

Abstract
Fast Photochemical Oxidation of Protein (FPOP), based on a pulsed KrF laser (248 nm) for free-radical generation, is a biophysical method that utilizes hydroxyl radicals to footprint proteins in solution. FPOP has been recognized for structural proteomics investigations, including epitope mapping, protein-aggregation characterization, protein-folding monitoring, and binding-affinity determination. The distinct merits of the platform are: i) the use of a scavenger to control radical lifetime and allow fast (“snapshot”) footprinting of solvent-accessible residues in a protein; ii) the employment of a flow system to enable single-shot irradiation of small plugs of the targeted sample; iii) the use of methionine and catalase after radical oxidation chemistry to prevent post-oxidation with residual oxidizing species; and iv) the utilization of mature mass spectrometry-based proteomic methods to afford detailed analysis. In addition to •OH, other reactive reagents (e.g., carbenes, iodide, sulfate radical anion, and trifluoromethyl radical) can be implemented on this platform to increase the versatility and scope. In this study, we further elaborate the use of FPOP platform to generate secondary radicals and establish a workflow to answer fundamental questions regarding the intrinsic selectivity and reactivity of radicals that are important in biology. Carbonate radical anion is the example we chose owing to its oxidative character and important putative pathogenic roles in inflammation. This systematic study with model proteins/peptides gives consistent results with a previous study that evaluated reactivity with free amino acids and shows that methionine and tryptophan are the most reactive residues with CO3−•. Other aromatic amino acids (i.e., tyrosine, histidine and phenylalanine) exhibit moderate reactivity, whereas, aliphatic amino acids are inert, unlike with •OH. The outcome demonstrates this approach to be appropriate for studying the fast reactions of radicals with proteins.
Graphical Abstract
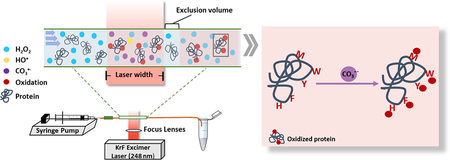
Introduction
Fast Photochemical Oxidation of Protein (FPOP), an alternative to radiolysis of water with synchrotron radiation to give HO• 1–2, was initially invented by Hambly and Gross3. Hydroxyl radicals, formed by a pulsed KrF laser irradiation of hydrogen peroxide, oxidize protein side chains in situ and footprint the exposed residues depending on their intrinsic reactivity and solvent accessibility. With employment of a scavenging reagent, radical lifetime can be tuned in the microsecond time frame to enable protein structural imprinting faster than most conformational changes. In addition, the irreversible modification by HO• allows comprehensive downstream proteomics sample handling, digestion, and mass spectrometry analysis. The FPOP approach, as a valuable biophysical tool in protein structure elucidation, has been recognized for: 1) mapping binding interfaces in protein-ligand interaction, including small molecules binding events4 and antigen-antibody interaction5–6; 2) revealing multistage protein aggregation7; 3) determining binding affinity; 4) identifying hidden conformational changes8, 5) characterizing overall protein dynamics9–10. In addition to •OH, other reactive reagents (e.g., sulfate radical anion11, carbene diradical12, iodide, and trifluoromethyl radical13) have been implemented on the FPOP platform to afford broader labeling opportunities, giving more comprehensive coverage of amino acid residues. Now, we wish to elaborate further the utility of the FPOP platform and to demonstrate its capacity to answer more fundamental questions in biological free-radical chemistry. To do this, we address the adaption of the FPOP platform to ascertain the selectivity and reactivity of the carbonate radical anion with model proteins.
In biological fluids, the bicarbonate anion exists at of 24 mM in serum and 14 mM in intracellular media equilibrated with approximately 1.3 mM carbon dioxide14. The radical anion can be formed from the abundant bicarbonate-carbon dioxide pair by superoxide dismutase (SOD)15, xanthine oxidase (XO)16, and other enzymes that can play pathogenic roles in many physiological conditions. As a key mediator, carbonate radical anion may cause oxidative damage that eventually leads to vascular disease and neurodegeneration14. Its best known metabolic function is to modulate peroxynitrite activity, a strong oxidant generated from nitric oxide and superoxide anion. The CO3− / NO2 couple, dissociated from nitrosuperoxycarbonate (ONOOCO2−), promotes protein nitration17. In this process, CO3− is believed to abstract H from amino acids, typically tyrosine, followed by NO2 addition to give a nitro-substituted residue.
The carbonate radical anion, a potent one electron oxidant (E0 = 1.59 V, pH = 7.0), can favorably oxidize electron-rich donors by electron transfer18. CO3− is a site-selective oxidant of guanine over other DNA bases as a damaging reagent19. The biological consequences of reactions of the carbonate radical anion on proteins are also of concern. Many investigations show, for example, that CO3− can oxidize tryptophan-rich lysozyme to afford a ditryptophan form20, can affect redox signaling pathways by oxidizing tyrosine-containing protein21 and can also cause activity loss of horseradish peroxidase22. Most of the research on CO3−• relies on a previous radiolysis study23 carried out in 1973 with free amino acids; this work shows tryptophan and tyrosine to be the preferred amino acids for reaction. The reactivity of amino acid residues, however, in peptides and proteins is certainly different, depending on context and solvent accessibility.
To provide more fundamental information and develop a method for determining the fast reactions of radicals with proteins, we describe a systematic study of the selectivity and specificity of CO3− with proteins/peptides as targets. To accomplish this, we extended the FPOP platform for generating secondary radicals. Incorporating hydrogen peroxide into sample aliquots, we generated by pulsed laser irradiation hydroxyl radicals, which then react with other reagents (here CO32-/HCO3−) to generate secondary radicals. The flow system we used overcomes potential protein damage of static laser irradiation, ensuring the homogeneity of each labeling event. Specifically, for proteins and peptides dissolved in sodium bicarbonate/bicarbonate buffer, we can successfully generate the carbonate radical anion and monitor the oxidation of selected residues on proteins and peptides, taking advantage of advanced mass-spectrometry-based analysis. For the first time, we successfully demonstrate the reactivity and selectivity of carbonate radical anion towards side chains of amino acid residues in proteins to advance significantly the previous studies of reactions with free amino acids. Moreover, the method and platform reported here can be generalized for other bio-relevant studies of free radicals, providing new possibilities for radical generation and subsequent fundamental investigations of protein/radical reactivity.
Principles
FPOP Platform
An FPOP platform (Figure 1) implements a 248 nm KrF excimer to photolyze cleavable reagents (e.g., hydrogen peroxide, persulfate). A silica capillary connects both a syringe pump and a collection tube that terminates the flow. The polyimide coating is removed along the tube to give a transparent window for the laser beam. The laser, typically generated at 7.4 Hz by an external pulse generator, is restricted by an iris and focused by two convex lenses to pass through the transparent window. Within the laser window, hydrogen peroxide is photolyzed into hydroxide radicals. These radicals under usual concentration conditions oxidize the protein sample usually by substitution of H by OH, but under the conditions of this study react with carbonate/bicarbonate anion to generate secondary carbonate radical anions. This approach should be adaptable to formation of other radicals that are important in biology.
Figure 1.
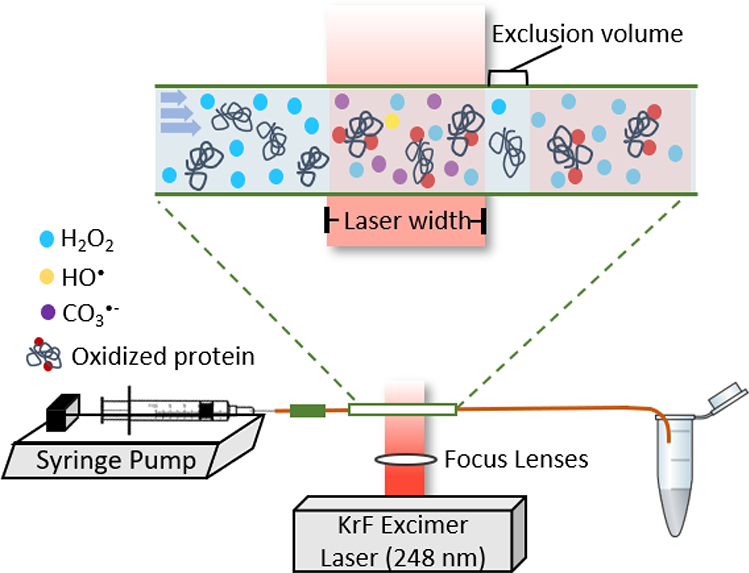
FPOP platform. A sample syringe, driven by a syringe pump, is connected to silica tubing, where part of the polyimide coating is removed to afford a transparent window. The other end of the capillary is inserted into a collection tube containing reagents to deactivate left-over oxidizing agents. A 248 nm KrF excimer laser is placed such that its beam is perpendicular to the flow system and has lenses to focus the beam on the flow tube. Sample proteins will react with the radicals within the laser window. The flow rate is determined such that between every two solution plugs exists an unexposed portion, termed an “exclusion volume”, that minimizes “double hits” on the protein solution.
To avoid over oxidation on the same solution plug, the flow rate on the syringe pump is carefully tuned. A small volume of solution portion is intentionally excluded to create a barrier between two exposed plugs, termed an exclusion volume. The flow-rate calculation is by equation 1, where the exclusion volume is typically ~ 20%. After the oxidation chemistry, the protein sample is collected in the tube containing methionine and catalase to prevent post-oxidation by excess hydrogen peroxide and any remaining oxidative species.
| (1) |
Numerical Simulation of CO3−• Oxidation Chemistry
Generation of the carbonate radical anion in the laboratory can be by pulse radiolysis of N2O-saturated aqueous solution containing sodium bicarbonate at alkaline pH22, 24 or UV photolysis of suitable metal complexes (e.g., [Co(NH3)4CO3]ClO420). It can also be formed from human superoxide dismutase (hSOD)25 in phosphate buffer with EPR spin-trapping detection or oxidation of bicarbonate with a sulfate radical anion generated by photolysis of persulfate anion and detected with UV spectroscopy19, 26. In the current study, we considered generating the carbonate radical anion as the secondary radical through a cascade process starting with sulfate radical anion, formed by photolysis of sodium persulfate. The persulfate anion, however, reacts moderately with amino acid residues (e.g., tryptophan and methionine) even in the absence of laser irradiation. In addition, the rate constant for carbonate radical anion production from the sulfate radical anion is at least 1000 times smaller than that of protein oxidation by the sulfate radical anion. Furthermore, the carbonate radical anion is a considerably weaker oxidizing agent (E0 = 1.59 V) than the sulfate radical anion (E0 = 2.43 V), rendering it difficult to make CO3−• the predominate oxidative species in the reaction regime. We anticipated these difficulties and chose instead to employ a weaker oxidant, hydroxyl radical (E0 = 2.30 V), from hydrogen peroxide photodissociation. To select appropriate conditions, we conducted numerical simulation of the cascade chemistry (for rate constants, see Table 1).
Table 1.
Reaction scheme and rate constants for generation of the carbonate radical anion by reactions of hydroxyl radicals from H2O2 with carbonate/bicarbonate in a buffer.
In a carbonate/bicarbonate aqueous buffer, hydroxyl radicals from photolysis of hydrogen peroxide upon pulsed laser irradiation (Table 1, Reaction 1) react with the carbonate (Table 1, Reaction 2) or bicarbonate anions (Table 1, Reaction 4) to form the carbonate radical anion. Both reactive radicals will decay through recombination (Table 1, Reactions 5, 6). Using these rate constants, we carried out a numerical simulation using Mathcad 14.0 to predict the outcome from second-order kinetics, as shown in Figure 2. We adopted the “Rkadapt” function in MathCad to solve the system of ordinary differential equations that arise from the network of second order reactions. The time interval from 0 to 10 ms was divided into 10 decades, each divided uniformly into 100 steps. The “RKadapt” solves to a “TOL” of 1 × 10−12.
Figure 2.
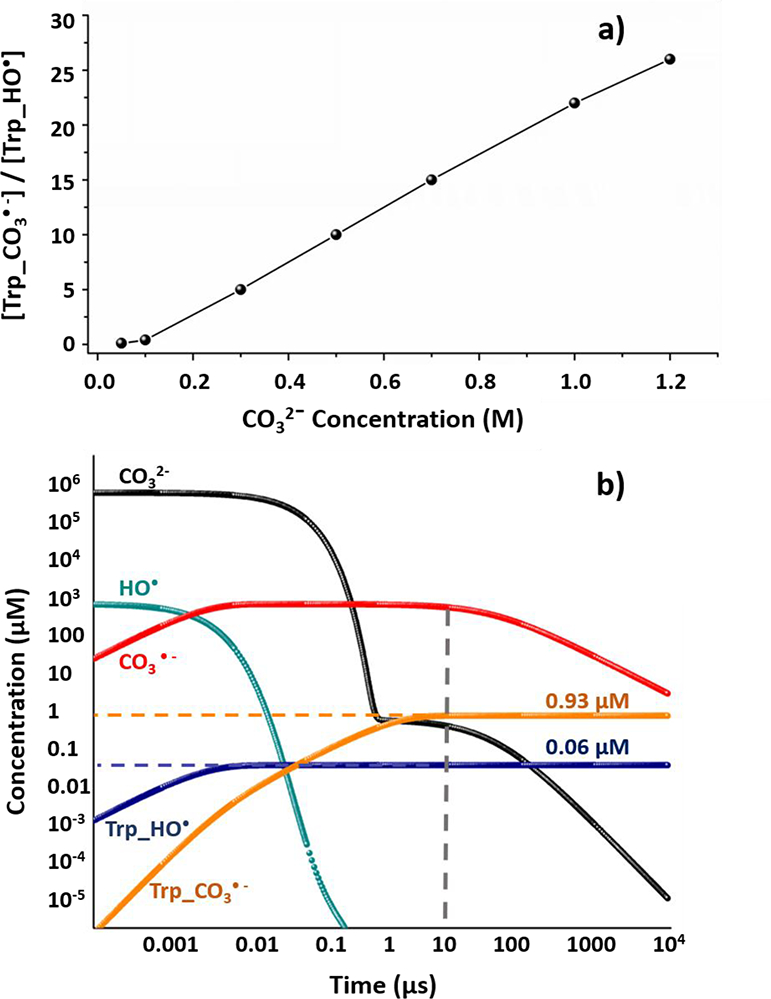
Numerical simulation of (a) CO32- concentration as a function of the ratio of [CO3−•] and [HO•] oxidized tryptophan concentration, where the amino acid tryptophan is taken as a model for reactions of amino acids with a target protein; (b) CO3−• and other related species as a function of time with 700 mM CO32-. Different curves, as denoted by different colors, are plotted on a log scale to represent time-dependent concentrations of each components. The products produced by oxidation of CO3−• and HO• are colored in orange and dark blue, respectively. The dashed line represents the time at which the chemistry is optimized (i.e., largely complete in 10 μs).
In the simulation, the initial concentration of hydrogen peroxide is 15 mM, and the starting concentration of HO• after laser irradiation is approximately 1 mM, which is the typical yield as measured previously29. In addition, we used the reaction with tryptophan as a model for the reaction of CO3−• with a target protein. Under the cascade chemistry regime (Figure 2a), the ratios of CO3−• and HO• oxidized tryptophan concentrations show corresponding increases when [CO32-] increases at pH = 10. With 1.2 M CO3 2- as buffer, CO3−• gives almost 26-fold more oxidized product than that by HO•, showing the best result. Taking the practical solubility of CO32- and HCO3− into consideration, however, we chose 700 mM carbonate anion as the condition for the detailed simulation shown in Figure 2b. As denoted by the green curve, the hydroxyl radical reacts with carbonate and bicarbonate anion, yielding nearly complete conversion to CO3−• in less than 1 μs. The concentration of the carbonate radical anion reaches a maximum at ~ 0.01 μs and then remains roughly constant until near 100 μs. Ultimately, CO3−• self quenches, showing a decrease as shown by the red curve. The vertical grey dash line represents approximately the timescale for oxidation chemistry (~ 10 μs). At and beyond this time, the CO3−• is already the predominant oxidative species as can be seen by comparing its underlying area with that of HO•, which has largely disappeared. The concentrations for CO3−• and HO• oxidized tryptophan after 10 μs is 0.93 μM and 0.06 μM, respectively. Although there are remaining hydroxyl radicals, the CO3−• oxidation contributes almost 15-fold more than HO•, allowing us to determine the reactivity and selectivity of CO3−• towards different amino acids contained in a protein.
Materials
Bombesin acetate salt hydrate, α-melanocyte stimulating hormone, angiotensin I bovine, leucine enkephalin, bradykinin, glycine, catalase, urea, L-histidine, L-methionine, 30% hydrogen peroxide, triethylammonium bicarbonate buffer (TEAB), HPLC-grade solvents, apo-myoglobin from equine skeletal muscle, ubiquitin from bovine erythrocytes (Millipore Sigma Co., St. Louis, MO, USA)
Sequencing grade trypsin (Promega Co., Madison, WI, USA)
Silica capillary (150 μm i.d., Polymicro Technologies, Pheonix, AZ, USA)
C18 reversed-phase desalting column (nanoViper, 100 μm × 2 cm, 5 μm,100 Å; Thermo Fisher Scientific, Waltham, MA, USA)
Analytical column with C18 reversed-phase material (Magic, 100 μm, 180 mm, 5 μm, 120 Å; Michrom Bioresources, Inc., Auburn, CA)
Solvent A (water with 0.1% formic acid by volume) and solvent B (80% acetonitrile with 0.1% formic acid by volume).
C18 NuTip (Glygen Co.)
Instrumentation
248nm KrF excimer laser (GAM Laser Inc., Orlando, FL, USA)
Syringe pump (Harvard Apparatus, Holliston, MA, USA)
External pulse generator (B&K Precision, Yorbal Linda, CA, USA)
Ultimate 3000 Rapid Separation system (Dionex, Thermo Fisher Scientific, Waltham, MA, USA)
Bruker MaXis 4G quadrupole time-of-flight mass spectrometer (Bruker, Co., Billerica, MA)
Thermo Q Exactive Plus orbitrap mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA)
Protocol
Preparation of buffer solution and stock solution
Dissolve 0.7819 g sodium carbonate and 0.5361 g sodium bicarbonate in 10 mL distilled water to constitute 700 mM carbonate/bicarbonate buffer to achieve a pH value of 10.0 ± 0.2.
Dissolve 0.376 g glycine, 0.12 g sodium hydroxide in 10 mL distilled water, then add 0.584 g sodium chloride and 0.745 g potassium chloride to constitute 700 mM glycine-NaOH buffer with the same ionic strength at pH = 10 ± 0.2.
Prepare histidine solution by dissolving 0.0233 g histidine in 1 mL glycine-NaOH buffer, then dilute into 50 mM histidine stock solution.
Make 70 mM methionine stock solution in both 1 mL carbonate/bicarbonate buffer and glycine-NaOH buffer with 0.0105 g methionine as scavenger.
Prepare 5 μM catalase solution in 1 mL distilled water by dissolving 0.0012 g catalase and diluting to 500 nM.
Dissolve 0.0050 g ubiquitin and 0.0085 g apo-myoglobin in two different buffers to afford 500 μM protein solutions. Dilute further to 50 μM stock solution. In addition, prepare 0.0010 g bombesin acetate salt hydrate, 0.0010 g α-melanocyte stimulating hormone, 0.0010 g angiotensin I bovine, 0.0010 g leucine enkephalin and 0.0010 g bradykinin solution in two different buffers with concentrations in the range 600 – 780 μM depending on its molecular weight. Dilute all solutions to ~ 500 μM and mix those in the same buffer to give 50 μM a ‘Peptide Cocktail’ stock solution.
Prepare 300 mM hydrogen peroxide solution immediately before the FPOP laser irradiation by mixing 30 μL 30% (v/v) hydrogen peroxide solution with 970 μL of two different buffers individually. Keep the hydrogen peroxide solution on ice throughout the experimental procedure to avoid decomposition.
Protein oxidation on FPOP
Oxidation achieved by carbonate radical anion
Warm the 248nm KrF laser and measure the laser energy with a sensor meter. Adjust the laser energy to 24 mJ/pulse at a frequency of 7.4 Hz. The laser width is measured by placing colored tape right behind the transparent silica tubing (width = 2.73 mm) and observing a burn mark. Choose a 20% exclusion volume, giving a flow rate of 26.8 μL/min, which can be calculated based on the equation 1.
Inject 10 μL of 70 mM methionine stock solution in carbonate/bicarbonate buffer and 1 μL 500 nM catalase solution to a low-bind Eppendorf tube to be used as the collection tube. Place the collection tube at the end of the silica tubing.
Mix 5 μL of 50 μM protein solution or ‘Peptide cocktail’ with 42 μL carbonate/bicarbonate buffer; then add 3 μL of 300 mM hydrogen peroxide solution. Transfer the sample solution to a syringe and insert it in the syringe pump (Figure 1). Start the pump and trigger the laser irradiation. The time window between the addition of hydrogen peroxide and laser irradiation is controlled as 20 s.
After laser irradiation, digest the protein sample with trypsin according to manufacturer’s protocol. Desalt the digested peptides or the ‘peptide cocktail’ by using C18 NuTip to eliminate excess salts and remaining reagents.
Oxidation by hydroxyl radicals
Adopt the same experimental procedure as in the above description with the reagents in glycine-NaOH buffer, except add 1 μL of 50 mM histidine stock solution into 46 μL protein before the final addition of hydrogen peroxide solution.
LC-MS/MS analysis
Dilute 5 μL digested protein sample into 45 μL water with 0.1% formic acid and centrifuge for 3 min. Then load the sample solution onto a C18 reversed-phase desalting column at 4 μL/min for 10 min. Use a custom-packed analytical column for sample separation on Ultimate 3000 Rapid Separation with C18 reversed-phase material in silica tubing. Use gradient solvents A (water with 0.1% formic acid by volume) and B (80% acetonitrile with 0.1% formic acid by volume). Control the flowrate at 400 nL/min with the following gradient: 2% B to 60% B in 60 min, increase to 90% in 2 min, maintain at 90% for 5 min, return to 2% B in 1 min and equilibrate at 2% B for 7 min. Use a Thermo Q Exactive Plus orbitrap mass spectrometer (or other suitable proteomics instrument) coupled with a Nanospray Flex source for downstream detection with 2.5 kV spray voltage at 250 °C. Acquire results in the data-dependent mode, where the 10 most abundant ions are selected for “higher energy” collisional dissociation (HCD).
Results and Discussion
Generation of Carbonate Radical Anion on FPOP Platform
To demonstrate the oxidative modification by carbonate radical anion, we chose apomyoglobin (aMb) as the model protein. We conducted the experiments with 15 mM hydrogen peroxide, as the radical precursor, at pH = 10 in 700 mM CO32-/HCO3− buffer to maximize CO3−• formation and in 700 mM glycine-NaOH buffer to give only HO• as a parallel control. In the negative control experiments, the solution composition, including ion strength of the buffer, was maintained as before. With no laser irradiation (Figure 3a), the unmodified protein signal (at charge state of +20) is the most intense one followed by a small signal for the oxidized species seen with a +15.9949 Da mass shift, showing a basal oxidation. Irradiation with the KrF laser under the same conditions generates CO3−• and oxidizes aMb to give more intense signals for singly (+15.9949 Da), doubly (+31.9898 Da) and triply oxidized species (+47.9847 Da), etc. (Figure 3b). As compared to the hydroxyl-radical oxidation in the glycine-NaOH buffer (Figure 3c), the two oxidation profiles show good similarity, demonstrating that CO3−• oxidation also results in the addition of ‘an oxygen’ to the target protein. This is consistent with an oxidation mechanism of CO3−• whereby the formation of protein radical occurs by hydrogen atom abstraction, followed by an addition of dioxygen from dissolved O2 in solution, reactions that are similar to those of hydroxyl radicals1.
Figure 3.
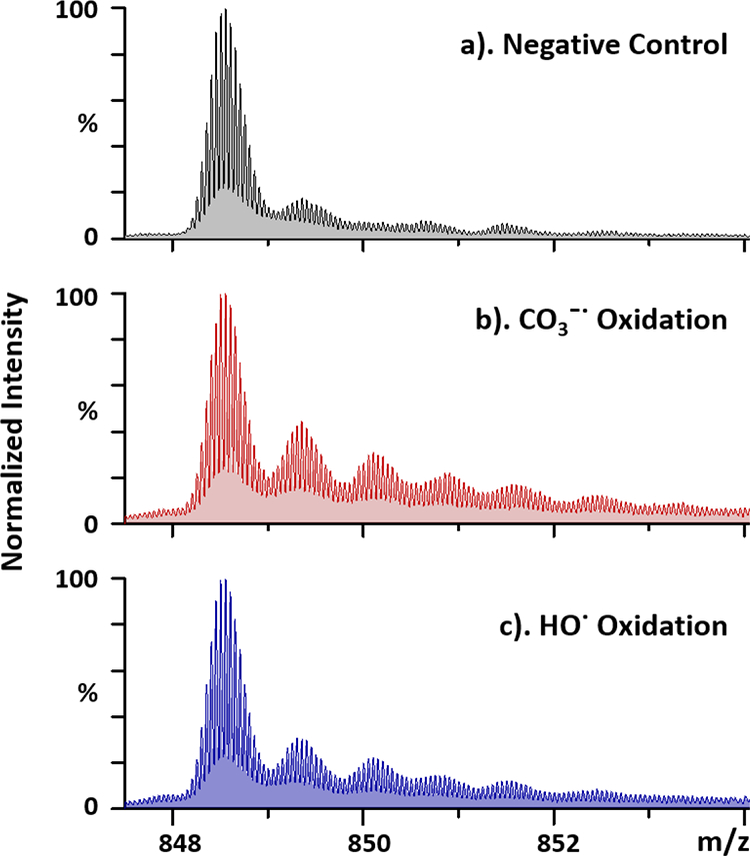
The quadrupole time-of-flight (QTOF) mass spectra of the +20 charge state of apomyoglobin(aMb) with labeling conditions of (a) in CO32-/HCO3− buffer without laser irradiation (negative control); (b) in CO32-/HCO3− buffer with laser irradiation; (c) in glycine-NaOH buffer with laser irradiation, oxidized by hydroxide radical and measured for the intact protein.
Selectivity and Reactivity of CO3−• with Model Peptides
Utilizing the conditions that favor formation of CO3−• initiation, we employed model peptides with little or no higher order structure for a study of its intrinsic residue-level specificity. Along with CO3−• chemistry, hydroxyl radical oxidation was also performed for batch control and for comparison purposes, as previously described. To examine peptides, we prepared a “peptide cocktail” containing bombesin acetate salt hydrate, α-melanocyte stimulating hormone (α-Mel), bovine angiotensin I, leucine enkephalin (Leu-enkephalin) and bradykinin and mixed the solutions with hydrogen peroxide in two different buffers at pH = 10. Oxidation percentages of the modified amino acids (Figure 4) are shown in the decreasing order upon the reactivity of CO3−•, and they were corrected by subtracting basal oxidation from negative control as shown in Figure 3a. Methionine and tryptophan are the most reactive amino acids for both radicals, where nearly 90% becomes oxidized by CO3−• and 60% by HO•. A similar phenomenon occurred for tyrosine where modification by CO3−• is almost 8-times higher than that by HO•. One possible reason is the weaker oxidative character leads to considerably longer lifetime for CO3−•. The more specific reactivity towards selected amino acids for CO3−• will likely give more oxidation of those reactive residues. Phenylalanine and histidine can also react with CO3−• with similar reactivity as for HO•. Oxidation of other aliphatic amino acids (e.g., proline on bradykinin) occurs with HO• treatment. For the CO3−• condition, however, only phenylalanine oxidation occurred. From all of the evidence, we conclude that CO3−• is a more selective oxidant than HO•, and the order of inherent reactivity of CO3−• is:
Figure 4.
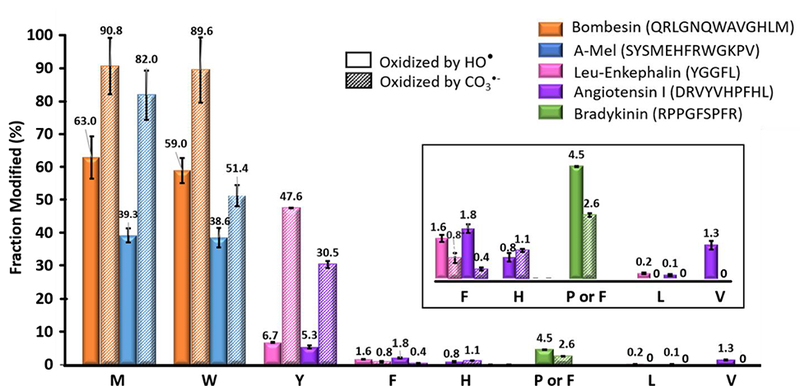
Comparison of the residue-level fraction modified (in percentage) of a ‘Peptide Cocktail’ by CO3−•˙ and HO•. Different colors represent different peptides, and the sequences are shown correspondingly. Solid bars denote the HO• oxidation extent and the patterned bars denote the CO3−• oxidation. The data were corrected with respect to the negative control. Error bars are the standard deviations of three independent runs. The inset is an enlarged portion of the figure. For bradykinin, only F undergoes oxidation with CO3−•, whereas F and P oxidation are both oxidized but not distinguishable because the chromatograms of the two modified peptides overlap.
M ≈ W > Y > H ≈ F.
The results are consistent with those of a previous investigation18 where the reactivity of free amino acids in solution was measured, and in which methionine, tryptophan and tyrosine were found to be the most reactive amino acids towards CO3−• with rate constants of 1.2×108 M−1s−1, 4.4×108 M−1s−1 and 2.9×108 M−1s−1, respectively. Although the reported rate constant for histidine is almost 100-fold greater than that for phenylalanine (i.e., rate constants of 7.0×106 M−1s−1 and 5.0×104 M−1s−1), the reactivities of the two residues are similar when they are part of proteins and peptides. It is worth noting that the electrostatic interaction between the negatively charged carbonate radical anion and the positive charged imidazole group at lower pH may contribute to an increased reactivity. Therefore, in real biological systems, the reaction rate with histidine may vary depending on the local pH.
Residue-Resolved Modification Measurement by LC-MS/MS
We quantified the oxidation extent at the residue level from the peak areas of the chromatograms. Extracted ion chromatograms (EICs) for each modified and unmodified species were exported from raw files with Qual Browser (Thermo Xcalibur 2.2 software) (Figure 5). The (+16 Da) angiotensin I oxidized by the carbonate radical anion (Figure 5b) and by the hydroxyl radical (Figure 5c) are listed below the unmodified peptides. Different colors represent different modified residues. The modification fraction for a residue is calculated with equation 2, where Aox is the peak area for the oxidized-species signal and Aun is the peak area for the unmodified. Chromatographic peaks are fitted by the Gaussian Function, and the averaged R2 is 0.97. For complex chromatograms with overlapping signals of oxidized residues (Figure 5c), the peak representing tyrosine oxidation (purple in Figure 5b) is scaled into the lower panel, and the oxidation extent of valine is represented by the grey gaussian peak. In addition, each production (MS/MS) spectrum (e.g., Figure 6) was manually validated to assure an accurate oxidizing residue assignment. Comparing the signals for the fragment ions, we see a clear +16 Da mass shift for b4 and subsequent b ions for the modified peptide as compared with the unmodified peptide, indicating that oxidation took place on the Y4 residue.
Figure 5.
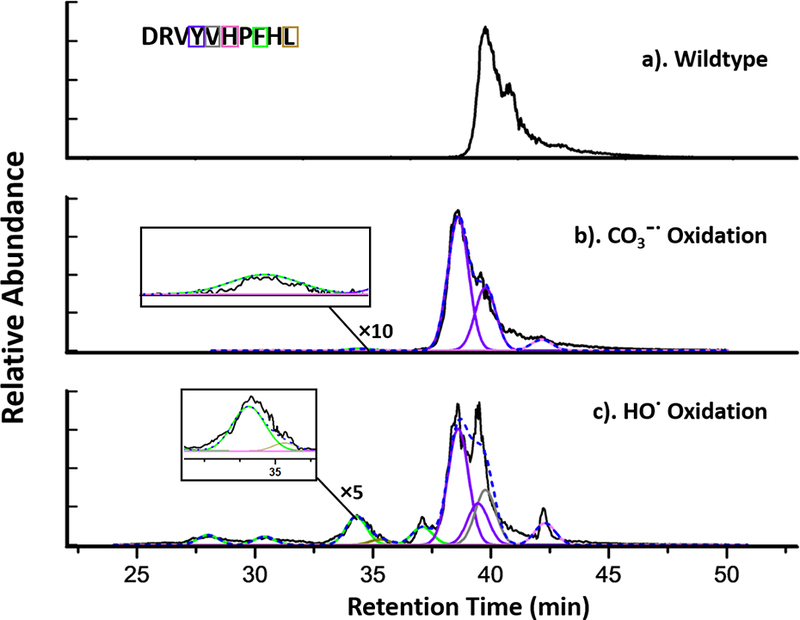
Extracted ion chromatogram (EICs) of angiotensin I wildtype (a), oxidized by CO3−• (b) and HO•. (c). Different colors (coded with the chromatogram colors) indicate differnet oxidized residues as determined by product-ion (MS/MS) spectra. Each peak is fitted by a Gaussian Function, and the overall fitting curve is presented as blue dotted line. The averaged R2 for the fitting is 0.97.
Figure 6.
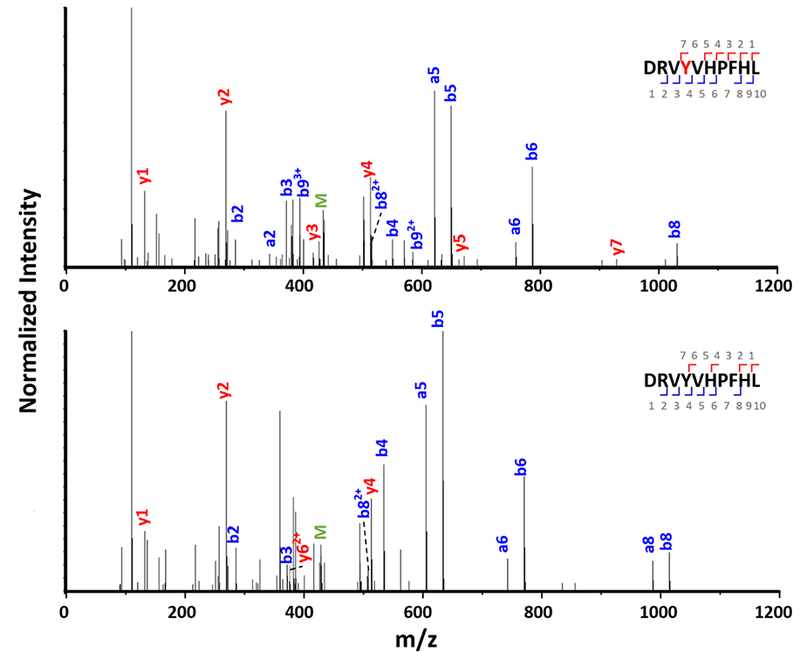
Product-ion (MS/MS) spectra of oxidized and wildtype angiotensin I.
| (2) |
From the chromatogram (Figure 5), the number of oxidized isomeric peptides by the CO3−• is significantly less than that by the HO•. Under the CO3−• oxidation conditions, we resolved at least four isomeric peptides, two of which undergo oxidation on Tyr, and two minor ones involve oxidation on His and Phe. For HO•. oxidation, however, a more complex pattern of oxidation occurs presenting at least six isomeric products, including oxidations on Val, Leu, Tyr, His and Phe with a greater discernible amounts.
Selectivity and Reactivity of CO3−• with Model Proteins
In addition to peptides mixture, we further investigated the selectivity and specificity of CO3−• with proteins, where apo-myoglobin and ubiquitin were chosen as the model systems. Experiments were carried out with CO3−• dominant condition and HO• governing condition individually, and the outcomes were analyzed by “bottom-up” proteomics. After integrating the signals obtained through LC-MS/MS, we can list the modified amino acids for each protein (Figure 7). From the ubiquitin data set (Figure 7a), CO3−• gives the most oxidation on methionine (M), as does HO•, underscoring their oxidation preference towards methionine. Other reactive residues include the aromatic amino acids phenylalanine (F4 and F45), tyrosine (Y59) and histidine (H68) undergo moderate oxidation by CO3−• as compared to HO•. Other amino acids that are reactive with HO• (e.g., valine (V), leucine (L), glutamine (Q), lysine (K), proline (P)) are unreactive with CO3−•.
Figure 7.
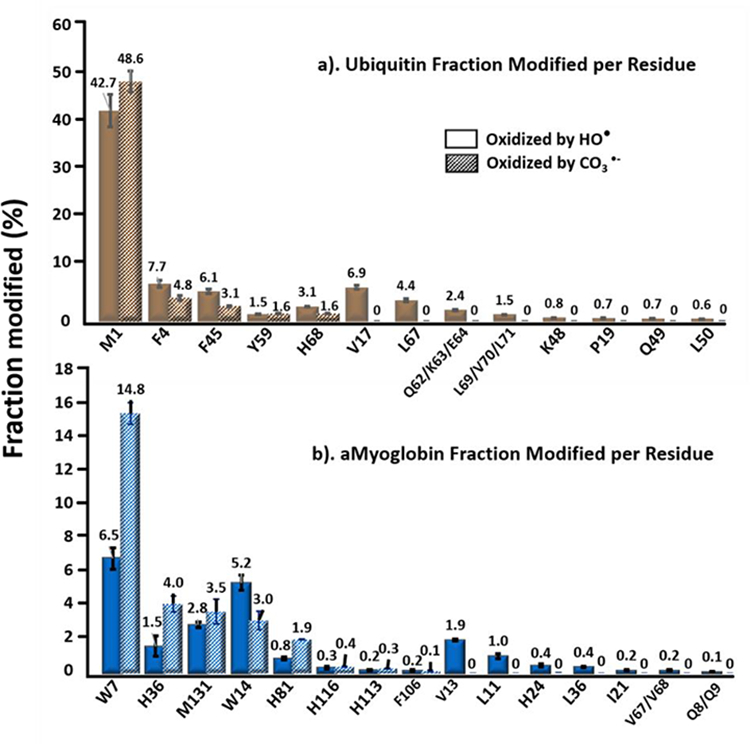
Comparison of residue-level fraction modified (in percentage) of (a) ubiquitin and (b) apo-myoglobin modified by carbonate radical (CO3−•) and hydroxyl radical (HO•). Solid bars denote the hydroxyl radical oxidation and the patterned bars denote the carbonate radical anion oxidation. Presented data were corrected with respect to the negative control. Error bars are standard deviations of three independent runs.
For apo-myoglobin (Figure 7b), tryptophan W7 exhibits the highest oxidation extent on by CO3−• showing a doubled fraction modified value as compared to that for HO•. Histidine also gives nearly doubled modification fraction by CO3−• oxidation, for example H36 and H81, indicating its considerable reactivity. The oxidation behavior towards methionine is consistent with the results for ubiquitin, where CO3−• oxidation slightly favors that of HO•, shown as M131. Other potentially reactive residues like H113, H116 and F106 undergo an oxidation extent of less than 0.5%, just as with HO•. The reason is likely the small solvent accessible area (SASA) for these sidechains on the protein surface. In addition, the size difference of CO3−• and HO• may contribute to the reaction extent on a given amino acid. For example, the oxidation percentage of W14 is 3%, which is nearly 5-times lower than that of W7 (14.8 %). The SASA of W7 and W14 is 15.3 Å2 and 6.8 Å2, respectively by calculation11. Thus, for the smaller HO•, the oxidation extent is greater than that for the larger CO3−•, which may have difficulty penetrating the surface to react with W14. Other residues like V13/67/68, L11/86, I21 and Q8/Q9 are relatively inert to CO3−• with no shown oxidation.
Compared to the results with the model peptides, the CO3−• oxidative preference towards Met and Trp over HO• in proteins is consistent and so is the negligible reactivity towards aliphatic amino acids. For tyrosine oxidation by CO3−• in ubiquitin (Y59 in Figure 7a), however, the extent is much less than that with model peptides, shown as 1.6% vs 30–50%. The difference arises from the location of the Y59 in the 3D structure and its limited SASA, emphasizing the importance of incorporating peptides with minimal higher order structure to eliminate conformational effect in the determination of residue-level specificity.
Advantages and caveats
We optimized the production of carbonate radical anion on a FPOP platform by using a reaction cascade scheme starting with the hydroxyl radical. This approach enables the study of peptides and proteins to determine the relative reactivities of various amino acid sites in those species. Although the results for the protein/peptides systems are largely consistent with those of an early study carried out with free amino acids, this new approach provides direct evidence for the radical reactions of proteins (and other biomolecules) with radicals, here the carbonate radical anion, demonstrating clearly those residues or regions of the biomolecule that are reactive.
The approach suggests that a general approach can be achieved by implementing the FPOP platform to generate either primary or secondary biologically relevant radicals (by cascade reactions) and to determine their fundamental reactivities with biomolecules. Primary HO• and SO4−• radicals can be formed from peroxy-compounds. Generation of secondary radicals (e.g., CO3−•, NO2•, halides) may be optimized by varying buffer contents, pH, and radical precursor and by involving a radical scavenger. In the current study, this was done by using kinetic simulations. For other radicals for which kinetic data do not exist, the conditions must be optimized by using mass spectrometry analysis. Other spectroscopy techniques (e.g., fluorescence microscopy) can also be incorporated in the post-modified identification.
Although we used no scavenging reagents in the current study for the generation of carbonate radical anion, incorporating such reagents can be done in future studies. The use of histidine as a scavenger in the hydroxyl-radical generation not only controls radical lifetime but also intercepts radicals and minimizes protein modification, a point that may be important for physiologically relevant studies. For example, biological systems respond to different oxidative stress, and the oxidizing potential can be varied on the FPOP platform. In addition, the FPOP flow design effectively avoids over-modification of a target and improves labeling homogeneity by controlling flow rate, exclusion volume, and radical lifetime. We suggest that this repurposed FPOP platform coupled with mass spectrometry-based analysis is a new option for studies of radical generation and their reactions with biomolecules.
Highlights:
Fast photochemical Oxidation of Protein (FPOP) platform established for generating and secondary radicals of biological interest.
Fundamental reactivity of biologically relevant radicals with peptides and proteins explored with FPOP coupled with mass spectrometry.
Selectivity and specificity of the carbonate radical anion (CO3−•) determined by analysis of modified proteins and peptides.
A promising platform now available for informative studies of site-specific reactivity of radicals with proteins sensitive to oxidative-stress.
Acknowledgments
The study was supported by National Institute of General Medical Sciences grant 5P41GM103422 and by 1S10OD016298 for instrumentation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference:
- 1.Xu GH; Chance MR, Hydroxyl radical-mediated modification of proteins as probes for structural proteomics. Chemical Reviews 2007, 107 (8), 3514–3543. [DOI] [PubMed] [Google Scholar]
- 2.Maleknia SD; Brenowitz M; Chance MR, Millisecond radiolytic modification of peptides by synchrotron X-rays identified by mass spectrometry. Analytical Chemistry 1999, 71 (18), 3965–3973. [DOI] [PubMed] [Google Scholar]
- 3.Hambly DM; Gross ML, Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. J Am Soc Mass Spectrom 2005, 16 (12), 2057–63. [DOI] [PubMed] [Google Scholar]
- 4.Li KS; Chen GD; Mo JJ; Huang RYC; Deyanova EG; Beno BR; O’Neil SR; Tymiak AA; Gross ML, Orthogonal Mass Spectrometry-Based Footprinting for Epitope Mapping and Structural Characterization: The IL-6 Receptor upon Binding of Protein Therapeutics. Analytical Chemistry 2017, 89 (14), 7742–7749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li J; Wei H; Krystek SR; Bond D; Brender TM; Cohen D; Feiner J; Hamacher N; Harshman J; Huang RYC; Julien SH; Lin Z; Moore K; Mueller L; Noriega C; Sejwal P; Sheppard P; Stevens B; Chen GD; Tyrniak AA; Gross ML; Schneeweis LA, Mapping the Energetic Epitope of an Antibody/Interleukin-23 Interaction with Hydrogen/Deuterium Exchange, Fast Photochemical Oxidation of Proteins Mass Spectrometry, and Alanine Shave Mutagenesis. Analytical Chemistry 2017, 89 (4), 2250–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yan YT; Chen GD; Wei H; Huang RYC; Mo JJ; Rempel DL; Tymiak AA; Gross ML, Fast Photochemical Oxidation of Proteins (FPOP) Maps the Epitope of EGFR Binding to Adnectin. J Am Soc Mass Spectr 2014, 25 (12), 2084–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li KS; Rempel DL; Gross ML, Conformational-Sensitive Fast Photochemical Oxidation of Proteins and Mass Spectrometry Characterize Amyloid Beta 1–42 Aggregation. J Am Chem Soc 2016, 138 (37), 12090–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hart KM; Ho CMW; Dutta S; Gross ML; Bowman GR, Modelling proteins’ hidden conformations to predict antibiotic resistance. Nat Commun 2016, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poor TA; Jones LM; Sood A; Leser GP; Plasencia MD; Rempel DL; Jardetzky TS; Woods RJ; Gross ML; Lamb RA, Probing the paramyxovirus fusion (F) protein-refolding event from pre- to postfusion by oxidative footprinting. Proc Natl Acad Sci U S A 2014, 111 (25), E2596–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen JW; Rempel DL; Gau BC; Gross ML, Fast Photochemical Oxidation of Proteins and Mass Spectrometry Follow Submillisecond Protein Folding at the Amino-Acid Level. Journal of the American Chemical Society 2012, 134 (45), 18724–18731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gau BC; Chen H; Zhang Y; Gross ML, Sulfate Radical Anion as a New Reagent for Fast Photochemical Oxidation of Proteins. Analytical Chemistry 2010, 82 (18), 7821–7827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang B; Rempel DL; Gross ML, Protein Footprinting by Carbenes on a Fast Photochemical Oxidation of Proteins (FPOP) Platform. J Am Soc Mass Spectrom 2016, 27 (3), 552–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng M; Zhang B; Cui W; Gross ML, Laser-Initiated Radical Trifluoromethylation of Peptides and Proteins: Application to Mass-Spectrometry-Based Protein Footprinting. Angew Chem Int Ed Engl 2017, 56 (45), 14007–14010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Medinas DB; Cerchiaro G; Trindade DF; Augusto O, The carbonate radical and related oxidants derived from bicarbonate buffer. Iubmb Life 2007, 59 (4–5), 255–262. [DOI] [PubMed] [Google Scholar]
- 15.Sankarapandi S; Zweier JL, Bicarbonate is required for the peroxidase function of Cu,Znsuperoxide dismutase at physiological pH. Journal of Biological Chemistry 1999, 274 (3), 1226–1232. [DOI] [PubMed] [Google Scholar]
- 16.Hodgson EK; Fridovich I, The mechanism of the activity-dependent luminescence of xanthine oxidase. Arch Biochem Biophys 1976, 172 (1), 202–5. [DOI] [PubMed] [Google Scholar]
- 17.Radi R, Nitric oxide, oxidants, and protein tyrosine nitration. P Natl Acad Sci USA 2004, 101 (12), 4003–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neta P; Huie RE; Ross AB, Rate Constants for Reactions of Inorganic Radicals in Aqueous-Solution. J Phys Chem Ref Data 1988, 17 (3), 1027–1284. [Google Scholar]
- 19.Shafirovich V; Dourandin A; Huang WD; Geacintov NE, The carbonate radical is a site-selective oxidizing agent of guanine in double-stranded oligonucleotides. Journal of Biological Chemistry 2001, 276 (27), 24621–24626. [DOI] [PubMed] [Google Scholar]
- 20.Paviani V; Queiroz RF; Marques EF; Di Mascio P; Augusto O, Production of lysozyme and lysozyme-superoxide dismutase dimers bound by a ditryptophan cross-link in carbonate radical-treated lysozyme. Free Radical Bio Med 2015, 89, 72–82. [DOI] [PubMed] [Google Scholar]
- 21.Surmeli NB; Litterman NK; Miller AF; Groves JT, Peroxynitrite Mediates Active Site Tyrosine Nitration in Manganese Superoxide Dismutase. Evidence of a Role for the Carbonate Radical Anion. Journal of the American Chemical Society 2010, 132 (48), 17174–17185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gebicka L; Didik J; Gebicki J, Reactions of heme proteins with carbonate radical anion. Res Chem Intermediat 2009, 35 (4), 401–409. [Google Scholar]
- 23.Chen SN; Hoffman MZ, Rate constants for the reaction of the carbonate radical with compounds of biochemical interest in neutral aqueous solution. Radiat Res 1973, 56 (1), 40–7. [PubMed] [Google Scholar]
- 24.Boccini F; Domazou AS; Herold S, Pulse radiolysis studies of the reactions of CO3 center dot- and NO2 center dot with nitrosyl(II)myoglobin and nitrosyl(II)hemoglobin. Journal of Physical Chemistry A 2006, 110 (11), 3927–3932. [DOI] [PubMed] [Google Scholar]
- 25.Zhang H; Joseph J; Crow J; Kalyanaraman B, Mass spectral evidence for carbonate-anion-radical-induced posttranslational modification of tryptophan to kynurenfne in human Cu, Zn superoxide dismutase. Free Radical Bio Med 2004, 37 (12), 2018–2026. [DOI] [PubMed] [Google Scholar]
- 26.Joffe A; Geacintov NE; Shafirovich V, DNA lesions derived from the site selective oxidation of Guanine by carbonate radical anions. Chem Res Toxicol 2003, 16 (12), 1528–38. [DOI] [PubMed] [Google Scholar]
- 27.Buxton GV; Elliot AJ, Rate-Constant for Reaction of Hydroxyl Radicals with Bicarbonate Ions. Radiat Phys Chem 1986, 27 (3), 241–243. [Google Scholar]
- 28.Schulz KG; Riebesell U; Rost B; Thoms S; Zeebe RE, Determination of the rate constants for the carbon dioxide to bicarbonate inter-conversion in pH-buffered seawater systems. Mar Chem 2006, 100 (1–2), 53–65. [Google Scholar]
- 29.Niu B; Zhang H; Giblin D; Rempel DL; Gross ML, Dosimetry Determines the Initial OH Radical Concentration in Fast Photochemical Oxidation of Proteins (FPOP). J Am Soc Mass Spectr 2015, 26 (5), 843–846. [DOI] [PMC free article] [PubMed] [Google Scholar]