

Abstract
Cadmium (Cd) causes acute and chronic lung toxicities at occupational exposure levels, yet the impacts of Cd exposure at low levels through dietary intake remain largely uncharacterized. Health concerns arise because humans do not have an effective Cd elimination mechanism, resulting in a long (10- to 35-y) biological half-life. Previous studies showed increased mitochondrial oxidative stress and cell death by Cd yet the details of mitochondrial alterations by low levels of Cd remain unexplored. In the current study, we examined the impacts of Cd burden at a low environmental level on lung metabolome, redox proteome, and inflammation in mice given Cd at low levels by drinking water (0, 0.2, 0.6 and 2.0 mg Cd/L) for 16 weeks. The results showed that mice accumulated lung Cd comparable to non-smoking humans and showed inflammation in lung by histopathology at 2 mg Cd/L. The results of high resolution metabolomics combined with bioinformatics showed that mice treated with 2 mg Cd/L increased levels of lipids in the lung, accompanied by disruption in mitochondrial energy metabolism. In addition, targeted metabolomic analysis showed that these mice had increased accumulation of mitochondrial carnitine and citric acid cycle intermediates. The results of redox proteomics showed that Cd at 2 mg/L stimulated oxidation of isocitrate dehydrogenase, malate dehydrogenase and ATP synthase. Taken together, the results showed impaired mitochondrial function and accumulation of lipids in the lung with a Cd dose response relevant to non-smokers without occupational exposures. These findings suggest that dietary Cd intake could be an important variable contributing to human pulmonary disorders.
Keywords: environmental toxicant, HRM, metal, MWAS, redox proteomics
Graphical Abstract:
1. Introduction
Cadmium (Cd) is a naturally occurring toxicant that causes multiple adverse health effects. High occupational Cd exposure can lead to multiple lung diseases like lung cancer, chronic obstructive pulmonary disease (COPD), and emphysema [1, 2]. In the general population, higher Cd levels were associated with lower lung function in current and former smokers [3, 4] and associated with COPD in male non-smokers [5]. However, less is known about the impact of low-level chronic Cd exposure in non-smokers, mostly obtained from dietary intake. Humans are exposed to Cd from diet at an intake level of approximately 19 – 21 μg per day [6] and have no effective mechanisms for Cd elimination [7, 8]. Thus, chronic exposure to low doses of Cd mainly via food consumption results in a progressive increase of Cd in human organs with age. In our recent studies using a mouse model, Cd burden at a level found in lungs of non-smokers showed enhanced severity of H1N1 influenza virus infection-induced lung inflammation [9]. These findings show the importance of elucidating effects of Cd at low dietary levels on lung metabolism and inflammation.
Early studies established that Cd exposure in the micromolar range disturbs mitochondrial respiratory functions and energy production in various cell types [10–16]. Additionally, Cd-induced apoptosis was attributed to selectively induced oxidation of thioredoxin (Trx) preferentially affecting mitochondrial Trx2 [17]. More recent studies on the redox proteome of human and mouse lung cells showed that low level Cd exposure (≤ 1 μM) caused oxidation of thiol/disulfide redox systems and protein redox states, resulting in disruption of the actin-cytoskeleton and stimulation of pro-inflammatory signaling [18, 19]. Research in liver mitochondria showed that Cd affected Cys and metabolites in branched chain amino acid and carnitine pathways [20]. These results underscore the importance of mitochondrial protein redox and metabolic function as potential targets for toxicity of low Cd exposure.
Accumulating evidence shows that perturbed mitochondrial signatures and function underlie the pathogenesis of numerous lung diseases [21]. In particular, mitochondrial function in type II alveolar epithelial cells (AECs) is critical to produce acetyl-CoA for de novo fatty acid (FA) synthesis, supporting the generation of phospholipid-rich surfactant. Metabolic alterations in FA synthesis and phospholipid production reflects lung mitochondrial stress, and disruption has profound pathogenic effects in deterioration of lung function and host defense against pathogens [21–25]. In fact, several mitochondrial derived damage-associated molecular patterns (DAMPs) cause pathological inflammation and irreversible lung injury, one of which is mitochondrial cardiolipins [25–27].
High-resolution metabolomics (HRM) measures hundreds of confirmed metabolites and, combined with bioinformatics and pathway enrichment analysis [28–33], simultaneously tests for changes in more than 100 metabolic pathways, thereby providing a global analysis of metabolic alterations at the molecular level [34]. Combination of HRM with redox proteomics, which identifies oxidatively modified proteins [35, 36], enables an integrated systems approach to investigate complex mechanisms of biological dysfunction during oxidative stress [20]. In the present study, we used HRM and redox proteomics of mouse lung to determine the pulmonary metabolic effects of Cd at low dose levels relevant to non-smoking humans without occupational exposures. The design included four low-dose groups of mice (0, 0.2, 0.6 and 2.0 mg Cd/L drinking water, 16 weeks), and Inductively-Coupled Plasma Mass Spectrometry (ICP-MS) to measure Cd content in lungs. The results showed that low level Cd disrupted mitochondrial metabolism, with the highest dose causing measurable lung inflammation.
2. Methods
2.1. Animals
Animal protocols were approved by the Institutional Animal Care and Use Committee at Emory University. Eight-week-old male C57BL/6J mice were purchased from Jackson Laboratory and housed under conventional conditions of 21 – 24 °C and 12 h light/dark cycle with commercial standard mouse diet (Laboratory Rodent Diet 5001, LabDiet, St. Louis, MO) containing 0.062 ppm Cd [37]. Thirty-two mice were randomly divided (n = 8 per group) and assigned to vehicle control [provided with purified water by reverse osmosis system, no detectable Cd (<0.0008 ppb)] and three Cd treatment groups exposed to 0.2, 0.6 and 2 mg/L Cd (in form of CdCl2, Sigma-Aldrich) in drinking water. For redox proteomic analysis, additional mice (n=3/group) were exposed to 2 mg/L Cd or vehicle control for 16 weeks. All mice were provided with mouse diet and water ad libitum and maintained for 16 weeks; subsets were used for sample collections for different assays as described below. Mice were group housed, and individual food or water consumption was not determined.
2.2. Histopathology
Mouse lungs (n = 4 per group) collected at the end of exposure were fixed with 10% formalin in phosphate buffered saline. After fixation, the tissues were transferred to 70% alcohol and processed using a Shandon Excelsior AS (Thermo Fisher Scientific) and a standard program of dehydration [38]. Finally, the tissue was soaked in paraffin for 2×30 min and then in the second paraffin bath for at least 1 h. Left lung tissue sections were stained with hematoxylin and eosin (H&E) to assess histopathological changes [38, 39]. Images were acquired with an Axiovert 100 (Zeiss, Oberkochen, Germany) at 100 × magnification. At least eight sections per mouse were obtained for histopathologic analysis. For numerical assessment of inflammation, vessels and interstitial space were scored on a scale of 0 to 3 by blinded observers with the severity scoring system as previously described [38, 39].
2.3. Quantification of Cd
114Cd were measured in mouse lung (n = 4 per group) with inductively-coupled plasma mass spectrometry (ICP-MS, iCap Q, ThermoFisher Scientific) following procedures to measure trace metals that conformed to accuracy (100 ± 10%) and precision standards (RSD < 12%) [40]. Briefly, 50 mg lung samples were homogenized in 500 μL purified water and digested in 2% nitric acid to a final volume of 10 mL.
2.4. High resolution metabolomics (HRM)
Metabolic extracts were analyzed as described previously [37, 40–42]. Mouse lung samples (n = 4 per group) were extracted at 0° C by homogenization in acetonitrile:water (2:1) containing a mixture of stable isotope-labeled internal standards [29]. The acetonitrile:water mixture was added to a weighed tissue sample (approximately 20 mg tissue) at a ratio of 15 μL/mg tissue. Following homogenization and incubation on ice for 30 min, extracts were centrifuged to remove protein [37, 40–42], randomized, and 10 μL aliquots were analyzed with three technical replicates using C18 chromatography or hydrophilic interaction liquid chromatography (HILIC) and mass spectrometry (85 – 1200 m/z) with positive electrospray ionization (ESI) on LTQ-Velos Orbitrap mass spectrometer (Thermo Fisher) [29, 30]. Additional HRM data was achieved using C18 chromatography with negative ESI on Q-Exactive HF mass spectrometer (Thermo Fisher) (85 – 1200 m/z) [43].
Mass spectrometry data were extracted using apLCMS [44] and xMSanalyzer [45]. Data were pre-filtered to retain only features with non-zero values in > 80% in all samples and >50% in each group. Linear regression model against the lung Cd content was used to select metabolic features associated with increasing Cd dose in metabolome-wide-association study (MWAS). One-way analysis of variance (ANOVA) using limma test was applied to compare metabolites among each condition. Significant features (raw P < 0.05 by limma test) were further studied by pathway enrichment analyses using mummichog [46]. This approach protects against type 2 statistical error by including all features at P < 0.05 and protects against type 1 statistical error by permutation testing in pathway enrichment analysis [47]. Hierarchical cluster analysis (HCA) and partial least square discriminant analysis (PLS-DA) were used for untargeted comparison of the significant features associated with Cd (raw P < 0.05 by limma test) across all groups.
2.5. Metabolite annotation
Metabolic features were annotated with xMSannotator [32]; confidence scores for annotation by xMSannotator are derived from a multistage clustering algorithm. Identities of selected metabolites were confirmed by co-elution and ion dissociation mass spectrometry (MS/MS) relative to authentic standards (Level 1 identification by criteria of Schymanski et al [48] or by MS/MS relative to METLIN spectral database [49] (Level 2 identification by criteria of Schymanski et al [48]). Additional annotations were made conservatively, i.e., those with high or medium confidence scores (≥ 2) with M+H adduct detected in positive mode or M-H detected in negative mode. Lower confidence annotations (Level 5 identification by criteria of Schymanski et al [48]) were made using HMDB (Human Metabolome Database [50]) and Metlin [51] mass spectrometry databases at 5 ppm tolerance.
2.6. Redox proteomic analysis
Redox proteomics was performed using redox isotope coded affinity tag (ICAT)-based mass spectrometry [20, 52, 53]. Briefly, freshly harvested lung tissue from mice exposed to Cd or vehicle control (n = 3/group) were homogenized in ice-cold 10% TCA. Protein precipitate was washed with ice-cold acetone, resuspended in denaturing buffer (50 mM Tris, 0.1% SDS, pH 8.5). Reduced cysteine (Cys) of proteins were labeled with the biotin-conjugated thiol reagent (Heavy isotopic [H-ICAT]). Proteins were then reprecipitated by ice-cold 10% TCA for 30 min on ice, pelleted, washed with acetone, and resuspended in denaturing buffer. Minimally oxidized (sulfenic acid) and disulfides in the proteins were then reduced by TCEP [tris-(2-carboxyethyl phosphine)] and labeled with another biotin-conjugated thiol reagent (Light isotopic [L-ICAT]). Samples including both H and L-ICAT-labeled Cys residues in proteins were digested with trypsin overnight, fractionated by cationic exchange followed by avidin purification, and analyzed by mass spectrometer as described below. ICAT-labeled Cys containing peptides (peptidyl Cys) were identified with an H to L ratio as a measure of the redox (reduced/oxidized) state of the protein, expressed as “% oxidation of protein”.
ICAT-labeled Cys peptides were analyzed by reverse-phase LC-MS/MS [18, 20, 54] using a LTQ-Orbitrap ion trap mass spectrometer (Thermo) (300–1600 m/z). The acquired MS/MS spectra were searched against a concatenated target-decoy mouse reference database of the National Center for Biotechnology Information using the SEQUEST Sorcerer algorithm [55]. The peptides were classified by charge state and tryptic state (fully and partial) and first filtered by mass accuracy (10 ppm for high-resolution MS) and then dynamically by increasing XCorr and Cn values to reduce protein false discovery rate (FDR) to less than 1%, according to the target-decoy strategy [56].
2.7. Enzyme activity assay
Fresh mitochondria were isolated from mouse lung homogenate via differential centrifugation in isolation buffer as described earlier [20]. Mitochondrial preparations (n = 5 per treatment) were incubated with control (0 μM Cd) or CdCl2 (0.001, 0.01 and 1 μM) in incubation buffer (220 mM mannitol, 2 mM HEPES, pH 7.0) for 20 min at room temperature. Excess Cd was then removed by centrifugation at 10,000 g for 1 min. Pelleted mitochondria were resuspended in assay buffer provided by the enzyme activity kit (Sigma-Aldrich, St. Louis, MO). Activity of malate dehydrogenase and isocitrate dehydrogenase was measured following manufacturer’s instructions and normalized to total protein content.
2.8. Statistics
Data are presented as the mean and standard error of the mean. Student’s t test (two-tailed with Welch’s correction for unequal variance) was used to test statistical significance between conditions. Linear regression was used to test the linearity of dose-response curve using SigmaPlot 13.0 (Systat Software, Inc.). Pathway enrichment analysis was performed in mummichog version 2.0 [46]; all other bioinformatics analysis were performed in RStudio version 1.1.447 (RStudio, Inc.). The significance level was P < 0.05 for all tests; Benjamini and Hochberg false discovery rate was used for multiple comparisons [57].
3. Results
3.1. Dose-dependent lung inflammation and Cd burden induced by dietary levels of Cd
Histologic evaluation of mouse lungs after 16 weeks of exposure to three low doses of Cd (0.2, 0.6 and 2.0 mg/L) showed an elevated inflammation, characterized by infiltration of inflammatory cells in interstitium, and around airway and blood vessels (Fig. 1A). Quantification of infiltration-positive area [39] showed a dose-dependent inflammation (linearity of dose-response relationship tested by regression, P < 0.05) (Fig. 1B). This Cd effect is consistent with previous observations showing low-dose Cd causes pulmonary inflammation and worsens infection-induced lung histopathlogy [58].
Fig. 1. Low-level Cd burden elevated inflammation in mouse lung histopathology.
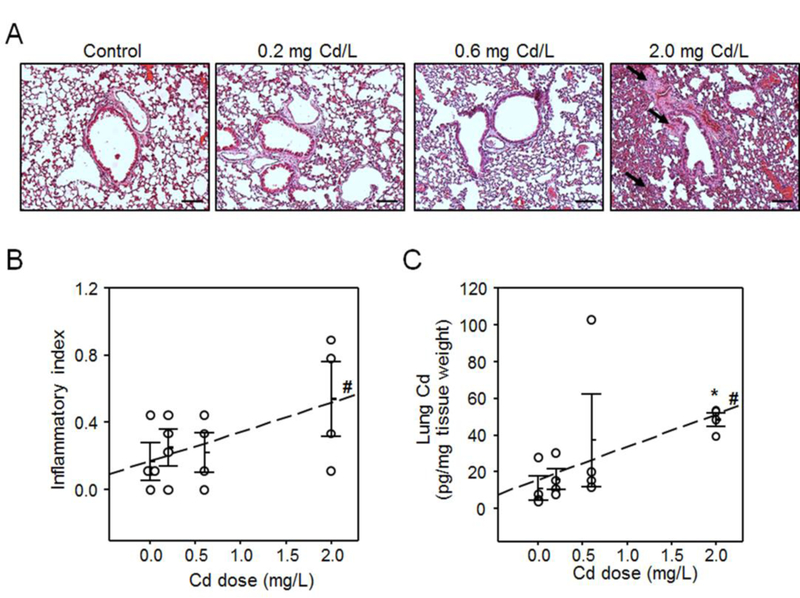
Collected lung tissues from individual mice (n = 4) from all four groups exposed to 0 (control), 0.2, 0.6 and 2 mg/L Cd in drinking water were examined for histopathology after staining with hematoxylin and eosin (H&E, A) and quantified (B) by cell infiltration around vessels and in interstitial spaces, and quantified Cd levels using ICP-MS (C). Arrows indicate representative inflamed airways, blood vessel and interstitium. Scale bars indicate 100 μm. Results are presented as mean ± SE. Asterisk indicates significant increase compared to control (*P < 0.05) and “#” indicates linearity of dose-response relationship tested by regression (#P < 0.05).
Cd content in mouse lung tissue was measured after exposure to the aforementioned doses of Cd. The results showed a dose-dependent increase of lung Cd burden (linearity of dose-response relationship tested by regression, P < 0.05) (Fig. 1C). The lung content in mice treated with 2.0 mg Cd /L was significantly higher (48 ± 3 pg Cd/mg tissue, Fig. 2A bottom) compared to control (11 ± 6 pg Cd/mg tissue). At this dose, the lung content in the Cd treated mice was in the same range as found for human lungs [37]. At the two lower doses (0.2 and 0.6 mg/L), the lung Cd content showed a dose-dependent trend (0.05 < P < 0.1) yet did not reach statistical significance (Fig. 2A bottom) even though our previous studies with these doses with larger n showed significance [37, 42].
Fig. 2. Metabolome-wide-association study (MWAS) of lung Cd content showed affected metabolome in mouse lung.
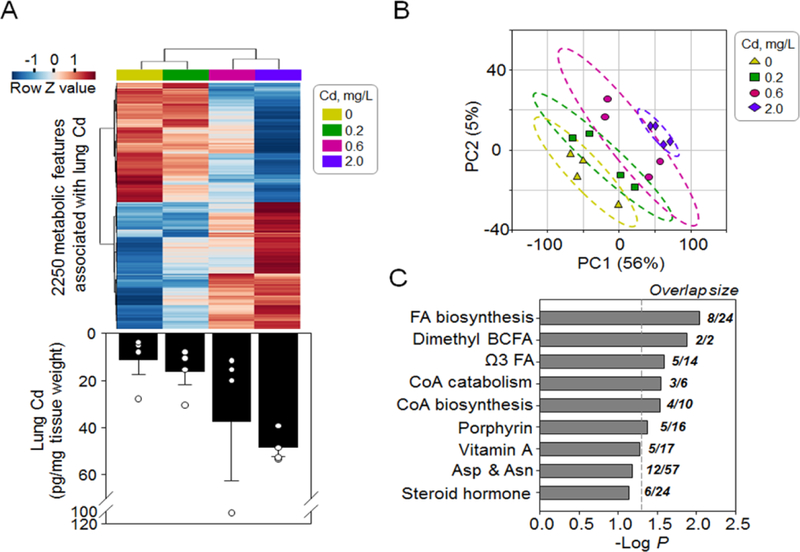
Normalized average levels of 2,250 metabolites (A, top) associated with (P < 0.05, linear regression) increased lung Cd content (A, bottom, a bar graph replotted from Fig 1C) were separated in a dose-dependent manner by hierarchical cluster analysis (A) and partial least square discriminant analysis (B). Pathway enrichment analysis (C) showed affected metabolic pathways enriched by the 2,250 metabolites. Asterisk indicates difference from control tested by student’s t test; * P < 0.05.
3.2. Metabolome wide association study (MWAS) of lung Cd burden showed enrichment in lipid metabolism
To study the global metabolic response to Cd within this low-dose range, we performed a metabolome wide association study (MWAS) of the untargeted HRM data from lung [37, 41] with the measured lung Cd content after 16 weeks of the experimental period. The results showed that out of the 15,975 metabolic features detected with the HILIC method in positive mode [37, 41], 2,250 features were associated with the lung Cd (P < 0.05, Fig. 2A top). Hierarchical cluster analysis (HCA) showed associations of metabolite levels and Cd doses (Fig. 2A top). Using the 2,250 features, the experimental groups were separated following a dose-dependent trajectory, with the highest dose (2.0 mg Cd/L) being most distinct from control (Fig. 2B). Pathway enrichment analysis with mummichog [46] showed that the 2,250 metabolic features were enriched in metabolic pathways related to fatty acid and lipid homeostasis, including fatty acid biosynthesis and metabolism, coenzyme A (CoA) biosynthesis and catabolism, and porphyrin metabolism (Fig. 2C, with details in Supplemental Table S1). This result suggested that low doses of Cd reflecting human dietary intake levels induced a metabolic alteration in pulmonary lipid homeostasis.
3.3. Low dose Cd causes increased lipid levels in mouse lung
We further examined the metabolic effects at each dose level by performing a one-way ANOVA and post hoc comparison. HCA showed that the 2.0 mg Cd/L dose induced a strong metabolic effect that was distinct from the control animals and the two lower doses (Supplemental Fig. 1A). Consistently, the 2 mg/L dose induced differences in a much greater number of metabolites (2,415 features, P < 0.05 post hoc vs control) than the other two doses (0.2 mg/L: 483 features; 0.6 mg/L: 715 features). Nevertheless, there was a substantial overlap of metabolic features among all three doses (Supplemental Fig. 1B).
We thus focused on the effects of the 2 mg/L dose. In addition to the HRM data from the HILIC method in positive ESI mode (HILIC+), we combined the data from the C18 method in both positive and negative ESI mode (C18+ and C18−) to obtain an enhanced coverage of metabolites [43, 59]. A number of metabolic pathways involving lipid metabolism were enriched by the Cd-affected HILIC+ metabolites (Fig. 3A, 2,415 out of 14,581 features, post hoc vs control) and C18+ metabolites (data not shown), while mitochondrial energy metabolism including fatty acid metabolism and citric acid cycle were enriched by the C18− metabolites (947 out of 6,503 features, post hoc vs control) (Fig. 3A).
Fig. 3. Low-dose Cd caused metabolic alterations in energy metabolism pathways and lipid abundance.
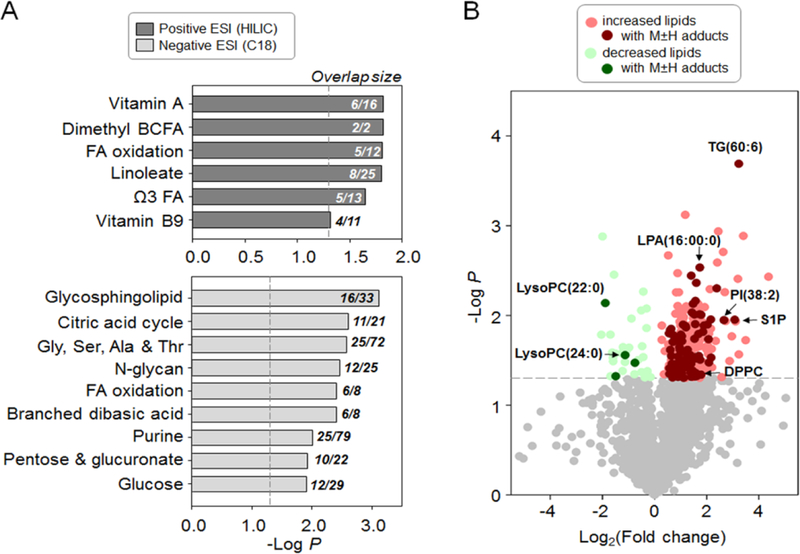
HRM data (detected by both positive and negative electron spray ionization [ESI]) were analyzed for alterations of metabolites (2 mg/L Cd vs control, limma test, P < 0.05) and metabolic pathway enrichment (A). Annotation of the HRM data using xMSanalyzer showed levels of 304 lipid were changed (B, colored circles, P < 0.05). Darker shade of colors indicates high-confident annotation with M+H or M−H adduct detected in positive or negative ESI, respectively. TG: triglyceride; LysoPC: lysophosphatidylcholine; LPA: lysophosphatidic acid; PI: phosphatidylinositol; DPPC: dipalmitoylphosphatidylcholine; S1P: sphingosine-1-phosphate.
We further annotated all the HRM data with xMSannotator, which assigns database matches into different confidence levels of annotation based on an isotope clustering algorithm [32]. By searching against HMDB (human metabolome database) [50] for matches of accurate mass m/z, we annotated 1,750 lipid species and found that 304 of them (Supplemental Table S2) were changed in abundance by Cd treatment at the 2.0 mg/L dose (P < 0.05). The large majority (n = 271) of the 304 lipids were increased, with a fold-change ranging from 1.2 to 20.7 relative to control (Fig. 3B). We conservatively annotated high confidence lipids (see Methods) including triglyceride, dipalmitoylphosphatidylcholine (DPPC), phosphatidylinositol (PI), and various other phospholipids that were increased by Cd (Supplemental Table S2) and different forms of lysoPC that were decreased by Cd (Fig. 3B). Additionally, two precursors of de novo fatty acid biosynthesis including acetyl-CoA and malonyl-CoA, as well as other lipid biosynthesis precursors, glycerol-3-phosphate and lysophosphatidyl acid were consistently elevated by Cd (Supplemental Table S2).
3.4. Low dose Cd caused disruption of mitochondrial metabolome and redox proteome
Following the results that mitochondrial metabolic pathways were changed by Cd (Fig. 2C and 3A), we further analyzed mitochondrial metabolites in a targeted approach. Most importantly, acetyl-CoA was increased 3-fold in the lung after 2 mg/L Cd exposure (P < 0.05, Fig. 4A). The increased abundance of metabolites followed a consistent trajectory at lower Cd doses (Fig 4) but was significant only at 2 mg/L Cd (Fig 1C and Fig 2A lower panel), Similar elevation at 2 mg/L Cd was found with other components of mitochondrial carnitine shuttle and fatty acid β-oxidation, i.e., CoA, carnitine and acetyl-carnitine (Fig. 4A). Accumulation of the downstream citric acid cycle intermediates also occurred, including succinate, malate, citrate/isocitrate, 2-oxogluatarate (Fig. 4B). Additional mitochondrial metabolites (Supplemental Table S3) showed a broad and dose-dependent impact of Cd on mitochondrial energy producing substrates.
Fig. 4. Low doses of Cd caused dose-dependent increase of metabolites in mitochondrial carnitine shuttle and citric acid cycle.
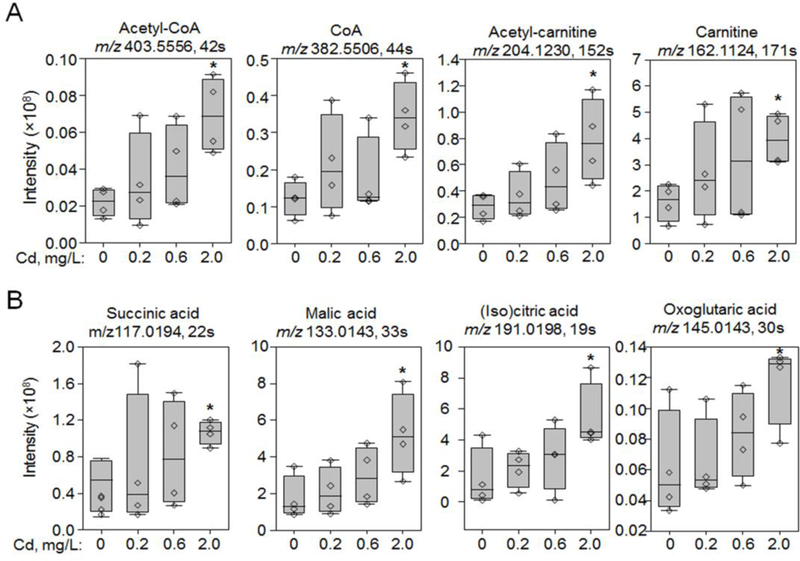
Only the most abundant adduct (M-2H or M−H for negative ESI or M+H for positive ESI) was presented for each metabolite. Acetyl-CoA and CoA were confirmed by MS/MS. Acetyl-carnitine, carnitine, succinic acid, malic acid and (iso)citric acid were confirmed by accurate m/z match and co-elution with authentic standards. Raw peak intensities of representative metabolites in mitochondria carnitine shuttle (A) and citric acid cycle (B) were presented as measurement of abundance in metabolites. Asterisk indicates difference from control tested by student’s t test; * P < 0.05. Additional information is presented in Supplemental Table S3.
Previous research showed Cd effects on oxidation of mitochondrial proteins, so we used redox proteomics to examine whether low-dose Cd caused oxidation of proteins/enzymes in citric acid cycle and ATP production. The results showed that Cd induced oxidation of mitochondrial enzymes catalyzing the citric acid cycle steps and other metabolism processes (Supplemental Table S4). These Cd-oxidized proteins included isocitrate dehydrogenase subunit γ1 (Idh3g), malate dehydrogenase (Mdh2), and a trend of oxidation (P < 0.1) of dihydrolipoamide dehydrogenase (Dld) (Fig. 5A–C). Importantly, ATP synthase subunit γ was oxidized by Cd (P < 0.05) suggesting that Cd disrupted the electron transport chain for ATP synthesis (Fig. 5D). We further confirmed Cd-impaired mitochondrial enzymes by measuring enzyme activity in isolated mitochondria from mouse lung. The activities of isocitrate dehydrogenase (IDH) and malate dehydrogenase (MDH) were inhibited by low doses Cd (Supplemental Figure S2). Results showed that MDH activity was inhibited by 15%, 14% and 31% at 1 nM, 10 nM and 1 μM Cd concentrations, respectively (P < 0.05). Activity of IDH was also inhibited by 7%, 22% and 20% at the same doses (P < 0.05) (Supplemental Figure S2).
Fig. 5. Low dose-Cd increased oxidation of proteins in citric acid cycle.
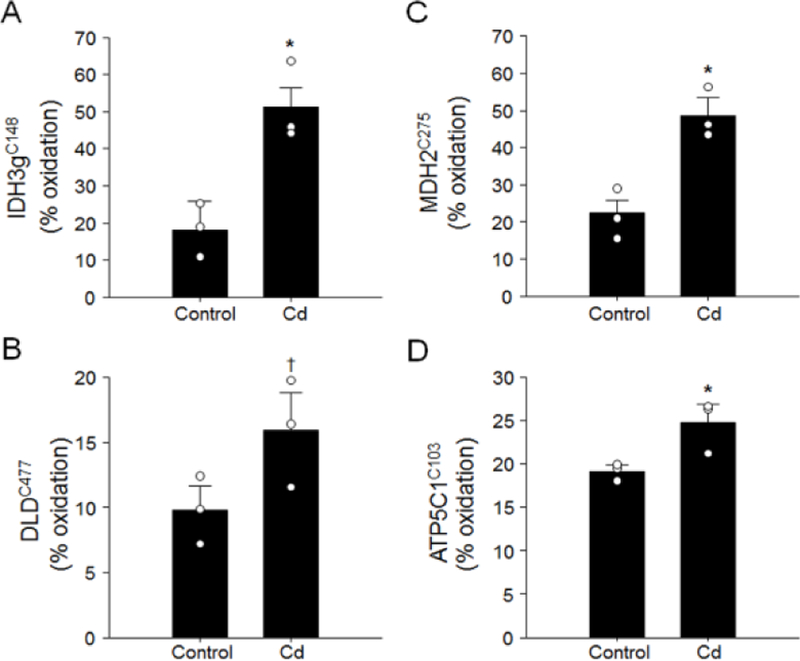
Lung collected from mouse treated with 2.0 mg Cd/L was analyzed for redox proteomics following procedures described previously [18, 20, 52]. Quantification of redox state of protein peptidyl cysteine (Cys) was performed by redox ICAT-based mass spectrometry (ICAT/MS). Results of % oxidation in Cys residue of isocitrate dehydrogenase subunit γ (Idh3g, A), dihydrolipoamide dehydrogenase (Dld, B), malate dehydrogenase (Mdh2, C) and ATP synthase subunit γ (Atp5C1, D) are presented as mean ± SE (* P < 0.05, † P < 0.01).
Cumulatively our results showed that low dose Cd oxidized and impaired activity of mitochondrial enzymes in citric acid cycle and ATP synthesis. Impaired function of enzyme leads to inefficient biochemical reactions that in turn cause the observed accumulation of citric acid cycle intermediates, and most importantly, acetyl-CoA. As a key regulator and precursor for fatty acid biosynthesis, acetyl-CoA facilitates de novo fatty acid biosynthesis and consequently accumulation of lipids. Dysregulation in lipids and other metabolites may impair the surfactant function [60, 61], dysregulate cell immune function [62] and activate inflammatory response [25, 63, 64]. This can ultimately contribute to Cd-induced inflammation (Fig. 6).
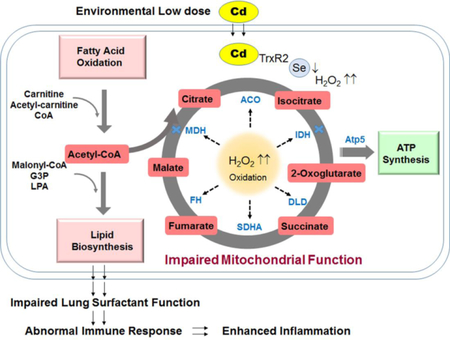
Figure 6. Proposed schematic diagram: Cd-induced mitochondrial dysfunction.
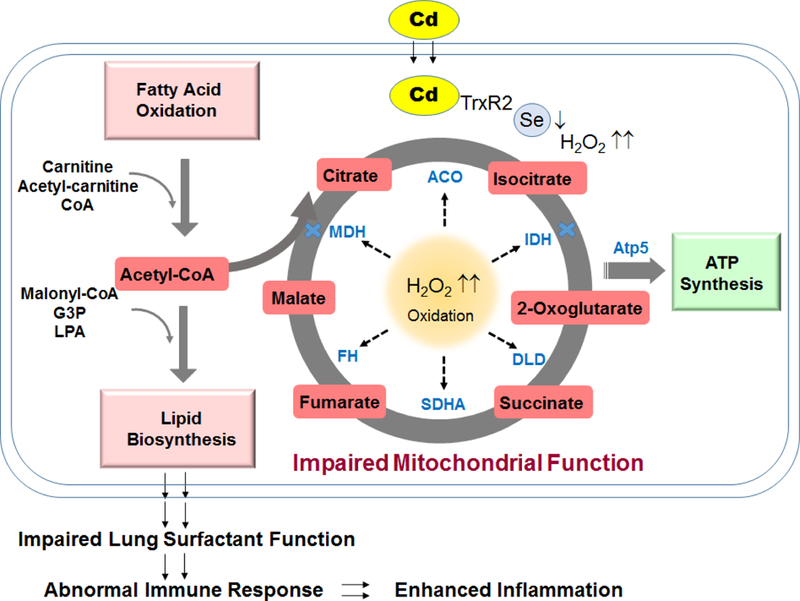
Lung Cd burden from low dose Cd exposures stimulates mitochondrial oxidation and metabolic disruption involving mechanism of increased H2O2 and inactivation of the major cellular redox controlling antioxidant such as selenocysteine- (Sec)-containing TrxR2. Oxidative mitochondrial environment results in oxidation of energy producing enzymes leading to impaired enzyme activity (indicated by blue cross) and inefficient reactions, and consequently accumulation of metabolic intermediates and elevation of lipids. Dysregulation of lipids and metabolites in the lung can contribute to impaired surfactant function and increased lung inflammation and abnormality.
4. Discussion
The present study was to examine impacts on lung metabolism of Cd burden from low-dose Cd exposures similar to human dietary intake. Relatively high-dose Cd from occupational exposures or cigarette smoking is a well-known cause of lung toxicities, yet the role of chronic low level exposure remains understudied. Mouse models to study low-dose Cd are complicated because of the low absorption rates of dietary Cd and 10- to 30-year biological half-life of Cd in humans. In the present studies, the data show that the Cd exposure at 2 mg/L in drinking water resulted in a lung burden of 48 ± 3 pg Cd/mg tissue. The human lung content reported previously by different instrumental methods is in the same range [37, 65], and our analyses of 31 human lung samples (48 ± 14 pg Cd/mg tissue) with the same instrumentation and analytic methods are also in this range. Thus, even though the precise modeling of non-smoking human lung Cd with a mouse model is not possible, the results indicate that the conditions used provide a useful approximation. With these conditions, the results showed that although the strongest effect occurred at 2.0 mg/L dose, the severity of responses and magnitude of alterations followed increasing trend at 0.2, 0.6 and 2 mg/L doses, indicating the presence of consistent effects even at the extremely low doses.
High dose Cd studies show pleiotropic toxicities dependent on the dosage and exposure time [66], with mitochondrial dysfunction appearing as an early and common event [13]. For instance, intraperitoneal injection of high dose Cd (3 mg/kg body weight) inhibited succinate dehydrogenase and cytochrome c oxidase activities in rat kidney, testis and lung mitochondria [15]. In vitro studies of rat hepatocytes (0.5 μM Cd) and isolated liver mitochondria (0.05 μM Cd) showed increased permeability of mitochondria accompanied by decrease in mitochondrial ATP. Mitochondrial dysfunction occurred earlier and at lower Cd dose than stimulation of lipid oxidation [13, 14]. The present research extends these studies to show effects on mitochondrial metabolism at relevant low Cd doses in vivo in mouse lung. Importantly, the results showed a broad impact on the citric acid cycle and other mitochondrial metabolism such as fatty acid oxidation, branched chain amino acid metabolism and porphyrin metabolism. Additional studies will be needed to more specifically address bioenergetics effects, such as effects on mitochondrial membrane potential and ATP levels.
Mechanistic effects of Cd on mitochondrial function have previously been linked to oxidative modifications in proteins. For instance, Cd inhibits thioredoxin reductase (TrxR), and this results in oxidation of Trx and other proteins [67] [68]. Study of subcellular compartmental redox systems additionally showed that mitochondria are more susceptible than cytoplasm and nucleus to oxidative stressors [17, 69] and environmental metals, including Cd [17]. The present research extends these findings to low dose Cd, identifying mitochondrial enzymes that were oxidized by Cd at 2.0 mg/L. Other research showed that a number of enzymes in the citric acid cycle are either sulfenylated or S-glutathionylated, leading to inhibition or inactivation of enzyme activity (reviewed in [70]), including isocitrate dehydrogenase [71], and ATP synthase α subunit [72] [73, 74]. The present research showed that inhibition of IDH and MDH occurred at Cd levels as low as 1 nM with isolated lung mitochondria, a dose 50 times lower than that disrupting energy production [10, 13–15]. Our findings support a sequence in which low dose Cd caused protein oxidation, inhibited activities of mitochondrial proteins, and resulted in associated alterations in lipids and metabolites.
Altered mitochondrial function is increasingly recognized to contribute to lung disease involving inflammation [21, 75] [76]. Mitochondrial proteins and metabolites (including mitochondria-specific cardiolipin as one of mitochondrial DAMPs [77]) interact with and regulate NLRP3 inflammasome and toll-like receptor signaling in lung [25, 78, 79]. The research directly links mitochondrial dysfunction to activation of pro-inflammatory cytokines in pulmonary pathogenesis [64, 80]. Cd also stimulates generation of pro-inflammatory cytokines [81–83] and exacerbates immune responses with increased immune cells and cytokines after lung H1N1 infection [9]. Thus, the data are converging to show that potentiation of lung injury by Cd may involve additive or synergistic effects on mitochondrial dysfunction and pro-inflammatory signaling.
The data also suggest that disruption of surfactant homeostasis could be a component of Cd- toxicity. Increased lipid-laden alveolar macrophages (“foam cells”) are frequently associated with human lung pathologies [84], and rodent inhalation exposures [85, 86] suggest a link between pulmonary pathogenesis and abnormal lipid accumulation induced by environmental stressors. Cd could exacerbate lung injury through these mechanisms, and our previous observations of Cd potentiation of virus-induced lung histopathology and inflammation [58] provide an example where impaired surfactant clearance could determine severity of response to H1N1 influenza virus. Although mechanisms are not clear, lung surfactant contains 90% lipids as the primary contributor to surface tension-reducing function. Type II alveolar epithelial cells rely on high mitochondria activity to produce acetyl-CoA, which is converted to malonyl-CoA in the rate limiting step [87] for synthesis of surfactant lipids [88]. Excess acetyl-CoA activates lipogenesis [89], and in the current study, we found that low dose Cd increased levels of acetyl-CoA, malonyl-CoA and glycerol-3-phosphate. Together with the upregulation of lipid biosynthesis enzymes (e.g. stearoyl-Coenzyme A desaturase and mevalonate kinase, data not shown) and increased lipid levels, the results indicate that Cd-dependent increase in surfactant generation coupled with impaired surfactant removal could potentially have far-reaching effects on inflammatory lung disease [62]. Additional studies are needed to determine whether dysregulation of lipids and other metabolites contribute to Cd-induced inflammation.
Our present results also suggest a potential therapeutic strategy to attenuate the deficiencies in Cd toxicity. Previously we found that co-treatment with equal molar of selenium (Se) prevented Cd-induced toxicity in lung transcriptome and metabolome [42], suggesting a protective role of Se in interaction with Cys residues. While more research on Cd-induced site-specific redox modification and interaction with Se is needed to better understand the molecular mechanisms, the results suggest that Se supplementation may provide an approach to ameliorate low-dose Cd effects on mitochondrial dysfunction.
In summary, Cd in a low-dose range relevant to non-occupational and non-smoking human exposure induced a dose-dependent increase in lung inflammation. MWAS showed that at these doses, Cd increased oxidation of enzymes catalyzing the citric acid cycle and ATP synthesis and caused metabolic dysregulation in fatty acids and lipids. The study shows that integrative metabolomics and redox proteomics is a powerful approach to advance translational research to understand complex biological responses of environmental exposures and disease. Finally, our findings suggest that metabolic alterations could be used for therapeutic development to protect against toxic effects of Cd burden in human lung disease.
Supplementary Material
Highlights.
Low environmental Cd caused lung inflammation in mice
Redox proteomics showed mitochondrial protein oxidation by low environmental Cd
Inhibited mitochondrial enzymes were associated with increased lipids and citric acid cycle intermediates
Combination of histopathology with data-driven omics supports analysis of toxicologic effects at low environmental Cd exposure
Acknowledgements
This study was supported by NIEHS Grants R01 ES023485 (DPJ and YMG), R21 ES025632 (DPJ and YMG), P30 ES019776 (DPJ) and U2C ES026560 (DPJ), and NIH S10 OD018006 (DPJ).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].ATSDR. Toxicological profile for cadmium 2012. [PubMed]
- [2].Hart B Response of the respiratory tract to cadmium 2000.
- [3].Lampe BJ; Park SK; Robins T; Mukherjee B; Litonjua AA; Amarasiriwardena C; Weisskopf M; Sparrow D; Hu H Association between 24-hour urinary cadmium and pulmonary function among community-exposed men: the VA Normative Aging Study. Environmental health perspectives 116:1226; 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Mannino DM; Holguin F; Greves H; Savage-Brown A; Stock A; Jones R Urinary cadmium levels predict lower lung function in current and former smokers: data from the Third National Health and Nutrition Examination Survey. Thorax 59:194–198; 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Oh C-M; Oh I-H; Lee J-K; Park YH; Choe B-K; Yoon T-Y; Choi J-M Blood cadmium levels are associated with a decline in lung function in males. Environmental Research 132:119–125; 2014. [DOI] [PubMed] [Google Scholar]
- [6].EFSA. Cadmium in food. The EFSA Journal 980:1–139; 2009. [Google Scholar]
- [7].Satarug S; Moore MR Adverse health effects of chronic exposure to low-level cadmium in foodstuffs and cigarette smoke. Environ Health Perspect 112:1099–1103; 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Suwazono Y; Kido T; Nakagawa H; Nishijo M; Honda R; Kobayashi E; Dochi M; Nogawa K Biological half-life of cadmium in the urine of inhabitants after cessation of cadmium exposure. Biomarkers 14:77–81; 2009. [DOI] [PubMed] [Google Scholar]
- [9].Chandler JD; Hu X; Ko E; Park S; Fernandes J; Lee Y-T; Orr M; Hao L; Smith MR; Nujahr D; Uppal K; Kang S-M; Jones D; Go Y-M Low-dose cadmium potentiates lung inflammatory response to 2009 pandemic H1N1 influenza virus in mice. bioRxiv; 2018. [DOI] [PMC free article] [PubMed]
- [10].Cross CE; Ibrahim AB; Ahmed M; Mustafa MG Effect of cadmium ion on respiration and ATPase activity of the pulmonary alveolar macrophage: A model for the study of environmental interference with pulmonary cell function. Environmental Research 3:512–520; 1970. [Google Scholar]
- [11].Jacobs EE; Jacob M; Sanadi DR; Bradley LB UNCOUPLING OF OXIDATIVE PHOSPHORYLATION BY CADMIUM ION. Journal of Biological Chemistry 223:147–156; 1956. [PubMed] [Google Scholar]
- [12].Järvisalo JO; Kilpiö J; Saris N-EL Toxicity of cadmium to renal mitochondria when administered in vivo and in vitro. Environmental Research 22:217–223; 1980. [DOI] [PubMed] [Google Scholar]
- [13].Müller L Consequences of cadmium toxicity in rat hepatocytes: Mitochondrial dysfunction and lipid peroxidation. Toxicology 40:285–295; 1986. [DOI] [PubMed] [Google Scholar]
- [14].Müller L; Ohnesorge FK Cadmium-induced alteration of the energy level in isolated hepatocytes. Toxicology 31:297–306; 1984. [DOI] [PubMed] [Google Scholar]
- [15].Prasada Rao PVV Effects of intraperitoneal cadmium administration on mitochondrial enzymes in rat tissues. Toxicology 27:81–87; 1983. [DOI] [PubMed] [Google Scholar]
- [16].Sato N; KAMADA T; SUEMATSU T; ABE H; FURUYAMA F; HAGIHARA B Cadmium toxicity and liver mitochondria: II. Protective effect of hepatic soluble fraction against cadmium-induced mitochondrial dysfunction. The Journal of biochemistry 84:127–133; 1978. [DOI] [PubMed] [Google Scholar]
- [17].Hansen JM; Zhang H; Jones DP Differential oxidation of thioredoxin-1, thioredoxin-2, and glutathione by metal ions*. Free Radical Biology and Medicine 40:138–145; 2006. [DOI] [PubMed] [Google Scholar]
- [18].Go YM; Orr M; Jones DP Actin cytoskeleton redox proteome oxidation by cadmium. Am J Physiol Lung Cell Mol Physiol 305:L831–843; 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Go YM; Orr M; Jones DP Increased nuclear thioredoxin-1 potentiates cadmium-induced cytotoxicity. Toxicol Sci 131:84–94; 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Go Y-M; Roede JR; Orr M; Liang Y; Jones DP Integrated Redox Proteomics and Metabolomics of Mitochondria to Identify Mechanisms of Cd Toxicity. Toxicological Sciences 139:59–73; 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Cloonan SM; Choi AMK Mitochondria in lung disease. The Journal of Clinical Investigation 126:809–820; 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Agarwal AR; Yin F; Cadenas E Short-term cigarette smoke exposure leads to metabolic alterations in lung alveolar cells. Am J Respir Cell Mol Biol 51:284–293; 2014. [DOI] [PubMed] [Google Scholar]
- [23].Otsubo C; Bharathi S; Uppala R; Ilkayeva OR; Wang D; McHugh K; Zou Y; Wang J; Alcorn JF; Zuo YY; Hirschey MD; Goetzman ES Long-chain Acylcarnitines Reduce Lung Function by Inhibiting Pulmonary Surfactant. Journal of Biological Chemistry 290:23897–23904; 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Pattle RE; Schock C; Dirnhuber P; Creasey JM Lamellar transformation of lung mitochondria under conditions of stress. Nature 240:468–469; 1972. [DOI] [PubMed] [Google Scholar]
- [25].Ray NB; Durairaj L; Chen BB; McVerry BJ; Ryan AJ; Donahoe M; Waltenbaugh AK; O’Donnell CP; Henderson FC; Etscheidt CA; McCoy DM; Agassandian M; Hayes-Rowan EC; Coon TA; Butler PL; Gakhar L; Mathur SN; Sieren JC; Tyurina YY; Kagan VE; McLennan G; Mallampalli RK Dynamic regulation of cardiolipin by the lipid pump, ATP8b1, determines the severity of lung injury in experimental bacterial pneumonia. Nature medicine 16:1120–1127; 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Nakahira K; Hisata S; Choi AMK The Roles of Mitochondrial Damage-Associated Molecular Patterns in Diseases. Antioxidants & Redox Signaling 23:1329–1350; 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhang Q; Raoof M; Chen Y; Sumi Y; Sursal T; Junger W; Brohi K; Itagaki K; Hauser CJ Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464:104; 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Go YM; Kim CW; Walker DI; Kang DW; Kumar S; Orr M; Uppal K; Quyyumi AA; Jo H; Jones DP Disturbed flow induces systemic changes in metabolites in mouse plasma: a metabolomics study using ApoE(−)/(−) mice with partial carotid ligation. Am J Physiol Regul Integr Comp Physiol 308:R62–72; 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Go Y-M; Sutliff RL; Chandler JD; Khalidur R; Kang B-Y; Anania FA; Orr M; Hao L; Fowler BA; Jones DP Low-dose cadmium causes metabolic and genetic dysregulation associated with fatty liver disease in mice. Toxicological Sciences 147:524–534; 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Go Y-M; Walker DI; Soltow QA; Uppal K; Wachtman LM; Strobel FH; Pennell K; Promislow DEL; Jones DP Metabolome-wide association study of phenylalanine in plasma of common marmosets. Amino Acids 47:589–601; 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Jones DP; Park Y; Ziegler TR Nutritional metabolomics: progress in addressing complexity in diet and health. Annual review of nutrition 32:183–202; 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Uppal K; Walker DI; Jones DP xMSannotator: An R Package for Network-Based Annotation of High-Resolution Metabolomics Data. Analytical Chemistry 89:1063–1067; 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Walker DI; Go Y-M; Liu K; Pennell KD; Jones DP, Population Screening for Biological and Environmental Properties of the Human Metabolic Phenotype: Implications for Personalized Medicine Elsevier.; 2016. [Google Scholar]
- [34].Karczewski KJ; Snyder MP Integrative omics for health and disease. Nature Reviews Genetics 19:299; 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Butterfield DA; Perluigi M Redox Proteomics: A Key Tool for New Insights into Protein Modification with Relevance to Disease. Antioxidants & Redox Signaling 26:277–279; 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Go Y-M; Jones DP The Redox Proteome. Journal of Biological Chemistry 288:26512–26520; 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Chandler JD; Wongtrakool C; Banton SA; Li S; Orr ML; Barr DB; Neujahr DC; Sutliff RL; Go YM; Jones DP Low‐dose oral cadmium increases airway reactivity and lung neuronal gene expression in mice. Physiological reports 4:e12821; 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ko E-J; Lee Y-T; Kim K-H; Jung Y-J; Lee Y; Denning TL; Kang S-M Effects of MF59 Adjuvant on Induction of Isotype-Switched IgG Antibodies and Protection after Immunization with T-Dependent Influenza Virus Vaccine in the Absence of CD4+ T Cells. Journal of Virology 90:6976–6988; 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hwang HS; Kwon Y-M; Lee JS; Yoo S-E; Lee Y-N; Ko E-J; Kim M-C; Cho M-K; Lee Y-T; Jung Y-J; Lee J-Y; Li J-D; Kang S-M Co-immunization with virus-like particle and DNA vaccines induces protection against respiratory syncytial virus infection and bronchiolitis. Antiviral Research 110:115–123; 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hu X; Chandler JD; Orr ML; Hao L; Liu K; Uppal K; Go Y-M; Jones DP Selenium Supplementation Alters Hepatic Energy and Fatty Acid Metabolism in Mice. The Journal of Nutrition 148:675–684; 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Chandler JD; Hu X; Ko E-J; Park S; Lee Y-T; Orr M; Fernandes J; Uppal K; Kang S-M; Jones DP; Go Y-M Metabolic pathways of lung inflammation revealed by high-resolution metabolomics (HRM) of H1N1 influenza virus infection in mice. American Journal of Physiology - Regulatory, Integrative and Comparative Physiology 311:R906–R916; 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hu X; Chandler JD; Fernandes J; Orr ML; Hao L; Uppal K; Neujahr DC; Jones DP; Go Y-M Selenium supplementation prevents metabolic and transcriptomic responses to cadmium in mouse lung. Biochimica et Biophysica Acta (BBA) - General Subjects; 2018. [DOI] [PMC free article] [PubMed]
- [43].Liu KH; Walker DI; Uppal K; Tran V; Rohrbeck P; Mallon TM; Jones DP High-resolution metabolomics assessment of military personnel: Evaluating analytical strategies for chemical detection. Journal of occupational and environmental medicine / American College of Occupational and Environmental Medicine 58:S53–S61; 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Yu T; Park Y; Johnson JM; Jones DP apLCMS—adaptive processing of high-resolution LC/MS data. Bioinformatics 25:1930–1936; 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Uppal K; Soltow QA; Strobel FH; Pittard WS; Gernert KM; Yu T; Jones DP xMSanalyzer: automated pipeline for improved feature detection and downstream analysis of large-scale, non-targeted metabolomics data. BMC Bioinformatics 14:15; 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Li S; Park Y; Duraisingham S; Strobel FH; Khan N; Soltow QA; Jones DP; Pulendran B Predicting Network Activity from High Throughput Metabolomics. PLOS Computational Biology 9:e1003123; 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Uppal K; Walker DI; Liu K; Li S; Go YM; Jones DP Computational Metabolomics: A Framework for the Million Metabolome. Chem Res Toxicol 29:1956–1975; 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Schymanski EL; Jeon J; Gulde R; Fenner K; Ruff M; Singer HP; Hollender J Identifying small molecules via high resolution mass spectrometry: communicating confidence. Environ Sci Technol 48:2097–2098; 2014. [DOI] [PubMed] [Google Scholar]
- [49]. http://metlin.scripps.edu/index.php.
- [50].Wishart DS; Tzur D; Knox C; Eisner R; Guo AC; Young N; Cheng D; Jewell K; Arndt D; Sawhney S; Fung C; Nikolai L; Lewis M; Coutouly MA; Forsythe I; Tang P; Shrivastava S; Jeroncic K; Stothard P; Amegbey G; Block D; Hau DD; Wagner J; Miniaci J; Clements M; Gebremedhin M; Guo N; Zhang Y; Duggan GE; Macinnis GD; Weljie AM; Dowlatabadi R; Bamforth F; Clive D; Greiner R; Li L; Marrie T; Sykes BD; Vogel HJ; Querengesser L HMDB: the Human Metabolome Database. Nucleic Acids Res 35:D521–526; 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Guijas C; Montenegro-Burke JR; Domingo-Almenara X; Palermo A; Warth B; Hermann G; Koellensperger G; Huan T; Uritboonthai W; Aisporna AE; Wolan DW; Spilker ME; Benton HP; Siuzdak G METLIN: A Technology Platform for Identifying Knowns and Unknowns. Analytical Chemistry 90:3156–3164; 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Go YM; Park H; Koval M; Orr M; Reed M; Liang Y; Smith D; Pohl J; Jones DP A key role for mitochondria in endothelial signaling by plasma cysteine/cystine redox potential. Free Radic Biol Med 48:275–283; 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Go Y-M; Roede JR; Walker DI; Duong DM; Seyfried NT; Orr M; Liang Y; Pennell KD; Jones DP Selective Targeting of the Cysteine Proteome by Thioredoxin and Glutathione Redox Systems. Molecular & Cellular Proteomics : MCP 12:3285–3296; 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Xu P; Duong DM; Peng J Systematical optimization of reverse-phase chromatography for shotgun proteomics. J Proteome Res 8:3944–3950; 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Eng JK; McCormack AL; Yates JR An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom 5:976–989; 1994. [DOI] [PubMed] [Google Scholar]
- [56].Elias JE; Gygi SP Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods 4:207–214; 2007. [DOI] [PubMed] [Google Scholar]
- [57].Benjamini Y; Hochberg Y Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society. Series B (Methodological) 57:289–300; 1995. [Google Scholar]
- [58].Chandler JD; Hu X; Ko E; Park S; Jolyn Fernandes J; Lee Y-T; Orr ML; Hao L; Smith MR; Neujahr DC; Uppal K; Kang S-M; Jones DP; Go Y-M Low-dose cadmium potentiates lung inflammatory response to 2009 pandemic H1N1 influenza virus in mice. bioRxiv 346866; 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Contrepois K; Jiang L; Snyder M Optimized Analytical Procedures for the Untargeted Metabolomic Profiling of Human Urine and Plasma by Combining Hydrophilic Interaction (HILIC) and Reverse-Phase Liquid Chromatography (RPLC)–Mass Spectrometry. Molecular & Cellular Proteomics : MCP 14:1684–1695; 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Vockeroth D; Gunasekara L; Amrein M; Possmayer F; Lewis JF; Veldhuizen RA Role of cholesterol in the biophysical dysfunction of surfactant in ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol 298:L117–125; 2010. [DOI] [PubMed] [Google Scholar]
- [61].Hiansen JQ; Keating E; Aspros A; Yao LJ; Bosma KJ; Yamashita CM; Lewis JF; Veldhuizen RA Cholesterol-mediated surfactant dysfunction is mitigated by surfactant protein A. Biochim Biophys Acta 1848:813–820; 2015. [DOI] [PubMed] [Google Scholar]
- [62].Fessler MB; Summer RS Surfactant Lipids at the Host–Environment Interface. Metabolic Sensors, Suppressors, and Effectors of Inflammatory Lung Disease. American Journal of Respiratory Cell and Molecular Biology 54:624–635; 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Iyer SS; He Q; Janczy JR; Elliott EI; Zhong Z; Olivier AK; Sadler JJ; Knepper-Adrian V; Han R; Qiao L; Eisenbarth SC; Nauseef WM; Cassel SL; Sutterwala FS Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39:311–323; 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Chen BB; Coon TA; Glasser JR; Zou C; Ellis B; Das T; McKelvey AC; Rajbhandari S; Lear T; Kamga C; Shiva S; Li C; Pilewski JM; Callio J; Chu CT; Ray A; Ray P; Tyurina YY; Kagan VE; Mallampalli RK E3 ligase subunit Fbxo15 and PINK1 kinase regulate cardiolipin synthase 1 stability and mitochondrial function in pneumonia. Cell Rep 7:476–487; 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Mari M; Nadal M; Schuhmacher M; Barbería E; García F; Domingo JL Human Exposure to Metals: Levels in Autopsy Tissues of Individuals Living Near a Hazardous Waste Incinerator. Biological Trace Element Research 159:15–21; 2014. [DOI] [PubMed] [Google Scholar]
- [66].Thevenod F Cadmium and cellular signaling cascades: to be or not to be? Toxicol Appl Pharmacol 238:221–239; 2009. [DOI] [PubMed] [Google Scholar]
- [67].Chrestensen CA; Starke DW; Mieyal JJ Acute cadmium exposure inactivates thioltransferase (Glutaredoxin), inhibits intracellular reduction of protein-glutathionyl-mixed disulfides, and initiates apoptosis. The Journal of biological chemistry 275:26556–26565; 2000. [DOI] [PubMed] [Google Scholar]
- [68].Lee SR; Bar-Noy S; Kwon J; Levine RL; Stadtman TC; Rhee SG Mammalian thioredoxin reductase: oxidation of the C-terminal cysteine/selenocysteine active site forms a thioselenide, and replacement of selenium with sulfur markedly reduces catalytic activity. Proc Natl Acad Sci U S A 97:2521–2526; 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Go YM; Jones DP Redox compartmentalization in eukaryotic cells. Biochim Biophys Acta 1780:1273–1290; 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Mailloux RJ; Jin X; Willmore WG Redox regulation of mitochondrial function with emphasis on cysteine oxidation reactions. Redox Biology 2:123–139; 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Kil IS; Park J-W Regulation of Mitochondrial NADP+-dependent Isocitrate Dehydrogenase Activity by Glutathionylation. Journal of Biological Chemistry 280:10846–10854; 2005. [DOI] [PubMed] [Google Scholar]
- [72].Gibbons C; Montgomery MG; Leslie AG; Walker JE The structure of the central stalk in bovine F(1)-ATPase at 2.4 A resolution. Nat Struct Biol 7:1055–1061; 2000. [DOI] [PubMed] [Google Scholar]
- [73].Garcia J; Han D; Sancheti H; Yap LP; Kaplowitz N; Cadenas E Regulation of mitochondrial glutathione redox status and protein glutathionylation by respiratory substrates. J Biol Chem 285:39646–39654; 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Wang S-B; Murray CI; Chung HS; Van Eyk JE Redox-regulation of mitochondrial ATP synthase. Trends in cardiovascular medicine 23:14–18; 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Piantadosi CA; Suliman HB Mitochondrial Dysfunction in Lung Pathogenesis. Annual Review of Physiology 79:495–515; 2017. [DOI] [PubMed] [Google Scholar]
- [76].Carraway MS; Suliman HB; Kliment C; Welty-Wolf KE; Oury TD; Piantadosi CA Mitochondrial Biogenesis in the Pulmonary Vasculature During Inhalational Lung Injury and Fibrosis. Antioxidants & Redox Signaling 10:269–276; 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Dela Cruz CS; Kang MJ Mitochondrial dysfunction and damage associated molecular patterns (DAMPs) in chronic inflammatory diseases. Mitochondrion 41:37–44; 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Gu X; Wu G; Yao Y; Zeng J; Shi D; Lv T; Luo L; Song Y Intratracheal administration of mitochondrial DNA directly provokes lung inflammation through the TLR9–p38 MAPK pathway. Free Radical Biology and Medicine 83:149–158; 2015. [DOI] [PubMed] [Google Scholar]
- [79].Ichinohe T; Yamazaki T; Koshiba T; Yanagi Y Mitochondrial protein mitofusin 2 is required for NLRP3 inflammasome activation after RNA virus infection. Proceedings of the National Academy of Sciences 110:17963–17968; 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Wiegman CH; Michaeloudes C; Haji G; Narang P; Clarke CJ; Russell KE; Bao W; Pavlidis S; Barnes PJ; Kanerva J; Bittner A; Rao N; Murphy MP; Kirkham PA; Chung KF; Adcock IM; Brightling CE; Davies DE; Finch DK; Fisher AJ; Gaw A; Knox AJ; Mayer RJ; Polkey M; Salmon M; Singh D Oxidative stress–induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. Journal of Allergy and Clinical Immunology 136:769–780; 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Odewumi C; Latinwo LM; Sinclair A; Badisa VL; Abdullah A; Badisa RB Effect of cadmium on the expression levels of interleukin-1alpha and interleukin-10 cytokines in human lung cells. Mol Med Rep 12:6422–6426; 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Kayama F; Yoshida T; Elwell MR; Luster MI Cadmium-induced renal damage and proinflammatory cytokines: possible role of IL-6 in tubular epithelial cell regeneration. Toxicol Appl Pharmacol 134:26–34; 1995. [DOI] [PubMed] [Google Scholar]
- [83].Olszowski T; Baranowska-Bosiacka I; Gutowska I; Chlubek D Pro-inflammatory properties of cadmium. Acta Biochim Pol 59:475–482; 2012. [PubMed] [Google Scholar]
- [84].Basset-Leobon C; Lacoste-Collin L; Aziza J; Bes JC; Jozan S; Courtade-Saidi M Cut-off values and significance of Oil Red O-positive cells in bronchoalveolar lavage fluid. Cytopathology 21:245–250; 2010. [DOI] [PubMed] [Google Scholar]
- [85].Morissette MC; Shen P; Thayaparan D; Stampfli MR Disruption of pulmonary lipid homeostasis drives cigarette smoke-induced lung inflammation in mice. Eur Respir J 46:1451–1460; 2015. [DOI] [PubMed] [Google Scholar]
- [86].Romero F; Shah D; Duong M; Penn RB; Fessler MB; Madenspacher J; Stafstrom W; Kavuru M; Lu B; Kallen CB; Walsh K; Summer R A pneumocyte-macrophage paracrine lipid axis drives the lung toward fibrosis. Am J Respir Cell Mol Biol 53:74–86; 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Pietrocola F; Galluzzi L; Bravo-San Pedro JM; Madeo F; Kroemer G Acetyl coenzyme A: a central metabolite and second messenger. Cell metabolism 21:805–821; 2015. [DOI] [PubMed] [Google Scholar]
- [88].Shi L; Tu BP Acetyl-CoA and the regulation of metabolism: mechanisms and consequences. Curr Opin Cell Biol 33:125–131; 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Saponaro C; Gaggini M; Carli F; Gastaldelli A The Subtle Balance between Lipolysis and Lipogenesis: A Critical Point in Metabolic Homeostasis. Nutrients 7:9453–9474; 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.