

Abstract
Meters of DNA wrap around histone proteins to form nucleosomes and fit inside the micron-diameter nucleus. For the genetic information encoded in the DNA to become available for transcription, replication and repair, the DNA-histone assembly must be disrupted. Experiment has indicated that the outer stretches of nucleosomal DNA “breathe” by spontaneously detaching from and reattaching to the histone core. Here, we report direct observation of spontaneous DNA breathing in atomistic molecular dynamics simulations, detailing a microscopic mechanism of the DNA breathing process. According to our simulations, the outer stretches of nucleosomal DNA detach in discrete steps involving five or ten base pairs, with the detachment process being orchestrated by the motion of several conserved histone residues. The inner stretches of nucleosomal DNA are found to be more stably associated with the histone core by more abundant nonspecific DNA-protein contacts, providing a microscopic interpretation of nucleosome unraveling experiments. The CG content of nucleosomal DNA is found to anticorrelate with the extent of unwrapping, supporting the possibility that AT-rich segments may signal the start of transcription by forming less stable nucleosomes.
Keywords: Nucleosomes, Chromatin, Molecular Dynamics, DNA
Graphical Abstract
1. Introduction
Eukaryotic DNA wraps around histone proteins to form arrays of nucleosomes, ultimately organizing into chromosomes. For DNA to become available for template-directed processes — transcription in particular — nucleosomes must be disrupted or displaced [1]. The mechanical stability of a nucleosome is determined by the strength of histone-DNA contacts and the mechanical properties of the nucleosomal DNA [2, 3]. Specifically, in vitro experiments [4–7], bioinformatics analysis [8], and structural studies [9] have identified CG content [5, 8] the placement of TA dinucleotides [5, 7], and the presence of CA=TG steps [9] as important determinants of nucleosome stability. Experimental studies have also shown that the outer stretches of DNA can spontaneously unwrap and rewrap from the histone core in a process known as “DNA breathing” [6, 7, 10–17]. Recent in vitro single-molecule studies revealed that nucleosomes unwrap asymmetrically under force, and that the preferred direction of unwrapping depends on the DNA sequence [7].
It is also important to consider what is not yet fully known concerning nucleosome occupancy and stability. Experimental and bioinformatics studies indicate that DNA sequences with greater CG content form more stable nucleosomes. However, nucleosomes have been shown to be depleted in so-called “CpG islands,” regions of elevated CG content and reduced methylation [18–20]. Furthermore, several specific DNA sequence motifs have also been proposed to be important, including ‘TA’ dinucleotides [4, 7, 21], poly-A tracts [22], and ‘TATA’ box regions [23]. The very mechanism of DNA unwrapping from a histone core is also under debate. Several different modes of nucleosome motion have been observed or proposed, including “DNA breathing” described above, the splitting of histone dimers of H2A and H2B from the histone core, which could accompany DNA unwrapping [14, 24, 25], the propagation of a loop [26] or twist defect [27], and the “gaping” between super-helical turns of DNA without unwrapping [28]. It is also possible that nucleosome repositioning in vivo may involve several different mechanisms.
Computational studies play an important role in examining nucleosome structure and dynamics, providing insight into microscopic details not readily accessible to experiment. Previous coarse-grained (CG) [29–34] and all-atom [35–44] molecular dynamics (MD) simulations have investigated the dynamics of nucleosomes and chromatin. CG studies have primarily been used to examine DNA positioning and superhelical sliding [45, 46], or the free energy associated with nucleosomal unwrapping [30–32], without commenting on how spontaneous detachment occurs at the molecular level. Previous all-atom MD simulations have characterized nucleosomal DNA’s primary modes of motion [35], discovered that the nu- cleosome behaves like a sponge absorbing water and ions [36], unwrapped nucleosomal DNA by force [38, 47], uncovered transient elements of structure in histone tails [37, 41, 43, 48–50], and found that histone variation may lead to enhanced flexibility [39, 40, 42, 51]. The spontaneous detachment of about one turn of DNA from a histone core has been observed in all-atom simulations [39, 40, 43], but those reports did not to attempt to describe the underlying mechanism. Multi-scale MD studies have combined all-atom and CG simulations [41, 52, 53] to form hypotheses on the potential role of histone tails in short-length (i.e. under 10 bp) nucleosomal DNA detachment [53] and in the mediation of nucleosome- nucleosome interactions [41]. Studies that use theory, or a combination of theory and simulation, have taken a more mathematical approach to investigating the possible effects of DNA sequence on nucleosome stability [54] and DNA flexibility [55]. Large-scale, all-atom simulations were also used to investigate how nucleic acids bend around nanoparticles designed to mimic histones [56]. Overall, a mechanistic account of spontaneous nucleosomal DNA detachment in atomistic detail has remained largely elusive.
Here, we report the molecular mechanism of spontaneous unraveling of nucleosome core particles observed in all-atom MD simulations. Our study examined nucleosomal DNA breathing at multiple ionic conditions, and for several DNA sequences. We identified a step-wise mechanism, involving a set of specific histone residues, that leads to the detachment of outer turns of nucleosomal DNA by observing many spontaneous detachment events in all-atom detail. Through another set of simulations, where DNA detachment was induced through partial rupture of the specific contacts, we confirmed the proposed mechanism and further probed the effects of DNA sequence. A mechanistic account of nucleosome unwrapping, and the effects of DNA sequence therein, is key to understanding fundamental biological processes, including transcription, replication, and repair.
2. Results and Discussion
Spontaneous and reversible nucleosomal unwrapping
Fig. 1a illustrates a typical simulation system considered in this work: an all-atom model of a nucleosome (PDB ID: 3LZ0 [57]) submerged in an electrolyte solution. Several simulation systems were constructed featuring a 601L nucleosome, differing by the electrolyte conditions: 0.15 M NaCl, 1.0 M MgCl2, and 3.0 M NaCl. The protein core of each model contained residues 16–118 for both copies of H2A, 29–121 for both copies of H2B, 39–134 for both copies of H3, and 25–102 for both copies of H4. The disordered N- and C-terminal tails of the core histones were not included in the nucleosome models. Based on the results of several experimental studies [58, 59], we anticipate that the removal of histone tails has accelerated the rate of spontaneous DNA unwrapping. See Materials and Methods for further details on the simulation protocol.
Figure 1:

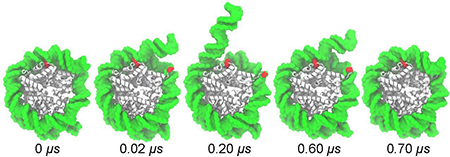
All-atom MD simulation of DNA breathing, (a) Simulation setup: a nucleosome particle composed of a histone core (white) and a 145-base pair (bp) fragment of DNA (green) is surrounded by electrolyte solution (semi-transparent blue), (b) The number of endpoint bp detached from the histone core as a function of time in the simulations of the Widom 601L nucleosome performed in 0.15 M NaCl (top), 1.0 M MgCl2 (middle), and 3.0 M NaCl (bottom). The number of endpoint bp detached was determined by finding all DNA-histone contacts, defining a contact as a pair of non-hydrogen atoms of the protein and DNA located within 4.5 A of each other, and identifying the contact-forming DNA bps closest to the entry and exit termini, the terminal contact bps. The total number of bp detached was calculated as the total number of bps in the nucleosomal DNA (i.e. 145) minus the number of bps located between the terminal contact bps. (c) Snapshots illustrating spontaneous unbinding and rebinding of nucleosomal DNA from and to the histone core observed in 1 M MgCl2 solution. Starting from a fully-bound state (0 μs), two DNA-histone contacts from the entry side break (0.02 μs, 0.20 μs) and later reform (0.60 μs, 0.70 μs). H2A Arg77 and H3 His39 are highlighted in red. (d) Reversibility of DNA detachment. Top and bottom panels illustrate the microscopic configurations of the two terminal fragments of DNA (first and last 25 bp, respectively) at the beginning (white) and after 0.8 μs (red and blue) of the MD simulation performed in 1.0 M MgCl2. The DNA fragment featured in the top panel unbinds from and rebinds to the histone core within the first 0.7 μs of the simulation, (e) Normalized distributions of the DNA fragments’ RMSD values for the [0.70, 0.85] μs segment of the 1.0 M MgCl2 trajectory. The RMSD values were computed with respect to the starting conformations using coordinates of all non-hydrogen DNA atoms, after alignment of the histone core backbone atoms.
For each nucleosome system considered, three independent replica simulations were performed separately, including minimization and structural relaxation, followed by approximately 0.35 μs of free equilibration at room temperature and standard pressure conditions. Initial velocities for replica simulations were generated using unique random seeds. The pairwise RMSD values at the beginning of free equilibration simulations ranged from 2.3 to 3.5 Å across all replica simulations, see Supplementary Fig. S1. In one of three replica simulations carried out for each 1.0 M MgCl2 and 3.0 M NaCl electrolyte condition, the nucleosomal DNA was observed to detach spontaneously within the first 0.35 μs. These two simulations were extended to 2 μs using DESRES Anton2 special purpose supercomputer [60]. Although no spontaneous DNA detachment was observed within the first 0.35 μs of the three 0.15 M NaCl simulations, one replica simulation was extended to 2 μs for comparison with the 1.0 M MgCl2 and 3.0 M NaCl systems.
In general agreement with salt-dissociation experiments [21, 61, 62], the structural stability of a nucleosome was found to depend on the electrolyte conditions used in an MD simulation. At physiological electrolyte concentration (0.150 M NaCl), the Widom 601L DNA remained stably bound to the histone core over the entire 2 μs simulation, Fig. 1b and Supplementary Movie 1. Such high integrity of the nucleosome structure is consistent with the experimentally-characterized timescale of 250 ms for the outer stretch of nucleosomal DNA to unwrap from a histone core at physiological conditions [63]. In a simulation carried out at high ionic strength electrolyte (1.0 M MgCl2), the outer turns of the 601L nucleosomal DNA unwrapped spontaneously from the histone core in a stepwise fashion, Fig. 1b; 601L DNA also became detached in another condition of high ionic strength (3.0 M NaCl). Fig. 1c and Supplementary Movie 2 illustrate the simulated DNA unbinding process in 1 M MgCl2, and Supplementary Movie 3 depicts a similar process in 3 M NaCl. Conditions of high ionic strength apparently reduce the electrostatic attraction between the negatively- charged DNA and the positively-charged histone core, thereby increasing the likelihood of DNA unwrapping. 3D density maps of Mg2+ and Na+, Supplementary Fig. S2a & b, indicate that both ion species can enter both minor and major grooves of the nucleosomal DNA, and that Na+ ions cover the DNA’s surface more evenly than Mg2+, matching the results of a previous study [64]. Note that, because the simulation time scale does not exceed single digit microseconds, we do not expect to observe complete unraveling of nucleosomal DNA, as observed in experiment performed at a much longer time scale [21, 62], Our simulations, thus, probe recovery of the system to equilibrium following the quench of ionic conditions, revealing the details of the nucleosome unraveling process.
To estimate the rate of unwrapping conditioned by the DNA elasticity alone, we simulated the Widom 601L DNA in the absence of the histone core starting from the conformation the DNA adopts in an intact nucleosome. The DNA was observed to unravel completely from its starting conformation within 300 ns of the simulation, see Supplementary Movie 4.
The simulated spontaneous unraveling of the nucleosomal particle was found to be reversible. In 3.0 M NaCl, about 13 bp of 601L DNA became detached around 0.03 μs, and then reattached to the histone core by 0.9 μs, see Fig. 1b and Supplementary Movie 4. In the simulation carried out at 1.0 M MgCl2, the first 25 bp of the nucleosomal DNA underwent two major detachment transitions, at 0.02 and 0.20 μs, but bound back to the histone core in two separate steps, at 0.60 and 0.70 μs, Fig. 1b & c. Furthermore, the DNA appears to bind back to the histone core in a conformation that is statistically indistinguishable from its initial state. To illustrate this point, we show in Fig. 1d the conformations of the terminal 25 bp of the nucleosomal DNA at the beginning of the simulation (0 μs) and after undergoing a reversible unbinding transition (0.7 μs). The overlay of the two conformations visually appear indistinguishable for the entry (first 25 bp) and exit (last 25 bp) termini of the nucleosomal DNA, although only the entry fragment underwent reversible unraveling while the exit fragment remained bound to the histone core. The distributions of the DNA fragments’ RMSD values with respect to their starting positions (both with primary peaks between 5.0 and 5.5 Å), Fig. 1e, illustrate the dynamic nature of these segments and the statistical similarity of their conformations upon reversible unbinding. The difference in the RMSD distributions arises from greater fluctuations of the initial 10 bp of the first 25 bp, compared to the final 10 bp of last 25 bp, Supplementary Fig. S3. Such spontaneous, reversible unwrapping of DNA was previously observed in experiment [6, 65] and was described as “DNA breathing.”
The effects of DNA sequence on nucleosome breathing
To examine the effects of the nucleotide sequence on DNA breathing, we repeated the simulations of the nucleosomal system at 1 M MgCl2 for the three additional variants of the nucleosomal DNA sequence: poly-AT (i.e. ATAT...; 0% CG), 5S mRNA gene (43% CG) [66], and poly-GC (i.e. GCGC...; 100% CG), which are known to form nucleosomes in vitro [66–68]. The nucleotide bases of the 601 DNA resolved in the X-ray structure were changed according to the target sequence (listed in Supplementary Table S1) while keeping the same conformation of the DNA backbone. Materials and Methods and Table S2 provide a detailed description of the structure building and simulation protocols. As done for 601L, three replica simulations were performed for the additional sequences in 1M MgCl2 for 0.35 μs. One replica of poly-AT, and one of 5S gene, underwent spontaneous detachment in that time period, and were extended to 2 μs. No replicas of poly-GC exhibited detachment within 0.35 μs, and one was extended to 2 μs for the sake of comparison.
Among the four systems (601L, poly-AT, 5S and poly-CG), the poly-AT system was found to be the most dynamic, Fig. 2a. Within the first ~0.8 μs of the simulation, about 45 bp detached from the histone core in four unbinding events occurring at both DNA termini, followed by the subsequent rebinding of about 20 bp; Supplementary Movie 5 illustrates this MD trajectory. The outcome of the 5S nucleosome simulation was similar to that of 601L: 25 bp ultimately became unwrapped within 2 μs, Fig. 2a and Supplementary Movie 6, which can be attributed to the similarity in CG content of their DNA sequences. On the other hand, none of the poly-GC bp unwrapped from the histone core, Fig. 2a and Supplementary Movie 7, despite considerable structural fluctuations. Thus, the extent of simulated DNA unraveling, Fig. 2b, appears to be correlated with the CG content: the poly-AT (0% CG) nucleosome unravels the most, the 5S (43% CG) and 601L (57% CG) nucleosomes display intermediate unraveling, whereas the poly-GC (100% CG) one does not unravel. Unlike the other DNA sequences considered, 5S is asymmetric: the 5’ side contains ~38% CG, and the 3’ side contains ~50% CG. As shown in Supplementary Fig. S4a, 5S DNA spontaneously unwraps from the 5’ side, the side smaller in CG content. This result is consistent with our overall finding that DNA sequences lower in CG content are less stably attached to a histone core. We also note that more 5S DNA unbinding events would need to be observed to definitively conclude that spontaneous unwrapping primarily occurs from one side.
Figure 2:

The effect of CG content on spontaneous unwrapping of a nucleosome. (a) The number of endpoint bp detached from the histone core as a function of simulation time for nucleosomes containing DNA of poly-AT (0% CG), 5S RNA gene (43% CG), Widom 601L (57% CG), and poly-CG (100% CG) nucleotide sequences. The 601L sequence trace is the same as in Fig. 1b. All nucleosome variants were simulated in 1.0 M MgCl2 electrolyte, (b) Representative conformations of the nucleosome systems corresponding to the states characterized by the maximum detachment of the nucleosomal DNA.
The results of our MD simulations suggest that a DNA molecule with greater CG content may form a more stable nucleosome, in agreement with the aforementioned bioinformatics analysis [4, 5, 8]. Our results are also consistent with a recent computational study that found nucleosomes to slide more slowly along poly-GC DNA, and more rapidly along poly- AT [69]. A set of independent simulations of 45 bp DNA fragments in 0.15 M NaCl and 1.0 M MgCl2 showed a statistically insignificant dependence of the dynamic persistence length [55] on the electrolyte conditions (see Supplementary Note 1 and Fig. S5), consistent with experimental studies that found only a small change in DNA’s persistence length when increasing from moderate to high salt concentrations [70, 71]. The possible implications of our results are also in agreement with a single-molecule experimental study that found a difference in the CG content of the left and right halves of nucleosomal DNA to determine, in part, the preferred direction of asymmetric unwrapping under tension [7]. To add more statistical weight to these results of spontaneous unbinding, we revisit the effects of DNA sequence below in computational assays where detachment is induced.
The stepwise mechanism of nucleosomal DNA unraveling
Close inspection of the MD trajectories reveals that a DNA-histone unbinding event is a two-step process, controlled by the conformation of several histone residues. These residues — H3 His39, H3 Arg40, H3 Arg49, H2A Arg30, H2A Arg77 — are all conserved for at least 84% of species that contained a functional equivalent of human histone proteins H3.1 and H2A.1 [72]. Fig. 3a & b depict a typical DNA-histone unbinding event, illustrating the distinct steps of the process. Initially, DNA binding to the histone core is stabilized by a charged side chain that is inserted into the minor groove of DNA and simultaneously forms salt bridges with two phosphate groups from different DNA strands, Fig. 3b i. The detachment begins after one of the salt bridges breaks, leaving the charged side chain interacting only with one of the DNA strands, Fig. 3b ii. After the second DNA-histone salt bridge breaks, one turn of DNA becomes detached from the histone core, Fig. 3b iii. For the example highlighted, the entire process leads to the detachment of about 10 bp, with the intermediate state (from ~0.18 to 0.20 μs) corresponding to the release of 5 bp. Supplementary Fig. S6 and Table S2 characterize the process of DNA unbinding for each major spontaneous detachment observed, showing that an intermediate partially-bound state occurred in each detachment event although the number of bp released during the intermediate step varied.
Figure 3:

Stepwise unraveling of nucleosomal DNA. (a) The number of endpoint bp detached (top) and the distance from the guanidinium group of H2A Arg77 to the nearest phosphorus atom of the I (red) and J (orange) strands of nucleosomal DNA (bottom) versus simulation time. These data derive from an MD simulation of the 601L nucleosome in 1 M MgCl2. (b) Snapshots illustrating the steps of DNA-histone unbinding. Nucleosomal DNA (green) transitions from being tightly bound to the histone core (white) by a charged residue (red) to only being partially bound to the core (i → ii); then DNA detaches completely from the core (ii → iii). A similar pattern of unbinding was observed for all detachment events, see Supplementary Table S2 and Fig. S6.
In seven out of eleven unbinding events, 4 or 5 bps were released at the intermediate step whereas no bp were released in the other four unbinding events, Supplementary Table S2. This observation is consistent with the results of a recent experimental study that found DNA to unwrap from the histone core predominantly in 10-bp steps that sometimes included an intermediate 5-bp step [73]. The proposed mechanism provides an explanation of why 5 or 10-bp unwrapping steps could occur. Our simulations revealed that the detachment of one turn of a DNA helix requires the sequential breaking of two salt bridges formed with the same histone residue. Because the nucleosomal DNA is in an over-twisted state, the phosphates that form these two interactions with the same histone residue are spaced about 5 bp apart on the opposite strands of the DNA duplex. Depending on which of the two salt bridges breaks first, the unwrapping can occur in two 5-bp steps or one 10-bp step. The rebinding of nucleosomal DNA to the histone core may follow a slightly different mechanism where a histone side chain slides along the DNA backbone before reinserting into the minor groove, see Supplementary Movie 2. As previously suggested by Kulic and Schiessel [74], we have encountered five instances of a 1-bp shift in the interaction between DNA phosphates and key histone residues out of 16 observed unwrapping or rewrapping events, Supplementary Fig. S7. Therefore, in our simulations, we have observed a combination of spontaneous nucleosome unwrapping and rewrapping, and the formation of twist defects that arise from a 1-bp shift in DNA-histone interaction.
To test the proposed mechanism of nucleosome breathing and its dependence on DNA sequence, we performed three independent replica simulations each of poly-AT, 5S gene, 601L, and poly-GC nucleosomes in 1.0 M MgCl2 where unwrapping was induced by partially rupturing essential DNA-histone contacts. The side chains of ten key histone residues were moved, and held, away from the three outermost locations of histone-DNA minor groove contacts at either termini of nucleosomal DNA, six such locations in total. Fig. 4a illustrates the extent of the initial forced side chain motion for one of the ten residues forming the DNA-histone contacts; Supplementary Methods 1, Fig. S8 and Table S3 describe the details of the simulation protocol.
Figure 4:

Test of the nucleosome dissociation mechanism. Specific DNA-histone core contacts were simultaneously broken by forcibly moving the side chains of the contact residues away from the DNA in a 10 ns MD simulation, (a) One of the ten DNA-histone contact forming residues before (blue) and after (red) the contact rupture; the DNA and histone core are shown as purple and white molecular surfaces, respectively, (b) The number of endpoint bp detached during simulations that followed the forc/. ijed contact rupture; the histone side chains were kept away from the DNA. Colored labels indicate the nucleosomal DNA sequences. Three independent replicas were run for each system. Simulations were performed in 1.0 M MgCl2 electrolyte. (c) The average number of endpoint bps detached from the histone core after 0.2 μs of holding key histone residues away from the DNA. Error bars represent standard deviations. DNA sequences are ordered from left to right by increasing CG content.
The partial rupture of the essential DNA-histone contacts was found to produce rapid unbinding of the DNA terminal fragments, Fig. 4b and Supplementary Movie 8. Unwrapping occurred for all DNA sequences considered within 0.2 μs, one-tenth of the timescale explored for spontaneous unraveling. The average number of bps detached at the end of these simulations is shown graphically in Fig. 4c. Poly-AT, containing 0% CG, had an average of 28.7 ± 4.1 endpoint bp detached after about 0.21 μs, the least CG and most bps detached on average. Over the same time period, an average of 26.0 ± 3.3 bp of 5S gene (43% CG) and 24.7 ± 9.9 bp of 601L (57% CG) became detached. Poly-GC, which is 100% CG, had an average of 16.7 ± 1.7 bp detached, the fewest of all sequences considered. The results of accelerated unwrapping when holding histone residues away from DNA demonstrate that greater CG content leads to a more stable nucleosome, expanding upon what was observed from simulations of spontaneous unraveling, Fig. 2. Furthermore, keeping the specific histone side chains held away prevented the nucleosomes from rewrapping. Across all systems in which histone-DNA contacts were ruptured, the DNA unraveling did not progress beyond 27 bp from either endpoint.
Central nucleosomal DNA more stably interacts with the histone core
Our results suggest that the outermost three turns of nucleosomal DNA unwrap more easily from the histone core than the inner DNA, implying that the latter is more strongly bound. Depicted in Fig. 5a, a nucleosome featuring only the central 85 bp of Widom 601 DNA (58% CG) was simulated to examine the stability of inner nucleosomal DNA in the absence of outer DNA. Over the first 0.75 μs of simulation, one brief DNA detachment was observed from the entry side, and one brief detachment from the exit side, shown in Fig. 5a and Supplementary Movie 9. For the rest of the time, the DNA remained stably bound to the histone core, with several short-lived partial detachments, indicating that inner DNA unwraps at a slower rate than outer DNA. Contact analysis of our MD trajectories demonstrates that the inner region of the nucleosomal DNA forms more contacts on average with the histone core than the outer region, Fig. 5b & c. Interestingly, the DNA sequence does not affect the average number of contacts for each region, Fig. 5c, indicating that the difference in the unbinding kinetics of the outer regions is caused by DNA mechanics, not DNA-protein contacts. Further analysis indicates that detachment of the outer stretches of DNA from the histone core does not influence the number of contacts that the inner stretches of DNA make with the histone core, Fig. S5. We note also that the number of DNA-histone contacts does indeed change over time throughout the 2 μs production MD simulations, see Supplementary Fig. S9a. In agreement with our observations, larger forces were required to unwrap inner stretches of nucleosomal DNA in single molecule-experiments [7, 73].
Figure 5:

Histone-DNA contacts enhance stability of the central region of nucleosomal DNA. (a) The number of endpoint bp detached as a function of time in an MD simulation of a nucleosome containing only the central 85 bp fragment of the Widom 601 DNA. The simulation was performed in 1 M MgCl2; initial structure shown on the left, (b) The inner and outer regions of DNA in a fully-assembled nucleosome. (c) Trajectory-average number of contacts that a histone core forms with the inner or the outer regions of the DNA per bp. A contact is defined as having a non-hydrogen protein atom within 4.5 Å of a non-hydrogen DNA atom. The bp detached from the histone core were not included in the calculation. Error bars represent standard deviations. The DNA sequence and electrolyte condition identified by label.
Interestingly, the DNA in 601L and 5S systems were found to detach spontaneously almost exclusively from one side in 1 M MgCl2 similar to the behavior of the 601L system in 3 M NaCl, Fig. S4a. On the other hand, poly-AT in 1 M MgCl2 is seen to detach from both ends simultaneously, Fig. S4a. After rupturing key histone-DNA contacts, or when considering a nucleosome system with only the central 85bp of DNA, detachment occurs fairly evenly from both sides, Fig. S4b & c. Thus, our simulations neither confirm nor rule out the possibility that detachment of nucleosomal DNA from opposite sides may be coordinated.
Conclusions
In this study, MD simulations were used to characterize nucleosome unwrapping in atomistic detail, uncovering the effects of ionic conditions (Fig. 1), DNA sequence (Fig. 2 and 4), and DNA position with respect to the pseudo-dyad (Fig. 5). By closely examining many major DNA-histone spontaneous unbinding events (Fig. 3a & b and Supplementary Fig. S6), we recognized a clear two-step mechanism of detachment: (1) periodically-placed DNA transitions from being fully-bound to a specific positively-charged residue of the histone core to being only partially-bound, and then (2) about one turn of DNA unbinds completely. This mechanism was then confirmed by inducing a partially-bound state for nucleosomal DNA at six major sites where the minor groove faces toward the histone core (one shown in Fig. 4a), and then observing accelerated DNA unwrapping (Fig. 4b & c). Together, spontaneous and induced nucleosome unwrapping of nucleosomes featuring four DNA sequences strongly suggest that greater CG content corresponds to more stable nucleosomes. A recent MD study of poly-lysine peptide binding to DNA suggests that histone tails could bind to CG-rich DNA more tightly than to AT-rich DNA, which would make the sequence dependence of nucleosome unraveling reported in this study even more pronounced [75].
Our results suggest that different mechanisms may govern the unwrapping of outer and inner nucleosomal DNA. Outer stretches of DNA, far from the pseudo-dyad, may rapidly unwrap and rewrap from the histone core spontaneously, while chaperones may be required to extend unwrapping to more central DNA. The sensitivity of unwrapping to nucleosomal DNA CG content observed here may be enhanced when forces or torques are applied to the DNA endpoints. The mutation or chemical modification of specific highly-conserved histone residues identified in this study to form important interactions with DNA could increase the rate of unwrapping or signal the initiation of transcription. Our study’s observation of transient nucleosome unwrapping and rewrapping is consistent with models of nucleosome energetics based on genome-wide maps, which indicate that nucleosomes often exist in partially wrapped states in a biological setting [76, 77]; and the results of this investigation could be used in the future to contribute an additional term to such a model. Along with bioinformatics [8, 78] and experimental [79–81] studies, our findings support the possibility of a mechanism whereby the initiation of transcription in yeast or plants is signaled by AT-rich segments of DNA that form less stable nucleosomes.
3. Materials and Methods
3.1. General simulation protocols
All-atom MD simulations were performed using the amber99SB-ILDN-PHI force field for proteins [82], the amber99-bsc0 variant for DNA [83], Young et al. [84] parameters for NaCl, the TIP3P water model [85], custom parameterization of Mg2+ ions [64] and the CUFIX corrections for non-bonded interactions between charged groups [86]. Periodic boundary conditions were employed in all simulations, and long-range electrostatics were calculated using the Particle Mesh Ewald method [87] over a 1.6 Å spaced grid. Non-bonded Coulomb and Lennard-Jones interactions were truncated at 12 Å, and the non-bonded list was updated every 10 steps with the Verlet cutoff-scheme [88]. Covalent bonds to hydrogen in water, and in non-water molecules, were constrained using SETTLE [89] and LINCS [90] algorithms, respectively. All minimization, structural relaxation, and initial unrestrained equilibration simulations of at least 50 ns were performed using the gromacs 5.0.4 MD package [91], in the constant number of particles N, pressure P and temperature T ensemble at 1.0 bar and 300 K maintained using the V-rescaled, modified Berendsen thermostat [92] with a 0.1 ps time-constant and the Parrinello-Rahman barostat [93] with a relaxation time of 2.0 ps. The 2-μs production runs were performed in the NPT ensemble on the D. E. Shaw Research Anton2 supercomputer [60]. The simulations on Anton2 employed a set of parameters equivalent to those listed above, except for using the Nosé-Hoover thermostat [94, 95], the Martyna-Tobias-Klein barostat [96], and the fc-space Gaussian split Ewald method [97] to calculate the electrostatic interactions.
3.2. All-atom models of nucleosome systems
A crystal structure of the canonical H3 nucleosome containing the Widom 601 [4] nucle-osomal DNA (PDB ID: 3LZ0 [57]) served as the basis for all our structural models. The structure contained a protein core made up of eight histone proteins—two copies of histones H2A, H2B, H3, H4, and a 145 base pair (bp) DNA fragment super-helically wrapped around the protein core. The protein model contained residues 16–118 for both copies of H2A; 29– 121 for both copies of H2B; 39–134 for both copies of H3; and 25–102 for both copies of H4. The disordered N- and C-terminal tails of the core histones were not included in the model. The structural starting point for our models, PDB ID 3LZ0 [57], contains 145 bp of DNA, with a single bp at its center. Using a structure with an even number of bp would introduce a minor shift in the DNA’s position. Overall, such a replacement would not lead to major changes, since 1 bp shifts were observed during spontaneous unwrapping and rewrapping, Supplementary Fig. S7.
Starting from the X-ray structure of the Widom 601 DNA nucleosome (PDB ID: 3LZ0 [57]), the pdb2gmx tool of gromacs was used to generate the computational model of the system. The protonation states of titratable residues were chosen to set the charge of Lys and Arg to +1e (where e is the charge of a proton), Asp and Glu to – le, and Gin and His to zero, the protonation state of all His residues was set to NE2. The resulting all-atom model was submerged in a cubic volume of water, producing a system of approximately 350,000 atoms, 150 Å on each side. The minimum distance between the periodic images of the nucleosomes was at least 30 Å. The all-atom model of the 601L nucleosome in physiological environment was produced by adding Na+ and Cl- ions to the system in the amount required to first neutralize the charge of the protein-DNA complex and then bring the ion concentration of the surrounding solvent to 0.15 M. The number of ions added to provide the desired concentrations c were determined using the expression Nion = 0.018 c Nwater, where Nion and Nwater are the numbers of ions per species and water molecules, respectively.
Computational models of the 601L nucleosome in high ionic strength environments were produced by adding (1) Mg2+ and Cl ions to neutralize the nucleosome charge and bring the ion concentration to 1 M MgCl2, and, separately, (2) Na+ and Cl- ions were introduced to neutralize the charge and reach an ionic concentration of 3 M NaCl. For simulations in magnesium, we chose to use the hydrated model of Mg2+ ions, where each Mg2+ ion forms a permanent cluster with six surrounding water molecules—magnesium hexahydrate [64]. This model was parametrized to reproduce the osmotic pressure of model compounds and was validated through simulations of DNA array systems [98]. The integrity of each Mg2+- hexahydrate complex was maintained by applying harmonic potentials between the magnesium and water oxygen atoms; the spring constant of the potential k= 500,000 kJ mol−1 nm−2 and the equilibrium distance was 1.94 Å.
Upon assembly, the systems were minimized using the steepest descent method [99], until the maximum force on any atom was less than 166 pN. Absolute position restraints (kpos = 1000 kJ mol−1 nm−2) were applied during the minimization to all non-hydrogen atoms of the protein and DNA to maintain their crystallographic coordinates. The minimized systems were equilibrated in several steps, see Supplementary Table S4, and Fig. S1. Briefly, a network of pairwise harmonic restraints was applied within and between the DNA backbone and histone core, and gradually reduced to 0 over the course of 3.5 ns. The final configuration obtained at the end of the multi-step structural relaxation process was used for a ~2 μsproduction simulation, which was performed in the absence of any restraints, in the NPT ensemble at 1.0 bar and 300 K. The coordinates, velocities, and energies were saved every 2 ps for further analysis.
The same steps were repeated to generate the all-atom models of nucleosomes containing DNA of other, non-601L, nucleotide sequences. First, the nucleotide sequence of target DNA was compared to the 601 sequence of the DNA resolved in the X-ray structure. The base moieties of bps that did not match were modified to conform to the target sequence using the psfgen tool of VMD [100]; the coordinates of the DNA backbone were not altered. The resulting conformations of newly generated bases were verified by computing the dihedral angle between the two atoms of the sugar backbone and the two of each base forming the glycosidic bond. The system featuring only the central 85 bp segment of Widom 601 DNA was built by removing 30 bps from each DNA terminus. The resulting structures were solvated; ions were added to bring the concentration of MgCl2 to 1 M. The EN of restraints and Watson-Crick base-paring restraints were generated for each system individually; the structural relaxation simulations followed exactly same protocols as for the 601L system. Supplementary Table S1 lists the nucleotide sequences of DNA used to build all nucleosome systems considered in this work. The system containing only the 601L DNA was built by removing the histone proteins from the X-ray structure and adding 1 M MgCl2 electrolyte. The production simulations of all systems were run for ~2 μs, except for the system featuring only the Widom 601L DNA (and no histone proteins), run for 0.3 μs, and simulations which forced the rupture of specific histone-DNA contacts (see Supplementary Methods 1, Table S3 and Fig. S8), which were run for 0.2 μs
Supplementary Material
Outer stretches of DNA spontaneously & reversibly detach in all-atom MD simulations
Each turn of DNA detaches after the sequential breaking of two salt bridges
Several highly-conserved histone residues orchestrate the process of DNA detachment
Less CG content was found to correlate with a greater nucleosome unraveling
4. Acknowledgements
This work was supported by grants from NSF (PHY-1430124) and NIH (P41 GM104601). Supercomputer time provided by XSEDE through allocation grant MCA05S028, the Blue Waters supercomputer at the University of Illinois, and the National Resource for Biomedical Supercomputing at the Pittsburgh Supercomputing Center through Anton2 allocation PSCA00052. The authors thank Drs. Jun Song and Hu Jin for valuable discussions, and Drs. Jejoong Yoo and Christopher Maffeo for their assistance in preparing jobs to run on Anton2.
Footnotes
Conflict of Interest Statement: The authors declare no conflicts of interest.
Appendix A. Supplementary Information
Supplementary data associated with this article can be found in the online version.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Lorch Y, LaPointe JW, Kornberg RD, Nucleosomes inhibit the initiation of transcription but allow chain elongation with the displacement of histones, Cell 49 (1987) 203–210. [DOI] [PubMed] [Google Scholar]
- [2].Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ, Crystal Structure of the Nucleosome Core Particle at 2.8 Å Resolution, Nature 389 (1997) 251–260. [DOI] [PubMed] [Google Scholar]
- [3].Davey CA, Sargent DF, Luger K, Maeder AW, Richmond TJ, Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 Å resolution, J. Mol. Biol 319 (2002) 1097–113. [DOI] [PubMed] [Google Scholar]
- [4].Lowary PT, Widom J, New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning, J. Mol. Biol 276 (1998) 19–42. [DOI] [PubMed] [Google Scholar]
- [5].Thåström A, Lowary P, Widlund H, Cao H, Kubista M, Widom J, Sequence motifs and free energies of selected natural and non-natural nucleosome positioning DNA sequences, J. Mol. Biol 288 (1999) 213–229. [DOI] [PubMed] [Google Scholar]
- [6].Li G, Levitus M, Bustamante C, Widom J, Rapid spontaneous accessibility of nu- cleosomal DNA, Nat. Struct. Mol. Biol 12 (2005) 46–53. [DOI] [PubMed] [Google Scholar]
- [7].Ngo TTM, Zhang Q, Zhou R, Yodh J, Ha T, Asymmetric Unwrapping of Nucleosomes under Tension Directed by DNA Local Flexibility, Cell 160 (2015) 1135–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tillo D, Hughes TR, G+C content dominates intrinsic nucleosome occupancy, BMC Bioinformatics 10 (2009) 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Richmond TJ, Davey CA, The structure of DNA in the nucleosome core, Nature 423 (2003) 145. [DOI] [PubMed] [Google Scholar]
- [10].Anderson JD, Lowaxy PT, Widom J, Effects of histone acetylation on the equilibrium accessibility of nucleosomal DNA target sites, J. Mol. Biol 307 (2001) 977–985. [DOI] [PubMed] [Google Scholar]
- [11].Tomschik M, Zheng H, van Holde K, Zlatanova J, Leuba SH, Fast, long-range, reversible conformational fluctuations in nucleosomes revealed by single-pair fluorescence resonance energy transfer, Proc. Natl. Acad. Sci. U. S. A 102 (2005) 3278–3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Koopmans W, Brehm A, Logie C, Schmidt T, Noort JV, Single-pair FRET microscopy reveals mononucleosome dynamics, J. Fluoresc 17 (2007) 785–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gansen A, Toth K, Schwarz N, Langowski J, Opposing roles of H3- and H4- cetylation in the regulation of nucleosome structurea FRET study, Nucleic Acids Res. 43 (2015) 1433–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Miyagi A, Ando T, Lyubchenko YL, Dynamics of nucleosomes assessed with time-lapse high-speed atomic force microscopy, Biochemistry 50 (2011) 7901–7908. [DOI] [PubMed] [Google Scholar]
- [15].North JA, Shimko JC, Javaid S, Mooney AM, Shoffner MA, Rose SD, et al. , Regulation of the nucleosome unwrapping rate controls DNA accessibility, Nucleic Acids Res. 40 (2012) 10215–10227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chen Y, Tokuda JM, Topping T, Sutton JL, Meisburger SP, Pabit SA, et al. , Revealing transient structures of nucleosomes as DNA unwinds, Nucleic Acids Res. 42 (2014) 8767–8776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chen Y, Tokuda JM, Topping T, Meisburger SP, Pabit SA, Gloss LM, et al. , Asymmetric unwrapping of nucleosomal DNA propagates asymmetric opening and dissociation of the histone core, Proc. Natl. Acad. Sci. U. S. A 114 (2017) 334–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bird AP, CpG-rich islands and the function of DNA methylation, Nature 321 (1986) 209–213. [DOI] [PubMed] [Google Scholar]
- [19].Gardiner-Garden M, Frommer M, CpG islands in vertebrate genomes, J. Mol. Biol 196 (1987) 261–282. [DOI] [PubMed] [Google Scholar]
- [20].Deaton AM, Bird A, CpG islands and the regulation of transcription, Genes and Devel. 25 (2011) 1010–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Chua EY, Vasudevan D, Davey GE, Wu B, Davey CA, The mechanics behind DNA sequence-dependent properties of the nucleosome, Nucleic Acids Res. 40 (2012) 6338–6352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Iyer V, Struhl K, Poly (dA: dT), a ubiquitous promoter element that stimulates transcription via its intrinsic DNA structure., EMBO J. 14 (1995) 2570–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sawadogo M, Roeder RG, Interaction of a gene-specific transcription factor with the adenovirus major late promoter upstream of the TATA box region, Cell 43 (1985) 165–175. [DOI] [PubMed] [Google Scholar]
- [24].Luger K, Dechassa ML, Tremethick DJ, New insights into nucleosome and chromatin structure: an ordered state or a disordered affair?, Nat. Rev. Mol. Cell Biol 13 (2012) 436–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bdhm V, Hieb AR, Andrews AJ, Gansen A, Rocker A, Toth K, et al. , Nucleosome accessibility governed by the dimer/tetramer interface, Nucleic Acids Res. 39 (2010) 3093–3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Schiessel H, Widom J, Bruinsma R, Gelbart W, Polymer reptation and nucleosome repositioning, Phys. Rev. Lett 86 (2001) 4414. [DOI] [PubMed] [Google Scholar]
- [27].Langst G, Becker PB, Nucleosome remodeling: one mechanism, many phenomena?, Biochim. Biophys. Acta - Gene Struct. Expression 1677 (2004) 58–63. [DOI] [PubMed] [Google Scholar]
- [28].Ngo TT, Ha T, Nucléosomes undergo slow spontaneous gaping, Nucleic Acids Res. 43 (2015) 3964–3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kang H, Yoon Y-G, Thirumalai D, Hyeon C, Confinement-induced glassy dynamics in a model for chromosome organization, Phys. Rev. Lett 115 (2015) 198102. [DOI] [PubMed] [Google Scholar]
- [30].Kenzaki H, Takada S, Partial unwrapping and histone tail dynamics in nucléosome revealed by coarse-grained molecular simulations, PLoS Comput. Biol 11 (2015) el004443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lequieu J, Cordoba A, Schwartz DC, de Pablo JJ, Tension-dependent free energies of nucleosome unwrapping, ACS Cent. Sci 2 (2016) 660–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zhang B, Zheng W, Papoian GA, Wolynes PG, Exploring the free energy landscape of nucléosomes, J. Am. Chem. Soc 138 (2016) 8126–8133. [DOI] [PubMed] [Google Scholar]
- [33].Pierro MD, Zhang B, Aiden EL, Wolynes PG, Onuchic JN, Transferable model for chromosome architecture, Proc. Natl. Acad. Sci. U. S. A (2016) 201613607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Korolev N, Lyubartsev AP, Nordenskiôld L, A systematic analysis of nucleosome core particle and nucleosome-nucleosome stacking structure, Sci. Reports 8 (2018) 1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bishop TC, Molecular dynamics simulations of a nucleosome and free DNA, J. Biomol. Struct. Dyn 22 (2005) 673–685. [DOI] [PubMed] [Google Scholar]
- [36].Materese CK, Savelyev A, Papoian GA, Counterion atmosphere and hydration patterns near a nucleosome core particle, J. Am. Chem. Soc 131 (2009) 15005–15013. [DOI] [PubMed] [Google Scholar]
- [37].Biswas M, Voltz K, Smith JC, Langowski J, Role of histone tails in structural stability of the nucleosome, PLoS Comput. Biol 7 (2011) el002279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ettig R, Kepper N, Stehr R, Wedemann G, Rippe K, Dissecting DNA-histone interactions in the nucleosome by molecular dynamics simulations of DNA unwrapping, Biophys. J 101 (2011) 1999–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kono H, Shirayama K, Arimura Y, Tachiwana H, Kurumizaka H, Two arginine residues suppress the flexibility of nucleosomal DNA in the canonical nucleosome core, PLoS One 10 (2015) e0120635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Winogradoff D, Zhao H, Dalai Y, Papoian GA, Shearing of the CENP-A dimerization interface mediates plasticity in the octameric centromeric nucleosome, Sci. Reports 5 (2015) 17038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Collepardo-Guevara R, Portella G, Vendruscolo M, Frenkel D, Schlick T, Orozco M, Chromatin unfolding by epigenetic modifications explained by dramatic impairment of internucleosome interactions: a multiscale computational study, J. Am. Chem. Soc 137 (2015) 10205–10215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Bowerman S, Wereszczynski J, Effects of macroH2A and H2A. Z on nucleosome dynamics as elucidated by molecular dynamics simulations, Biophys. J 110 (2016) 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Shaytan AK, Armeev GA, Goncearenco A, Zhurkin VB, Landsman D, Panchenko AR, Coupling between Histone Conformations and DNA Geometry in Nucléosomes on a Microsecond Timescale: Atomistic Insights into Nucleosome Functions, J. Mol. Biol 428 (2016) 221–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Pasi M, Lavery R, Structure and dynamics of DNA loops on nucleosomes studied with atomistic, microsecond-scale molecular dynamics, Nucleic Acids Res. 44 (2016) 5450–5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lequieu J, Schwartz DC, de Pablo JJ, In silico evidence for sequence-dependent nucleosome sliding, Proc. Natl. Acad. Sci. U. S. A (2017) 201705685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Niina T, Brandani GB, Tan C, Takada S, Sequence-dependent nucléosome sliding in rotation-coupled and uncoupled modes revealed by molecular simulations, PLoS Comput. Biol 13 (2017) el005880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Maffeo C, Yoo J, Comer J, Wells DB, Luan B, Aksimentiev A, Close Encounters with DNA, J. Phys.: Condens. Matter 26 (2014) 413101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Potoyan DA, Papoian GA, Energy Landscape Analyses of Disordered Histone Tails Reveal Special Organization of Their Conformational Dynamics, J. Am. Chem. Soc 133 (2011) 7405–7415. [DOI] [PubMed] [Google Scholar]
- [49].Potoyan DA, Papoian GA, Regulation of the H4 tail binding and folding landscapes via Lys-16 acetylation, Proc. Natl. Acad. Sci. U. S. A 109 (2012) 17857–17862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Winogradoff D, Echeverria I, Potoyan DA, Papoian GA, The Acetylation Landscape of the H4 Histone Tail: Disentangling the Interplay between the Specific and Cumulative Effects, J. Am. Chem. Soc 137 (2015) 6245–53. [DOI] [PubMed] [Google Scholar]
- [51].Zhao H, Winogradoff D, Bui M, Dalai Y, Papoian GA, Promiscuous histone mis-assembly is actively prevented by chaperones, J. Am. Chem. Soc 138 (2016) 13207–13218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Sharma S, Ding F, Dokholyan NV, Multiscale modeling of nucleosome dynamics, Biophys. J 92 (2007) 1457–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Voltz K, Trylska J, Calimet N, Smith JC, Langowski J, Unwrapping of nucleosomal DNA ends: a multiscale molecular dynamics study, Biophys. J 102 (2012) 849–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Culkin J, De Bruin L, Tompitak M, Phillips R, Schiessel H, The role of DNA sequence in nucleosome breathing, Eur. Phys. J. E 40 (2017) 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Mitchell JS, Glowacki J, Grandchamp AE, Manning RS, Maddocks JH, Sequence-dependent persistence lengths of DNA, J. Chem. Theory Comput 13 (2017) 1539–1555. [DOI] [PubMed] [Google Scholar]
- [56].Nash JA, Singh A, Li NK, Yingling YG, Characterization of nucleic acid compaction with histone-mimic nanoparticles through all-atom molecular dynamics, ACS Nano 9 (2015) 12374–12382. [DOI] [PubMed] [Google Scholar]
- [57].Vasudevan D, Chua EY, Davey CA, Crystal structures of nucleosome core particles containing the ‘601’ strong positioning sequence, J. Mol. Biol 403 (2010) 1–10. [DOI] [PubMed] [Google Scholar]
- [58].Ferreira H, Somers J, Webster R, Flaus A, Owen-Hughes T, Histone tails and the H3 αN helix regulate nucleosome mobility and stability, Mol. Cell. Biol 27 (2007) 4037–4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Bintu L, Ishibashi T, Dangkulwanich M, Wu Y-YY, Lubkowska L, Kashlev M, et al. , Nucleosomal Elements That Control the Topography of the Barrier to Transcription, Cell 151 (2012) 738–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Shaw DE, Chao JC, R Eastwood M, Gagliardo J, R Grossman J, Ho CR, et al. , Anton, a Special-purpose Machine for Molecular Dynamics Simulation, Comm. ACM 51 (2008) 91. [Google Scholar]
- [61].Yager TD, McMurray CT, Holde KV, Salt-induced release of DNA from nucleosome core particles, Biochemistry 28 (1989) 2271–2281. [DOI] [PubMed] [Google Scholar]
- [62].Gansen A, Valeri A, Hauger F, Felekyan S, Kalinin S, Tóth K, et al. , Nucleosome disassembly intermediates characterized by single-molecule FRET, Proc. Natl. Acad. Sci. U. S. A 106 (2009) 15308–15313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Tims HS, Gurunathan K, Levitus M, Widom J, Dynamics of nucleosome invasion by DNA binding proteins, J. Mol. Biol 411 (2011) 430–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Yoo J, Aksimentiev A, Improved Parametrization of Li+, Na+, K+, and Mg2+ Ions for All-Atom Molecular Dynamics Simulations of Nucleic Acid Systems, J. Phys. Chem. Lett 3 (2012) 45–50. [Google Scholar]
- [65].Polach K, Widom J, Mechanism of protein access to specific DNA sequences in chromatin: a dynamic equilibrium model for gene regulation, J. Mol. Biol 254 (1995) 130–149. [DOI] [PubMed] [Google Scholar]
- [66].Simpson RT, Stafford DW, Structural features of a phased nucleosome core particle, Proc. Natl. Acad. Sci. U. S. A 80 (1983) 51–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Rhodes D, Nucleosome cores reconstituted from poly (dA-dT) and the octamer of histones, Nucleic Acids Res. 6 (1979) 1805–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Simpson RT, Kiinzler P, Chromatin and core particles formed from the inner histones and synthetic polydeoxyribonucleotides of defined sequence, Nucleic Acids Res. 6 (1979) 1387–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Brandani GB, Niina T, Tan C, Takada S, DNA sliding in nucleosomes via twist defect propagation revealed by molecular simulations, Nucleic Acids Res. 46 (2018) 2788–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Baumann CG, Smith SB, Bloomfield VA, Bustamante C, Ionic effects on the elasticity of single DNA molecules, Proc. Natl. Acad. Sci. U. S. A 94 (1997) 6185–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Manning GS, The persistence length of DNA is reached from the persistence length of its null isomer through an internal electrostatic stretching force, Biophys. J 91 (2006) 3607–3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].McMillan LE, Martin AC, Automatically extracting functionally equivalent proteins from SwissProt, BMC Bioinformatics 9 (2008) 418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Hall MA, Shundrovsky A, Bai L, Fulbright RM, Lis JT, Wang MD, High- resolution dynamic mapping of histone-DNA interactions in a nucleosome, Nat. Struct. Mol. Biol 16 (2009) 124–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Kulić I, Schiessel H, Chromatin dynamics: nucleosomes go mobile through twist defects, Phys. Rev. Lett 91 (2003) 148103. [DOI] [PubMed] [Google Scholar]
- [75].Kang H, Yoo J, Sohn B-K, Lee S-W, Lee HS, Ma W, et al. , Sequence-dependent DNA condensation as a driving force of DNA phase separation, Nucleic Acids Res. (2018), Published online, doi: 10.1093/nar/gky639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Locke G, Tolkunov D, Moqtaderi Z, Struhl K, Morozov AV, High-throughput sequencing reveals a simple model of nucleosome energetics, Proc. Natl. Acad. Sci. U. S. A 107 (2010) 20998–21003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Chereji RV, Morozov AV, Ubiquitous nucleosome crowding in the yeast genome, Proc. Natl. Acad. Sci. U. S. A Ill (2014) 5236–5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Kaplan N, Moore IK, Fondufe-Mittendorf Y, Gossett AJ, Tillo D, Field Y, et al. , The DNA-encoded nucleosome organization of a eukaryotic genome, Nature 458 (2009) 362–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Struhl K, Naturally occurring poly (dA-dT) sequences are upstream promoter elements for constitutive transcription in yeast, Proc. Natl. Acad. Sci. U. S. A 82 (1985) 8419–8423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Russell DW, Smith M, Cox D, Williamson VM, Young ET, DNA sequences of two yeast promoter-up mutants, Nature 304 (1983) 652–654. [DOI] [PubMed] [Google Scholar]
- [81].Tjaden G, Coruzzi GM, A novel AT-rich DNA binding protein that combines an HMG I-like DNA binding domain with a putative transcription domain, Plant Cell 6 (1994) 107–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Lindstrom S, Andersson-Svahn H, Overview of single-cell analyses: Microdevices and Applications, Lab Chip 10 (2010) 3363–3372. [DOI] [PubMed] [Google Scholar]
- [83].Perez A, Marchan I, Svozil D, Sponer J, Cheatham TE, Laughton CA, et al. , Refinement of the AMBER Force Field for Nucleic Acids: Improving the Description of α /γ Conformers, Biophys. J 92 (2007) 3817–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Joung IS, Cheatham TE, Determination of Alkali and Halide Monovalent Ion Parameters for Use in Explicitly Solvated Biomolecular Simulations, J. Phys. Chem. B 112 (2008) 9020–9041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML, Comparison of Simple Potential Functions for Simulating Liquid Water, J. Chem. Phys 79 (1983) 926–935. [Google Scholar]
- [86].Yoo J, Aksimentiev A, New tricks for old dogs: improving the accuracy of biomolecular force fields by pair-specific corrections to non-bonded interactions, Phys. Chem. Chem. Phys 20 (2018) 8432–8449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Darden TA, York D, Pedersen L, Particle mesh Ewald: An N log(N) method for Ewald sums in large systems, J. Chem. Phys 98 (1993) 10089–92. [Google Scholar]
- [88].Pall S, Hess B, A flexible algorithm for calculating pair interactions on SIMD architectures, Comput. Phys. Commun 184 (2013) 2641–2650. [Google Scholar]
- [89].Miyamoto S, Kollman PA, SETTLE: An Analytical Version of the SHAKE and RATTLE Algorithm for Rigid Water Molecules, J. Comput. Chem 13 (1992) 952–962. [Google Scholar]
- [90].Hess B, Bekker H, Berendsen HJC, Fraaije JGEM, LINCS: A Linear Constraint Solver for Molecular Simulations, J. Comput. Chem 18 (1997) 1463–72. [Google Scholar]
- [91].Abraham MJ, Murtola T, Schulz R, Pall S, Smith JC, Hess B, et al. , GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers, SoftwareX 1 (2015) 19–25. [Google Scholar]
- [92].Bussi G, Donadio D, Parrinello M, Canonical sampling through velocity rescaling, J. Chem. Phys 126 (2007) 014101. [DOI] [PubMed] [Google Scholar]
- [93].Parrinello M, Rahman A, Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method, J. Appl. Phys 52 (1981) 7182–90. [Google Scholar]
- [94].Nose S, A unified formulation of the constant temperature molecular dynamics methods, J. Chem. Phys 81 (1984) 511–519. [Google Scholar]
- [95].Hoover WG, Canonical Dynamics: Equilibrium Phase-Space Distributions, Phys. Rev. A 31 (1985) 1695–1697. [DOI] [PubMed] [Google Scholar]
- [96].Martyna GJ, Tobias DJ, Klein ML, Constant pressure molecular dynamics algorithms, J. Chem. Phys 101 (1994) 4177–4189. [Google Scholar]
- [97].Shan Y, Klepeis JL, Eastwood MP, Dror RO, Shaw DE, Gaussian split Ewald: A fast Ewald mesh method for molecular simulation, J. Chem. Phys 122 (2005) 054101. [DOI] [PubMed] [Google Scholar]
- [98].Yoo J, Aksimentiev A, Improved Parameterization of Amine-Carboxylate and Amine-Phosphate Interactions for Molecular Dynamics Simulations Using the CHARMM and AMBER Force Fields, J. Chem. Theory Comput 12 (2016) 430–443. [DOI] [PubMed] [Google Scholar]
- [99].Fletcher R, Powell MJ, A rapidly convergent descent method for minimization, Comput. J 6 (1963) 163–168. [Google Scholar]
- [100].Humphrey W, Dalke A, Schulten K, VMD: Visual molecular dynamics, J. Mol. Graphics 14 (1996) 33–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.