

Abstract
Type VII secretion systems (ESX) are responsible for transport of multiple proteins in mycobacteria. How different ESX systems achieve specific secretion of cognate substrates remains elusive. In the ESX systems, the cytoplasmic chaperone EspG forms complexes with heterodimeric PE-PPE substrates that are secreted from the cells or remain associated with the cell surface. Here we report the crystal structure of the EspG1 chaperone from the ESX-1 system determined using a fusion strategy with T4 lysozyme. EspG1 adopts a quasi 2-fold symmetric structure that consists of a central β-sheet and two α-helical bundles. Additionally, we describe the structures of EspG3 chaperones from four different crystal forms. Alternate conformations of the putative PE-PPE binding site are revealed by comparison of the available EspG3 structures. Analysis of EspG1, EspG3 and EspG5 chaperones using small-angle X-ray scattering (SAXS) reveals that EspG1 and EspG3 chaperones form dimers in solution, which we observed in several of our crystal forms. Finally, we propose a model of the ESX-3 specific EspG3-PE5-PPE4 complex based on the SAXS analysis.
Keywords: Mycobacterium tuberculosis, protein export, small-angle X-ray scattering, PE-PPE proteins
Introduction
The most deadly bacterial pathogen worldwide is Mycobacterium tuberculosis (Mtb), which causes tuberculosis (TB). While many infectious diseases can be controlled by vaccination, TB lacks an effective vaccine and even prior infection with Mtb does not provide lasting immunity. Moreover, standard anti-TB therapy requires the use of a combination of drugs for six months, which leads to poor compliance and to emergence of drug resistance [1]. Even more threatening is the global increase in extensively drug-resistant Mtb and the emergence of extremely drug-resistant Mtb. Anti-virulence drugs targeting mycobacterial secretion have the potential to become a valuable alternative to classical antibiotics [2]. During infection, pathogenic mycobacteria use several related protein secretion pathways designated ESX systems [3–5]. The Mtb genome encodes five such secretion systems, ESX-1 through ESX- 5. Each consists of ATPases, membrane proteins, a protease, accessory proteins, and secreted substrates [6, 7]. Four conserved components of the ESX-5 system ‒ EccB5, EccC5, EccD5 and EccE5 ‒ form a platform complex with six-fold symmetry that is embedded in the mycobacterial inner membrane [8, 9]. The ESX-1 core complex is composed of paralogous components [10], which suggests that all ESX systems assemble into similar complexes. Many ESX-secreted substrates are interdependent on each other for secretion, suggesting that they might be a part of the ESX secretion machinery [11, 12]. The most abundant class of ESX substrates is represented by the so-called PE and PPE proteins [13]. These proteins generally form alpha-helical heterodimers that are probably secreted in a folded conformation [14]. Several PE/PPE proteins are major antigens for TB diagnostic and vaccine development [15–19]. Importantly, PE/PPE proteins are secreted specifically by their cognate ESX secretion systems [20–24] raising the question of how various ESX systems discriminate among PE/PPE substrates.
Previously it was demonstrated that PE/PPE protein secretion in Mycobacterium marinum is impaired upon disruption of the espG gene encoded within its respective ESX gene locus leading to accumulation of substrates in the bacterial cytosol [25]. The crystal structure of the heterotrimeric EspG5-PE25–PPE41 protein complex revealed that EspG5 interacts with a PE25–PPE41 heterodimer by binding to a hydrophobic patch at the tip of PPE41 [26, 27]. The general YxxD/E secretion motif [28, 29] at the distal end of PE25 is free to interact with the ESX-5 secretion machinery in the inner membrane, probably by interaction with the Ftsk/SpoIIIE-like ATPase EccC5 [30, 31]. In addition, EspG5 was reported to improve solubility of aggregation-prone PE–PPE pairs upon co-expression [26, 32]. Thus, EspG acts as a disaggregase of ESX substrates in the cytosol prior to secretion. Moreover, substrate specificity is determined by the EspG-binding domain of PPE proteins as demonstrated by substrate re-routing experiments [33]. While structures of the EspG5 chaperone in complexes with PE–PPE substrates and a monomeric EspG3 chaperone have been reported [26, 27, 34], structural information on EspG1 is lacking.
In this study, we report the first crystal structure of an EspG1 chaperone from Mycobacterium kansasii and four crystal structures of EspG3 from M. smegmatis and M. marinum. We analyze here the available atomic structures of EspG chaperones and present a thorough study of the conformational variability of EspG proteins in apo and substrate- bound forms (EspG-PE–PPE) using small-angle X-ray scattering (SAXS). In addition, we characterize the SAXS-based rigid-body structure of the EspG3-PE5–PPE4 protein complex in solution and compare it to the atomic structure of EspG5-PE25–PPE41 in order to obtain further insights into protein flexibility and substrate recognition. Our study shows that the EspG chaperones are capable of adopting multiple conformational states, likely a key determinant of their ability to recognize multiple PE–PPE substrates.
Results
The crystal structure of M. kansasii EspG1
Initial attempts to crystallize M. tuberculosis EspG1 (EspG1mtu) were not successful; therefore we screened several homologs of EspG1 from other mycobacterial species. We obtained microcrystals using an optimized construct of M. kansassi EspG1 (EspG1mka) that has 80% sequence identity with EspG1mtu (Supplementary Fig. S1). However, extensive optimization of these crystals did not lead to diffraction quality crystals. To overcome these difficulties, we utilized a fusion approach using maltose binding protein or T4 lysozyme (T4L) as the N- terminal fusions. Whereas maltose binding protein fusion did not crystallize, crystals of the T4L-EspG1mka fusion could be readily optimized and diffracted to 2.27 Å resolution. The structure of T4L-EspG1mka was solved by molecular replacement and refined to Rwork 0.214 and Rfree 0.251 with good geometry (Table 1). The structure contains two molecules in the asymmetric unit (Fig. 1a) with an extensive interface between the T4L moieties (2250 Å2 buried surface area). Surprisingly, part of the TEV cleavage sequence at the N-terminus of T4L is ordered in the structure and contributes both to the T4L dimer interface (849 Å2 buried surface area corresponding to 38% of the T4L–T4L interface) and the intra-subunit contacts between T4L and EspG1mka. The conformations of the two copies of EspG1mka in the asymmetric unit are very similar and superimpose with a root-mean-square deviation (RMSD) of 1.0 Å over 231 Cα atoms (Supplementary Fig. S2). EspG1mka has a typical EspG fold characterized by a central anti-parallel β-sheet and two α-helical bundles (Fig. 1b). Several parts of the EspG1mka structure did not have interpretable electron density and were not modeled, including the loop preceding the α2 helix (chains A and B), the β2–β3 loop (chain A), part of the β6 strand (chain B), and the α6 helix (chains A and B) (Fig. 1a). The residues corresponding to the C-terminal helical bundle displayed higher B factors compared to other parts of the structure (Supplementary Fig. S2b). The C-terminal helical bundle likely forms part of the substrate recognition site and could become more ordered upon binding of a cognate PE–PPE dimer. The N-terminal and C-terminal subdomains of EspG1mka are related by a quasi two-fold symmetry, have 10% sequence identity, and can be superimposed with a RMSD of 2.7 Å over 71 Cα atoms (Supplementary Fig. S3).
Table 1.
Data collection and refinement statistics.
| T4L-EspG1 M. kansasii PDB 5VBA |
EspG3 M. marinum PDB 5DLB |
EspG3 M. smegmatis PDB 4L4W |
EspG3 M. smegmatis PDB 4RCL |
EspG3 M. smegmatis PDB 5SXL |
|
|---|---|---|---|---|---|
| Data collection | |||||
| Wavelength (Å) | 1.0000 | 1.0702 | 0.9793 | 1.0000 | 1.0000 |
| Space group | P212121 | C2 | C2221 | P43212 | P3221 |
| Cell dimensions: | |||||
| a, b, c (Å) | 64.14, 81.69, | 112.21, 46.02, | 47.08, 123.69, | 92.68, 92.68, 158.85 | 59.15, 59.15, |
| 160.02 | 58.01 | 180.85 | 183.1 | ||
| α, β, γ (°) | 90, 90, 90 | 90, 92.24, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 120 |
| Resolution (Å) | 59.54–2.27 (2.33– 2.27)a |
57.96–1.77 (1.87– 1.77) |
45.21–2.04 (2.09– 2.04) |
46.34–2.70 (2.77–2.70) | 39.24–2.46 (2.52–2.46) |
| Rsym | 0.097 (1.110) | 0.066 (0.827) | 0.087 (0.809) | 0.087 (1.600) | 0.075 (0.882) |
| CC1/2b | 99.7 (58.3) | 99.5 (65.9) | 99.7 (64.6) | 99.9 (76.2) | 99.8 (79.3) |
| I / σI | 11.1 (1.74) | 12.6 (1.40) | 12.2 (1.55) | 19.8 (1.92) | 12.2 (1.62) |
| Completeness (%) | 99.8 (100.0) | 89.0 (90.0) | 97.6 (79.5) | 99.8 (98.5) | 99.6 (99.5) |
| Multiplicity | 6.3 (6.5) | 3.2 (3.4) | 5.5 (3.1) | 11.1 (11.2) | 5.4 (5.5) |
| Refinement | |||||
| Resolution (Å) | 59.54–2.27 | 57.96–1.77 | 45.21–2.04 | 46.34–2.70 | 39.24–2.46 |
| No. reflections (total / free) | 39576 / 1882 | 24426 / 1310 | 33548 / 1682 | 19765 / 1008 | 13963 / 707 |
| Rwork / Rfree | 0.214 / 0.251 | 0.209 / 0.245 | 0.186 / 0.231 | 0.230 / 0.278 | 0.206 / 0.247 |
| Number of atoms: | |||||
| Protein | 6288 | 1944 | 4003 | 3709 | 2078 |
| Ligand/ion | 10 | 140 | |||
| Water | 56 | 65 | 225 | 14 | 17 |
| B-factors: | |||||
| Protein | 61.6 | 37.8 | 37.9 | 92.1 | 81.7 |
| Ligand/ion | 68.1 | 72.2 | |||
| Water | 48.5 | 47.1 | 39.3 | 40.6 | 63.4 |
| All atoms | 61.5 | 40.4 | 38.0 | 91.9 | 81.6 |
| Wilson B | 47.8 | 38.0 | 37.2 | 74.7 | 67.6 |
| R.m.s. deviations: | |||||
| Bond lengths (Å) | 0.004 | 0.019 | 0.008 | 0.003 | 0.002 |
| Bond angles (°) | 0.657 | 2.130 | 1.166 | 0.722 | 0.494 |
| Ramachandran distributionc (%): |
|||||
| Favored | 97.73 | 94.16 | 97.32 | 96.07 | 95.97 |
| Outliers | 0.13 | 0.39 | 0 | 0.21 | 0.37 |
Values in parentheses are for the highest-resolution shell.
CC1/2 correlation coefficient as defined in Karplus & Diederichs [42] and calculated by XSCALE [41].
Calculated using the MolProbity server (http://molprobity.biochem.duke.edu) [76].
Figure 1. Crystal structure of M. kansasii EspG1 (EspG1mka).
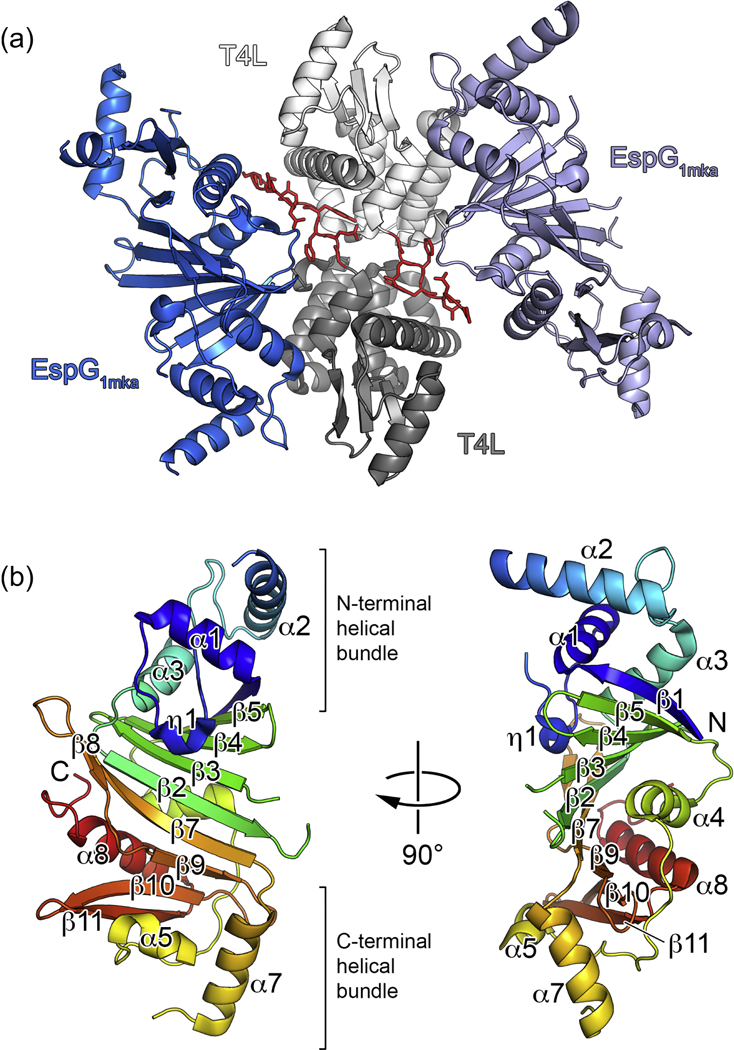
(a) View of the two subunits of the T4L-EspG1mka fusion protein in the asymmetric unit. Chain A is shown in light grey (T4L) and light blue (EspG1mka), and chain B is shown in dark grey (T4L) and dark blue (EspG1mka). Residues corresponding to the TEV cleavage sequence are shown in red with side chains in stick representation. (b) A monomer of EspG1mka is shown in ribbon representation colored in rainbow colors from N-terminus (blue) to C-terminus (red). The N- terminal T4L fusion moiety is not shown for clarity.
The EspG fold is conserved in ESX-1, ESX-3 and ESX-5 systems
In order to extend the structural knowledge of the EspG-substrate interaction and the differences between these interactions in ESX-1, ESX-3 and ESX-5 secretion, we determined additional crystal structures of the EspG chaperones from the ESX-1 and ESX-3 systems (Table 1). Despite the fact that EspG chaperones display the lowest level of protein sequence similarity (13–23% sequence identity) of all the core components of the ESX systems, the EspG1, EspG3 and EspG5 structures have a highly similar fold (Fig. 2, Fig. 3, Supplementary Figs. S4 and S5 and Supplementary Table S1). RMSD of the aligned atoms for EspG superposition is 2.4 Å for EspG1mka vs. EspG5mtu over 224 Cα atoms, 2.4 Å for EspG3mma vs. EspG5mtu over 238 Cα atoms and 2.4 Å for EspG1mka vs. EspG3mma over 234 Cα atoms. Despite the high overall structural similarity, the C-terminal helical bundles of EspG1mka and EspG3 structures have a distinct conformation, when compared to the EspG5mtu structure bound to the PE25–PPE41 dimer, which appears to be incompatible with substrate binding (Fig. 3). Another significant difference is the length and the conformation of the β2–β3 loop. In the EspG5mtu-PE25–PPE41–structure (PDB ID 4KXR [26]), it extends 23 amino acid residues (Gly92-Asn114) and interacts strongly with the PE25–PPE41 dimer, whereas, for example, in the EspG3msm (PDB ID 4L4W) structure the loop consists of only 12 amino acid residues (Ser87-Leu98). These structural differences could be explained by conformational changes in EspG5mtu induced by PE–PPE binding, suggesting that binding of EspG1 and EspG3 to their cognate PPE protein partners is different from the EspG5-PPE41 interaction observed in the EspG5mtu-PE25–PPE41–structure.
Figure 2. Structural comparison between EspG1mka and EspG5mtu.
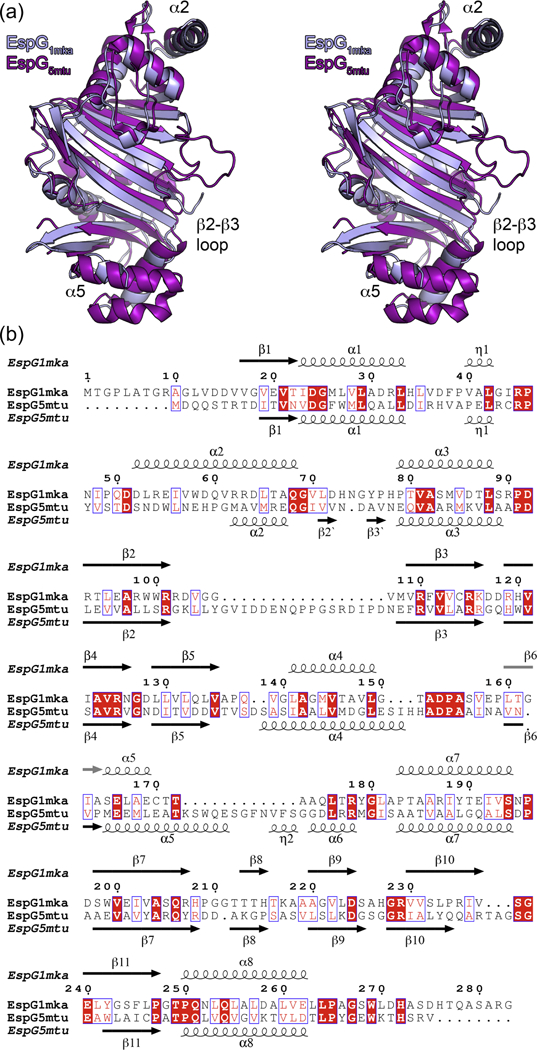
(a) Stereo view of superposed EspG1mka and EspG5mtu crystal structures. The structure of the EspG5mtu monomer is derived from the trimeric EspG5mtu-PE25–PPE41–complex (PDB ID 4KXR, [26]). (b) Structure-based sequence alignment of EspG1mka and EspG5mtu. Secondary structure elements corresponding to the EspG1mka structure (PDB ID 5VBA) and EspG5mtu structure (PDB ID 4KXR) are displayed above and below the alignment.
Figure 3. Crystal structures of EspG3 chaperones display variations in their putative PE-PPE binding region.
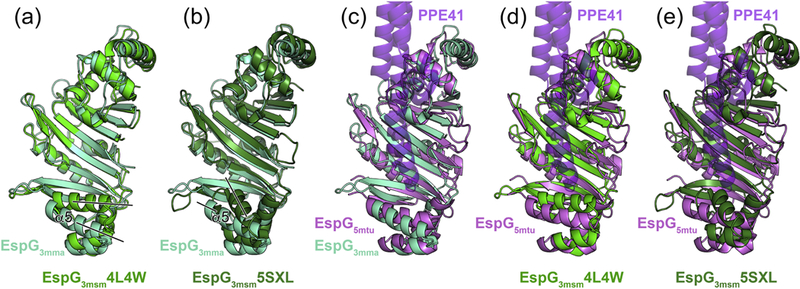
(a) Structural superposition of EspG3mma (aquamarine) and EspG3msm (PDB ID 4L4W, green). Black lines indicate differences in the orientation of the α5 helix. A stereo version is available as Supplementary Figure 4a. (b) Structural superposition of EspG3mma and EspG3msm (PDB ID 5SXL, dark green). A stereo version is available as Supplementary Figure 4b. (c,d,e) Structural superposition of EspG3mma, EspG3msm (PDB ID 4L4W), and EspG3msm (PDB ID 5SXL) with EspG5mtu (PDB ID 4W4I [27], violet) derived from the heterotrimeric EspG5mtu-PE25-PPE41 structure (PDB ID 4KXR [26]). PPE41 (purple) is shown in semi-transparent ribbon representation. PE25 is omitted for clarity as it is not in contact with EspG5mtu. Stereo versions of (c,d,e) are available as Supplementary Figure 5.
Variation of quaternary structure within the EspG protein family
Analysis of crystal packing in the available EspG3 structures revealed a number of possible quaternary arrangements in addition to the monomeric state. Firstly, a “wing-shaped dimer” was found in the asymmetric unit of the EspG3msm (PDB ID 4L4W) structure with 1892 Å2 buried surface area (Fig. 4). The dimeric interface is mediated by residues from the C- terminal helical bundles and strands β6, β10, β11 and helix α8. In contrast, the asymmetric unit of the EspG3msm (PDB ID 4RCL) structure contains a dimer in front-to-front orientation with 1761 Å2 buried surface area. The β8 strands from the two subunits are located at the core of the interface and form an inter-subunit β-sheet. This dimeric conformation is further referred to as a “β8-mediated dimer”. However, in addition a “wing-shaped dimer” similar to the EspG3msm (PDB ID 4L4W) structure is present in the crystal lattice, with 2054 Å2 buried surface area. Furthermore, the asymmetric unit of the EspG3msm (PDB ID 4W4J [27]) structure also contains a similar wing-shaped dimer with substantial buried surface area as well as an β8-mediated dimer (Fig. 4).
Figure 4. Cartoon representation of the common dimer structures observed in crystal forms of EspG3.
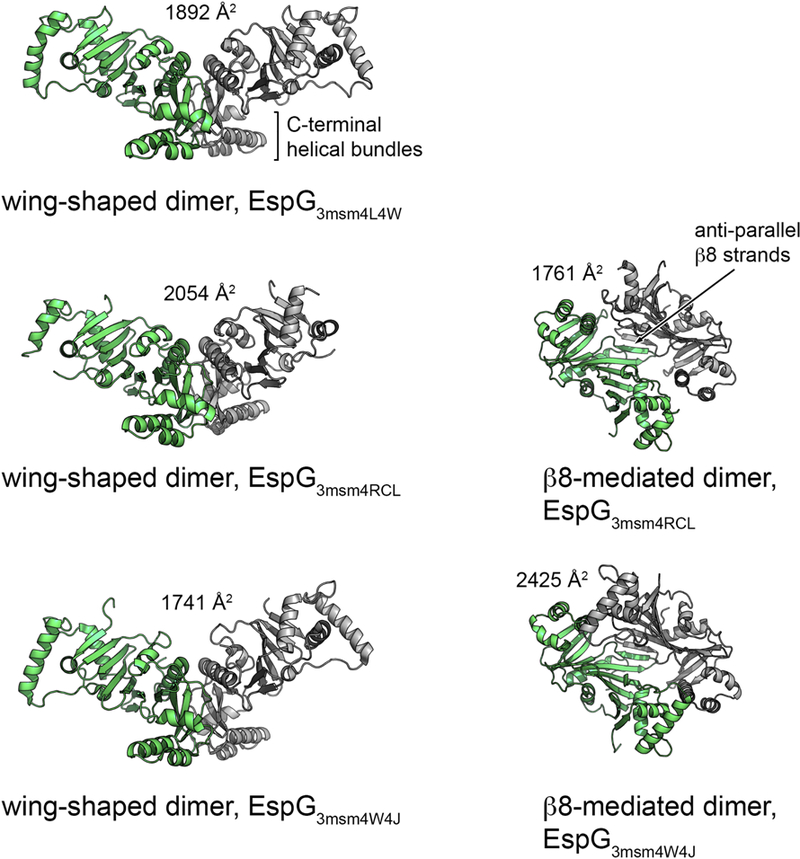
Superimposed subunits are in green with the buried surface area of the dimer interface indicated above the structure. The wing-shaped dimers are present in the asymmetric unit of EspG3msm (PDB ID 4L4W) and EspG3msm (PDB ID 4W4J) or generated by crystallographic symmetry in EspG3msm (PDB ID 4RCL). The β8-mediated dimer is present in the asymmetric unit in EspG3msm (PDB ID 4RCL) and generated by crystallographic symmetry in EspG3msm (PDB ID 4W4J [27]).
Altogether, EspG3msm wing-shaped dimers are observed in three independent crystal structures, and β8-mediated dimers are seen in two crystal structures. The dimer interfaces are highly similar, with the subunits rotated relative to each other by 12 degrees in the wing- shaped dimers and 5 degrees in the β8-mediated dimers (Supplementary Fig. S6). The different quaternary structures of EspG chaperones reflect the variability that exists within this protein family (Table 2). As previously proposed [26], EspG likely acts as a chaperone that maintains PE/PPE secretion targets in the cytosol in a soluble state. Further experiments will be required to elucidate whether the dimerization of EspG3msm plays a role in the function of the chaperone in vivo. To assess whether EspG proteins of ESX-1 and ESX-5 display similar oligomerization behavior in solution, we performed a structural analysis of several EspG proteins in solution.
Table 2.
Overview of EspG crystal structures.
| Chaperone | PDB ID | Structure | Oligomerization | Species | Reference |
|---|---|---|---|---|---|
| EspG1 | 5VBA |  |
Monomer (dimerisation of T4L) |
M. kansasii | This work |
| EspG3 | 4L4W |  |
Wing-shaped dimer |
M. smegmatis | This work |
| 4RCL |  |
β8-mediated | M. smegmatis | This work | |
| 5SXL |  |
Monomer | M. smegmatis | This work | |
| 4W4J |  |
Wing-shaped dimer |
M. smegmatis | [27] | |
| 4W4I |  |
Monomer | M. tuberculosis | [27] | |
| 5DLB |  |
Monomer | M. marinum | This work | |
| EspG5 | 4KXR |  |
Complex with PE25-PPE41 |
M. tuberculosis | [26] |
| 4W4L |  |
Complex with PE25-PPE41 |
M. tuberculosis | [27] | |
| 5XFS |  |
Complex with PE8-PPE15 |
M. tuberculosis | [34] |
Concentration-dependent oligomerization of EspG chaperones
We studied solution structures of EspG proteins from ESX-1 (EspG1mma), ESX-3 (EspG3mma, EspG3mtu, EspG3msm) and ESX-5 (EspG5mtu) secretion systems using SAXS (Table 3). The EspG proteins from different secretion systems have diverged significantly although the EspG3 homologs from different species have a high degree of sequence identity. The Guinier analysis of the obtained SAXS profiles confirmed that the proteins were not aggregated allowing further analysis of their structures and oligomeric states (Table 3). The dependencies of the molecular weight estimates and the excluded volume of the hydrated particles on concentration were studied for all proteins. Prior structural information of the monomeric form existed for all of the studied proteins, but experimental models of the dimeric state were only available for EspG3msm.
Table 3.
Collected SAXS data
| Data collection parameters | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Instrument | P12 at EMBL/DESY, storage ring PETRA III, Germany | ||||||||
| Beam geometry (mm2) | 0.2×0.12 | ||||||||
| Wavelength (Å) | 1.24 | ||||||||
| q-range (Å−1) | 0.0023–0.47 | ||||||||
| Exposure time (ms) | 20×50 | ||||||||
| Temperature (K) | 283 | ||||||||
| Instrument | B21 at Diamond Light Source, United kingdom | ||||||||
| Beam geometry (mm2) | 1.0×5.0 | ||||||||
| Wavelength (Å) | 1.0 | ||||||||
| q-range (Å−1) | 0.0038–0.42 | ||||||||
| Exposure time (ms) | 60×100 | ||||||||
| Temperature (K) | 283 | ||||||||
| Structural parameters | |||||||||
| Sample | Conc. (mg ml−1) |
Rg, Guinier (nm) |
Rg, Pr (nm) |
Dmax (nm) |
MMtheor (kDa) |
MMSAXS (kDa) |
MMPOROD (kDa) |
MMDAM (kDa) |
Ab initio resolution, (Å) |
| EspG1mma | 1.0 | 2.7 | 2.9 | 9.7 | 29.8 | 25.0 | 43.0 | 45.0 | 42±3 |
| 2.0 | 3.2 | 3.1 | 11.0 | 29.8 | 32.0 | 55.0 | |||
| 4.2 | 3.4 | 3.5 | 11.0 | 29.8 | 37.0 | 65.0 | |||
| 6.0 | 3.5 | 3.6 | 12.0 | 29.8 | 40.0 | 72.0 | |||
| EspG3mtu | 1.1 | 2.5 | 2.6 | 9.0 | 31.6 | 23.0 | 41.0 | 41.0 | 34±3 |
| 2.0 | 2.8 | 3.0 | 10.0 | 31.6 | 30.0 | 47.0 | |||
| 4.0 | 3.2 | 3.1 | 10.0 | 31.6 | 32.0 | 56.0 | |||
| 6.1 | 3.5 | 3.5 | 12.0 | 31.6 | 39.0 | 68.0 | |||
| EspG3mma | 1.1 | 2.3 | 2.5 | 8.0 | 32.0 | 20.0 | 39.0 | 34.0 | 23±2 |
| 2.1 | 2.3 | 2.4 | 8.0 | 32.0 | 22.0 | 35.0 | 35.0 | ||
| 4.0 | 2.3 | 2.4 | 8.0 | 32.0 | 21.0 | 40.0 | 38.0 | ||
| 6.0 | 2.3 | 2.3 | 8.0 | 32.0 | 22.0 | 44.0 | 36.0 | ||
| EspG3msm | 0.8 | 2.5 | 2.6 | 8.6 | 31.6 | 20.5 | 39.0 | 48.0 | 36±3 |
| 1.8 | 2.5 | 2.5 | 8.8 | 31.6 | 21.4 | 32.0 | 41.0 | ||
| 3.9 | 2.6 | 2.6 | 9.3 | 31.6 | 22.1 | 39.0 | 44.0 | ||
| 6.2 | 2.8 | 2.7 | 9.7 | 31.6 | 24.0 | 39.0 | 47.0 | ||
| EspG3msm Se-Met |
0.9 | 2.5 | 2.5 | 9.2 | 31.6 | 22.5 | 38.0 | 43.0 | 39±3 |
| 2.1 | 2.5 | 2.6 | 9.2 | 31.6 | 22.5 | 38.0 | |||
| 3.8 | 2.6 | 2.6 | 9.2 | 31.6 | 24.3 | 40.0 | |||
| EspG5mtu | 0.9 | 2.3 | 2.5 | 8.0 | 32.4 | 23.0 | 41.0 | 47.0 | 25±2 |
| 1.7 | 2.4 | 2.4 | 8.0 | 32.4 | 22.0 | 42.0 | |||
| 4.1 | 2.4 | 2.5 | 8.0 | 32.4 | 21.0 | 43.0 | |||
| 6.7 | 2.4 | 2.4 | 8.0 | 32.4 | 22.0 | 41.0 | |||
| EspG5mtu- PE25- PPE41 |
0.5 | 4.0 | 4.0 | 13.0 | 65.0 | 76.0 | 46.0 | 76.0 | 37±3 |
| EspG3msm- PE5-PPE4 |
1.2 | 4.0 | 4.2 | 14.2 | 95.0 | 83.0 | 52.0 | 83.0 | 38±3 |
SAXS measurements of EspG1mma at four different protein concentrations resulted in distinct scattering profiles. The molecular weight (based on the Porod volume) increased with increasing concentration from 43 to 72 kDa at 1.0 mg/mL and 6.0 mg/mL, respectively (Table 3). A similar trend was observed for the Rg and Dmax values. The oligomer analysis of the concentration-dependent SAXS data was carried out using theoretical scattering profiles based on the monomeric EspG1mka (PDB ID 5VBA) structure and two different dimeric structural models. The β8-mediated dimer observed in the EspG3msm (PDB ID 4RCL) structure and the wing-shaped dimer structure of EspG3msm (PDB ID 4L4W) were used as templates to generate EspG1mma dimer models. The corresponding theoretical scattering profiles were exploited to decompose the experimental data (Table 4). The OLIGOMER analysis showed that EspG1mma is predominantly a monomer at low protein concentrations, while the fraction of dimeric protein increases up to 50% at the highest concentration measured. However, the goodness-of-fit (χ2) of the theoretical scattering based on linear combinations of theoretical monomer/dimer scattering profiles to the experimental SAXS data varied significantly between the two dimer models (Table 4). The SAXS data could not be interpreted successfully using the β8-mediated dimeric arrangement (Table 4). Additional evidence that the β8-mediated dimer is a non-physiological crystallographic dimer is the substantial structural clashes observed when the β8-mediated EspG1 dimer is superimposed onto the EspG5mtu structure derived from the heterotrimeric EspG5mtu-PE25–PPE41 crystal structure (Fig. 3). The wing-shaped dimer conformation based on the EspG3msm (PDB ID 4L4W) crystal structure yields better fits (χ2 –values between 0.79 and 1.27, Table 4) to the measured scattering data, which strongly indicates that EspG1 dimers adopt the wing- shaped conformation in solution.
Table 4.
Oligomer analysis using the crystallographic monomer and dimer structures of EspG proteins.
| Sample | Template (PDB ID) |
Concentration (mg ml−1) |
Monomeric Fraction |
Dimeric fraction |
Goodness-of- fit,χ2 |
|---|---|---|---|---|---|
| EspG1mma | β8-mediated dimer (4RCL) |
1.0 | 0.94 | 0.06 | 0.80 |
| 2.0 | 0.74 | 0.26 | 0.93 | ||
| 4.2 | 0.58 | 0.42 | 1.44 | ||
| 6.0 | 0.50 | 0.50 | 1.90 | ||
| Wing-shaped dimer (4L4W) |
1.0 | 0.93 | 0.07 | 0.79 | |
| 2.0 | 0.72 | 0.28 | 0.86 | ||
| 4.2 | 0.54 | 0.46 | 1.01 | ||
| 6.0 | 0.43 | 0.57 | 1.27 | ||
| EspG3mtu | Wing-shaped dimer (4L4W) |
1.1 | 0.537 | 0.462 | 0.89 |
| 2.0 | 0.364 | 0.636 | 0.90 | ||
| 4.0 | 0.113 | 0.887 | 0.95 | ||
| 6.1 | 0.0 | 0.999 | 0.89 | ||
| EspG3mma | Wing-shaped dimer (4L4W) |
1.1 | 0.531 | 0.468 | 0.82 |
| 2.1 | 0.195 | 0.805 | 0.88 | ||
| 4.0 | 0.000 | 1.000 | 1.05 | ||
| 6.0 | 0.00 | 1.000 | 1.69 | ||
| EspG3msm (Native) |
Wing-shaped dimer (4L4W) |
0.78 | 0.589 | 0.410 | 0.90 |
| 1.75 | 0.654 | 0.346 | 1.32 | ||
| 3.94 | 0.575 | 0.425 | 1.35 | ||
| 6.24 | 0.449 | 0.550 | 2.03 | ||
| EspG3msm (SetMet) |
Wing-shaped dimer (4L4W) |
0.90 | 0.70 | 0.30 | 0.92 |
| 2.14 | 0.70 | 0.30 | 1.80 | ||
| 3.79 | 0.59 | 0.41 | 2.50 |
The program OLIGOMER was also used to fit a set of theoretical scattering profiles of monomeric and dimeric EspG3 models to the experimental EspG3mtu SAXS data. A dimer model was constructed based on the wing-shaped dimer structure of EspG3msm (PDB ID 4L4W). All fits at different concentrations provided good χ2 values indicating that our wing- shaped dimer EspG3mtu model based on the EspG3msm structure is appropriate. Analysis of the volume distributions of the oligomeric states showed that even at the lowest concentration (1.1 mg/mL), 46% of the protein was in dimeric form. The dimeric protein fraction increased to 100% at the highest measured concentration (6.1 mg/mL) (Table 4). Likewise, the volume fractions of the oligomeric states were calculated for EspG3mma. Again the dimeric experimental structure of EspG3msm (PDB ID 4L4W) was used as the template to model the dimeric form of EspG3mma. EspG3mma showed a similar oligomerization pattern as EspG3mtu. The volume fraction of the dimeric form of the protein is above 46% even at the lowest protein concentration (Table 4).
We measured the SAXS profiles of two different EspG3msm protein preparations, native and selenomethionine (SeMet) incorporated forms. Interestingly, the two proteins showed distinct oligomerization behaviors. The native EspG3msm sample had a significant fraction of the dimeric form present already at the lowest measured concentration and the fraction stayed stable as the protein concentration increased (Table 4). On the contrary, the SeMet-labeled form remained monomeric over the whole concentration range (Table 4). The CRYSOL fit of the monomeric structure (PDB ID 4L4W) to the scattering data from the SeMet-labeled construct provided a χ2 value of 0.93. This enabled us to use the SAXS data from the SeMet-labeled protein for ab initio modeling, which requires a monodisperse sample (Supplementary Fig. S7a). The SAXS-based ab initio model calculated with DAMMIF and the monomeric structure of SeMet-labeled EspG3msm (PDB ID 4L4W) are in a good agreement (normalized spatial discrepancy (NSD) = 1.1).
The SAXS profiles of the native form of EspG5mtu, the only member of the EspG5 family that was analyzed, do not show any concentration dependence over the measured range (0.9 to 6.7 mg/mL) (Table 3). The constant molecular mass estimates suggest that EspG5mtu is monomeric in solution. We used the program CRYSOL to fit the theoretical scattering profile based on the crystal structure of the EspG5mtu monomer from the EspG5mtu–PE25–PPE41 structure (PDB ID 4KXR) to the experimental SAXS data. A significant misfit was observed in the range of momentum transfer of 1.8 – 2.0 nm−1 (Fig. 5a). In order to evaluate whether flexibility of the EspG5mtu β2–β3 loop (Gly92-Asn114) might cause this discrepancy, we employed molecular dynamics (MD) simulations to assign flexible amino acid residues and ensemble optimization method (EOM) to fit the measured SAXS data. The amino acid residue Root-Mean-Square-Fluctuations (RMSF) monitored during a 2 ns production run indicated high flexibility of the β2–β3 loop region and other shorter loop segments (Fig. 5b). Thus, we introduced flexibility in all loop regions of EspG5 with the program EOM and also modeled the 24 amino acid residues that were missing from the crystallographic structure (Fig. 5c). In the EOM approach, the scattering profile is fitted by a linear combination of scattering profiles from several structural models co-existing in solution. The resulting EOM models where the flexible β2–β3 loop exhibits the largest conformational changes compared to the crystallographic structure provide an excellent fit with the experimental SAXS data (Fig. 5a) (χ2 = 0.99). In contrast to the original crystallographic structure in which the β2–β3 loop interacts with the PE–PPE heterodimer in an extended conformation, all the EOM structures of EspG5mtu in solution have more compact forms. More specifically, the β2–β3 loop of EspG5mtu folds closely onto the protein core in the SAXS-refined solution structures (Fig. 5c). In addition, the monodisperse EspG5mtu SAXS data were employed for ab initio modeling using DAMMIF (Supplementary Fig. S7b). The structural alignment yielded a NSD value of 0.93 indicating excellent agreement.
Figure 5. Solution structure of EspG5mtu.
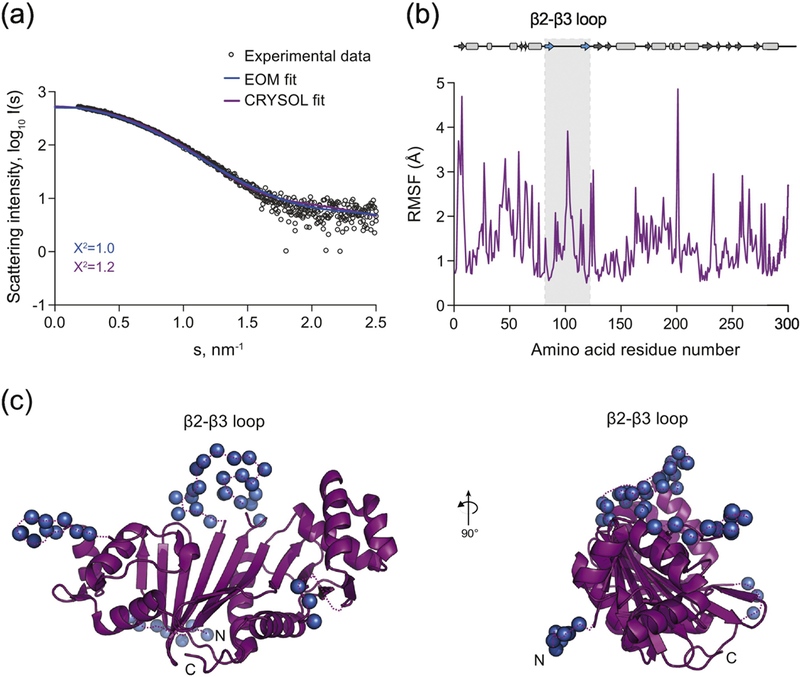
(a) Experimental SAXS data (black circles) and computed fits (solid lines) with respective discrepancy values (χ2). The theoretical SAXS curve of the EspG5mtu crystal structure when in complex with PE25/PPE41 as calculated using CRYSOL (purple) (PDB ID 4KXR, chain C) fits the experimental SAXS data with a χ2 of 1.2 and an apparent misfit around s = 1.8 – 2.0 nm-1. To calculate the EOM fit, the 24 amino acid residues that were missing from the crystallographic structure of EspG5mtu were added at the N-terminus and flexibility of all loops was allowed. This procedure resulted in an improvement of the fit to a χ2 value of 0.99 (blue). (b) The amino acid residue root-mean- square-fluctuation (RMSF) of EspG5mtu (starting structure PDB ID 4KXR, chain C) during a molecular dynamics simulation run. The secondary structure assignment of the crystallographic structure is shown above the RMSF plot for reference, where β-sheets and α-helices are represented by arrows and rectangles, respectively. (c) The flexible loop regions and the amino acid residues missing from the crystallographic structure of EspG5mtu crystal structure when in complex with PE25-PPE41 (purple) are modeled as dummy residues (blue spheres).
Taken together, these data show clear differences in the oligomerization trends of each EspG ortholog from different ESX systems, indicating that this could be another level of system specificity involved in the secretion of PE–PPE proteins via ESX systems.
Model of the EspG3-PE5–PPE4 trimer structure adopts an extended conformation
To further analyze substrate recognition by different EspG proteins, we measured solution scattering of EspG3–PE5–PPE4 and EspG5–PE25–PPE41 complexes from M. tuberculosis. Model-free parameters derived from the SAXS data indicate that EspG3msm–PE5–PPE4 has a more extended open overall conformation (Dmax =14.5 nm / Rg = 4.25 nm) than the crystallographic structure of the EspG5mtu–PE25–PPE41 complex (Dmax = 13.0 nm / Rg = 3.93 nm) (Fig. 6a). Ab initio modeling in P1 symmetry using SAXS data provided independent information about the overall shapes. The EspG3msm complex reveals a more open structure than the EspG5mtu complex, which is consistent with the model-free parameters. In addition, the overall shape of the EspG5mtu complex ab initio structure is in agreement with the crystallographic structure showing a compact structure (PDB ID 4KXR; NSD = 1.7, data not shown). The theoretical scattering of the crystallographic structure of the EspG5mtu–PE25– PPE41 complex fits the measured SAXS data very well (χ2 = 0.95) (Fig. 6b). This indicates that, in solution, the EspG5mtu β2–β3 loop is in the extended conformation seen in the crystal structure of the EspG5mtu–PE25–PPE41 heterotrimer. Thus, the SAXS data for EspG5mtu alone and in complex with PE25–PPE41 indicates that the β2–β3 loop undergoes a significant conformational change upon binding.
Figure 6. Comparison of EspG5-PE25-PPE41 and EspG3-PE5-PPE41−178 complexes structures.
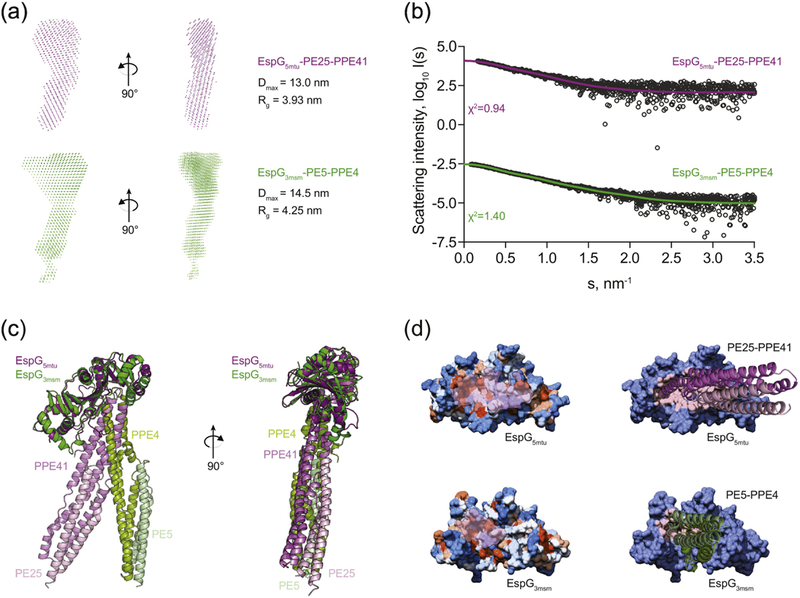
(a) SAXS-based ab initio models of the EspG5mtu-PE25-PPE41 complex (purple) and the EspG3msm-PE5–PPE41−178 complex (green) reveal a more extended conformation of the EspG3msm-PE5–PPE41−178 complex, as indicated by the maximum diameter (Dmax) and radius of gyration (Rg) of each complex. (b) Experimental SAXS data (black circles) with the computed fits (solid lines) and the respective discrepancy values (χ2) for EspG5mtu-PE25- PPE41 and EspG3msm-PE5-PPE4 protein complexes. The theoretical SAXS curve, calculated with CRYSOL from the EspG5mtu-PE25–PPE41 complex structure (PDB ID 4KXR), fits the experimental SAXS data with a goodness-of-fit (χ2) of 0.95 (purple). The theoretical SAXS curve calculated from the EspG3msm rigid-body model in complex with a homology model of PE5-PPE41−178 fits the experimental SAXS data (green) with a goodness- of-fit values (χ2) of 1.40. (c) Superposition of the crystal structure of EspG5mtu-PE25-PPE41 (purple-lilac-light pink) and the SAXS-derived rigid body model of EspG3msm-PE5-PPE41−178 (dark green-light green-light blue). (d) Hydrophobicity surface representation of EspG proteins with PPE-interacting surface highlighted in transparent pink (left panel). Interaction interface of EspG5mtu-PPE41 and EspG3msm-PPE4, with PPE-PE proteins represented as cartoon and EspG proteins as surface with the contact residues in the interface colored in light pink in each EspG protein (right panel).
In order to produce an atomic rigid body model of the EspG3msm–PE5–PPE4 complex, 300 decoy structures were generated using a molecular docking approach [35] and the crystallographic EspG3msm (PDB ID 4L4W, chain B) structure together with homology models of a complex of full length PE5 and the N-terminal domain of PPE4 (residues 1–178) from M. smegmatis generated with SWISS-MODEL [36]. The decoy structures were then ranked by the goodness-of-fit values of their theoretical scattering compared to the experimental scattering data. Fourteen complex structures were selected for further analysis on the basis of their chi2–values (χ2 < 1.7). In addition, all structures had acceptable fits according to p- values provided by the correlation map approach [37]. A representative model with the lowest χ2–value was selected for the comparison with the EspG5mtu–PE25–PPE41 complex (Fig. 6b-d).
Comparison of the crystallographic structure of EspG5mtu–PE25–PPE41 (PDB ID 4KXR, [26]) and the SAXS-based rigid-body model of EspG3msm–PE5–PPE4 shows similarities in the binding interfaces of the two complexes, but also significant differences related to the overall binding orientation (Fig. 6c). As expected, the interface between EspG3msm and the PE5– PPE4 heterodimer was found to be mostly comprised of hydrophobic amino acid residues allowing interaction with the hh-motif of PPE4 [26] (Fig. 6d). Analogous to the EspG5mtu– PE25–PPE41 complex, the loop between helices α4 and α5 of PPE4 (Ala125-lIe134) interacts with the central β-sheet of EspG3. The structure suggests a hydrogen bonding network between EspG3 and PPE4 formed by several hydrophobic, polar and charged amino acid residues Trp27, Glu196, Glu211, Ser215 of EspG3msm and residues Thr126, Phe128, Gly130, Asn132, Thr133, Ile134 of PPE4 from M. smegmatis. To validate our SAXS model and the interface of EspG3msm and PPE4 in particular, we constructed two mutants of EspG3msm: E196R and S215Y. Substitution E196R reverses the charge of a conserved E196 residue in the EspG3 orthologs, whereas mutation S215Y introduces a bulky residue that would sterically prevent binding of PPE4. Pull-down experiments showed that E196R and S215Y variants of EspG3msm could not bind PE5–PPE4 dimers (Supplementary Fig. S8). Based on the SAXS data and models, we suggest that the more open and flexible structure of EspG3–PE5–PPE4 is due to differences in the EspG3 β2–β3 loop region. The EspG5mtu β2–β3 loop is comprised of 23 amino acid residues (Gly92-Asn114) and its interaction with PE25–PPE41 forms an extended interface extending from the central β-sheet of EspG5mtu. However, the homologous loop region in the EspG3msm structure is significantly shorter (12 amino acid residues; Val87 to Leu98) thus an analogous interaction is missing in the EspG3msm–PE5–PPE4 rigid-body model. Given that ESX-5 is the major secretion system for export of PE/PPE proteins [22–24, 32], the length and flexibility of the EspG5mtu β2-β3 loop could be an important structural feature allowing EspG5mtu to interact specifically with many different PE–PPE heterodimers.
Conclusions
Transport of multiple proteins across the mycobacterial cell envelope is facilitated by the ESX system and cytoplasmic EspG chaperones [38]. However, the precise recognition mechanism of the cognate substrates by ESX system-specific chaperones is not yet fully understood. In this work we present structural analyses of EspG1, EspG3 and EspG5 and their complexes with PE–PPE secretion substrates using a combination of experimental and in silico methods. The solution scattering data together with novel X-ray crystallographic structures allows us to hypothesize about the substrate specificity of EspG chaperones and provides insight into chaperone-substrate binding mechanisms. This study demonstrates that the β2–β3 loop of EspG plays an important role in PE–PPE binding and is a major differentiation factor between the EspG chaperones of orthologous ESX secretion systems. Our results also suggest that EspG dimerization may play a role in substrate recognition.
Materials and methods
Cloning, expression and purification of M. kansasii EspG1
The DNA sequence corresponding to the full-length EspG1mka was PCR amplified using primers G1mka_F1Nde, 5’-GATACATATGACCGGTCCGCTCGCTAC and G1mka_R283Hind, 5’-CTCAAGCTTAGCCTCGGGCGGAGGCTTG, and genomic DNA of M. kansasii ATCC 12478. The PCR product was digested with NdeI/HindIII and ligated into the corresponding sites of a modified pET-28b vector to create an N-terminal His6-tag with a tobacco etch virus (TEV) cleavage site. In efforts to optimize initial crystals, the Cys114Ala and Cys170Ala mutations were introduced using the QuikChange protocol (Stratagene). A truncated DNA fragment corresponding to residues 17–271 was PCR amplified using primers G1mka_F17Nco, 5’-GATTCCATGGTCGGCGTCGAGGTCACC and G1mka_R271Hind, 5’- CTCAAGCTTCAATCTAACCAGGAGCCCGC and cloned into a pET-based vector containing an N-terminal His6-tag followed by a TEV cleavage site and T4L sequence (residues 2–162). The T4L sequence corresponds to a cysteine-less variant with Cys54Thr and Cys97Ala mutations [39, 40]. T4L-EspG1mka was expressed in E. coli Rosetta2(DE3) cells in LB supplemented with 50 μg mL−1 kanamycin and 34 μg mL−1 chloramphenicol. Cells were grown at 37ºC and expression was induced with 0.5 mM IPTG at A600 of 0.6. Cells were harvested by centrifugation after 3 h, resuspended in lysis buffer (20 mM Tris-HCl pH 8.5, 300 mM NaCl, 10 mM imidazole) and lysed using an EmulsiFlex C5 homogenizer (Avestin). EspG3msm was purified via Ni-NTA metal affinity chromatography. The His6-tag was cleaved using TEV protease followed by a second Ni-NTA purification step to remove uncleaved T4L-EspG1mka and His6-tagged TEV protease. Size-exclusion chromatography was performed using a Superdex 200 column (GE Biosciences) equilibrated in buffer containing 20 mM HEPES pH 7.5, 300 mM NaCl.
Crystallization and structure solution of T4L-EspG1mka
Crystals of T4L-EspG1mka were obtained by the hanging drop vapor diffusion method using crystallization solution containing 0.1 M Bicine pH 9.0, 1.0 M LiCl, 10% PEG6000. The crystals were cryoprotected in crystallization solution supplemented with 20% glycerol and flash cooled in liquid nitrogen before data collection. Data were collected at the SER-CAT beamline 22-ID at the Advanced Photon Source, Argonne National Laboratory. Data were processed and scaled using XDS and XSCALE [41]. CC1/2 value of 0.5 for the outer shell was used to determine the resolution [42]. The T4L-EspG1mka structure was solved by molecular replacement using Phaser [43], with the T4L structure (PDB 4GBR) [44] and poly- Ala EspG3msm (PDB ID 4L4W) structure as search models. Two copies of T4L-EspG1mka were located in the asymmetric unit. Density modification was performed using Parrot [45], and the molecular replacement model was re-built using Buccaneer [46, 47], ARP/wARP [48] and manual building in Coot [49]. The iterative rounds of refinement and re-building were performed using phenix.refine [50] and Coot. Non-crystallographic symmetry (NCS) restraints were applied throughout the refinement. Statistics for data collection, refinement, and model quality are listed in Table 1.
Cloning, expression, and purification of M. marinum EspG3
The gene (MMAR_0548) encoding EspG3mma was PCR-amplified from M. marinum M genomic DNA with Phusion DNA polymerase (New England Biolabs) using gene specific primers (MMAR_0548.For, 5’-AACCTGTATTTCCAGAGTATGGAGTCAATGCCCAACG and MMAR_0548.Rev, 5’-ttcgggctttgttagcagttaGGAGGGTTGACTCGAGAAATCT) and was cloned into a modified pET28 vector, pMAPLe4 [51], using the Gibson ISO assembly procedure [52]. The DNA sequence of the construct was verified by DNA sequencing (Genewiz). EspG3mma was expressed from pMAPLe4 as a maltose binding protein (MBP) fusion which was cleaved in vivo from MBP via a tobacco vein mottling virus (TVMV) protease cleavage site situated between the two moieties; a His6 affinity tag and a tobacco etch virus (TEV) protease cleavage site is encoded in the linker between the TVMV protease cleavage site and the N-terminus of the target protein. EspG3mma was expressed in E. coli BL21-Gold (DE3) (Agilent Technologies) using Terrific broth media and protein expression was induced with 1 mM IPTG at 18°C overnight. Harvested cells were resuspended in lysis buffer (20 mM Tris, pH 8.0, 300 mM NaCl, 10% glycerol, 10 mM imidazole) supplemented with β-mercaptoethanol (2 mM), DNase I, lysozyme, and Complete protease inhibitor cocktail (Roche) and lysed by sonication. The lysate was centrifuged (39,000 g, 30 minutes, 4°C) and EspG3mma was purified from the clarified supernatant using Ni-NTA resin (Thermo Fisher Scientific) equilibrated in lysis buffer. The bound protein was eluted with lysis buffer containing 300 mM imidazole and further purified by size exclusion chromatography (SEC) using a HiLoad 16/60 Superdex 75 column (GE Healthcare) equilibrated with 20 mM Tris, pH 8.0, 300 mM NaCl, 10% glycerol. EspG3mma eluted from the column in a single symmetrical peak which was concentrated to 21.5 mg mL−1 for crystallization screening.
Crystallization and structure determination of EspG3mma
Small crystals of EspG3mma were grown using the hanging drop vapor-diffusion method by mixing 1 µL of protein with 0.5 µL of reservoir solution (1.4 M ammonium sulfate, 200 mM lithium sulfate, 60 mM CAPS, pH 10.5). These small crystals were used to streak seed other drops that had been equilibrated for a week but showed no signs of crystal growth. Large crystals were found in seeded drops with a reservoir solution containing 1.45 M ammonium sulfate, 200 mM lithium sulfate, 70 mM CAPS, pH 10.5 after 5 weeks. For phase determination and cryoprotection crystals were soaked for 30 minutes at room temperature in a solution containing approximately 4 mM platinum potassium thiocyanate, 1.17 M ammonium sulfate, 160 mM lithium sulfate, 56 mM CAPS pH 10.5, and 15.5% glycerol. Diffraction data were collected at the Advanced Photon Source at Argonne National Laboratory on beamline 24-ID-C. The data were processed with XDS [41], and the structure was solved by single wavelength anomalous dispersion using HKL2MAP [53], in the SHELX suite of programs [54], which determined the position of seven platinum atoms in the K2Pt(SCN)6–soaked crystal. An initial model was built using SHELXE [55] which was improved through iterative rounds of manual model building using Coot [49] interspersed with refinement using REFMAC5 [56].
Cloning, expression and purification of M. smegmatis EspG3
The gene msmeg_0622 encoding EspG3msm was PCR amplified from genomic DNA of M. smegmatis mc2155 using primers MsmG3_F1Nde, 5’- GAGACATATGGGGCCTAACGCTGTTG, and MsmG3_R293Hind, 5’- CTCAAGCTTACTAGTCATGCTTTCTGGGTTCTTCTCTG. The PCR product was digested with NdeI/HindIII and ligated into the corresponding sites of a modified pET-28b vector to create TEV protease-cleavable N-terminal His6-tag fusion. The construct was verified by DNA sequencing (Eurofins Genomics). EspG3msm was expressed and purified using procedures similar to the T4L-EspG1mka fusion, except the final SEC step was performed in buffer containing 20 mM HEPES pH 7.5, 100 mM NaCl.
Crystallization and structure solution of EspG3msm
Crystals of EspG3msm in space group P3221 were obtained by hanging drop vapor diffusion method with crystallization solutions containing 0.1 M Tris-HCl pH 7.0, 0.2 M Mg acetate, 1.8 M NaCl (SeMet substituted EspG3msm) and 0.1 M HEPES pH 7.4, 1.0 M LiCl, 10% PEG6000 (native EspG3msm). Crystals were transferred to crystallization solutions supplemented with 20–25% glycerol and flash-cooled in liquid nitrogen prior to data collection. Data for native and SeMet substituted EspG3msm crystals were collected at the SER-CAT beamline 22-ID at the Advanced Photon Source, Argonne National Laboratory. Data were processed and scaled using XDS and XSCALE [41]. The EspG3msm structure was solved by SeMet-SAD. The initial selenium positions were found with SHELXD [54] using HKL2MAP interface [53]. Phasing, density modification and initial model building was performed using autoSHARP [57]. A partial model was refined against a native dataset and rebuilt using REFMAC5 [56], ARP/wARP [48] and AutoBuild within PHENIX [58]. The model was completed by iterative rounds of refinement and rebuilding using phenix.refine [50] and Coot [49].
Crystals of SeMet substituted EspG3msm in space group C2221 were grown using crystallization solution containing 0.1 M Tris-HCl pH 8.5, 1.0 M LiCl, 20% PEG6000. Crystals were transferred into crystallization solution supplemented with 20% glycerol and flash cooled in liquid nitrogen. The structure was solved by molecular replacement using Phaser [43] with EspG3msm (PDB ID 5SXL) structure as a search model. Two EspG3msm molecules were located in the asymmetric unit. Following density modification using Parrot [45], the molecular replacement model was rebuilt using Buccaneer [46, 47]. The model was further improved using Coot, ARP/wARP and AutoBuild within PHENIX. The final model was refined using phenix.refine.
Crystals of EspG3msm in space group P43212 were grown using crystallization solution containing 0.1 M Na cacodylate pH 6.0, 15% PEG200, 5% PEG3350. The crystals were cryoprotected in solution containing 0.1 M Na cacodylate pH 6.0, 35% PEG200, 5% PEG3350 and flash cooled in liquid nitrogen. Molecular replacement using Phaser and EspG3msm (PDB ID 4L4W) structure as a search model located 2 molecules in the asymmetric unit. The model was refined using REFMAC5 and rebuilt using AutoBuild within PHENIX. The iterative rounds of refinement and rebuilding were performed using phenix.refine and Coot. NCS restraints were applied in early rounds of refinement and were later omitted as the model quality improved. The last several rounds of refinement were performed using 4 translation/libration/screw (TLS) groups, identified by the TLSMD server [59, 60], per protein chain.
Sample preparation for SAXS measurements
The gene MMAR_5441 encoding EspG1mma was PCR amplified from genomic DNA of M. marinum E11 using primers DF018, 5’- ATATATAGATCTACCGGTCCGCTCGCTACCGG’,and DF019, 5’- ATATATATGCGGCCGCTTAACCTCGGGCGGTGGCGTCG’. The PCR product was digested with BglII/NotI. The gene MMAR_0548 encoding EspG3mma was PCR amplified from genomic DNA of M. marinum E11 using primers DF020, 5’- ATATATACCGGTGGAATGGAGTCAATGCCCAACGC’, and DF021,5’- ATATATATGCGGCCGCTTAGGAGGGTTGACTCGAGAA’. The PCR product was digested with AgeI/NotI. Clones containing genes Rv0289 and Rv1794 encoding EspG3mtu and EspG5mtu were further digested with BglII/NotI and AgeI/NotI. Digested fragments were ligated into the corresponding sites of pETM11-SUMO3 to create SENP-2 protease- cleavable N-terminal His6-SUMO3-tag fusions.
His6-SUMO3-EspG1mma, His6-SUMO3-EspG3mma, His6-SUMO3-EspG3mtu and His6-SUMO3- EspG5mtu proteins for SAXS measurements were purified as follows: cells were resuspended in lysis buffer 20 mM HEPES pH 7.5, 150 mM NaCl, 10 mM imidazole, 0.25 mM tris(2- carboxyethyl)phosphine (TCEP), 10% (w/v) glycerol (pH 7.5) containing 1/100 protease inhibitor mix HP (Serva), DNAse I (10 μg/ml) and disrupted by lysozyme treatment followed by sonication. The protein was purified via Ni-NTA (Qiagen) affinity chromatography. His6- SUMO3-tag was cleaved by SenP2 protease and further purified using a Phenyl Sepharose HP column (GE Biosciences), followed by SEC using a Superdex 200 16/60 column (GE Biosciences) pre-equilibrated with 20 mM HEPES pH 7.5, 150 mM NaCl, 0.25 mM TCEP, 10% (w/v) glycerol.
The purification procedure for the EspG5mtu-PE25-PPE41-His6 complex was the same as for the His6-SUMO3-EspG proteins described above with the exception that the cleavage of the His6 tag was performed by addition of TEV protease. For final aggregated protein removal, the complex was concentrated and injected into a Superdex 200 16/60 size-exclusion chromatography column (GE Biosciences) pre-equilibrated with 20 mM HEPES pH 7.5, 150 mM NaCl, 0.25 mM TCEP, 10% (w/v) glycerol. EspG3msm–PE5–PPE4 complex was obtained as described in [26], and further purified using a Superdex 200 column equilibrated in buffer containing 20 mM HEPES pH 7.5, 100 mM NaCl. All samples used for SAXS experiments were concentrated to the appropriate protein concentrations ranging from ~1–7 mg ml-1.
Mutations E196R and S215Y were introduced into EspG3msm using Gibson mutagenesis protocol and following primers: G3msm_E196R_F5’- GGTCGGCGCACCTACGTCCGTATCGTCGCGGGCGAGCAT,G3msm_E196R_R5’-ATGCTCGCCCGCGACGATACGGACGTAGGTGCGCCGACC,G3msm_S215Y_F5’-CACCACCGAGGTGGGGGTCTACATCATCGACACCCCACAC,G3msm_S215Y_R5’-GTGTGGGGTGTCGATGATGTAGACCCCCACCTCGGTGGTG,IsoKan_15’-GACAATTACAAACAGGAATCGAATGCandIsoKan_25’-GCATTCGATTCCTGTTTGTAATTGTC. The pull-down experiments were performed as described in [26].
SAXS measurements
SAXS measurements were carried out at beamline P12 (EMBL/DESY, Hamburg) [61] at the PETRA-III storage ring using a Pilatus 2M detector (Dectris). Measurements for the purified proteins were made at several concentrations (Table 4). For each measurement twenty 50 ms exposure frames were collected and averaged using a sample volume of 30 µl at a temperature of 10°C. The SAXS camera was set to a sample-detector distance of 3.1 m, covering the momentum transfer range 0.008 Å-1 < s < 0.47 Å−1 (s = 4π sin(θ)/λ where 2θ is the scattering angle and λ=1.24 Å is the X-ray wavelength). Prior to and following each sample exposure, a buffer control was measured to allow for background subtraction.
SAXS data analysis using model-free parameters
Radius of gyration Rg and forward scattering intensity I(0), were independently determined using Guinier analysis [62] and the indirect Fourier transformation approach of the program GNOM [63]. Additionally, the maximum particle dimension Dmax was obtained from the latter approach. Molecular masses of protein constructs (MMSAXS) were calculated by comparing the extrapolated forward scattering intensities with that of a reference BSA sample (MMref = 66 kDa) together with concentration information. The excluded volume of the hydrated protein Vp was obtained with DATPOROD [64] and used to extract an independent estimate of molecular mass (MMPOROD). For globular proteins, hydrated protein volumes in Å3 are approximately 1.7 times the molecular masses in Dalton.
SAXS-based structural modeling
Ab initio models were reconstructed from the scattering data using the simulated annealing based bead-modeling program DAMMIF [65]. Ten independent reconstructions were averaged to generate a representative model with the program DAMAVER [66]. In addition, the average DAMMIF ab initio model was used to calculate an excluded volume of the particle, VDAM, from which an additional independent MM estimate can be derived (empirically, MMDAM ~ VDAM/2). The resolutions of the model ensembles were estimated by a Fourier Shell correlation approach [67].
Theoretical scattering profiles from available high-resolution crystallographic structural models were calculated using the program CRYSOL [68] and used to determine the fit of these models to the experimental scattering data. Given the atomic coordinates of a structural model, CRYSOL minimizes the discrepancy between the experimental and theoretical scattering intensities by adjusting the excluded volume of the particle and the contrast of the hydration layer. Rigid-body modeling was performed using the ZDOCK docking approach [35] to generate decoy structures and complexes were ranked based on their fit to the experimental scattering data.
The scattering profile from a molecular mixture can be decomposed into a linear combination of individual contributions Ii(s) from the different species. If the structures of the components are known or their individual scattering profiles can be measured, the volume fractions of the species that fit the SAXS data can be determined by the program OLIGOMER utilizing nonnegative least-squares fitting [69]. Dimeric structures of EspG3 from M. tuberculosis and M. marinum were generated from their monomeric crystallographic structures (PDB IDs 4W4I and 5DBL, respectively) using the M. smegmatis EspG3 dimer structure (PDB ID 4L4W) as an interaction template. We also used the program OLIGOMER [69] for fitting of experimental scattering profiles of EspG3mtu and EspG3mma by weighted combinations of theoretical scattering profiles from the monomeric crystallographic structures and dimeric models. In the case of EspG3msm, theoretical scattering profiles based on the dimeric and monomeric crystallographic structures (PDB ID 4L4W) were used as inputs for OLIGOMER. For EspG1mma, two different dimeric structures were tested: the first dimer structure is based on the structure of the EspG1mka dimer (PDB ID 5VBA) while the second model for a EspG1mma dimer was constructed using the EspG3msm dimer structure (PDB ID 4L4W) as a template.
Flexibility analyses of protein structures in solution were conducted using their crystallographic structures as starting points for the ensemble optimization method (EOM). This approach seeks to best fit the experimental scattering profile with an ensemble of conformations [70, 71]. Possible conformations of loop regions were modeled with the program RANCH producing 10,000 random configurations, while the rest of the protein was kept fixed. A genetic algorithm was employed to find the set of conformations best fitting the SAXS data. The structures selected from the random pool of structures were analyzed with respect to the Rg distribution.
Molecular dynamics simulations
The program NAMD was employed with the CHARMM27 force field for description of the protein and the TIP3P solvent model for water. Constant particle number, constant pressure and constant temperature (NpT) ensembles were assumed [72–74]. Langevin dynamics were used to maintain constant temperature. Pressure was controlled using a hybrid Nose-Hoover Langevin piston method. An in-house computational pipeline for high-throughput MD simulations and the visualization program VMD were used to prepare input files and to analyze the simulation trajectories [75].
Supplementary Material
Highlights.
The crystal structure of EspG1 reveals the common architecture of the type VII secretion system chaperones
Structures of EspG3 chaperones display a number of conformations that could reflect alternative substrate binding modes
EspG3 chaperones dimerize in solution
A model of EspG3 in complex with its substrate PE-PPE dimer is proposed based on SAXS data
Acknowledgements
We thank Marcel Behr for providing M. kansasii genomic DNA, Wilbert Bitter for providing M. marinum genomic DNA and Carlo Carolis for the espG3mtu construct. We thank the staff of the UCLA-DOE Institute Protein Expression Technology Center, supported by the U.S. Department of Energy, Office of Biological and Environmental Research (BER) program under Award Number DE-FC02–02ER63421, and the UCLA Crystallization Core for assistance in protein purification and crystallization screening. Authors thank staff members of the Northeastern Collaborative Access Team (NE-CAT) and Southeast Regional Collaborative Access Team (SER-CAT) at the Advanced Photon Source, Argonne National Laboratory, for assistance during data collection. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31–109-Eng-38. We acknowledge the Sample Preparation and Characterization (SPC) facility of EMBL at PETRA3 (DESY, Hamburg) for technical support. Work performed in the laboratory of D.E. is supported by the Howard Hughes Medical Institute and National Institutes of Health grants 23616–002-06 F3:02, TBSGC P01 (AI068135), and TBSGC P01 (AI095208). Research reported in this publication was partially supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant numbers P20GM103486 and P30GM110787, and by the National Institute of Allergy and Infectious Diseases grant number R01AI119022 to KVK. A.T.T. was supported by the EMBL EIPOD program under Marie Curie COFUND actions and by the Bundesministerium für Bildung and Forschung (BMBF) project BIOSCAT (grant 05K12YE1).
Abbreviations used:
- EOM
ensemble optimization method
- MD
molecular dynamics
- RMSD
root-mean-square deviation
- NSD
normalized spatial discrepancy
- RMSF
root-mean-square-fluctuations
- SAXS
small-angle X-ray scattering
- SEC
size-exclusion chromatography
- SeMet
selenomethionine
- TEV
tobacco etch virus
- TCEP
tris(2-carboxyethyl)phosphine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Accession numbers
The structure factors and atomic coordinates have been deposited in the Protein Data Bank under accession codes 5VBA (T4L-EspG1mka), 5DLB (EspG3mma), 4L4W, 4RCL and 5SXL (EspG3msm). The SAXS data were deposited in the SASBDB under accession codes SASDDQ2, SASDDR2, SASDDS2, SASDDT2, SASDDU2, SASDDV2, SASDDW2, SASDDX2.
References
- [1].Pai M, Behr MA, Dowdy D, Dheda K, Divangahi M, Boehme CC, et al. Tuberculosis. Nat Rev Dis Primers 2016;2:16076. [DOI] [PubMed] [Google Scholar]
- [2].Cole ST. Inhibiting Mycobacterium tuberculosis within and without. Philosophical transactions of the Royal Society of London Series B, Biological sciences 2016;371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Stanley SA, Raghavan S, Hwang WW, Cox JS. Acute infection and macrophage subversion by Mycobacterium tuberculosis require a specialized secretion system. Proc Natl Acad Sci U S A 2003;100:13001–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lewis KN, Liao R, Guinn KM, Hickey MJ, Smith S, Behr MA, et al. Deletion of RD1 from Mycobacterium tuberculosis mimics bacille Calmette-Guerin attenuation. J Infect Dis 2003;187:117–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Guinn KM, Hickey MJ, Mathur SK, Zakel KL, Grotzke JE, Lewinsohn DM, et al. Individual RD1-region genes are required for export of ESAT-6/CFP-10 and for virulence of Mycobacterium tuberculosis. Mol Microbiol 2004;51:359–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Houben EN, Korotkov KV, Bitter W. Take five - Type VII secretion systems of Mycobacteria. Biochim Biophys Acta 2014;1843:1707–16. [DOI] [PubMed] [Google Scholar]
- [7].Groschel MI, Sayes F, Simeone R, Majlessi L, Brosch R. ESX secretion systems: mycobacterial evolution to counter host immunity. Nat Rev Microbiol 2016;14:677–91. [DOI] [PubMed] [Google Scholar]
- [8].Houben EN, Bestebroer J, Ummels R, Wilson L, Piersma SR, Jimenez CR, et al. Composition of the type VII secretion system membrane complex. Mol Microbiol 2012;86:472–84. [DOI] [PubMed] [Google Scholar]
- [9].Beckham KS, Ciccarelli L, Bunduc CM, Mertens HD, Ummels R, Lugmayr W, et al. Structure of the mycobacterial ESX-5 type VII secretion system membrane complex by single-particle analysis. Nat Microbiol 2017;2:17047. [DOI] [PubMed] [Google Scholar]
- [10].van Winden VJ, Ummels R, Piersma SR, Jimenez CR, Korotkov KV, Bitter W, et al. Mycosins Are Required for the Stabilization of the ESX-1 and ESX-5 Type VII Secretion Membrane Complexes. mBio 2016;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998;393:537–44. [DOI] [PubMed] [Google Scholar]
- [12].Abdallah AM, Gey van Pittius NC, Champion PA, Cox J, Luirink J, Vandenbroucke- Grauls CM, et al. Type VII secretion--mycobacteria show the way. Nat Rev Microbiol 2007;5:883–91. [DOI] [PubMed] [Google Scholar]
- [13].Gey van Pittius NC, Sampson SL, Lee H, Kim Y, van Helden PD, Warren RM. Evolution and expansion of the Mycobacterium tuberculosis PE and PPE multigene families and their association with the duplication of the ESAT-6 (esx) gene cluster regions. BMC evolutionary biology 2006;6:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Strong M, Sawaya MR, Wang S, Phillips M, Cascio D, Eisenberg D. Toward the structural genomics of complexes: crystal structure of a PE/PPE protein complex from Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 2006;103:8060–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Delogu G, Brennan MJ. Comparative immune response to PE and PE_PGRS antigens of Mycobacterium tuberculosis. Infect Immun 2001;69:5606–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Brennan MJ, Delogu G, Chen Y, Bardarov S, Kriakov J, Alavi M, et al. Evidence that mycobacterial PE_PGRS proteins are cell surface constituents that influence interactions with other cells. Infect Immun 2001;69:7326–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Banu S, Honore N, Saint-Joanis B, Philpott D, Prevost MC, Cole ST. Are the PE-PGRS proteins of Mycobacterium tuberculosis variable surface antigens? Mol Microbiol 2002;44:9–19. [DOI] [PubMed] [Google Scholar]
- [18].Li Y, Miltner E, Wu M, Petrofsky M, Bermudez LE. A Mycobacterium avium PPE gene is associated with the ability of the bacterium to grow in macrophages and virulence in mice. Cellular microbiology 2005;7:539–48. [DOI] [PubMed] [Google Scholar]
- [19].Sayes F, Sun L, Di Luca M, Simeone R, Degaiffier N, Fiette L, et al. Strong immunogenicity and cross-reactivity of Mycobacterium tuberculosis ESX-5 type VII secretion: encoded PE-PPE proteins predicts vaccine potential. Cell Host Microbe 2012;11:352–63. [DOI] [PubMed] [Google Scholar]
- [20].Fortune SM, Jaeger A, Sarracino DA, Chase MR, Sassetti CM, Sherman DR, et al. Mutually dependent secretion of proteins required for mycobacterial virulence. Proc Natl Acad Sci U S A 2005;102:10676–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sani M, Houben EN, Geurtsen J, Pierson J, de Punder K, van Zon M, et al. Direct visualization by cryo-EM of the mycobacterial capsular layer: a labile structure containing ESX-1-secreted proteins. PLoS Pathog 2010;6:e1000794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Abdallah AM, Verboom T, Hannes F, Safi M, Strong M, Eisenberg D, et al. A specific secretion system mediates PPE41 transport in pathogenic mycobacteria. Mol Microbiol 2006;62:667–79. [DOI] [PubMed] [Google Scholar]
- [23].Abdallah AM, Verboom T, Weerdenburg EM, Gey van Pittius NC, Mahasha PW, Jimenez C, et al. PPE and PE_PGRS proteins of Mycobacterium marinum are transported via the type VII secretion system ESX-5. Mol Microbiol 2009;73:329–40. [DOI] [PubMed] [Google Scholar]
- [24].Bottai D, Di Luca M, Majlessi L, Frigui W, Simeone R, Sayes F, et al. Disruption of the ESX-5 system of Mycobacterium tuberculosis causes loss of PPE protein secretion, reduction of cell wall integrity and strong attenuation. Mol Microbiol 2012;83:1195–209. [DOI] [PubMed] [Google Scholar]
- [25].Daleke MH, van der Woude AD, Parret AH, Ummels R, de Groot AM, Watson D, et al. Specific chaperones for the type VII protein secretion pathway. J Biol Chem 2012;287:31939–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Korotkova N, Freire D, Phan TH, Ummels R, Creekmore CC, Evans TJ, et al. Structure of the Mycobacterium tuberculosis type VII secretion system chaperone EspG5 in complex with PE25-PPE41 dimer. Mol Microbiol 2014;94:367–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ekiert DC, Cox JS. Structure of a PE-PPE-EspG complex from Mycobacterium tuberculosis reveals molecular specificity of ESX protein secretion. Proc Natl Acad Sci U S A 2014;111:14758–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Daleke MH, Ummels R, Bawono P, Heringa J, Vandenbroucke-Grauls CM, Luirink J, et al. General secretion signal for the mycobacterial type VII secretion pathway. Proc Natl Acad Sci U S A 2012;109:11342–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Poulsen C, Panjikar S, Holton SJ, Wilmanns M, Song YH. WXG100 protein superfamily consists of three subfamilies and exhibits an alpha-helical C-terminal conserved residue pattern. PLoS One 2014;9:e89313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ramsdell TL, Huppert LA, Sysoeva TA, Fortune SM, Burton BM. Linked domain architectures allow for specialization of function in the FtsK/SpoIIIE ATPases of ESX secretion systems. J Mol Biol 2015;427:1119–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Rosenberg OS, Dovala D, Li X, Connolly L, Bendebury A, Finer-Moore J, et al. Substrates Control Multimerization and Activation of the Multi-Domain ATPase Motor of Type VII Secretion. Cell 2015;161:501–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Daleke MH, Cascioferro A, de Punder K, Ummels R, Abdallah AM, van der Wel N, et al. Conserved Pro-Glu (PE) and Pro-Pro-Glu (PPE) protein domains target LipY lipases of pathogenic mycobacteria to the cell surface via the ESX-5 pathway. J Biol Chem 2011;286:19024–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Phan TH, Ummels R, Bitter W, Houben EN. Identification of a substrate domain that determines system specificity in mycobacterial type VII secretion systems. Sci Rep 2017;7:42704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Chen X, Cheng HF, Zhou J, Chan CY, Lau KF, Tsui SK, et al. Structural basis of the PE-PPE protein interaction in Mycobacterium tuberculosis. J Biol Chem 2017;292:16880–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Pierce BG, Wiehe K, Hwang H, Kim BH, Vreven T, Weng Z. ZDOCK server: interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 2014;30:1771–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, et al. SWISS- MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 2014;42:W252–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Franke D, Jeffries CM, Svergun DI. Correlation Map, a goodness-of-fit test for one- dimensional X-ray scattering spectra. Nature methods 2015;12:419–22. [DOI] [PubMed] [Google Scholar]
- [38].Phan TH, Houben ENG. Bacterial secretion chaperones: the mycobacterial type VII case. FEMS Microbiol Lett 2018;365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Matsumura M, Becktel WJ, Levitt M, Matthews BW. Stabilization of phage T4 lysozyme by engineered disulfide bonds. Proc Natl Acad Sci U S A 1989;86:6562–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, et al. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science 2007;318:1266–73. [DOI] [PubMed] [Google Scholar]
- [41].Xds Kabsch W.. Acta Crystallogr D Biol Crystallogr 2010;66:125–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Karplus PA, Diederichs K. Linking crystallographic model and data quality. Science 2012;336:1030–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr 2007;40:658–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zou Y, Weis WI, Kobilka BK. N-terminal T4 lysozyme fusion facilitates crystallization of a G protein coupled receptor. PLoS One 2012;7:e46039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Cowtan K Recent developments in classical density modification. Acta Crystallogr D Biol Crystallogr 2010;66:470–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Cowtan K The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr D Biol Crystallogr 2006;62:1002–11. [DOI] [PubMed] [Google Scholar]
- [47].Cowtan K Fitting molecular fragments into electron density. Acta Crystallogr D Biol Crystallogr 2008;64:83–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Langer G, Cohen SX, Lamzin VS, Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat Protoc 2008;3:1171–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 2010;66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr 2012;68:352–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Arbing MA, Chan S, Harris L, Kuo E, Zhou TT, Ahn CJ, et al. Heterologous expression of mycobacterial Esx complexes in Escherichia coli for structural studies is facilitated by the use of maltose binding protein fusions. PLoS One 2013;8:e81753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Gibson DG. Enzymatic assembly of overlapping DNA fragments. Methods Enzymol 2011;498:349–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Pape T, Schneider TR. HKL2MAP: a graphical user interface for phasing with SHELX programs. J Appl Cryst 2004;37:843–4. [Google Scholar]
- [54].Sheldrick GM. A short history of SHELX. Acta Crystallogr A 2008;64:112–22. [DOI] [PubMed] [Google Scholar]
- [55].Sheldrick GM. Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr D Biol Crystallogr 2010;66:479–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr 2011;67:355–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Vonrhein C, Blanc E, Roversi P, Bricogne G. Automated structure solution with autoSHARP. Methods Mol Biol 2007;364:215–30. [DOI] [PubMed] [Google Scholar]
- [58].Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 2010;66:213–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr D Biol Crystallogr 2006;62:439–50. [DOI] [PubMed] [Google Scholar]
- [60].Painter J, Merritt EA. TLSMD web server for the generation of multi-group TLS models. J Appl Cryst 2006;39:109–11. [Google Scholar]
- [61].Blanchet CE, Spilotros A, Schwemmer F, Graewert MA, Kikhney A, Jeffries CM, et al. Versatile sample environments and automation for biological solution X-ray scattering experiments at the P12 beamline (PETRA III, DESY). J Appl Crystallogr 2015;48:431–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Guinier A La diffraction des rayons X aux très petits angles : application à l’étude de phénomènes ultramicroscopiques. Ann Phys 1939;11:161–237. [Google Scholar]
- [63].Svergun DI. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Cryst 1992;25:495–503. [Google Scholar]
- [64].Petoukhov MV, Franke D, Shkumatov AV, Tria G, Kikhney AG, Gajda M, et al. New developments in the ATSAS program package for small-angle scattering data analysis. J Appl Crystallogr 2012;45:342–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Franke D, Svergun DI. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J Appl Crystallogr 2009;42:342–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Volkov VV, Svergun DI. Uniqueness of ab initio shape determination in small-angle scattering. J Appl Cryst 2003;36:860–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Tuukkanen AT, Kleywegt GJ, Svergun DI. Resolution of ab initio shapes determined from small-angle scattering. IUCrJ 2016;3:440–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Svergun D, Barberato C, Koch MHJ. CRYSOL - a program to pvaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J Appl Cryst 1995;28:768–73. [Google Scholar]
- [69].Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J Appl Cryst 2003;36:1277–82. [Google Scholar]
- [70].Bernado P, Mylonas E, Petoukhov MV, Blackledge M, Svergun DI. Structural characterization of flexible proteins using small-angle X-ray scattering. J Am Chem Soc 2007;129:5656–64. [DOI] [PubMed] [Google Scholar]
- [71].Tria G, Mertens HD, Kachala M, Svergun DI. Advanced ensemble modelling of flexible macromolecules using X-ray solution scattering. IUCrJ 2015;2:207–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, et al. Scalable molecular dynamics with NAMD. J Comput Chem 2005;26:1781–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, et al. All- atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B 1998;102:3586–616. [DOI] [PubMed] [Google Scholar]
- [74].Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys 1983;79:926–35. [Google Scholar]
- [75].Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph 1996;14:33–8, 27–8. [DOI] [PubMed] [Google Scholar]
- [76].Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 2010;66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.