

Abstract
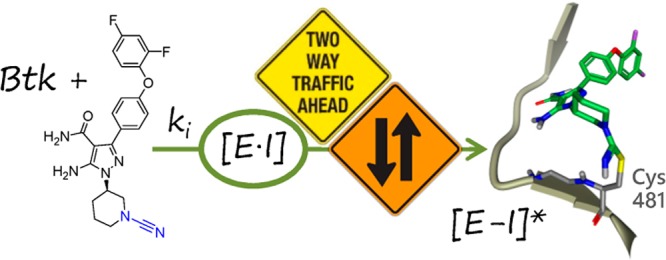
Potent covalent inhibitors of Bruton’s tyrosine kinase (BTK) based on an aminopyrazole carboxamide scaffold have been identified. Compared to acrylamide-based covalent reactive groups leading to irreversible protein adducts, cyanamide-based reversible-covalent inhibitors provided the highest combined BTK potency and EGFR selectivity. The cyanamide covalent mechanism with BTK was confirmed through enzyme kinetic, NMR, MS, and X-ray crystallographic studies. The lead cyanamide-based inhibitors demonstrated excellent kinome selectivity and rat pharmacokinetic properties.
Keywords: BTK, EGFR, irreversible covalent inhibitor, reversible covalent inhibitor
Bruton’s tyrosine kinase (BTK) is a nonreceptor tyrosine kinase belonging to the Tec family of kinases.1 BTK is broadly expressed in hematopoietic cells, especially B lymphocytes and myeloid cells, but notably not in T cells. In B cells, BTK is essential for competent signaling through the B cell antigen receptor (BCR) and is required for B cell development and differentiation.2 The critical role BTK plays in B cell development is evident by the phenotype of patients suffering from X-linked agammaglobulinemia (XLA), a hereditary immunodeficiency disease that results from mutations in the gene encoding BTK.3−5 XLA patients have very few circulating mature B cells as a result of a developmental arrest at the pre-B cell stage in the bone marrow and the further failure of immature B cells that do reach the peripheral compartment to mature.6 As a consequence, XLA patients display a compromised humoral immune response leading to low levels of immunoglobulin and a failure to develop antibody responses. BTK is also an essential component to enable signaling through the Ig Fc receptors (FcεR and FcγR) and specific toll-like receptors in myeloid cells.7 Signaling through BTK in these circumstances regulates inflammatory cytokine and chemokine production by macrophages and can lead to degranulation of mast cells.
The inhibition of BTK activity is an attractive therapeutic approach to treat autoimmune diseases where B cells and myeloid cells play an important role such as rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE).8−12 BTK is also a promising target in cancer chemotherapy since it is required for the survival and proliferation of leukemic B cells, and myeloid cells are important components of the tumor microenvironment.13 Ibrutinib (1, Figure 1) and acalabrutinib have been approved as second-line therapy for mantle cell lymphoma (MCL). Ibrutinib has also been approved for the treatment of chronic lymphocytic leukemia (CLL) and Waldenström macroglobulinemia (WM). Both of these agents are considered targeted, covalent-irreversible inhibitors of the kinase.14−16 They react covalently with a noncatalytic cysteine residue (Cys481) in the ATP binding site to form an irreversible protein adduct. Covalent inhibitors have drawn interest for their potential to offer (a) improved biochemical efficiency, (b) improved selectivity versus targets lacking a reactive amino acid residue, and (c) a prolonged pharmacodynamic-driven duration of action compared to reversible, substrate competitive inhibitors.17 However, the concern over indiscriminant covalency leading to idiosyncratic adverse drug reactions (IADR) has largely limited the application of this drug class to life threatening indications.18 In response to the latter concern, reversible inhibitors of BTK have also advanced into clinical trials19,20 as well as a third class, reversible-covalent inhibitors.21 Inhibitors that form a reversible covalent bond with the cysteine residue of the kinase offer the advantage of prolonged residence time on the target but theoretically lack the long-lived protein adduct that may be a trigger for drug hypersensitivity liabilities.
Figure 1.
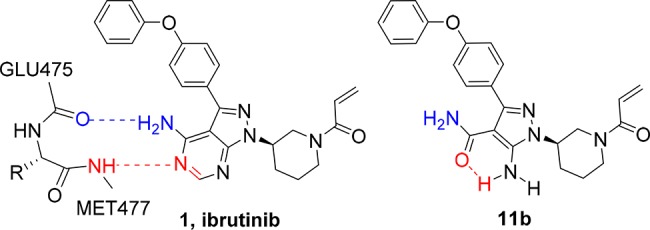
Structure of BTK inhibitor ibrutinib (1) and aminopyrazole carboxamide 11b.
Although compound 1 is a potent inhibitor of BTK, its incomplete off-target selectivity does not fully exploit the potential of covalent design. Indeed it potently inhibits several other kinases that also have an equivalent cysteine in their active site such as epidermal growth factor receptor (EGFR) and interleukin-2-inducible T-cell kinase (ITK).15 It is also a potent reversible inhibitor of SRC family kinases such as SRC, LCK, and LYN. Inhibition of wild-type EGFR leads to skin rash and gastrointestinal side effects, while inhibition of ITK and SRC family kinases risks expanded immune suppression beyond the B and myeloid cell compartments. Herein, we describe the discovery of potent BTK inhibitors based upon an aminopyrazole carboxamide kinase hinge binding scaffold. Through fine-tuning of the covalent reactive group (CRG) and structural discrimination in noncovalent kinome affinity, highly selective inhibitors of BTK have been identified spanning irreversible to reversible-covalent modalities.
As revealed in the cocrystal structure of 1 with the human BTK kinase domain,22 the aminopyrimidine ring forms two hydrogen bond contacts with the kinase hinge residues, specifically (a) the amino substituent and the backbone carbonyl of Glu475 and (b) the N5 ring nitrogen atom with the backbone NH of Met477. We hypothesized that comparable hinge interactions could be achieved by excising the pyrimidine ring and replacing the 4-aminopyrazolopyrimidine scaffold by a 5-aminopyrazole 4-carboxamide as in compound 11b, Figure 1. A proposed intramolecular hydrogen bond between the carbonyl of the carboxamide and the 5-amino substituent would maintain the rigidity provided by the pyrimidine ring in compound 1, while the amino group may facilitate further hinge contacts. This approach was indeed successful with compound 11b demonstrating comparable potency to 1 for BTK inhibition in a Lanthascreen assay (IC50 = 0.18 nM compared to 0.13 nM, 1). Unfortunately, the undesired attributes of EGFR and SRC kinase inhibition (IC50 = 15 and 72 nM, respectively) were maintained. In order to explore the potential of the aminopyrazole carboxamide scaffold to deliver improved kinome selectivity, we undertook a matrix survey of two regions of the molecule. These included (1) replacement of the acrylamide with alternative CRGs possessing different electronic or steric factors and (2) modification of the terminal phenoxy substituent through substitution or pyridine replacement. The desired aminopyrazole carboxamides were prepared through a convergent route (see Supporting Information).
A series of compounds with various CRGs was profiled in Lanthascreen assays for inhibition of BTK, EGFR, and SRC, as well as in time dependent kinetics experiments against the former two kinases, Table 1. The electrophilicity index (ω) was calculated for each as an approximation of the intrinsic CRG reactivity. Potency against the BTK C481S mutant, incapable of forming the covalent adduct, was measured to approximate the intrinsic binding (Ki) component. The unsubstituted acrylamide 9b was found to be the most potent BTK inhibitor of the series; however, it was also a potent inhibitor of EGFR. Consistent with the dominant role of covalency in potency, compound 9b was 68-fold less potent against the BTK C481S mutant (2 h incubation). Other CRG derivatives showed modestly weaker inhibition toward the mutant suggesting that the sterics or electronics of the CRG can influence the reversible binding component of the two step inhibition mechanism. The CRG choice did not have a significant influence on SRC kinase inhibition with the exception of dimethylaminobutenamide 9i owing to the selection of the phenoxy substituents (vide infra). Substitution of the acrylamide C1-position by methyl (9c) was the most detrimental to BTK potency and led to a compound lacking time dependent inhibition. This can be attributed to a 1,3-strain interaction disfavoring coplanarity of the carbonyl and olefin π-systems. Crotyl analog 9d also suffered significant erosion of BTK potency presumably due to reduced electrophilicity and steric clash with the cysteine nucleophile. Increasing the electrophilicity of the reactive carbon center by fluorine substitution of the terminal methyl as in 9g and 9h recovered BTK potency. The substituted crotyl analogs displayed a significantly reduced time dependent kinetics with respect to EGFR compared to acrylamide 9b with compounds 9f and 9g showing the best overall BTK potency and selectivity. The structurally distinct cyanamide 9a also showed excellent BTK potency and superior EGFR selectivity to the Michael acceptor-based analogs.
Table 1. Impact of Covalent Reactive Group Selection on Compound Pharmacologya.

| IC50c (nM) |
kinact/Kid (1/(M·s)) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| compd | X | Y | ωb | BTK WT | BTK C481S | EGFR | SRC | BTK WTe | EGFRf | BTK T1/2e,g (h) | THLE IC50 (μM) |
| 1 | 0.83 | 0.1 | 3.4 | 18 | 38 | 226 000 | 45 040 | >24 | 11 | ||
| 9a | 0.44 | 1.5 | 43 | 16 800 | 1 630 | 123 900 | 56 | 6 | 137 | ||
| 9b | H | H | 0.83 | 0.25 | 17 | 26 | 4 030 | 206 500 | 25 110 | >24 | 15 |
| 9c | CH3 | H | 0.48 | 247 | 176 | >20 000 | 3 350 | <1h | i | i | i |
| 9d | H | CH3 | 0.60 | 61 | 66 | >20 000 | 3 410 | 4 910 | i | >24 | 149 |
| 9e | F | H | 0.69 | 5.3 | 170 | 570 | 6 480 | 35 300 | 435 | >24 | 67 |
| 9f | H | CH2OH | 0.61 | 2.3 | 33 | 2 910 | 4 610 | 26 650 | 213 | >24 | 43 |
| 9g | H | CH2F | 0.65 | 2.7 | 90 | 2 180 | 3 140 | 85 770 | 332 | >24 | 49 |
| 9h | H | CHF2 | 0.81 | 0.41 | 115 | 274 | 5 840 | 80 500 | i | >24 | 8 |
| 9i | H | CH2NMe2 | 0.59 | 3.7 | 65 | 149 | 370 | 19 180 | 514 | >24 | 15 |
All values are the mean of two or more independent assays.
Electrophilicity index calculated using DMol3 at PBE/TNP/COSMO level of theory in water with LowModeMD.
Lanthascreen assay, [ATP] = Km.
For reversible inhibitors, the analogous second-order rate constant is given.
TR-FRET binding assay.
Caliper enzymatic assay.
Apparent dissociation rate.
Kinetics consistent with reversible inhibition; time dependence not observed.
Not determined.
Notably, compared to the acryl- and crotylamide analogs, which were effectively irreversible-covalent inhibitors of BTK (dissociation T1/2 > 24 h), cyanamide 9a displayed a finite residence time on BTK (T1/2 = 6 h), Table 1. Therefore, cyanamide 9a would be considered a reversible-covalent inhibitor of BTK. This finding is consistent with cyanamide-based covalent inhibitors of the cathepsins previously reported.23,24 The lack of a long-lived covalent adduct with 9a was attractive to us for the application to non-life-threatening disease indications. Also, compared to the other potent CRGs studied, compound 9a was less cytotoxic to THLE cells, a potential risk indicator of IADR in addition to hapten generation.25 Consequently, lead identification was directed toward the cyanamide as the preferred CRG.
In order to confirm that cyanamide 9a formed a discrete protein adduct, three biophysical approaches were examined. First, compound 9a was incubated with recombinant human BTK kinase domain, and the resulting product was characterized by mass spectroscopy. A shift in the parent protein mass consistent with the mass of 9a was observed supporting the formation of a 1:1 (BTK/9a) adduct, Figure S1 (see Supporting Information). Second, compound 9a was isotopically labeled with a 13C atom at the cyanamide carbon. Upon incubation with BTK protein, the intensity of the parent 13C signal (119.3 ppm) was diminished resulting in a broad signal at 167.2 ppm (25 Hz line width) consistent with formation of the isothiourea adduct, Figure S2 (see Supporting Information).23 Lastly, compound 9a was cocrystallized with the kinase domain of mouse BTK, which confirmed adduct formation between the cyanamide carbon and the thiol of Cys481, Figure 2. As anticipated, the primary carboxamide of 9a formed hydrogen bond contacts with hinge residues Glu475 and Met477. The latter also forms a favorable contact with the amino substituent of 9a.
Figure 2.
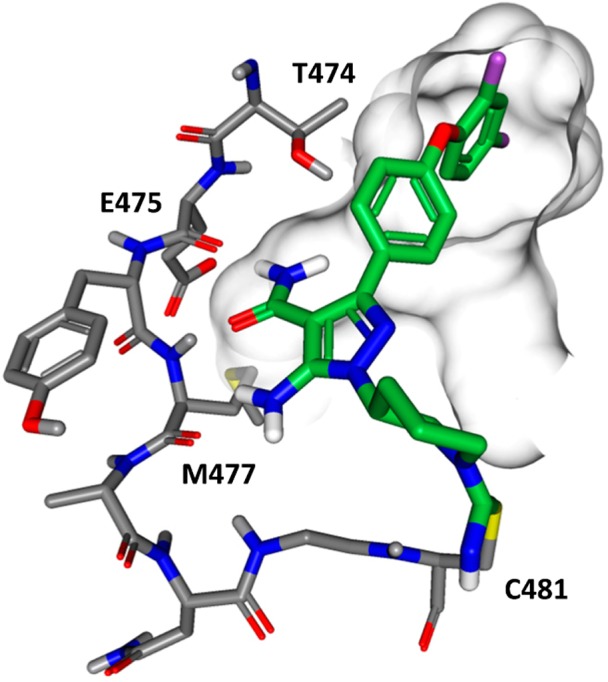
Cocrystal structure of the covalent adduct between compound 9a and the mouse BTK kinase domain.
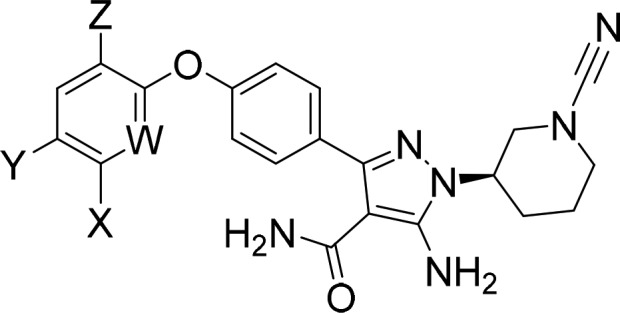
A representative survey of the terminal phenoxy substituent in the cyanamide series is depicted in Table 2. Irrespective of the terminal substituent, all of the cyanamides demonstrated excellent selectivity against EGFR with very weak time dependent inhibition. As with 9a, all were reversible-covalent inhibitors of BTK with dissociation rates from the recombinant enzyme of 3–7 h. The unsubstituted phenoxy analog 11a was the most potent inhibitor of both wild-type BTK and the C481S mutant. However, as with acrylamide-based inhibitors 1 and 11b also having the phenoxy substituent, 11a was a very potent inhibitor of SRC kinase. On the other hand, substituted phenyl (9a and 10) or pyridinyl (20a–c) ethers showed significantly weaker inhibition of SRC kinase. Substitution also had a detrimental effect on the reversible binding (Ki) component of BTK inhibition as shown by their reduced potency against the BTK C481S mutant. For example, compound 9a was 15-fold less potent than 11a. However, SRC kinase was more sensitive to this modification yielding a 388-fold reduction. The size of the phenoxy pocket is largely governed by the conformational state of the C-helix (in versus out). The observed selectivity may suggest greater accommodation of ligands through the inactive C-helix out conformation for BTK. Indeed, in an analysis of PDB structures by Möbitz,26 BTK structures were predominantly in a C-helix out conformation while SRC-family kinases predominately were C-helix in. Since SRC kinase lacks the homologous cysteine residue to BTK, this noncovalent intrinsic selectivity is further amplified when the covalent nature of the inhibitors is realized (e.g., 1290-fold, 9a).
Table 2. Impact of Aryl Ether Substituent on Pharmacology of Reversible-Covalent Cyanamidesa.
| IC50b (nM) |
kinact/Kic (1/(M·s)) |
BTK T1/2d,f (h) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| compd | W | X | Y | Z | BTK WT | BTK C481S | EGFR | SRC | BTK WTd | EGFRe | |
| 9a | CH | H | F | F | 1.5 | 43 | 16 800 | 1 630 | 123 900 | 56 | 6 |
| 10 | CH | H | Cl | H | 0.64 | 48 | 3 890 | 1 400 | 118 300 | 87 | 6 |
| 11a | CH | H | H | H | 0.37 | 2.8 | 1 710 | 4.2 | 135 400 | 17 | 7 |
| 20a | N | H | Cl | H | 6.2 | 92 | 15 400 | >20 000 | 24 370 | 121 | 7 |
| 20b | N | CF3 | H | H | 2.6 | 36 | >20 000 | 14 400 | 55 870 | 7 | 3 |
| 20c | N | H | Cl | F | 7.6 | 230 | >20 000 | >20 000 | 60 970 | 155 | 5 |
All values are the mean of two or more independent assays.
Lanthascreen assay, [ATP] = Km.
For reversible inhibitors, the analogous second-order rate constant is given.
TR-FRET binding assay.
Caliper enzymatic assay.
Apparent dissociation rate.
Selective cyanamide-based BTK inhibitors were further profiled in cellular in vitro assays, Table 3. All compounds potently inhibited B cell proliferation while displaying minimal inhibition of T cell proliferation. Potent inhibition of histamine release from Mast cells was observed in a human whole blood assay. Similar inhibition was observed toward anti-IgD-mediated CD69 upregulation on B cells. The substituted cyanamide analogs also displayed favorable ADME properties with low in vitro human microsomal clearance (HLM CL < 8 μL/(min·mg)) and good permeability (RRCK cells, >10 × 10–6 cm/s). After IV dosing to rats, the compounds showed low to moderate in vivo clearance with the phenyl analogs (9a and 10) having the longest half-lives, Table S1 (see Supporting Information). All compounds demonstrated excellent oral bioavailability following oral dosing in rats (76–93%).
Table 3. Cellular Potency of Cyanamidesa.
| IC50 (nM) |
||||
|---|---|---|---|---|
| proliferation |
||||
| compd | B cell | T cell | HWB histamine | CD69 expression |
| 9a | 2.7 | >10 000 | 64 | 53 |
| 10 | 1.7 | 8 150 | 21 | 46 |
| 20a | 1.6 | >10 000 | 95 | 45 |
| 20b | 14.9 | 10 100 | 42 | 33 |
| 20c | 19.1 | >10 000 | 50 | 89 |
All values are the mean of two or more independent assays.
Compound 9a showed high kinome specificity when profiled against a panel of 51 kinases (1 mM ATP, 1 μM compound), Table S2 (see Supporting Information). Dose response curves were obtained for the 11 kinases that share a homologous cysteine residue to BTK C481 and representative SRC family kinases (IC50, 1 mM ATP), Table S3 (see Supporting Information). Significant inhibition by 9a was only observed against a subset of the kinases with a shared cysteine (BMX, TEC, and TXK; 5–29-fold BTK). BTK and all three other kinases share a common gatekeeper residue (Thr) as well as a common Cys+3 residue (Asn). The nature of the Cys+3 residue has been postulated to strongly influence the pKa of the cysteine thiol and consequently its nucleophilicity.27 The pKa for the reactive cysteine thiol in BTK and other family members sharing an Asn residue has been estimated to be the most acidic (i.e., most nucleophilic at physiological pH) of the 11 kinases.16 The pKa was calculated to be higher (i.e., less nucleophilic) for other members in the family having an Asp or Glu residue with EGFR being one of the highest. In contrast, the calculated electrophilicity of the cyanamide was significantly lower than the corresponding acrylamide, Table 1. Consequently, the greater differential in CRG electrophilicity and cysteine nucleophilicity is a plausible hypothesis driving the excellent EGFR selectivity seen with the cyanamide analogs such as 9a. In addition, a weak stabilizing interaction between the isothiourea of the protein adduct and the Cys+3 Asn residue may contribute to the selectivity observed.
The identification of compound 9a as a potent and selective BTK inhibitor demonstrates the importance of considering reversible ligand affinity and CRG selection in covalent inhibitor design. Aligning the steric environment of the CRG reactive site and its degree of electrophilicity with the nature of the reactive cysteine residue, especially nucleophilicity (pKa), were found to be important factors for achieving selectivity among covalently amenable targets. As shown here, the acceptance of diminished reversible binding affinity toward the covalent target (9a vs 11a, Table 2) in favor of greater affinity loss against an off-target lacking covalent enablement is a useful tactic to maximize target selectivity of covalent inhibitors. The potential advantage of reversible covalency toward safety must be balanced with the need for more sustained pharmacokinetic exposure since BTK enzyme turnover (T1/2 > 24 h)28 is no longer the dominant pharmacodynamics determinant. Fortunately, we were able to incorporate good metabolic stability into the cyanamide series as evident by the low clearance and high oral bioavailability in rat especially for 9a. The success of the covalent-reversible strategy in the case of BTK is further supported by the excellent in vivo efficacy of 9a that we have previously described in the murine NZBxW lupus model29 and suggests that cyanamides may have broad applicability in covalent drug design.
Acknowledgments
We thank Yuchuan Wu, Anne Akin, Joe Collins, James Doom, Ning Pan, Laurence Philippe, and Frank Riley for synthesis and chiral separations, Brian Samas for X-ray crystallography analysis, Jeffrey Hirsch for assay development guidance, and Suvit Thaisrivongs for helpful discussions.
Glossary
Abbreviations
- ADME
absorption, distribution, metabolism, and excretion
- BTK
Bruton’s tyrosine kinase
- CRG
covalent reactive group
- IARD
idiosyncratic adverse drug reactions
- THLE
SV40 T-antigen-immortalized human liver epithelial cells
- XLA
X-linked agammaglobulinemia.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00461.
Synthetic procedures and characterization for final analogs and intermediates, single crystal determination supporting absolute configuration assignment, protein cocrystallography and MS adduct characterization methods, and pharmacology assay procedures (PDF)
Accession Codes
PDB accession code 6MNY (9a). Authors will release the atomic coordinates and experimental data upon article publication.
All procedures performed on animals were in accordance with regulations and established guidelines and were reviewed and approved by the Pfizer Institutional Animal Care and Use Committee. Compound 9a (PF-06250112) is commercially available via MilliporeSigma (catalog no. PZ0220).
The authors declare no competing financial interest.
Supplementary Material
References
- Mohamed A. J.; Yu L.; Bäckesjö C.-M.; Vargas L.; Faryal R.; Aints A.; Christensson B.; Berglöf A.; Vihinen M.; Nore B. F.; Smith C. I. E. Bruton’s tyrosine kinase (Btk): function, regulation, and transformation with special emphasis on the PH domain. Immunol. Rev. 2009, 228, 58–73. 10.1111/j.1600-065X.2008.00741.x. [DOI] [PubMed] [Google Scholar]
- Corneth O. B. J.; Wolterink R. G. J. K.; Hendriks R. W. BTK signaling in B cell differentiation and autoimmunity. Curr. Top. Microbiol. Immunol. 2015, 393, 67–105. 10.1007/82_2015_478. [DOI] [PubMed] [Google Scholar]
- Khan W. N. Colonel Bruton’s kinase defined the molecular basis of X-linked agammaglobulinemia, the first primary immunodeficiency. J. Immunol. 2012, 188, 2933–2935. 10.4049/jimmunol.1200490. [DOI] [PubMed] [Google Scholar]
- Tsukada S.; Saffran D. C.; Rawlings D. J.; Parolini O.; Allen R. C.; Klisak I.; Sparkes R. S.; Kubagawa H.; Mohandas T.; Quan S.; Belmont J. W.; Cooper M. D.; Conley M. E.; Witte O. W. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell 1993, 72, 279–290. 10.1016/0092-8674(93)90667-F. [DOI] [PubMed] [Google Scholar]
- Vetrie D.; Vořechovský I.; Sideras P.; Holland J.; Davies A.; Flinter F.; Hammarström L.; Kinnon C.; Levinsky R.; Bobrow M.; Smith C. I. E.; Bentley D. R. The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nature 1993, 361, 226–233. 10.1038/361226a0. [DOI] [PubMed] [Google Scholar]
- Conley M. E. B cells in patients with X-linked agammaglobulinemia. J. Immunol. 1985, 134, 3070–3074. [PubMed] [Google Scholar]
- López-Herrera G.; Vargas-Hernández A.; González-Serrano M. E.; Berrón-Ruiz L.; Rodríguez-Alba J. C.; Espinosa-Rosales F.; Santos-Argumedo L. Bruton’s tyrosine kinase–an integral protein of B cell development that also has an essential role in the innate immune system. J. Leukocyte Biol. 2014, 95, 243–250. 10.1189/jlb.0513307. [DOI] [PubMed] [Google Scholar]
- Crofford L. J.; Nyhoff L. E.; Sheehan J. H.; Kendall P. L. The role of Bruton’s tyrosine kinase in autoimmunity and implications for therapy. Expert Rev. Clin. Immunol. 2016, 12, 763–773. 10.1586/1744666X.2016.1152888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dörner T.; Radbruch A.; Burmester G. R. B-cell-directed therapies for autoimmune disease. Nat. Rev. Rheumatol. 2009, 5, 433–441. 10.1038/nrrheum.2009.141. [DOI] [PubMed] [Google Scholar]
- Boumans M. J. H.; Thurlings R. M.; Gerlag D. M.; Vos K.; Tak P. P. Response to rituximab in patients with rheumatoid arthritis in different compartments of the immune system. Arthritis Rheum. 2011, 63, 3187–3194. 10.1002/art.30567. [DOI] [PubMed] [Google Scholar]
- Hui-Yuen J. S.; Nguyen S. C.; Askanase A. D. Targeted B cell therapies in the treatment of adult and pediatric systemic lupus erythematosus. Lupus 2016, 25, 1086–1096. 10.1177/0961203316652491. [DOI] [PubMed] [Google Scholar]
- Hahn B. H. Belimumab for systemic lupus erythematosus. N. Engl. J. Med. 2013, 368, 1528–1535. 10.1056/NEJMct1207259. [DOI] [PubMed] [Google Scholar]
- Hendriks R. W.; Yuvaraj S.; Kil L. P. Targeting Bruton’s tyrosine kinase in B cell malignancies. Nat. Rev. Cancer 2014, 14, 219–232. 10.1038/nrc3702. [DOI] [PubMed] [Google Scholar]
- Pan Z.; Scheerens H.; Li S.-J.; Schultz B. E.; Sprengeler P. A.; Burrill L. C.; Mendonca R. V.; Sweeney M. D.; Scott K. C. K.; Grothaus P. G.; Jeffery D. A.; Spoerke J. M.; Honigberg L. A.; Young P. R.; Dalrymple S. A.; Palmer J. T. Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. ChemMedChem 2007, 2, 58–61. 10.1002/cmdc.200600221. [DOI] [PubMed] [Google Scholar]
- Honigberg L. A.; Smith A. M.; Sirisawad M.; Verner E.; Loury D.; Chang B.; Li S.; Pan Z.; Thamm D. H.; Miller R. A.; Buggy J. J. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B cell activation and is efficacious in models of autoimmune disease and B cell malignancy. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 13075–13080. 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barf T.; Covey T.; Izumi R.; van de Kar B.; Gulrajani M.; van Lith B.; van Hoek M.; de Zwart E.; Mittag D.; Demont D.; Verkaik S.; Krantz F.; Pearson P. G.; Ulrich R.; Kaptein A. Acalabrutinib (ACP-196): A covalent Bruton tyrosine kinase inhibitor with a differentiated selectivity and in vivo potency profiles. J. Pharmacol. Exp. Ther. 2017, 363, 240–252. 10.1124/jpet.117.242909. [DOI] [PubMed] [Google Scholar]
- Singh J.; Petter R. C.; Baillie T. A.; Whitty A. The resurgence of covalent drugs. Nat. Rev. Drug Discovery 2011, 10, 307–317. 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- Cho T.; Uetrecht J. How reactive metabolites induce an immune response that sometimes leads to an idiosyncratic drug reaction. Chem. Res. Toxicol. 2017, 30, 295–314. 10.1021/acs.chemrestox.6b00357. [DOI] [PubMed] [Google Scholar]
- Crawford J. J.; Johnson A. R.; Misner D. L.; Belmont L. D.; Castanedo G.; Choy R.; Coraggio M.; Dong L.; Eigenbrot C.; Erickson R.; Ghilardi N.; Hau J.; Katewa A.; Kohli P. B.; Lee W.; Lubach J. W.; McKenzie B. S.; Ortwine D. F.; Schutt L.; Tay S.; Wei B.; Reif K.; Liu L.; Wong H.; Young W. B. Discovery of GDC-0853: A potent, selective, and noncovalent Bruton’s tyrosine kinase inhibitor in early clinical development. J. Med. Chem. 2018, 61, 2227–2245. 10.1021/acs.jmedchem.7b01712. [DOI] [PubMed] [Google Scholar]
- Watterson S. H.; De Lucca G. V.; Shi Q.; Langevine C. M.; Liu Q.; Batt D. G.; Beaudoin Bertrand M.; Gong H.; Dai J.; Yip S.; Li P.; Sun D.; Wu D.-R.; Wang C.; Zhang Y.; Traeger S. C.; Pattoli M. A.; Skala S.; Cheng L.; Obermeier M. T.; Vickery R.; Discenza L. N.; D’Arienzo C. J.; Zhang Y.; Heimrich E.; Gillooly K. M.; Taylor T. L.; Pulicicchio C.; McIntyre K. W.; Galella M. A.; Tebben A. J.; Muckelbauer J. K.; Chang C.; Rampulla R.; Mathur A.; Salter-Cid L.; Barrish J. C.; Carter P. H.; Fura A.; Burke J. R.; Tino J. A. Discovery of 6-fluoro-5-(R)-(3-(S)-(8-fluoro-1-methyl-2,4-dioxo-1,2-dihydroquinazolin-3(4H)-yl)-2-methylphenyl)-2-(S)-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-1H-carbazole-8-carboxamide (BMS-986142): A reversible inhibitor of Bruton’s tyrosine kinase (BTK) conformationally constrained by two locked atropisomers. J. Med. Chem. 2016, 59, 9173–9200. 10.1021/acs.jmedchem.6b01088. [DOI] [PubMed] [Google Scholar]
- Bradshaw J. M.; McFarland J. M.; Paavilainen V. O.; Bisconte A.; Tam D.; Phan V. T.; Romanov S.; Finkle D.; Shu J.; Patel V.; Ton T.; Li X.; Loughhead D. G.; Nunn P. A.; Karr D. E.; Gerritsen M. E.; Funk J. O.; Owens T. D.; Verner E.; Brameld K. A.; Hill R. J.; Goldstein D. M.; Taunton J. Prolonged and tunable residence time using reversible covalent kinase inhibitors. Nat. Chem. Biol. 2015, 11, 525–531. 10.1038/nchembio.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender A. T.; Gardberg A.; Pereira A.; Johnson T.; Wu Y.; Grenningloh R.; Head J.; Morandi F.; Haselmayer P.; Liu-Bujalski L. Ability of Bruton’s tyrosine kinase inhibitors to sequester Y551 and prevent phosphorylation determines potency for inhibition of Fc receptor but not B-cell receptor signaling. Mol. Pharmacol. 2017, 91, 208–219. 10.1124/mol.116.107037. [DOI] [PubMed] [Google Scholar]
- Falgueyret J.-P.; Oballa R. M.; Okamoto O.; Wesolowski G.; Aubin Y.; Rydzewski R. M.; Prasit P.; Riendeau D.; Rodan S. B.; Percival M. D. Novel, nonpeptidic cyanamides as potent and reversible inhibitors of human cathepsins K and L. J. Med. Chem. 2001, 44, 94–104. 10.1021/jm0003440. [DOI] [PubMed] [Google Scholar]
- Deaton D. N.; Hassell A. M.; McFadyen R. B.; Miller A. B.; Miller L. R.; Shewchuk L. M.; Tavares F. X.; Willard D. H.; Wright L. L. Novel and potent cyclic cyanamide-based cathepsin K inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 1815–1819. 10.1016/j.bmcl.2005.02.033. [DOI] [PubMed] [Google Scholar]
- Thompson R. A.; Isin E. M.; Li Y.; Weidolf L.; Page K.; Wilson I.; Swallow S.; Middleton B.; Stahl S.; Foster A. J.; Dolgos H.; Weaver R.; Kenna J. G. In vitro approach to assess the potential for risk of idiosyncratic adverse reactions caused by candidate drugs. Chem. Res. Toxicol. 2012, 25, 1616–1632. 10.1021/tx300091x. [DOI] [PubMed] [Google Scholar]
- Möbitz H. The ABC of protein kinase conformations. Biochim. Biophys. Acta, Proteins Proteomics 2015, 1854, 1555–1566. 10.1016/j.bbapap.2015.03.009. [DOI] [PubMed] [Google Scholar]
- Zapf C. W.; Gerstenberger B. S.; Xing L.; Limburg D. C.; Anderson D. R.; Caspers N.; Han S.; Aulabaugh A.; Kurumbail R.; Shakya S.; Li X.; Spaulding V.; Czerwinski R. M.; Seth N.; Medley Q. G. Covalent inhibitors of interleukin-2 inducible T cell kinase (Itk) with nanomolar potency in a whole-blood assay. J. Med. Chem. 2012, 55, 10047–10063. 10.1021/jm301190s. [DOI] [PubMed] [Google Scholar]
- Evans E. K.; Tester R.; Aslanian S.; Karp R.; Sheets M.; Labenski M. T.; Witowski S. R.; Lounsbury H.; Chaturvedi P.; Mazdiyasni H.; Zhu Z.; Nacht M.; Freed M. I.; Petter R. C.; Dubrovskiy A.; Singh J.; Westlin W. F. Inhibition of Btk with CC-292 provides early pharmacodynamic assessment of activity in mice and humans. J. Pharmacol. Exp. Ther. 2013, 346, 219–228. 10.1124/jpet.113.203489. [DOI] [PubMed] [Google Scholar]
- Rankin A. L.; Seth N.; Keegan S.; Andreyeva T.; Cook T. A.; Edmonds J.; Mathialagan N.; Benson M. J.; Syed J.; Zhan Y.; Benoit S. E.; Miyashiro J. S.; Wood N.; Mohan S.; Peeva E.; Ramaiah S. K.; Messing D.; Homer B. L.; Dunussi-Joannopoulos K.; Nickerson-Nutter C. L.; Schnute M. E.; Douhan J. Selective inhibition of BTK prevents murine lupus and antibody-mediated glomerulonephritis. J. Immunol. 2013, 191, 4540–4550. 10.4049/jimmunol.1301553. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.