

Abstract
Over the last two decades there has been an exponential rise in the number of patients receiving deep brain stimulation (DBS) to manage debilitating neurological symptoms in conditions such as Parkinson's disease, essential tremor, and dystonia. Novel applications of DBS continue to emerge including treatment of various psychiatric conditions (e.g. obsessive‐compulsive disorder, major depression) and cognitive disorders such as Alzheimer's disease. Despite widening therapeutic applications, our understanding of the mechanisms underlying DBS remains limited. In addition to modulation of local and network‐wide neuronal activity, growing evidence suggests that DBS may also have important neuroprotective effects in the brain by limiting synaptic dysfunction and neuronal loss in neurodegenerative disorders. In this review, we consider evidence from preclinical and clinical studies of DBS in Parkinson's disease, Alzheimer's disease, and epilepsy that suggest chronic stimulation has the potential to mitigate neuronal loss and disease progression.
Introduction
Deep brain stimulation (DBS) involves the implantation of electrodes into targeted regions of the brain for chronic delivery of electrical stimulation from an implantable pulse generator. Continuous high‐frequency stimulation of subcortical structures such as the subthalamic nucleus (STN) and globus pallidus pars interna (GPi) has become a well‐established treatment of motor symptoms in patients with Parkinson's disease (PD).1 Encouraged by this success, researchers have since found new neuroanatomical targets of DBS to treat other motor disorders (e.g. freezing of gait, tardive dyskinesia, secondary dystonia), epilepsy, psychiatric conditions (e.g. obsessive‐compulsive disorder, major depression) and more recently, Alzheimer's disease.2
Despite more patients receiving DBS surgery, the mechanisms underlying its therapeutic effect remain unclear. At the site of electrode implantation, DBS may modulate local neuronal activity by direct stimulation of axons and dendrites.3 Alterations in local firing patterns could also have important effects on the synchronization of neuronal networks by disrupting pathological oscillatory activity in diseased brain regions (e.g. excessive β oscillations in the basal ganglia of PD patients).3 This electrical modulation relieves motor symptoms within seconds of current onset, as shown by the immediate relief of essential tremor when current is delivered through a DBS electrode in the ventral intermediate nucleus of the thalamus (VIM).4 However, growing evidence suggests that DBS is more than just a “neuromodulatory switch” to control debilitating motor symptoms. Chronic DBS has been shown to induce gradual reorganization of neuronal circuits through enhanced synaptic plasticity and neurogenesis.4 In addition, recent studies in preclinical animal models and humans suggest that DBS may also protect neurons from disease‐related neurotoxicity in certain conditions. This raises the exciting possibility that DBS may have the unanticipated benefit of slowing rates of disease progression or even improving long‐term survival in some patients.
Here, we review evidence from preclinical and clinical studies of DBS in Parkinson's disease, Alzheimer's disease, and refractory epilepsy to determine whether chronic neuromodulation could have neuroprotective properties with clinical relevance. We have limited our review to these disorders, since they are all characterized by a progressive neurodegenerative phenotype culminating in neuronal loss. For a treatment to be considered “neuroprotective” or “disease modifying,” it will be important to demonstrate that it: (1) has an effect on disease pathogenesis; (2) reduces the rate of neuronal loss and (3) is associated with slower symptom progression or an improvement in survival.
Methods
The authors searched the online PubMed database for peer‐reviewed articles published in English between 1 January 1987 and 1 May 2018. The search term “deep brain stimulation” combined with either “Parkinson's disease,” “Alzheimer's disease,” or “epilepsy” was used. Articles for in‐depth review were next identified by the authors by searching the article title, abstract, and keywords for mention of “neuroprotection,” “neuroprotective,” “disease modifying,” “survival,” “neuronal loss,” “apoptosis,” “synaptic dysfunction,” “synaptic loss,” or “neurotoxicity.” Additional articles were identified by screening reference lists of recent review articles focusing on mechanisms of DBS.
Parkinson's Disease
Parkinson's disease (PD) is the most common neurodegenerative movement disorder, affecting an estimated 2–3% of adults over the age of 65 years.5 It is characterized by intracellular accumulation of misfolded forms of α‐synuclein and progressive loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc).5 Evidence from clinical‐pathological correlation studies suggests that early depletion of these dopaminergic neurons is the likely cause of characteristic motor features, such as bradykinesia & rigidity.5 Neuronal loss has also been reported in various other brain regions including the locus ceruleus, nucleus basalis of Meynert, raphe nucleus, pedunculopontine nucleus, dorsal motor nucleus of the vagus, amygdala, and hypothalamus.5 In the absence of disease‐modifying therapies, current treatment is primarily focused on controlling motor symptoms using drugs that enhance intracerebral dopamine concentrations (e.g. levodopa, selegiline) or stimulate dopamine receptors (e.g. pramipexole). In the long‐term, complications of dopaminergic treatment, including on/off motor fluctuations and dyskinesias, frequently occur.5 DBS of subcortical structures such as the STN, GPi, and occasionally VIM, may be offered as an established treatment for well‐selected patients with levodopa‐responsive motor symptoms who experience disabling medication‐related side effects.6 The mechanism underlying DBS‐mediated motor symptom control remains uncertain, however it is likely to involve both local effects on neuronal firing patterns and network‐wide effects on pathological oscillatory activity.3, 7 In addition to effective relief of motor symptoms, emerging evidence from preclinical models suggests that long‐term DBS may have the potential to protect against neuronal loss and limit motor dysfunction. However it remains uncertain whether this occurs by mitigating pathological oscillatory activity or through other mechanisms.
Neuroprotection in PD animal models
Early evidence that DBS could have neuroprotective properties arose from studies of STN‐DBS in preclinical models of PD. Chronic STN stimulation in rats was shown to protect dopaminergic neurons in the SNpc from the toxic effects of intrastriatal 6‐hydroxydopamine (6‐OHDA) administered 5–7 days previously.8, 9 To exclude an effect on uptake or metabolism of 6‐OHDA, a later study by Spieles‐Engemann et al. delayed STN‐DBS to 2 weeks after 6‐OHDA administration, once significant neuronal loss had already occurred.10 Rats treated with STN‐DBS again showed increased survival of dopaminergic neurons in the SNpc suggesting a bona fide neuroprotective effect. In another study, stimulation of the rodent analogue of the GPi, the entopeduncular nucleus, did neither mitigate 6‐OHDA‐mediated behavioral deficits nor loss of dopaminergic neurons, suggesting neuroprotective effects could be restricted to certain stimulation targets.11 Alongside rodent models, DBS has also been shown to protect against neurotoxicity in nonhuman primate models of PD. Following 1‐methyl‐4‐phenyl‐1, 2, 3, 6‐ tetrahydropyridine (MPTP) administration in monkeys, STN‐DBS was shown to limit dopaminergic neuronal loss in the SNpc12, 13 and periaqueductal grey matter (PAG).13 Interestingly, less neuronal rescue was observed in animals with more severe MPTP lesions,12 suggesting that DBS may only protect neurons from milder forms of neurotoxicity and that there may be a threshold of severity of dopaminergic neuronal loss after which neuronal rescue and associated behavioral rescue may no longer be possible.
Despite being widely used in preclinical research, toxin models of PD have limited translational value due to the acute nature of MPTP or 6‐OHDA lesions and the absence of key neuropathological features, most notably α‐synuclein aggregates.14 To mirror the clinical phenotype more closely, two recent studies have examined the effect of STN‐DBS in non‐toxicant‐based PD animal models. Mussachio et al. studied the effect of chronic STN‐DBS in rats with viral vector‐mediated nigrostriatal overexpression of human A53T α‐synuclein.15 As in PD patients, these animals develop a progressive neurodegenerative phenotype with accumulation of insoluble α‐synuclein aggregates, dopaminergic neuronal loss in the SNpc and motor impairment.16 To mimic a moderate stage of human PD, chronic STN‐DBS was commenced 3 weeks after mutant α‐synuclein expression, at which point striatal dopaminergic fiber loss and motor deficits were already established. High frequency STN stimulation reversed motor deficits and was accompanied by higher numbers of tyrosine‐hydroxylase‐(TH) expressing SNpc neurons compared with unstimulated rats.15 Motor improvements were sustained 24 h after stimulation was terminated, suggesting a disease‐modifying effect of STN‐DBS.15 Despite rescue of TH‐positive SNpc neurons and improved motor function, striatal dopamine levels remained depleted in rats treated with chronic STN stimulation.15 This suggests that dopaminergic terminals in the striatum may be particularly vulnerable to A53T α‐synuclein‐mediated toxicity, resulting in early synaptic loss before the onset of DBS treatment.
A later study by Fischer and colleagues studied the effect of chronic STN‐DBS in a different PD rat model characterized by viral vector‐mediated nigrostriatal overexpression of human wild‐type α‐synuclein.17 Despite low levels of transgene expression and a relatively mild clinical phenotype, STN‐DBS did not protect against forelimb akinesia, striatal denervation, or SNpc neuronal loss. These findings are in contrast to those of Mussachio et al., which could partly be explained by differences in mechanisms of neurotoxicity arising from mutant and wild‐type alpha‐synuclein,18 as well as variation in stimulation currents used. This highlights the need for further research to evaluate the reproducibility of the above findings across different preclinical PD models, including transgenic α‐synuclein mice and viral vector‐based rodent and nonhuman primate α‐synuclein expression models. In addition, it will be important to evaluate the effect of different stimulation parameters (i.e. frequency, pulse width, current, voltage, and duration) on the neuroprotective potential of STN‐DBS.
The mechanisms by which DBS may mitigate the loss of dopaminergic neurons are not well understood. Several studies have proposed that STN‐DBS may protect SNpc neurons by reducing excitotoxicity arising from overactive glutamatergic projections which originate in the STN.12, 19, 20 This hypothesis is based on the observation that STN‐DBS has very similar effects to STN lesions in toxin models of PD.12, 21, 22, 23 Experimental data supporting this proposed neuroprotective mechanism are currently lacking. Indeed there is electrophysiological evidence that white matter tracts surrounding the STN may also be simultaneously stimulated by DBS thus making it difficult to parse out if the observed effect is only due to reduced glutamatergic outflow from the STN.3 In addition to limiting excitoxicity, STN‐DBS may promote neuronal survival by stimulating the release of neurotrophic factors. STN‐DBS has been shown to increase levels of brain‐derived neurotrophic factor (BDNF) in the nigrostriatal system and primary motor cortex of 6‐OHDA lesioned rats.10, 24 Following release, BDNF binds the transmembrane receptor tropomyosin‐related kinase type B (trkB) resulting in activation of three intracellular signalling cascades: (1) mitogen‐activated protein kinase/extracellular signal related‐kinase (MAPK/ERK) which promotes protein synthesis; (2) phosphatidylinositol 3‐kinase/protein kinase B (PI3K/Akt) which regulates protein translation/trafficking and inhibits apoptosis; (3) phospholipase Cγ/protein kinase C (PLCγ/PKC) which is involved in the regulation of synaptic plasticity.25 Using the 6‐OHDA rat model, Fischer et al. recently demonstrated that chronic STN‐DBS results in phosphorylation of Akt and ribosomal protein S6 in SNpc neurons, suggesting activation of the BDNF/trkB signalling pathway.26 In addition, STN‐DBS‐mediated rescue of SNpc neurons was abrogated by selective pharmacological blockade of the trkB receptor.26 Taken together, these findings identify BDNF/trkB signalling as a possible mechanism for STN‐DBS‐mediated neuroprotection (Fig. 1). Since activity‐dependent BDNF release is believed to occur at dendrites,27 STN‐DBS may have greater protective effects if initiated at an early stage of disease when synaptic degeneration is less advanced.
Figure 1.
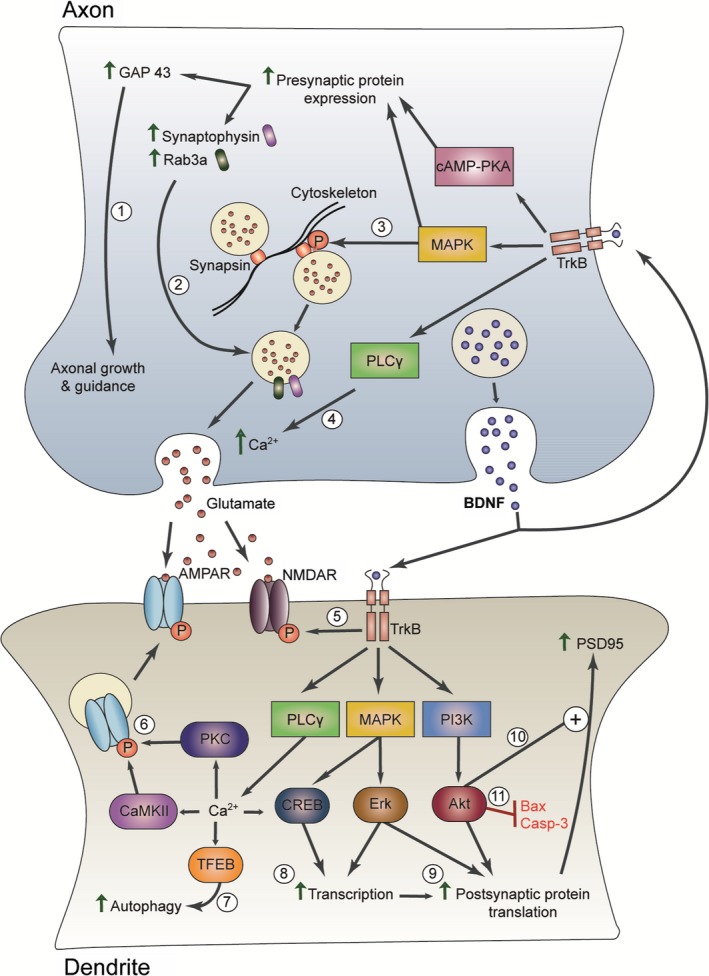
Model of neuroprotective effects of DBS at the synapse. DBS stimulates increased BDNF release which induces structural and functional changes at the synapse by binding pre‐ and postsynaptic TrkB receptors. At the presynaptic terminal, activation of the cAMP‐PKA and MAPK pathways promotes increased presynaptic gene expression including key proteins involved in axonal growth and guidance (e.g. GAP 43) (1) and neurotransmitter vesicle release (e.g. synaptophysin, Rab3a) (2).90, 91, 92, 93, 94 MAPK phosphorylation of synapsin also releases neurotransmitter vesicles from cytoskeleton‐bound pools (3).95 TrkB‐mediated activation of PLC γ increases intracellular Ca2+ concentration leading to increased presynaptic neurotransmitter release (4).90 At the postsynaptic terminal, TrkB activation leads to tyrosine phosphorylation of NMDA‐type glutamate receptor (NMDAR) subunits, increasing conductance (5).96, 97 Activation of the PLC γ pathway induces a rise in intracellular Ca2+ concentration and subsequent activation of Ca2+/calmodulin‐dependent kinase II (CaMKII) and protein kinase C (PKC), which phosphorylate AMPA‐type glutamate receptors (AMPAR) subunits and increase their delivery to the postsynaptic terminal (6).98 Elevated Ca2+ levels may also activate transcription factor EB (TFEB), which is the major regulator of autophagy and could enhance clearance of toxic misfolded proteins (7).70, 99 TrkB‐mediated activation of cAMP response element binding protein (CREB) and Erk enhances gene transcription (8).25 BDNF‐TrkB signaling also enhances postsynaptic protein translation through both MAPK/Erk and PI3K/Akt pathways (9).25 The PI3K/Akt pathway is also involved in regulating the trafficking of proteins (e.g. PSD95) to the postsynaptic terminal (10)25 and inhibition of apoptosis (11).100, 101
Does DBS modify disease progression in patients?
Despite promising reports of DBS‐mediated neuroprotection in PD animal models, there is limited evidence from clinical studies to support a similar disease‐modifying effect in patients. Follow‐up studies of STN‐DBS patients over a 3–10 year period have revealed sustained symptomatic benefit in terms of levodopa‐responsive motor symptoms (e.g. tremor, rigidity) and dyskinesias.28 Most longitudinal cohort studies have reported stabilization or a trend towards improvement in off‐medication/off‐stimulation motor scores compared with preoperative baseline.28 Despite evidence of slowed progression of motor deficits, a recent 18F‐fluorodopa positron emission tomography (PET) study reported continuous decline in dopaminergic function in patients with advanced PD despite STN‐DBS.29 Furthermore, a postmortem analysis of PD patients who received long‐term STN‐DBS revealed no rescue of SNpc dopaminergic neurons or reduction in α‐synuclein burden.30 The discrepancy between preserved motor scores and continued dopaminergic neurodegeneration could be explained by a prolonged stimulation “wash‐out” period, in which DBS has a lasting effect in disrupting pathological oscillatory activity in the basal ganglia after being switched off. In contrast to motor symptoms, patients receiving chronic STN‐DBS display continued progression of levodopa‐unresponsive features such as axial symptoms, speech disturbance, and cognitive dysfunction, suggesting ongoing progression of the disease in nondopaminergic regions of the brain.28, 31, 32, 33
A limited number of studies have investigated the effect of STN‐DBS on survival. In a nonrandomized trial, Ngoga et al. showed improved survival in PD patients who received DBS surgery compared to controls treated with medication alone;34 however this could be an indirect effect of improved motor function in reducing complications such as aspiration pneumonia. Another study by Lilleeng et al. reported no difference in mortality rates between a cohort of PD patients treated with STN‐DBS and a control group treated only with medication.35 These results must be interpreted with caution since the authors identified control subjects from historical records, raising the possibility that confounding environmental factors affected mortality rates in the two groups.
In the absence of a clear neuroprotective effect in patients, it is important to consider possible limitations of the long‐term studies of STN‐DBS to date. At the stage of subject enrollment, PD has been treated as a single disorder, despite marked genotypic, and phenotypic heterogeneity between patients. Variation in rates of disease progression between trial subjects creates significant “noise” which can mask treatment effects, particularly within the short time scale of most clinical trials.36 As an alternative, PD patients could be recruited in stratified cohorts with similar predicated rates of disease progression based on validated biomarkers. A biomarker is defined as “a characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or pharmacological responses to a therapeutic intervention”.37 However, despite intensive study of DNA, RNA, biofluid samples, peripheral tissue sampling, and imaging, no reliable biomarkers have been identified which can predict the trajectory of disease progression in PD patients.36 In their absence, clinical trials seeking to determine the neuroprotective potential of STN‐DBS must compensate for phenotypic heterogeneity by recruiting larger numbers of patients to ensure that there is sufficient statistical power to detect disease‐modifying effects.
Another limitation of subject selection in long‐term STN‐DBS studies to date has been the recruitment of patients with advanced PD when nigral cell death is already well‐established. The majority of dopaminergic terminals are lost within 4 years of PD diagnosis,38 and yet the average STN‐DBS surgery takes place 12–15 years after diagnosis.39 It is therefore unsurprising that evidence of a neuroprotective effect of DBS on SNpc neurons has been lacking.
In choosing clinical trial endpoints to assess the long‐term efficacy of STN‐DBS, to date there has been no consensus in what constitutes neuroprotection or a disease‐modifying effect. To demonstrate that an intervention has modified the natural course of PD, an effect on disease pathogenesis would be expected, such as a reduction in the rate of neuronal loss or alpha‐synuclein burden. In the absence of reliable biomarkers which can predict the extent of neurodegeneration premortem, trials to date have instead had to rely on clinical assessments of symptom severity.40 For example, many trials have employed symptomatic rating scales such as the Unified PD Rating Scale (UPDRS) as clinical endpoints. The UPDRS investigates motor symptom severity, behavior, mood, mentation, and ability to perform activities of daily living. The scale is vulnerable to both intra‐ and inter‐rater variability.41 In addition, any stabilization of the UPDRS in patients receiving DBS is likely to be confounded by improvements in motor symptoms. Other endpoints used to assess PD progression in trials include time taken for a drug‐naïve patient to require dopaminergic therapy, as well as stability of the levodopa equivalent dose (LED) over years.40, 41 However, response to dopaminergic therapy is often considered in determining candidacy for DBS and thus cannot serve as an endpoint to assess if DBS is neuroprotective.40
Due to ethical concerns, it will not be possible to compare STN‐DBS “on” and “off” conditions using a long‐term randomized control trial study design. Instead, long‐term follow‐up of patients treated with STN‐DBS from an early stage of PD41, 42 could give important insights into the neuroprotective potential of DBS in patients with a more intact nigrostriatal system. In the absence of validated biomarkers, it will be necessary to recruit larger numbers of subjects and attempt stratification of subjects into cohorts by their baseline clinical phenotype. Instead of symptom rating scales, future STN‐DBS neuroprotection trials would benefit from employing more objective measures of disease progression (e.g. 18F‐fluorodopa PET, serial structural magnetic resonance imaging).
Alzheimer's Disease
An estimated 46.8 million people worldwide are living with dementia, with an associated global cost of approximately $818 billion.43 Alzheimer's disease (AD) is the most common form of dementia, characterized by the widespread accumulation of extracellular amyloid plaques and intraneuronal neurofibrillary tangles (NFTs). Amyloid plaques are composed of an aggregated 40‐ to 42‐residue peptide called beta‐amyloid (Aβ 1–40 and Aβ 1–42), while the main component of NFTs is a hyperphosphorylated form of tau protein.44 Recent investigation of DBS as a potential novel treatment for AD was prompted by the serendipitous discovery that stimulation of the fornix resulted in episodic memory recall during DBS electrode implantation for treatment of morbid obesity.45 Located in the medial diencephalon, the fornix is a critical component of the well‐known circuit of Papez. A phase I trial of forniceal DBS in patients with mild AD revealed an improved temporoparietal hypometabolism and a reduction in the expected rate of cognitive decline at 6 and 12 months in some patients.46 A 1‐year follow‐up structural MRI study showed an overall reduction in the rate of hippocampal atrophy in DBS‐treated AD patients compared to matched AD controls not receiving DBS.47 The later phase II multi‐center double blind randomized controlled ADvance trial of 42 patients with mild AD examined the safety and efficacy of forniceal DBS.48 Whilst both electrode implantation surgery and subsequent stimulation were well tolerated, no significant difference in primary clinical outcome at 12 months was observed, as measured by AD Assessment Scale cognitive subscale (ADAS‐Cog13) and Clinical Dementia Rating scale sum of boxes (CDR‐SB) cognitive scores. Patients receiving stimulation displayed elevated glucose metabolism at 6 months, but this effect was not seen at a later 12 month time point. Post‐hoc analysis revealed a significant interaction between age and clinical outcome, with trends toward improvement in patients over the age of 65 years and worsening in younger patients. These early studies of forniceal DBS suggest that neuromodulation of memory circuits is feasible, well tolerated, and may have the potential to slow disease progression in a subgroup of AD patients over 65 years of age. A planned phase III international multi‐centre trial will investigate the potential benefits of forniceal DBS in this age group further.
In addition to the fornix, DBS of the nucleus basalis of Meynert (NBM) in the basal forebrain is being considered as a therapeutic target in patients with AD. The NBM is an important source of acetylcholine supply to the neocortex, and neuronal loss within this region correlates closely with cortical cholinergic deficits and the degree of cognitive impairment in patients with AD.49 Low‐frequency stimulation of the NBM aims to excite residual cholinergic neurons, leading to increased cholinergic transmission to the neocortex.49 In an initial safety study, four out of six AD patients who received low‐frequency NBM‐DBS remained stable or showed improvement in cognitive assessment scores after 1 year.50 Interestingly, patients who respond to NBM‐DBS appear to have higher baseline cognitive function51 and more limited fronto‐parieto‐temporal cortical atrophy.52 This suggests that NBM‐DBS may slow cognitive decline in patients with early AD by stimulating increased transmission from remaining cholinergic neurons.
Despite early evidence to suggest that forniceal and NBM‐DBS may stabilize or even improve cognitive scores in certain patients with mild AD, confirmation of a neuroprotective effect is still lacking. As in PD, electrical stimulation of brain regions involved in memory function may lead to symptomatic improvement in patients with mild AD by temporarily enhancing the formation or retrieval of memories. Whether forniceal or NBM‐DBS are associated with lasting cognitive benefits on cessation of stimulation or can limit the rate of disease progression at a neuropathological level remains unclear. In the first instance, it will be important to identify which stimulation parameters are associated with the sustained memory benefit, if any, before assessing any long‐term impact on disease progression. Future clinical studies may be guided by evidence from studies in laboratory animals, which have started to yield important insights into the neuroprotective potential of DBS as a treatment for AD, as discussed below.
Neuroprotection in animal models
Electrical stimulation of the NBM increases cortical acetylcholine release53 and improves memory function in wild‐type rats.54, 55, 56 Increased cholinergic input to the neocortex in response to NBM‐DBS induces secretion of nerve growth factor (NGF) by target cortical neurons.57 This is known to be critical for the survival and function of cholinergic basal forebrain neurons.58 In addition to enhanced neurotrophic support, NBM‐DBS has been shown to increase synaptic plasticity in the cortex of wild‐type rats.59 Further studies are required to determine whether NBM‐DBS has similar neuroprotective effects in rodents with established features of AD neuropathology.
Forniceal DBS has also been shown to have beneficial effects on cognitive function in rodents, with reported improvements in spatial,60, 61, 62, 63 contextual,60 and recognition63 memory. DBS of the entorhinal cortex (EC) has also been shown to improve spatial64, 65 and contextual65 memory, suggesting that the pro‐cognitive effects of DBS may reflect generic activation of the circuit of Papez. Consistent with this theory, forniceal DBS upregulates activity‐dependent cFos gene expression in connected remote areas of the hippocampus within 2–2.5 h of stimulation onset.66, 67 Despite early changes in gene expression, studies of EC‐DBS in mice show that improvements in memory can take 3–6 weeks to emerge after a single episode of acute stimulation.64, 65 This temporal lag could reflect gradual structural changes in the hippocampus (e.g. synaptic remodelling, neurogenesis) in response to stimulation‐induced neurotrophic factor release.10, 24, 66 In support of this hypothesis, DBS of the fornix,60 EC,64 and anterior thalamic nucleus68, 69 promote neurogenesis in the dentate gyrus. Moreover, forniceal DBS increases hippocampal expression of synaptophysin and GAP43 in wild‐type rats66 and EC‐DBS rescues synaptophysin levels in the triple transgenic mouse model of AD.70 Both synaptic proteins have important roles in axonal growth and guidance, as well as synaptic plasticity. Akwa and colleagues reported that EC‐DBS limits synaptic loss in an AD mouse model by stimulating autophagic‐lysosomal clearance of pathological forms of tau protein.70 These findings are of particular relevance in AD, where synaptic failure is one of the earliest hallmarks of disease and correlates closely with the degree of memory impairment.71 In addition to tau protein, amyloid plaque load has been shown to be reduced with EC‐DBS if initiated at early stages of memory impairment in an AD mouse model.65
Early animal studies support a potential neuroprotective role for DBS in the treatment of AD by promoting synaptic plasticity, hippocampal neurogenesis, and increased clearance of misfolded protein conformers. It is, however, important to note some key limitations of these studies. Results from studies of wild‐type animals61, 66, 67 or pharmacological models of AD62, 63 may not be translatable to AD patients who have evidence of amyloid deposition, widespread synaptic dysfunction, and established neuronal loss at time of diagnosis. In addition, all animal studies to date have employed acute stimulation paradigms (e.g. single 1 h episode), rather than the continuous stimulation protocol currently used in patients. Future studies using standardised chronic stimulation parameters in validated AD animal models may yield important insights into how the neuroprotective effects of DBS could be optimized in future clinical trials.
Epilepsy
Epilepsy is a common neurological disorder characterized by a predisposition to recurrent seizures. Many forms of epilepsy can have profound structural and functional effects on the brain if left untreated. For instance, longitudinal MRI studies of patients with temporal lobe epilepsy (TLE), the most common type of epilepsy in adults, have revealed hippocampal volume loss which correlates with the frequency of generalized seizures, suggesting seizure‐associated hippocampal damage.72, 73 TLE is resistant to antiepileptic medications in approximately one third of cases.74 Surgical resection of an epileptogenic focus can prevent disabling seizures in approximately 65% of these patients.75 In patients who lack a single epileptogenic focus or have significant medical comorbidities, DBS of the anterior thalamic nucleus (ANT) can be considered as an alternative to resective surgery.76, 77 Whilst long‐term follow‐up studies of ANT‐DBS in patients are currently limited, early evidence from animal models suggests that DBS may have important neuroprotective properties in epilepsy, in addition to improved seizure control.
Neuroprotection in animal models
Kainic acid (KA), a potent agonist of kainic receptors and α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazole propionate (AMPA)‐type receptors, is frequently used to model human TLE due to its ability to induce seizures and associated hippocampal cell death.78 Chen and colleagues reported that high‐frequency hippocampal stimulation reduced seizure frequency and protected against neuronal loss following KA injection in a macaque model of epilepsy.79 Neuronal rescue was associated with reduced levels of the pro‐apoptotic factors Bax and activated caspase‐3, and increased levels of the antiapoptotic protein Bcl‐2.79 A similar reduction in seizure frequency and hippocampal neuronal loss was also reported following DBS targeting the ANT region in KA‐induced epileptic monkeys.80 Several mechanisms have been proposed which could explain this neuroprotective effect. Human and animal models of epilepsy are characterized by acutely and chronically elevated levels of pro‐inflammatory cytokines (e.g. TNF‐α, IL‐1β and IL‐6) in affected brain regions.81 Recent studies by Chen et al. demonstrated that ANT‐DBS attenuated this pro‐inflammatory cytokine response and reduced neuronal injury following KA administration in rats.82, 83 Dampening neuroinflammation could help to reduce neurotoxicity and limit apoptosis.
In addition to anti‐inflammatory effects, DBS may protect neurons by reducing neurotransmitter excitotoxicity. Excessive glutamate levels are believed to contribute to neurotoxicity and neuronal loss in epilepsy.84 Recently, Shi et al. used microdialysis techniques to show that chronic ANT‐DBS reduces glutamate levels and increases levels of the inhibitory neurotransmitters GABA and taurine in KA‐induced epileptic monkeys while stimulation was switched on.85 By reversing the imbalance in neurotransmitter levels, ANT‐DBS may help to limit glutamate‐induced excitotoxicity in epileptic neuronal networks.
Taken together, early preclinical evidence suggests that ANT‐DBS has the potential to limit neurotoxicity associated with recurrent seizures and protect against progressive hippocampal neuronal loss. However, it is important to acknowledge some key technical limitations of the existing studies. Firstly, there has been a lack of consistency in the duration of stimulation (e.g. 12 h vs. 6 months) and species (e.g. rat vs. macaque) used, making it challenging to corroborate findings and to delineate common underlying mechanisms of ANT‐DBS. Secondly, reported alterations in cytokine or neurotransmitter levels were measured while ANT stimulation was switched on. As a result, it is not possible to determine whether any reduction in neurotoxicity was a direct effect of stimulation, or secondary to a reduction in recent seizure frequency. Lastly, the promising effects of ANT‐DBS on hippocampal neuronal counts must be interpreted with caution since none of the above mentioned studies employed unbiased stereological methods, which are considered to be the “gold standard” in quantitative neuropathology.
Summary and Future Directions
In addition to delivering substantial clinical benefit, evidence from preclinical models suggests that DBS may have a wide range of neuroprotective effects including stimulation of neurotrophic factor release, synaptic remodeling, inhibition of apoptosis, dampening of neuroinflammation, reduction in glutamate excitotoxicity, and enhanced clearance of toxic misfolded proteins. BDNF was recently identified as a critical mediator of the neuroprotective response to DBS26 and could link many of these pleiotropic effects (Fig. 1). Despite mounting evidence to support a disease‐modifying role of DBS in preclinical models of AD, PD, and epilepsy, as discussed above significant variation in the animal species, genetic backgrounds, and stimulation conditions used makes it challenging to draw any definitive conclusions about a potential underlying mechanism. Furthermore given the significant differences between neurodegenerative diseases and respective preclinical models there may be pathology specific variations that determine whether there will be a response to stimulation and by which specific mechanism. Since studies of DBS‐mediated neuroprotection in patients necessitate a long follow‐up period, it will be critical to continue to study DBS in validated animal models to guide selection of neuroanatomical targets and stimulation parameters associated with optimal neuroprotective properties.
Support for the neuroprotective theory of DBS amongst clinicians has been lacking due to limited evidence of disease‐modifying effects in human trials to date. In the clinical setting, DBS is often deployed late in the disease course and thus the “window of opportunity” for neuroprotection may be lost. The therapeutic benefit of DBS is thought to depend on modulation of remote brain regions connected in a neural network with the stimulation site.86 Since synaptic loss has been implicated in neural network dysfunction,87, 88 the therapeutic potential of DBS is likely to decline with progression of neurodegenerative conditions such as PD and AD. As a result, randomized controlled trials at early stages of disease are required to assess the neuroprotective potential of DBS before significant synaptic loss has occurred. Consistent with this approach, a phase III multicenter randomized double‐blind placebo controlled trial evaluating the effect of DBS in early PD is currently underway.41, 89 In addition to studying the effects of DBS on earlier stages of disease, future trials would benefit from the discovery of reliable disease biomarkers, which can predict the extent of disease progression (e.g. biofluid or serial structural/volumetric MRI markers). These could help to minimize heterogeneity between subjects at the time of trial recruitment and could also be used as trial endpoints to determine if DBS can slow the rate of disease progression, independent of any effect on symptom severity scores.
Disclosures
CH and SKK have received speaker honoraria from Medtronic. AML is a consultant to Medtronic, St Jude and Boston Scientific as well as Chairman of SAB and Director, Functional Neuromodulation with IP in neuromodulation for AD. AML is funded by a Tier 1 Canada Research Chair. LVK and SKK are funded by CIHR.
Conflict of Interest
None.
Funding Information
This research did not receive any specific grant from funding agencies in the public, commercial or not‐for‐profit sectors.
References
- 1. Fasano A, Lozano AM. Deep brain stimulation for movement disorders: 2015 and beyond. Curr Opin Neurol 2015;28:423–436. [DOI] [PubMed] [Google Scholar]
- 2. Tierney TS, Sankar T, Lozano AM. Deep brain stimulation emerging indications. Prog Brain Res 2011;194:83–95. [DOI] [PubMed] [Google Scholar]
- 3. Lozano AM, Hutchison WD, Kalia SK. What have we learned about movement disorders from functional neurosurgery? Annu Rev Neurosci 2017;40:453–477. [DOI] [PubMed] [Google Scholar]
- 4. Herrington TM, Cheng JJ, Eskandar EN. Mechanisms of deep brain stimulation. J Neurophysiol 2016;115:19–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kalia LV, Lang AE. Parkinson's disease. Lancet 2015;386:896–912. [DOI] [PubMed] [Google Scholar]
- 6. Fox SH, Katzenschlager R, Lim S‐Y, et al. The movement disorder society evidence‐based medicine review update: treatments for the motor symptoms of Parkinson's disease. Mov Disord Off J Mov Disord Soc 2011;26(Suppl 3):S2–S41. [DOI] [PubMed] [Google Scholar]
- 7. Ashkan K, Rogers P, Bergman H, Ughratdar I. Insights into the mechanisms of deep brain stimulation. Nat Rev Neurol 2017;13:548–554. [DOI] [PubMed] [Google Scholar]
- 8. Temel Y, Visser‐Vandewalle V, Kaplan S, et al. Protection of nigral cell death by bilateral subthalamic nucleus stimulation. Brain Res 2006;1120:100–105. [DOI] [PubMed] [Google Scholar]
- 9. Harnack D, Meissner W, Jira JA, et al. Placebo‐controlled chronic high‐frequency stimulation of the subthalamic nucleus preserves dopaminergic nigral neurons in a rat model of progressive Parkinsonism. Exp Neurol 2008;210:257–260. [DOI] [PubMed] [Google Scholar]
- 10. Spieles‐Engemann AL, Behbehani MM, Collier TJ, et al. Stimulation of the rat subthalamic nucleus is neuroprotective following significant nigral dopamine neuron loss. Neurobiol Dis 2010;39:105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fischer DL, Collier TJ, Cole‐Strauss A, et al. High‐frequency stimulation of the rat entopeduncular nucleus does not provide functional or morphological neuroprotection from 6‐hydroxydopamine. PLoS ONE 2015;10:e0133957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wallace BA, Ashkan K, Heise CE, et al. Survival of midbrain dopaminergic cells after lesion or deep brain stimulation of the subthalamic nucleus in MPTP‐treated monkeys. Brain J Neurol 2007;130(Pt 8):2129–2145. [DOI] [PubMed] [Google Scholar]
- 13. Shaw VE, Keay KA, Ashkan K, et al. Dopaminergic cells in the periaqueductal grey matter of MPTP‐treated monkeys and mice; patterns of survival and effect of deep brain stimulation and lesion of the subthalamic nucleus. Parkinsonism Relat Disord 2010;16:338–344. [DOI] [PubMed] [Google Scholar]
- 14. Koprich JB, Kalia LV, Brotchie JM. Animal models of α‐synucleinopathy for Parkinson disease drug development. Nat Rev Neurosci 2017;18:515–529. [DOI] [PubMed] [Google Scholar]
- 15. Musacchio T, Rebenstorff M, Fluri F, et al. Subthalamic nucleus deep brain stimulation is neuroprotective in the A53T α‐synuclein Parkinson's disease rat model. Ann Neurol 2017;81:825–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Koprich JB, Johnston TH, Reyes MG, et al. Expression of human A53T alpha‐synuclein in the rat substantia nigra using a novel AAV1/2 vector produces a rapidly evolving pathology with protein aggregation, dystrophic neurite architecture and nigrostriatal degeneration with potential to model the pathology of Parkinson's disease. Mol Neurodegener 2010;5:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fischer DL, Manfredsson FP, Kemp CJ, et al. Subthalamic nucleus deep brain stimulation does not modify the functional deficits or axonopathy induced by nigrostriatal α‐synuclein overexpression. Sci Rep 2017;7:16356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lu J, Sun F, Ma H, et al. Comparison between α‐synuclein wild‐type and A53T mutation in a progressive Parkinson's disease model. Biochem Biophys Res Commun 2015;464:988–993. [DOI] [PubMed] [Google Scholar]
- 19. Rodriguez MC, Obeso JA, Olanow CW. Subthalamic nucleus‐mediated excitotoxicity in Parkinson's disease: a target for neuroprotection. Ann Neurol 1998;44(3 Suppl 1):S175–S188. [DOI] [PubMed] [Google Scholar]
- 20. Maesawa S, Kaneoke Y, Kajita Y, et al. Long‐term stimulation of the subthalamic nucleus in hemiparkinsonian rats: neuroprotection of dopaminergic neurons. J Neurosurg 2004;100:679–687. [DOI] [PubMed] [Google Scholar]
- 21. Nakao N, Nakai E, Nakai K, Itakura T. Ablation of the subthalamic nucleus supports the survival of nigral dopaminergic neurons after nigrostriatal lesions induced by the mitochondrial toxin 3‐nitropropionic acid. Ann Neurol 1999;45:640–651. [PubMed] [Google Scholar]
- 22. Chen L, Liu Z, Tian Z, et al. Prevention of neurotoxin damage of 6‐OHDA to dopaminergic nigral neuron by subthalamic nucleus lesions. Stereotact Funct Neurosurg 2000;75:66–75. [DOI] [PubMed] [Google Scholar]
- 23. Piallat B, Benazzouz A, Benabid AL. Subthalamic nucleus lesion in rats prevents dopaminergic nigral neuron degeneration after striatal 6‐OHDA injection: behavioural and immunohistochemical studies. Eur J Neurosci 1996;8:1408–1414. [DOI] [PubMed] [Google Scholar]
- 24. Spieles‐Engemann AL, Steece‐Collier K, Behbehani MM, et al. Subthalamic nucleus stimulation increases brain derived neurotrophic factor in the nigrostriatal system and primary motor cortex. J Parkinson's Dis 2011;1:123–136. [PMC free article] [PubMed] [Google Scholar]
- 25. Yoshii A, Constantine‐Paton M. Postsynaptic BDNF‐TrkB signaling in synapse maturation, plasticity, and disease. Dev Neurobiol 2010;70:304–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fischer DL, Kemp CJ, Cole‐Strauss A, et al. Subthalamic nucleus deep brain stimulation employs trkB signaling for neuroprotection and functional restoration. J Neurosci Off J Soc Neurosci 2017;37:6786–6796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Matsuda N, Lu H, Fukata Y, et al. Differential activity‐dependent secretion of brain‐derived neurotrophic factor from axon and dendrite. J Neurosci 2009;29:14185–14198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Krack P, Martinez‐Fernandez R, DelAlamo M, Obeso JA. Current applications and limitations of surgical treatments for movement disorders. Mov Disord Off J Mov Disord Soc 2017;32:36–52. [DOI] [PubMed] [Google Scholar]
- 29. Hilker R, Portman AT, Voges J, et al. Disease progression continues in patients with advanced Parkinson's disease and effective subthalamic nucleus stimulation. J Neurol Neurosurg Psychiatry 2005;76:1217–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pal GD, Ouyang B, Serrano G, et al. Comparison of neuropathology in Parkinson's disease subjects with and without deep brain stimulation. Mov Disord Off J Mov Disord Soc 2017;32:274–277. [DOI] [PubMed] [Google Scholar]
- 31. Rodriguez‐Oroz MC, Moro E, Krack P. Long‐term outcomes of surgical therapies for Parkinson's disease. Mov Disord Off J Mov Disord Soc 2012;27:1718–1728. [DOI] [PubMed] [Google Scholar]
- 32. Fasano A, Romito LM, Daniele A, et al. Motor and cognitive outcome in patients with Parkinson's disease 8 years after subthalamic implants. Brain J Neurol 2010;133:2664–2676. [DOI] [PubMed] [Google Scholar]
- 33. Castrioto A, Lozano AM, Poon Y‐Y, et al. Ten‐year outcome of subthalamic stimulation in Parkinson disease: a blinded evaluation. Arch Neurol 2011;68:1550–1556. [DOI] [PubMed] [Google Scholar]
- 34. Ngoga D, Mitchell R, Kausar J, et al. Deep brain stimulation improves survival in severe Parkinson's disease. J Neurol Neurosurg Psychiatry 2014;85:17–22. [DOI] [PubMed] [Google Scholar]
- 35. Lilleeng B, Brønnick K, Toft M, et al. Progression and survival in Parkinson's disease with subthalamic nucleus stimulation. Acta Neurol Scand 2014;130:292–298. [DOI] [PubMed] [Google Scholar]
- 36. Chen‐Plotkin AS, Albin R, Alcalay R, et al. Finding useful biomarkers for Parkinson's disease. Sci Transl Med 2018;10:eaam6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Biomarkers Definitions Working Group . Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther 2001;69:89–95. [DOI] [PubMed] [Google Scholar]
- 38. Kordower JH, Olanow CW, Dodiya HB, et al. Disease duration and the integrity of the nigrostriatal system in Parkinson's disease. Brain J Neurol 2013;136(Pt 8):2419–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Volkmann J. Deep brain stimulation for the treatment of Parkinson's disease. J Clin Neurophysiol Off Publ Am Electroencephalogr Soc 2004;21:6–17. [DOI] [PubMed] [Google Scholar]
- 40. Athauda D, Foltynie T. The ongoing pursuit of neuroprotective therapies in Parkinson disease. Nat Rev Neurol 2015;11:25–40. [DOI] [PubMed] [Google Scholar]
- 41. Charles D, Konrad PE, Neimat JS, et al. Subthalamic nucleus deep brain stimulation in early stage Parkinson's disease. Parkinsonism Relat Disord 2014;20:731–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schuepbach WMM, Rau J, Knudsen K, et al. Neurostimulation for Parkinson's disease with early motor complications. N Engl J Med 2013;368:610–622. [DOI] [PubMed] [Google Scholar]
- 43. World Alzheimer Report 2015. The global impact of dementia|Alzheimer's disease international [Internet]. [date unknown];[cited 2017 Oct 14 ] Available from: https://www.alz.co.uk/research/world-report-2015
- 44. Schellenberg GD, Montine TJ. The genetics and neuropathology of Alzheimer's disease. Acta Neuropathol 2012;124:305–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hamani C, McAndrews MP, Cohn M, et al. Memory enhancement induced by hypothalamic/fornix deep brain stimulation. Ann Neurol 2008;63:119–123. [DOI] [PubMed] [Google Scholar]
- 46. Laxton AW, Tang‐Wai DF, McAndrews MP, et al. A phase I trial of deep brain stimulation of memory circuits in Alzheimer's disease. Ann Neurol 2010;68:521–534. [DOI] [PubMed] [Google Scholar]
- 47. Sankar T, Chakravarty MM, Bescos A, et al. Deep brain stimulation influences brain structure in Alzheimer's disease. Brain Stimul 2015;8:645–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lozano AM, Fosdick L, Chakravarty MM, et al. A phase II study of fornix deep brain stimulation in mild Alzheimer's disease. J Alzheimers Dis 2016;54:777–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gratwicke J, Kahan J, Zrinzo L, et al. The nucleus basalis of Meynert: a new target for deep brain stimulation in dementia? Neurosci Biobehav Rev 2013;37(10 Pt 2):2676–2688. [DOI] [PubMed] [Google Scholar]
- 50. Kuhn J, Hardenacke K, Lenartz D, et al. Deep brain stimulation of the nucleus basalis of Meynert in Alzheimer's dementia. Mol Psychiatry 2015;20:353–360. [DOI] [PubMed] [Google Scholar]
- 51. Hardenacke K, Hashemiyoon R, Visser‐Vandewalle V, et al. Deep brain stimulation of the nucleus basalis of Meynert in Alzheimer's dementia: potential predictors of cognitive change and results of a long‐term follow‐up in eight patients. Brain Stimul 2016;9:799–800. [DOI] [PubMed] [Google Scholar]
- 52. Baldermann JC, Hardenacke K, Hu X, et al. Neuroanatomical characteristics associated with response to deep brain stimulation of the nucleus basalis of meynert for Alzheimer's disease. Neuromodulation J Int Neuromodulation Soc 2017;21(2):184–190. [DOI] [PubMed] [Google Scholar]
- 53. Rasmusson DD, Clow K, Szerb JC. Frequency‐dependent increase in cortical acetylcholine release evoked by stimulation of the nucleus basalis magnocellularis in the rat. Brain Res 1992;594:150–154. [DOI] [PubMed] [Google Scholar]
- 54. Montero‐Pastor A, Vale‐Martínez A, Guillazo‐Blanch G, Martí‐Nicolovius M. Effects of electrical stimulation of the nucleus basalis on two‐way active avoidance acquisition, retention, and retrieval. Behav Brain Res 2004;154:41–54. [DOI] [PubMed] [Google Scholar]
- 55. McLin DE, Miasnikov AA, Weinberger NM. Induction of behavioral associative memory by stimulation of the nucleus basalis. Proc Natl Acad Sci USA 2002;99:4002–4007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Boix‐Trelis N, Vale‐Martínez A, Guillazo‐Blanch G, et al. Effects of nucleus basalis magnocellularis stimulation on a socially transmitted food preference and c‐Fos expression. Learn Mem 2006;13:783–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hotta H, Kagitani F, Kondo M, Uchida S. Basal forebrain stimulation induces NGF secretion in ipsilateral parietal cortex via nicotinic receptor activation in adult, but not aged rats. Neurosci Res 2009;63:122–128. [DOI] [PubMed] [Google Scholar]
- 58. Whittemore SR, Seiger A. The expression, localization and functional significance of beta‐nerve growth factor in the central nervous system. Brain Res 1987;434:439–464. [DOI] [PubMed] [Google Scholar]
- 59. Kilgard MP, Merzenich MM. Cortical map reorganization enabled by nucleus basalis activity. Science 1998;279:1714–1718. [DOI] [PubMed] [Google Scholar]
- 60. Hao S, Tang B, Wu Z, et al. Forniceal deep brain stimulation rescues hippocampal memory in Rett syndrome mice. Nature 2015;526:430–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hescham S, Temel Y, Schipper S, et al. Fornix deep brain stimulation induced long‐term spatial memory independent of hippocampal neurogenesis. Brain Struct Funct 2017;222:1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hescham S, Lim LW, Jahanshahi A, et al. Deep brain stimulation of the forniceal area enhances memory functions in experimental dementia: the role of stimulation parameters. Brain Stimul 2013;6:72–77. [DOI] [PubMed] [Google Scholar]
- 63. Zhang C, Hu W‐H, Wu D‐L, et al. Behavioral effects of deep brain stimulation of the anterior nucleus of thalamus, entorhinal cortex and fornix in a rat model of Alzheimer's disease. Chin Med J 2015;128:1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Stone SSD, Teixeira CM, Devito LM, et al. Stimulation of entorhinal cortex promotes adult neurogenesis and facilitates spatial memory. J Neurosci Off J Soc Neurosci 2011;31:13469–13484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Xia F, Yiu A, Stone SS, et al. Entorhinal cortical deep‐brain stimulation rescues memory deficits in both young and old mice genetically engineered to model Alzheimer's disease. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol 2017;42(13):2493–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gondard E, Chau HN, Mann A, et al. Rapid modulation of protein expression in the rat hippocampus following deep brain stimulation of the fornix. Brain Stimul 2015;8:1058–1064. [DOI] [PubMed] [Google Scholar]
- 67. Hescham S, Jahanshahi A, Schweimer JV, et al. Fornix deep brain stimulation enhances acetylcholine levels in the hippocampus. Brain Struct Funct 2016;221:4281–4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Toda H, Hamani C, Fawcett AP, et al. The regulation of adult rodent hippocampal neurogenesis by deep brain stimulation. J Neurosurg 2008;108:132–138. [DOI] [PubMed] [Google Scholar]
- 69. Encinas JM, Hamani C, Lozano AM, Enikolopov G. Neurogenic hippocampal targets of deep brain stimulation. J Comp Neurol 2011;519:6–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Akwa Y, Gondard E, Mann A, et al. Synaptic activity protects against AD and FTD‐like pathology via autophagic‐lysosomal degradation. Mol Psychiatry 2017;23(6):1530–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Selkoe DJ. Alzheimer's disease is a synaptic failure. Science 2002;298:789–791. [DOI] [PubMed] [Google Scholar]
- 72. Briellmann RS, Berkovic SF, Syngeniotis A, et al. Seizure‐associated hippocampal volume loss: a longitudinal magnetic resonance study of temporal lobe epilepsy. Ann Neurol 2002;51:641–644. [DOI] [PubMed] [Google Scholar]
- 73. Fuerst D, Shah J, Shah A, Watson C. Hippocampal sclerosis is a progressive disorder: a longitudinal volumetric MRI study. Ann Neurol 2003;53:413–416. [DOI] [PubMed] [Google Scholar]
- 74. Halpern CH, Samadani U, Litt B, et al. Deep brain stimulation for epilepsy. Neurotherapeutics 2008;5:59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. West S, Nolan SJ, Cotton J, et al. Surgery for epilepsy. Cochrane Database Syst Rev 2015;(7):CD010541. [DOI] [PubMed] [Google Scholar]
- 76. Velasco M, Velasco F, Velasco AL. Centromedian‐thalamic and hippocampal electrical stimulation for the control of intractable epileptic seizures. J Clin Neurophysiol Off Publ Am Electroencephalogr Soc 2001;18:495–513. [DOI] [PubMed] [Google Scholar]
- 77. Fisher R, Salanova V, Witt T, et al. Electrical stimulation of the anterior nucleus of thalamus for treatment of refractory epilepsy. Epilepsia 2010;51:899–908. [DOI] [PubMed] [Google Scholar]
- 78. Ben‐Ari Y. Limbic seizure and brain damage produced by kainic acid: mechanisms and relevance to human temporal lobe epilepsy. Neuroscience 1985;14:375–403. [DOI] [PubMed] [Google Scholar]
- 79. Chen N, Gao Y, Yan N, et al. High‐frequency stimulation of the hippocampus protects against seizure activity and hippocampal neuronal apoptosis induced by kainic acid administration in macaques. Neuroscience 2014;256:370–378. [DOI] [PubMed] [Google Scholar]
- 80. Yang A‐C, Shi L, Li L‐M, et al. Potential protective effects of chronic anterior thalamic nucleus stimulation on hippocampal neurons in epileptic monkeys. Brain Stimul 2015;8:1049–1057. [DOI] [PubMed] [Google Scholar]
- 81. van Vliet EA, Aronica E, Vezzani A, Ravizza T. Review: neuroinflammatory pathways as treatment targets and biomarker candidates in epilepsy: emerging evidence from preclinical and clinical studies. Neuropathol Appl Neurobiol 2018;44:91–111. [DOI] [PubMed] [Google Scholar]
- 82. Chen Y‐C, Zhu G‐Y, Wang X, et al. Deep brain stimulation of the anterior nucleus of the thalamus reverses the gene expression of cytokines and their receptors as well as neuronal degeneration in epileptic rats. Brain Res 2017;1657:304–311. [DOI] [PubMed] [Google Scholar]
- 83. Chen Y‐C, Zhu G‐Y, Wang X, et al. Anterior thalamic nuclei deep brain stimulation reduces disruption of the blood‐brain barrier, albumin extravasation, inflammation and apoptosis in kainic acid‐induced epileptic rats. Neurol Res 2017;39(12):1103–1113. [DOI] [PubMed] [Google Scholar]
- 84. Cho C‐H. New mechanism for glutamate hypothesis in epilepsy [Internet]. Front Cell Neurosci. 2013;7(127): 1–2 [cited 2017 Oct 17 ] Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3741557/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Shi L, Yang A‐C, Li J‐J, et al. Favorable modulation in neurotransmitters: effects of chronic anterior thalamic nuclei stimulation observed in epileptic monkeys. Exp Neurol 2015;265:94–101. [DOI] [PubMed] [Google Scholar]
- 86. Lozano AM, Lipsman N. Probing and regulating dysfunctional circuits using deep brain stimulation. Neuron 2013;77:406–424. [DOI] [PubMed] [Google Scholar]
- 87. Canter RG, Penney J, Tsai L‐H. The road to restoring neural circuits for the treatment of Alzheimer's disease. Nature 2016;539:187–196. [DOI] [PubMed] [Google Scholar]
- 88. Zhai S, Tanimura A, Graves SM, et al. Striatal synapses, circuits, and Parkinson's disease. Curr Opin Neurobiol 2017;48:9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Charles PD, Dolhun RM, Gill CE, et al. Deep brain stimulation in early Parkinson's disease: enrollment experience from a pilot trial. Parkinsonism Relat Disord 2012;18:268–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Park H, Poo M. Neurotrophin regulation of neural circuit development and function. Nat Rev Neurosci 2012;14:nrn3379. [DOI] [PubMed] [Google Scholar]
- 91. Gupta SK, Mishra R, Kusum S, et al. GAP‐43 is essential for the neurotrophic effects of BDNF and positive AMPA receptor modulator S18986. Cell Death Differ 2009;16:624–637. [DOI] [PubMed] [Google Scholar]
- 92. Fournier AE, Beer J, Arregui CO, et al. Brain‐derived neurotrophic factor modulates GAP‐43 but not T alpha1 expression in injured retinal ganglion cells of adult rats. J Neurosci Res 1997;47:561–572. [PubMed] [Google Scholar]
- 93. Tartaglia N, Du J, Tyler WJ, et al. Protein synthesis‐dependent and ‐independent regulation of hippocampal synapses by brain‐derived neurotrophic factor. J Biol Chem 2001;276:37585–37593. [DOI] [PubMed] [Google Scholar]
- 94. Coffey ET, Akerman KE, Courtney MJ. Brain derived neurotrophic factor induces a rapid upregulation of synaptophysin and tau proteins via the neurotrophin receptor TrkB in rat cerebellar granule cells. Neurosci Lett 1997;227:177–180. [DOI] [PubMed] [Google Scholar]
- 95. Jovanovic JN, Czernik AJ, Fienberg AA, et al. Synapsins as mediators of BDNF‐enhanced neurotransmitter release. Nat Neurosci 2000;3:323–329. [DOI] [PubMed] [Google Scholar]
- 96. Suen PC, Wu K, Levine ES, et al. Brain‐derived neurotrophic factor rapidly enhances phosphorylation of the postsynaptic N‐methyl‐D‐aspartate receptor subunit 1. Proc Natl Acad Sci USA 1997;94:8191–8195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Lin SY, Wu K, Levine ES, et al. BDNF acutely increases tyrosine phosphorylation of the NMDA receptor subunit 2B in cortical and hippocampal postsynaptic densities. Brain Res Mol Brain Res 1998;55:20–27. [DOI] [PubMed] [Google Scholar]
- 98. Caldeira MV, Melo CV, Pereira DB, et al. Brain‐derived neurotrophic factor regulates the expression and synaptic delivery of alpha‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazole propionic acid receptor subunits in hippocampal neurons. J Biol Chem 2007;282:12619–12628. [DOI] [PubMed] [Google Scholar]
- 99. Medina DL, Di Paola S, Peluso I, et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol 2015;17:288–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Yamaguchi H, Wang HG. The protein kinase PKB/Akt regulates cell survival and apoptosis by inhibiting Bax conformational change. Oncogene 2001;20:7779–7786. [DOI] [PubMed] [Google Scholar]
- 101. Pérez‐Navarro E, Gavaldà N, Gratacòs E, Alberch J. Brain‐derived neurotrophic factor prevents changes in Bcl‐2 family members and caspase‐3 activation induced by excitotoxicity in the striatum. J Neurochem 2005;92:678–691. [DOI] [PubMed] [Google Scholar]