | 9.0 | 5 | 4 |
| 28 | i-Bu | n-Bu | H | H | 1 | 8.1 | 6.9 | ||
| 29 | i-Bu | n-Bu | H | H | 0 | 7.5 | 7.7 | 32 | 1.9 |
| 30 | Et | PrO | H | H | 1 | 8.6 | 7.3 | 65 | 1.9 |
| 31 | Et | PrO | H | H | 0 | 7.7 | 9 | 23 | 3.8 |
| 32 | Et | EtO | H | H | 1 | 8.9 | 7.5 | 67 | 2.9 |
| 33 | Et | EtO | H | H | 0 | 8.2 | 9 | 57 | 3.6 |
| 34 | Et | EtO | F | H | 1 | 9.1 | 7.8 | 85 | 1.8 |
| 35 | Et | EtO | F | H | 0 | 8.9 | 9.2 | 76 | 3.5 |
| 36 | Et | EtO | H | F | 1 | 8.9 | 7.1 | ||
| 37 | Et | EtO | H | F | 0 | 8.3 | 8.5 | 57 | 3.3 |
| 38 | Et | EtO | F | F | 0 | 8.3 | 7.8 |
To determine the in vivo activity of these novel dual pharmacology molecules, a rat pharmacodynamic (PD) assay was developed to simultaneously measure AT1 antagonism via inhibition of the angiotensin-II-evoked pressor response and NEP inhibition via potentiation of ANP-induced elevation of urinary cyclic guanosine monophosphate (cGMP) output relative to vehicle.20 Compounds were dosed IV in the PD assay to determine the intrinsic activity of the parent molecule without oral absorption playing a role. In general, in vivo activity correlated with in vitro activity (Table 2). Compounds with pKi values >8.0 at AT1 inhibited the angiotensin-II blood pressure response to a similar extent as losartan. With respect to NEP inhibition, pIC50 values of >8.0 generally produced more than 3-fold increases in urinary cGMP relative to vehicle. None of the compounds were as active as omapatrilat in this assay, in keeping with their reduced in vitro potency relative to omapatrilat.
On the basis of its activity in the PD assay and rat pharmacokinetics (see Supporting Information), compound 35 (Scheme 2) was advanced to a dose–response study in the spontaneously hypertensive rat (SHR) model, which is known to be sensitive to ARBs.21 In this model, telemetry is used to monitor blood pressure for a period of 24 h after oral dosing of the compound. Efficacy was determined by the peak percent fall in mean arterial pressure (MAP) from baseline, while duration of action was reflected by the vehicle-adjusted % change in AUC over 24 h. Administration of sequential escalating doses of 3, 10, 30, and 100 mg/kg of 35 at 24 h intervals between the successive doses produced dose-dependent reductions in MAP (Figure 3). At doses of 10 mg/kg and above, the duration of effect was sustained for 24 h. In comparison to vehicle treatment the effect was statistically significant at 10 mg/kg and higher doses (p < 0.05, two way ANOVA with Bonferroni post hoc analysis).
Scheme 2. Synthesis of 35 (TD-0212).

Reagents and conditions: (a) SEMCl, DIPEA, DCM, rt; (b) KOt-Bu, EtOH, reflux; (c) DMF, n-BuLi, THF, −78 °C; (d) TFA, 0 °C; (e) K2CO3, DMF; (f) Pd(PPh3)4, (C2H3)Sn(n-Bu)3, DMF, 90 °C; (g) NH2OH.HCl, pyridine, H2O; (h) H2, Pd(OH)2, EtOH; (i) NaBH3CN, TiCl3, NH4OAc, MeOH, 0 °C; (j) (S)-2-(acetylthio)-4-methylpentanoic acid, HATU, DIPEA, DMF, rt; (k) TFA, DCM, rt; (l) NaOH, MeOH, H2O, rt.
Figure 3.
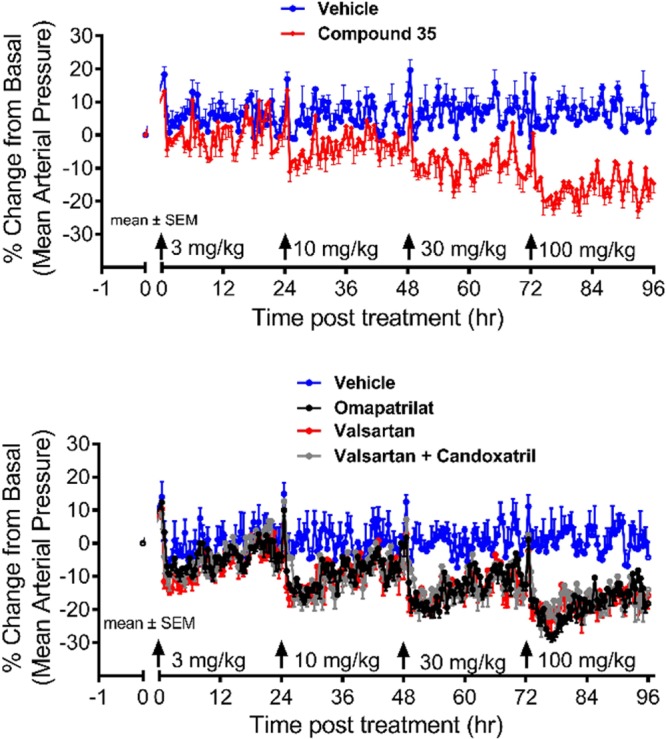
Comparison of 24 h average MAP reductions following oral administration of 35, omapatrilat, valsartan, or a valsartan/candoxatril combination in conscious SHR.
Omapatrilat and valsartan produced a dose-dependent reduction in MAP similar in magnitude to 35. The addition of the NEP inhibitor prodrug candoxatril (at a dose of 100 mg/kg) to valsartan did not result in further blood pressure lowering in the SHR model relative to valsartan alone, which is consistent with its non-RAS action. The estimated ED10 (dose required to produce an average 10% reduction in the 24 h average MAP) for 35 (ED10 = 13 mg/kg) was comparable to that of omapatrilat, valsartan, and the valsartan/candoxatril combination (ED10 = 15, 17, and 15 mg/kg respectively).
In vivo NEP inhibition after oral dosing was determined using the deoxycorticosterone acetate (DOCA) salt rat model of hypertension. The DOCA model is insensitive to ARBs and therefore considered a model for low-renin hypertension. Compound 35 produced a dose-dependent reduction of blood pressure in the DOCA model (Figure 4). The potency and duration of effect of 35 (ED10 = 44 mg/kg) were comparable to omapatrilat (ED10 = 66 mg/kg).
Figure 4.
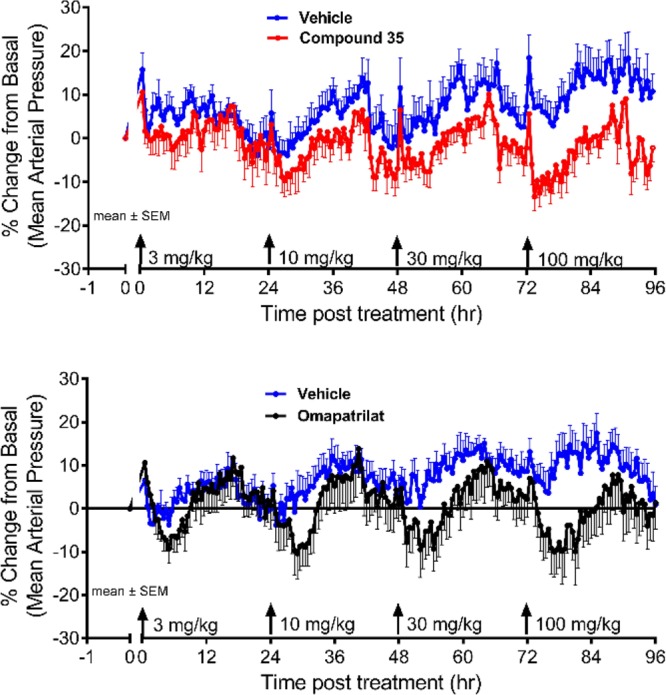
Comparison of antihypertensive efficacy and duration following oral administration of 35 or omapatrilat in conscious DOCA-salt hypertensive rats.
Additional in vitro studies confirmed that 35 is a potent, competitive antagonist of the AT1 receptor, with a selectivity for AT1 over AT2 of approximately 2000-fold (Table 3). In contrast to losartan, which is classified as a surmountable AT1 antagonist,2235 was found to be partially insurmountable in an inositol phosphate accumulation assay (<5% vs 87% accumulation, respectively). Compound 35 is also a potent, selective, and competitive inhibitor of NEP, exhibiting 120-fold selectivity for inhibition of human recombinant NEP over human recombinant ECE-1, and no measurable activity at human ACE or APP at a concentration of 10 μM.
Table 3. Selectivity Profile of Compound 35, Valsartan, and Omapatrilat against Related Targets.
| potency |
|||
|---|---|---|---|
| target | 35 | valsartan | omapatrilat |
| AT2a | 5.6 | <5.0 | ND |
| ACEb | <5.0 | ND | 10 |
| APPb | <5.0 | ND | 6.7 |
| ECE-1b | 7.1 | ND | ND |
pKi.
pIC50.
In order to assess the risk of angioedema with 35 relative to that with omapatrilat, a rat tracheal plasma extravasation (TPE) model was developed based on a method described by Sulpizio et al.23 In this model, TPE was used as a surrogate to assess the propensity of compounds to promote upper airway angioedema by measuring the extent of Evans Blue dye leakage into peritracheal tissue at different antihypertensive doses. Omapatrilat produced a robust increase in TPE at low oral subactive doses of 0.3 and 3 mg/kg. In contrast, 35 had no effect at doses of up to 100 mg/kg (Figure 5).
Figure 5.
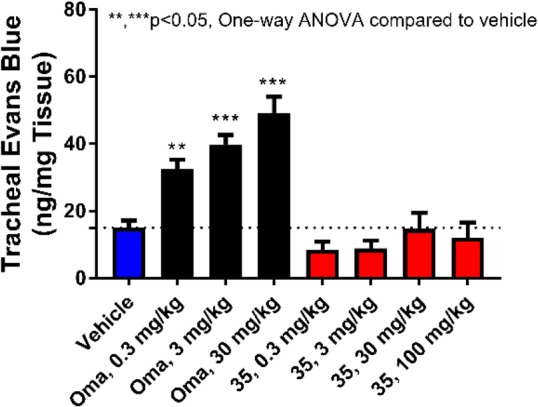
Comparison of tracheal plasma extravasation following administration of vehicle, omapatrilat, or 35 in normotensive rats.
The combined results from in vivo pharmacology studies indicate that 35 is as effective as omapatrilat in terms of antihypertensive activity. Unlike omapatrilat, 35 does not increase tracheal plasma extravasation in rats, which is indicative of a low risk for causing angioedema. Taken together, 35 can be described as an ARB-equivalent of omapatrilat with a lower risk of angioedema. Additional studies with 35 will be reported in future publications.
Acknowledgments
The authors would like to thank David Beattie, Venkat Thalladi, and Zhengtian Gu for assistance preparing this manuscript.
Glossary
ABBREVIATIONS
- AT1
angiotensin II type 1 receptor
- ACE
angiotensin-converting enzyme
- NEP
neprilysin
- TPE
tracheal plasma extravasation
- RAS
renin angiotensin-system
- ARB
AT1 receptor antagonist
- ANP
atrial natriuretic peptide
- BNP
brain-derived natriuretic peptide
- CNP
C-type natriuretic peptide
- CNPS
cardiac natriuretic peptide system
- APP
aminopeptidase P
- DPP-IV
Dipeptidyl Peptidase IV
- CHF
chronic heart failure
- SHR
spontaneously hypertensive rat
- MAP
mean arterial pressure
- DOCA
deoxycorticosterone acetate
- cGMP
guanosine monophosphate
- ED10
dose required to produce an average 10% reduction in the 24h average MAP
- ECE
endothelin converting enzyme 1
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00462.
In vitro and in vivo assay procedures; representative synthetic procedures and compound characterization data (PDF)
Author Contributions
The manuscript was written through contributions of all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Timmermans P. B.; Wong P. C.; Chiu A. T.; Herblin W. F.; Benfield P.; Carini D. J.; Lee R. J.; Wexler R. R.; Saye J. A.; Smith R. D. Angiotensin II receptors and angiotensin II receptor antagonists. Pharmacol. Rev. 1993, 45, 201–251. [PubMed] [Google Scholar]
- Hall J. E. The renin-angiotensin system: renal actions and blood pressure regulation. Compr. Ther 1991, 17, 8–17. [PubMed] [Google Scholar]
- De Mello W. C.; Danser A. H. J. Angiotensin II and the Heart. Hypertension 2000, 35, 1183–1188. 10.1161/01.HYP.35.6.1183. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Unal H.; Desnoyer R.; Han G. W.; Patel N.; Katritch V.; Karnik S. S.; Cherezov V.; Stevens R. C. Structural Basis for Ligand Recognition and Functional Selectivity at Angiotensin Receptor. J. Biol. Chem. 2015, 290, 29127–29139. 10.1074/jbc.M115.689000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ram C. V. Angiotensin receptor blockers: current status and future prospects. Am. J. Med. 2008, 121, 656–663. 10.1016/j.amjmed.2008.02.038. [DOI] [PubMed] [Google Scholar]
- Schiering N.; D’Arcy A.; Villard F.; Ramage P.; Logel C.; Cumin F.; Ksander G. M.; Wiesmann C.; Karki R. G.; Mogi M. Structure of neprilysin in complex with the active metabolite of sacubitril. Sci. Rep. 2016, 6, 27909. 10.1038/srep27909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roques B. P.; Noble F.; Daugé V.; Fournié-Zaluski M. C.; Beaumont A. Neutral endopeptidase 24.11: structure, inhibition, and experimental and clinical pharmacology. Pharmacol Rev. 1993, 45, 87–146. [PubMed] [Google Scholar]
- Turner A. J.Neprilysin. In Handbook of Proteolytic Enzymes; Barrett A. J., Rawlings D., Woessner J. F., Eds.; Elsevier: London, 2004; pp 419–426. [Google Scholar]
- Kostis J. B.; Packer M.; Black H. R.; Schmieder R.; Henry D.; Levy E. Omapatrilat and enalapril in patients with hypertension: the Omapatrilat Cardiovascular Treatment vs. Enalapril (OCTAVE) trial. Am. J. Hypertens. 2004, 17, 103–111. 10.1016/j.amjhyper.2003.09.014. [DOI] [PubMed] [Google Scholar]
- Rouleau J. L.; Pfeffer M. A.; Stewart D. J.; Isaac D.; Sestier F.; Kerut E. K.; Porter C. B.; Proulx G.; Qian C.; Block A. J. Comparison of vasopeptidase inhibitor, omapatrilat, and lisinopril on exercise tolerance and morbidity in patients with heart failure: IMPRESS randomised trial. Lancet 2000, 356, 615–620. 10.1016/S0140-6736(00)02602-7. [DOI] [PubMed] [Google Scholar]
- Fryer R. M.; Segreti J.; Banfor P. N.; Widomski D. L.; Backes B. J.; Lin C. W.; Ballaron S. J.; Cox B. F.; Trevillyan J. M.; Reinhart G. A.; von Geldern T. W. Effect of bradykinin metabolism inhibitors on evoked hypotension in rats: rank efficacy of enzymes associated with bradykinin-mediated angioedema. Br. J. Pharmacol. 2008, 153, 947–55. 10.1038/sj.bjp.0707641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packer M.; McMurray J. J.; Desai A. S.; Gong J.; Lefkowitz M. P.; Rizkala A. R.; Rouleau J. L.; Shi V. C.; Solomon S. D.; Swedberg K.; Zile M.; Andersen K.; Arango J. L.; Arnold J. M.; Bělohlávek J.; Böhm M.; Boytsov S.; Burgess L. J.; Cabrera W.; Calvo C.; Chen C. H.; Dukat A.; Duarte Y. C.; Erglis A.; Fu M.; Gomez E.; Gonzàlez-Medina A.; Hagège A. A.; Huang J.; Katova T.; Kiatchoosakun S.; Kim K. S.; Kozan Ö.; Llamas E. B.; Martinez F.; Merkely B.; Mendoza I.; Mosterd A.; Negrusz-Kawecka M.; Peuhkurinen K.; Ramires F. J.; Refsgaard J.; Rosenthal A.; Senni M.; Sibulo A. S. Jr; Silva-Cardoso J.; Squire I. B.; Starling R. C.; Teerlink J. R.; Vanhaecke J.; Vinereanu D.; Wong R. C. PARADIGM-HF Investigators and Coordinators. Angiotensin receptor neprilysin inhibition compared with enalapril on the risk of clinical progression in surviving patients with heart failure. Circulation 2015, 131, 54–61. 10.1161/CIRCULATIONAHA.114.013748. [DOI] [PubMed] [Google Scholar]
- Morphy R.; Rankovic Z. Designed Multiple Ligands. An Emerging Drug Discovery Paradigm. J. Med. Chem. 2005, 48, 6523–6543. 10.1021/jm058225d. [DOI] [PubMed] [Google Scholar]
- Carini D. J.; Duncia J. V.; Aldrich P. E.; Chiu A. T.; Johnson A. L.; Pierce M. E.; Price W. A.; Santella J. B.; Wells G. J.; Wexler R. R.; Wong P. C.; Yoo S. – E.; Timmermans P. B. M. W. M. Nonpeptide angiotensin II receptor antagonists: The Discovery of a Series of N-(Biphenylylmethyl)imidazoles as Potent, Orally Active Antihypertensives. J. Med. Chem. 1991, 34, 2525–2547. 10.1021/jm00112a031. [DOI] [PubMed] [Google Scholar]
- Breschi M. C.; Calderone V.; Digiacomo M.; Macchia M.; Martelli A.; Martinotti E.; Minutolo F.; Rapposelli S.; Rossello A.; Testai L.; Balsamo A. New NO-Releasing Pharmacodynamic Hybrids of Losartan and Its Active Metabolite: Design, Synthesis, and Biopharmacological Properties. J. Med. Chem. 2006, 49, 2628–2639. 10.1021/jm0600186. [DOI] [PubMed] [Google Scholar]
- Garcia G.; Rodriguez-Puyol M.; Alajarin R.; Serrano I.; Sánchez-Alonso P.; Griera M.; Vaquero J. J.; Rodriguez-Puyol D.; Alvarez-Builla J.; Diez-Marqués M. L. Losartan-Antioxidant Hybrids: Novel Molecules for the Prevention of Hypertension-Induced Cardiovascular Damage. J. Med. Chem. 2009, 52, 7220. 10.1021/jm9003957. [DOI] [PubMed] [Google Scholar]
- Garcia G.; Serrano I.; Sánchez-Alonso P.; Rodriguez-Puyol M.; Alajarin R.; Griera M.; Vaquero J. J.; Rodriguez-Puyol D.; Alvarez-Builla J.; Diez-Marqués M. L. New losartan-hydrocaffeic acid hybrids as antihypertensive-antioxidant dual drugs: Ester, amide and amine linkers. Eur. J. Med. Chem. 2012, 50, 90–101. 10.1016/j.ejmech.2012.01.043. [DOI] [PubMed] [Google Scholar]
- Meyer M.; Foulquier S.; Dupuis F.; Flament S.; Grimaud L.; Henrion D.; Lartaud I.; Monard G.; Grillier-Vuissoz I.; Boisbrun M. Synthesis and evaluation of new designed multiple ligands directed towards both peroxisome proliferator-activated receptor-γ and angiotensin II type 1 receptor. Eur. J. Med. Chem. 2018, 158, 334–352. 10.1016/j.ejmech.2018.08.082. [DOI] [PubMed] [Google Scholar]
- Jaffe I. A. Adverse effects profile of sulfhydryl compounds in man. Am. J. Med. 1986, 80, 471–6. 10.1016/0002-9343(86)90722-9. [DOI] [PubMed] [Google Scholar]
- Hegde L. G.; Yu C.; Renner T.; Thibodeaux H.; Armstrong S. R.; Park T.; Cheruvu M.; Olsufka R.; Sandvik E. R.; Lane C. E.; Budman J.; Hill C. M.; Klein U.; Hegde S. S. Concomitant angiotensin AT1 receptor antagonism and neprilysin inhibition produces omapatrilat-like antihypertensive effects without promoting tracheal plasma extravasation in the rat. J. Cardiovasc. Pharmacol. 2011, 57, 495–504. 10.1097/FJC.0b013e318210fc7e. [DOI] [PubMed] [Google Scholar]
- Li X. C.; Widdop R. E. Angiotensin type 1 antagonists CV-11974 and EXP 3174 cause selective renal vasodilatation in conscious spontaneously hypertensive rats. Clin. Sci. 1996, 91, 147–154. 10.1042/cs0910147. [DOI] [PubMed] [Google Scholar]
- Vanderheyden P. M.; Fierens F. L.; De Backer J. P.; Fraeyman N.; Vaquelin G. Distinction between surmountable and insurmountable selective AT1 receptor antagonists by use of CHO-K1 cells expressing human angiotensin II AT1 receptors. Br. J. Pharmacol. 1999, 126, 1057–1065. 10.1038/sj.bjp.0702398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulpizio A. C.; Pullen M. A.; Edwards R. M.; Louttit J. B.; West R.; Brooks D. P. Mechanism of vasopeptidase inhibitor-induced plasma extravasation: comparison of omapatrilat and the novel neutral endopeptidase 24.11/angiotensin-converting enzyme inhibitor GW796406. J. Pharmacol. Exp. Ther. 2005, 315, 1306–13. 10.1124/jpet.105.084749. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.