

Abstract
DNA methylation is a dynamic epigenetic modification with a prominent role in determining mammalian cell development, lineage identity and transcriptional regulation. Primarily linked to gene silencing, novel technologies have expanded the ability to measure DNA methylation on a genome-wide scale and uncover context-dependent regulatory roles. The immune system is a prototypic model for studying how DNA methylation patterning modulates cell type- and stimulus-specific transcriptional programs. Preservation of host defense and organ homeostasis depends on fine-tuned epigenetic mechanisms controlling myeloid and lymphoid cell differentiation and function, which shape innate and adaptive immune responses. Dysregulation of these processes can lead to human immune system pathology as seen in blood malignancies, infections and autoimmune diseases. Identification of distinct epigenotypes linked to pathogenesis carries the potential to validate therapeutic targets in disease prevention and management.
Keywords: DNA methylation, immune system, epigenetics, transcription, hematopoiesis, leukemia, autoimmunity, DNA methyltransferase inhibitors
Introduction
Initially coined by embryologist Conrad Waddington, epigenetics refers to the study of heritable changes in gene function that do not alter the DNA sequence yet give rise to distinct phenotypes during development [1]. Fine-tuned epigenetic mechanisms reshape the topology of the genome to influence gene expression and regulate cell development, differentiation and phenotypic plasticity in response to environmental cues. Epigenetic phenomena include non-coding RNAs along with DNA methylation and post-transcriptional modifications of nucleosomal histones that alter chromatin accessibility and recruitment of the transcriptional machinery to modulate gene expression [2]. Because of their inherently plastic and cue-sensing nature, epigenetic mechanisms play a prominent role in governing the transcriptional programs of the immune system in health and disease.
DNA methylation is a widely studied chromatin modification given its pivotal role in gene silencing, X-chromosome inactivation, genomic stability and imprinting [3]. In mammalian cells, methylation occurs almost exclusively at the fifth carbon position of cytosines in the context of CpG dinucleotides (5’ – cytosine – phosphate – guanine – 3’). Although prokaryotes classically display non-CG methylation (mCH, in which H is any non-guanine residue), a recent study revealed the presence of non-canonical (non-CG) methylation in multiple human tissues [4]. There are over 28 million CpG dinucleotides in the human genome, and 60–80% display methylation in any given cell. Conversely, CpG dinucleotide-enriched regions known as CpG islands, mostly located near gene promoter sequences, are predominantly hypomethylated, linking de novo methylation marks to gene expression [5].
DNA methylation patterns are established through the function of a family of DNA methyltransferases (DNMTs) that catalyze the covalent addition of a methyl group to the 5-carbon of the substrate base cytosine. The product of the DNA methyltransferase reaction is 5-methylcytosine (5mC). In mammals, DNMT3A and DNMT3B catalyze de novo methylation, and the maintenance DNA methyltransferase DNMT1, which binds hemi-methylated DNA during cell division, copies the parental strand CpG methylation pattern to the daughter strand [6]. A group of methylated-DNA binding proteins, including methyl-CpG-binding domain proteins, zinc finger proteins and certain transcription factors, act as readers of methylation marks to link DNA methylation and gene expression changes [7, 8]. Alterations to the DNA methylation maintenance machinery over successive rounds of cell division can result in passive DNA demethylation. Additionally, active DNA demethylation plays an important role during development, cell differentiation and function. The ten-eleven translocation (TET) family of enzymes bears primary responsibility for active DNA demethylation [9]. Fig 1 outlines the biochemistry of DNA methylation dynamics.
Figure 1. DNA methylation chemistry.
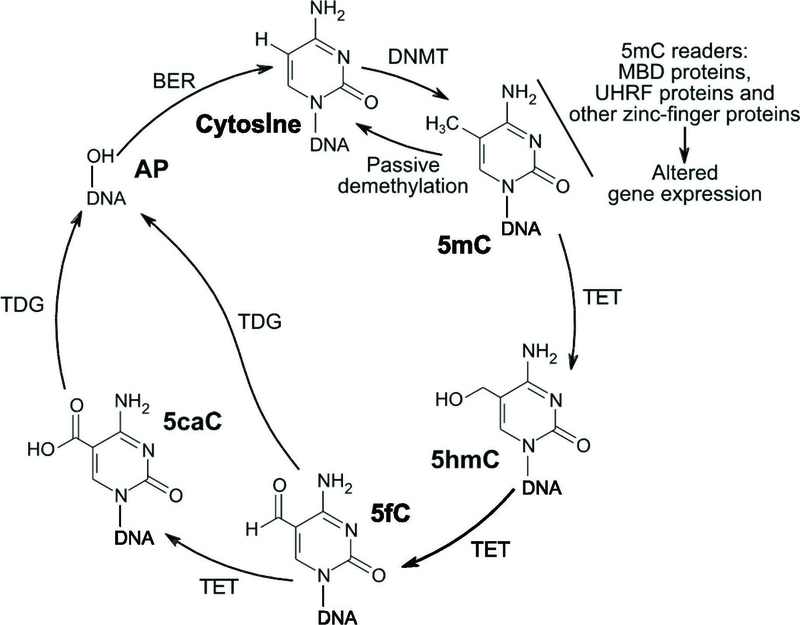
DNA methyltransferases (DNMT) use S-adenosylmethionine as a methyl donor to catalyze the addition of a methyl group to the 5-carbon position of cytosine, resulting in 5-methylcytosine (5mC). 5mC can be read by multiple nuclear proteins that lead to changes in gene expression, including methyl-CpG-binding domain (MBD) proteins; ubiquitin-like, containing PHD and RING finger domains (UHRF) proteins; and other zinc-finger proteins. Demethylation of 5mC back to cytosine can occur passively during cell division. Active demethylation can occur via the ten-eleven translocation (TET) enzymes, which use oxygen, 2-oxoglutarate, and ferrous iron to catalyze the conversion of 5mC to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC). Each reaction generates an oxidized ferric iron, succinate, and carbon dioxide. G/T-mismatch-specific thymine DNA glycosylase (TDG) can then excise 5fC and 5caC, resulting in an apyrimidinic (AP) site. These AP sites can then undergo base excision repair (BER), which replaces cytosine at that position. Not shown are other demethylation pathways including deamination of 5mC or 5hmC by activation-induced deaminase/apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like (AID/APOBEC) family members to form 5-methyluracil (5mU) or 5-hydroxymethyluracil (5hmU), respectively, which can be catalyzed to cytosine via the TDG/BER pathway.
In order to better understand the influence of DNA methylation on gene expression (the transcriptome), numerous technologies have sought to accurately profile DNA methylation on a genome-wide scale (Table 1). Because DNA methylation marks are erased by molecular biology techniques used for whole-genome DNA sequencing, such as PCR amplification and cloning of DNA fragments into bacterial vectors, different tools were developed in order to identify methylated DNA through endonuclease digestion, immunoprecipitation or bisulfite conversion [10]. Integration of these tools with array-based and next-generation sequencing technologies allows for genome-scale DNA methylation profiling.
Table 1. Principal methods for genome-wide DNA methylation analysis.
There is an extensive variety of DNA methylation profiling techniques available to survey the whole genome. Selection of a specific method requires appropriate understanding of their respective advantages and disadvantages. The type and quantity of available sample combined with the desired depth of genome coverage and resolution provides guidance to select an approach. Methyl-DNA immunoprecipitation (MeDIP), chromatin immunoprecipitation (ChIP), methyl-CpG binding domain (MBD), reduced representation bisulfite sequencing (RRBS), whole-genome bisulfite sequencing (WGBS).
| Method (ref) |
Description | Advantages | Disadvantages |
|---|---|---|---|
|
MeDIP [140] |
Utilizes specific antibodies capable of immuno-capturing single-stranded methylated cytosines followed by analysis through tiling arrays (MeDIP- ChIP) or next-generation sequencing (MeDIP-seq). |
Increade Sensitivity for identifying regions of low CpG density (gene bodies and shores) when compared to MBD |
Similar to MBD. this method has a strong bias toward identification of hypermethylated DNA regions and lacks single- CpG dinucleotide resolution Due to enrichment technique for identification of methylated DNA regions followed by their subsequent amplification and sequencing, there is a bias for analyzing densely methylated regions over hypomethylated regions. |
| Cost-efficient method Increased sensitivity for identifying regions of high CpG density when compared to MeDIP. | |||
|
MBD [141] |
Affinity-based capture method using beads coated with the DNA-binding protein MBD, which specifically binds double-stranded methylated CpGs. This is followed by a salt fractionation step that allows DNA methylation density assessment and fragment separation. Analysis is then performed through profiling with tiling microarrays (MBD-ChIP) or next- generation sequencing (MBD-seq). |
||
| Ionic strength modulates MBD affinity for methylated DNA therefore, changes in salt elution permit isolation of DNA based on methylation density | |||
| Lack of power to discern DNA methylation changes at single-CpG dinucleotide resolution. | |||
| Cost-efficient method Identifies nearly every cytosine residue on a genome-wide scale, making this method the gold standard in DNA methylome analysis. | |||
|
WGBS [142] |
Treatment of genomic DNA with sodium bisulfite selectively deaminates unmethylated cytosine residues into uracil at a much faster rate than methylated cytosines. Uracil bases are then converted to thymidine bases after PCR amplification while unmethylated cytosines remain unchanged. The resulting library undergoes next- generation sequencing. |
Genome-wide sequencing requirements result in high costs. |
|
| A modification to the bisulfite sequencing chemistry also permits assessment of 5hmC marks. Reduces sequence redundancy resulting from bisulfite treatment by selecting specific regions for sequencing |
DNA degradation due to bisulfite conversion. |
||
| Captures the majority, but not all CpG islands and promoters | |||
| Combines the use of restriction enzyme digestion (e.g., MspI) to select for CpG-enriched regions followed by bisulfite conversion and next-generation sequencing | |||
| Limited coverage of other genomic regions (e.g., distal regulatory elements and shores). | |||
|
RRBS [103,143] |
Requires low amounts of DNA, which translates into fewer reads needed for accurate sequencing, thereby reducing cost |
||
| DNA degradation due to bisulfite conversion. |
The immune system is responsible for host defense, a primordial function of living organisms, with the goal of preserving tissue and organismal homeostasis. Fine-tuned immunological mechanisms allow the host to develop a nearly unlimited repertoire to respond to a variety of pathogens and environmental cues while providing a long-lasting memory to fight recurrent triggers (immunological memory) and at the same time mitigate damage to its own tissues (as seen in cancer and autoimmunity). Two distinct branches divide the immune response into innate and adaptive immunity. Innate immunity exhibits germline inheritance and corresponds to a fast-paced, non-specific response against pathogens coordinated by myeloid phagocytes, toll-like receptors and the complement system. Adaptive immunity is a more sophisticated, highly specific and long-lasting response orchestrated by B and T lymphocytes that relies on antigen recognition through a very large set of lymphocyte-specific receptors [11]. Epigenetic modifications, including DNA methylation, tightly regulate immune system development, differentiation and function (Fig 2).
Figure 2.

DNA methylation state during innate and adaptive immune cell development, differentiation and function. Epigenetic modifications, including DNA methylation, modulate the self-renewal capacity of the hematopoietic stem cell pool throughout the lifespan and tightly regulate the hierarchical differentiation process of immune cells. DNA methylation patterning is specific to immune cell types during distinct maturation stages and correlate with a cell’s gene regulatory and functional programs. DNA methyltransferase (DNMT), multipotent progenitors (MPP), common lymphoid progenitors (CLP), common myeloid progenitors (CMP), 5-hydroxymethylcytosine (5hmC), conserved noncoding sequence 2 (CNS2).
In this review, we summarize the role distinct DNA methylation patterns play in regulating the transcriptomic landscape of the immune system. We will examine how DNA methylation shapes early immune cell development and differentiation, immune function in host defense mechanisms and immune dysregulation exhibited during blood malignancies and autoimmune diseases. Our review concludes with an overview of epigenetic therapeutic approaches in malignant immune system disorders.
DNA methylation patterning during immune system progenitor development, differentiation and lineage commitment
Hematopoiesis constitutes an ideal system to study the intricate gene regulatory networks controlling multi-lineage cellular differentiation, identity and function. During the early 20th century, Alexander Maximov introduced the unitarian theory of hematopoiesis, in which distinct subpopulations of mature blood cells are generated from a single precursor cell known as the hematopoietic stem cell [12]. Hematopoietic stem cells are long-lived progenitors that display self-renewal capacity as well as the ability to differentiate into intermediate progenitors through a stepwise fate restriction process that ultimately confers lineage identity. DNA methylation regulates self-renewal and differentiation processes in the hematopoietic system and is specific to individual cell types that retain epigenetic memory of their lineage trajectory. Analysis of cell type-specific DNA methylation patterns therefore allows hierarchical reconstruction of a cell type’s developmental history [13].
Early studies established a pivotal role for de novo DNA methylation in enhancing hematopoietic stem cell self-renewal capacity without altering hematopoietic cell differentiation [14, 15]. Experimental models using hypomorphic Dnmt1 mice and hematopoietic stem cells revealed that constitutive maintenance DNA methylation is not only required for self-renewal capacity of the hematopoietic stem cell pool, but also plays a crucial role in regulating myeloerythroid versus lymphoid lineage differentiation. Transcriptomic analysis of hypomethylated murine hematopoietic stem cells showed increased expression of signature genes encoding for lineage-specific transcription factors such as Gata1, Id2 and Cebpa, which are involved in myeloerythroid differentiation [16, 17]. Array-based analysis of genome-wide DNA methylation patterns in hematopoietic progenitors revealed marked epigenetic plasticity during myeloid and lymphoid differentiation. Hematopoietic progenitor cell differentiation from multipotent progenitors (MPP) into common lymphoid and myeloid progenitors (CLP and CMP) involved DNA methylation of CMP-specific genomic regions. Specifically, lymphoid cells demonstrated increased DNA methylation at several key regulatory transcription factors of the myeloid lineage, such as Gata2, Tal1 and Lmo2 [18]. Moreover, in an in vitro assay in which MPP were treated with the hypomethylating agent 5-aza-2’deoxycytidine (decitabine), the overall percentage of CMP increased at the expense of CLP, further confirming that reduced methylation favors myeloid as opposed to lymphoid differentiation [19].
The advent of next-generation sequencing technologies such as whole-genome bisulfite sequencing made possible a more comprehensive and unsupervised analysis of the DNA methylome at single-CpG dinucleotide resolution. Implementation of this novel approach to the study of hematopoietic stem cell differentiation uncovered that functional and dynamic methylation changes were located outside of CpG-dense regions, overlapping with cis-regulatory sites (such as enhancers and promoters) and inversely correlating with gene expression profiles [20]. For example, DNA methylation affects nucleosome dynamics and binding of CCCTC-binding factor (CTCF), a DNA-binding protein with insulator properties best known for its function as a linker between nuclear architecture and gene expression [21, 22]. Nucleosome positioning is widely inconsistent across the genome with the exception of chromatin surrounding CTCF-binding sites. Therefore, analysis of these sites has the potential to understand the oscillatory relationship between DNA methylation and nucleosome positioning (i.e., peaks of DNA methylation correlating to valleys of nucleosome density). Recently, investigators have shown that during B and T lymphocyte development, CTCF-binding sites demonstrate an increased oscillatory methylation pattern as opposed to a less divergent pattern during myeloid differentiation, indicating a dissimilar usage of DNA methylation between immune cell lineages [23].
The adaptive immune system is mainly comprised of B and T lymphocytes. The expression of specific surface receptors allows these cells to recognize, differentiate, proliferate and acquire lineage-specific effector functions in order to respond to diverse pathogens, tumors and environmental cues. Hematopoietic progenitors of the T cell lineage enter the thymus as double-negative cells (CD4-CD8-, DN), differentiate into double-positive cells (CD4+CD8+, DP) and ultimately, through sequential maturation stages, become either naïve CD4+ or CD8+ T cells as they egress from the thymus. Lee and colleagues showed that Cre-mediated deletion of Dnmt1 in DN thymocytes led to a significant reduction of DP thymocytes and peripheral naïve CD4+ and CD8+ T cells, demonstrating a key role for methylation in thymopoiesis [24]. Interestingly, Dnmt1 deletion at the DP stage caused a significant increase in cytokine production by naïve T cells, suggesting that methylation prevents premature activation of differentiation and functional programs that could lead to immune pathology. Global methylation analysis during human intrathymic and peripheral T cell differentiation revealed an increased frequency of TET-mediated demethylation events often associated with gene expression programs controlling lymphopoiesis [25, 26]. Investigators demonstrated 5hmC enrichment at active cell-specific enhancers and genes encoding key lineage-specifying transcription factors (for example, ThPOK, Gata3 and Runx3) at distinct maturation stages, positively linking DNA methylation kinetics with transcriptional regulatory networks during T cell development [27]. Taken together, these studies demonstrate a functional role for active loss of DNA methylation during early thymic and peripheral T cell development, which determines their capacity to further differentiate and acquire lineage-specific functions.
Upon antigenic stimulation, naïve CD4+ T cells differentiate into distinct helper T (Th) cells whose phenotypic characterization is defined by the expression of several signature cytokines (for example, IL-2, IL-10, IFN-γ and IL-17)—a process also regulated by active DNA demethylation along with histone modifications [28]. These differentiated cells go on to orchestrate immune function through enhancement of B cell-mediated antibody production, macrophage and CD8+ T cell effector function as well as maintenance of immunological memory and self-tolerance.
Cytotoxic CD8+ T cells are essential for clearance of intracellular pathogens and tumor cells. After stimulation of naïve CD8+ T cells with lymphocytic choriomeningitis virus, Scharer and colleagues demonstrated that differentially methylated regions were enriched for transcription factors that regulated effector cytolytic functions in CD8+ T cells, suggesting that methylation patterns are not only key for T cell differentiation but also for the establishment of cellular immunity [29]. After successful clearance of a pathogen, most effector T cells undergo programmed cell death, except for a persistent fraction of memory T cells. This subset of long-lived lymphocytes is able to regain effector function after recognition of the pathogen that initially triggered their activation and therefore are important in maintaining long-term immunity as conferred after vaccination [30]. Interestingly, methylation profiling of terminal effector versus memory precursor CD8+ T cell subsets showed that the subset of cells that give rise to memory cells acquired repressive de novo DNA methylation at genes expressed by naïve cells. Effector genes became demethylated, further demonstrating that these cells are poised and capable of initiating an effective cellular immune response without needing further differentiation [31].
Non-canonical DNA methylation has been described in pluripotent stem cell development and differentiation and has been found in multiple human tissues during development. In this context, mCH is anti-correlated with gene expression and decreases during cell differentiation [4, 32]. Shuyler and colleagues described prominent levels of mCH in myeloid leukemia and naïve T cells compared to lymphoid neoplasms and myeloid cells with a subsequent disappearance of this mark with T cell lineage development. Moreover, lymphoid neoplasms showed reduced levels of CpG methylation in comparison with myeloid malignancies. These observations have important clinical relevance, given that demethylating agents commonly used to treat myeloid leukemias primarily target canonical (CG context) DNA methylation and could potentially demonstrate divergent therapeutic efficacy in myeloid neoplasms as opposed to lymphoid-derived malignancies [23].
Overall, studying mammalian blood formation offers the opportunity to identify the different layers of dynamic epigenetic modifications, including DNA methylation along with its respective functional genomic annotations, which control cell fate decisions, renewal capacity and identity. Isolation and methylation analysis of cellular intermediates targeted by blood malignancy-related mutations could provide links between epigenotypes and disease development, as well as novel therapeutic targets for drug discovery.
Dysregulated DNA methylation in immune system and hematological malignancies
Disruption of DNA methylation has long been associated with the pathobiology of cancers of the immune system and hematopoietic progenitors. Investigators initially reported global DNA hypomethylation in multiple cancer cell lines and tumors when compared with normal tissues [33, 34]. In contrast, hypermethylation of CpG islands at the promoter regions of tumor-suppressor genes has also been linked tightly to tumorigenesis [35]. Because epigenetic modifications are pivotal in determining cell-fate decisions during hematopoiesis, investigators predicted that alterations of this gene regulatory machinery would play a key role in the development of hematological malignancies.
Accumulation of somatic mutations leading to clonal expansion of progenitor cells in the aging population can drive cancer evolution [36]. Identification of recurrent mutations, many of which affect the epigenetic machinery, are usually present years before the clinical onset of cancer. Known pre-malignant states include monoclonal gammopathy of unknown significance in multiple myeloma [37], monoclonal B cell lymphocytosis in chronic lymphocytic leukemia [38], and more recently clonal hematopoiesis of indeterminate potential (CHIP) in myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) [39]. Interestingly, the most common mutations found in CHIP are in the DNMT3A locus [36, 40, 41]. DNMT3A mutations have also been observed in patients with MDS and myeloproliferative neoplasms [42, 43] and have been linked to secondary AML, suggesting these mutations might represent an early event in leukemic transformation [44]. With the use of an unsupervised deep sequencing approach, tumor sampling of adult patients with AML uncovered that close to 20% of study subjects carried DNMT3A mutations, which were associated with an unfavorable prognosis [45, 46]. DNA methylation profiling identified distinct clusters of AML patients associated with specific mutations. Samples with DNMT3A, MLL fusions, NMP1 and FLT3 mutations were linked to extensive loss of DNA methylation, while samples with isocitrate dehydrogenase 1 and 2 mutations (IDH1 and IDH2) demonstrated a substantial gain of methylation marks (Table 2). Collectively, these data show that profiling of the DNA methylome is a powerful tool for clinical stratification and development of targeted therapeutic strategies [47–49].
Table 2. DNA methylation profiling of common genetic mutations associated with acute myeloid leukemia.
DNA methylation patterning identifies specific methylation signatures in genes associated with the development of AML. Since epigenetic mechanisms are linked to gene regulation in leukemogenesis, they constitute a novel tool to enhance our understanding of AML pathogenesis, classification, and development of targeted therapies. DNA methyltransferase 3A (DNMT3A), ten-eleven translocation 2 (TET2), isocitrate dehydrogenase 1 and 2 (IDH 1 and IDH 2), nucleophosmin 1 (NMP1), mixed lineage leukemia (MLL), acute myeloid leukemia (AML), α-ketoglutarate (α-KG), 2-hydroxyglutarate (2-HG).
| Genetic mutation | DNA menthylation pattern | Oncogenic mechanism in AML (ref) |
|---|---|---|
| DNMT3A | Primarily hypomethylated | Mutation of the DNMTA3 residue Arg882 (R882) leads to potentiation of aberrant stemness genes linked to AML development [144]. |
| TET2 | Hypermethylation | Loss of TET2 leads to a genome- wide enhancer hypermethylation state including downregulation of putative tumor suppressor genes linked to AML development [145]. |
| IDH 1 and 2 | Marked hypermethylation | Hypermethylation leads to loss of IDH enzymatic activity and α-KG production accompanied by an increase in the oncometabolite 2- HG [61]. |
| NMP1 | Primarily hypomethylated | Unclear mechanism. NMP1 is known to be important for nucleolar integrity (ribosome biogenesis) and function (DNA-repair processes). Involved in regulating activity of tumor suppressor genes (e.g., p53) [146] |
| MLL-fusion proteins | Marked hypomethylation | Gain of function of MLL fusion proteins leads to increased levels of MEIS1 and HOXA gene expression which have been linked to leukemogenesis [147]. |
Active DNA demethylation has a well-established role in the maintenance of hematopoietic homeostasis, differentiation of granulomonocytic progenitors and leukemogenesis [48, 50–54]. In fact, the discovery of active DNA demethylation by the TET family of enzymes was first described through the cloning of TET1 as a fusion partner of MLL (mixed lineage leukemia gene on 11q23, which frequently harbors cytogenetic abnormalities related to AML) in AML patients with t(10;11) (q22;q23) translocations [55–57]. Deletion of TET2 in the hematopoietic compartment can lead to the development of distinct myeloid disorders including chronic myelomonocytic leukemia, MDS, myeloproliferative neoplasms and AML [58–60]. Additionally, metabolic changes can lead to oncogenic disruption of the active demethylation machinery. Isocitrate dehydrogenase 1 and 2 (IDH 1 and IDH 2) are metabolic enzymes that convert isocitrate to α-ketoglutarate (α-KG) during the Krebs cycle. DNA sequencing in AML patients identified different IDH mutations that result in neomorphic enzymatic activity leading to conversion of α-KG into 2-hydroxyglutarate (2-HG) [61–63]. Subsequent studies demonstrated that 2-HG is an oncometabolite that exerts its oncogenic effect through competitive inhibition of α-KG-dependent TET proteins, thereby phenocopying a TET2 mutation methylation profile [64, 65].
Altogether, the past decade has seen discoveries into the role played by epigenetic modifier-related mutations in the development of myeloid malignancies. These revelations have come in contrast to the prior long-held dogma that the pathogenesis of myeloid malignancies was the result of class I and class II mutations, which are associated with dysregulated cell growth (activation of signaling pathways, such as signal transducer and activators of transcription) and impaired cellular differentiation (altered expression of key transcription factors), respectively. This paradigm shift has resulted in the development of novel therapeutic agents targeting epigenetic modifiers such as ivosidenib (an IDH1 inhibitor), which has recently demonstrated clinical efficacy with durable and complete remission in patients with refractory AML [66].
DNA methylation dynamics as a modulator of immune system function and host-pathogen interactions
The mammalian immune system is comprised of two distinct branches: innate and adaptive immunity. Along with the epithelium, myeloid cells of the innate immune branch constitute the first line of host defense against invading pathogens. Macrophages are specialized phagocytes that orchestrate this initial immune response through antigen presentation, cytokine release and pathogen clearance. The interplay of transcriptional and epigenetic mechanisms tightly regulates macrophage identity, function and differentiation. Earlier studies have mainly focused on the interaction of pioneer, lineage-specifying tissue factors such as PU.1 with cell-specific enhancers, as they modulate and reshape the chromatin landscape during macrophage development and activation [67–71]. The combination of the well-known chemical stability of DNA methylation as an epigenetic mark and the fast-changing nature of innate immune responses initially limited the study of DNA methylation in innate myeloid immune cells. However, Pacis and colleagues showed that Mycobacterium tuberculosis infection of dendritic cells induces rapid loss of DNA methylation within 24 hours at distal enhancers that activate master immune transcription factors (including Nuclear Factor-kB and members of the Interferon Regulatory Factor family), suggesting an important role for DNA methylation in regulating innate immune responses [72]. More recently, Wallner and colleagues demonstrated that a gene regulatory network controlling macrophage structure and phagocytosis undergoes TET-mediated demethylation during monocyte-to-macrophage differentiation [73]. Furthermore, both de novo and maintenance DNA methylation can modulate macrophage polarization in murine models of chronic inflammatory diseases, such as obesity and atherosclerosis [74, 75]. Overall these data show that epigenetic modifications not only exert a pivotal role during immune cell development but also modulate functional immune programs during disease, offering an opportunity for the development of targeted therapies.
Epigenetic landscape of host-pathogen interactions
The interaction of infectious pathogens with host cells is a dynamic relationship that carries clinical relevance as we aim to target the host response to infection. In response to infection, the host can undergo epigenetic reprogramming, provoking a drastic change of gene expression and phenotype leading to worsening or de novo human pathology [76]. The initial immune response that a host mounts to overcome a particular pathogen must be tightly balanced in order to ensure an effective defense accompanied by minimal tissue damage. Although immunological memory has traditionally been ascribed to the adaptive immune system, the characterization of trained immunity and endotoxin tolerance as innate-type memory responses displayed by innate immune cells has challenged this long-held dogma [77]. Trained immunity explains how the initial priming of innate immune cells can lead to an enhanced response upon re-stimulation. Lipopolysaccharide (LPS)-mediated macrophage stimulation expands the enhancer landscape governing gene expression, which then confers an epigenetic footprint that strengthens immune responses upon secondary challenges [71]. Conversely, endotoxin tolerance consists of a partially hypo-responsive state displayed by macrophages after recurrent LPS exposures aimed at mitigating tissue damage during ongoing inflammation (e.g., sepsis). Tolerized macrophages undergo functional reprogramming through distinct patterning of histone modifications resulting in an immunocompromised state that can portend a high risk for secondary nosocomial infections in septic patients [78, 79]. Although immunologic memory can be useful to overcome and tolerate infections, it can also have deleterious consequences. For example, rewiring of the monocytic epigenetic landscape leading to induction of trained immunity and endotoxin tolerance has been associated with the development of autoimmune diseases and chronic bacterial colonization in cystic fibrosis patients, respectively [80, 81].
Epigenetic reprogramming of host gene expression profiles in response to infection can also lead to pathogen persistence and dissemination, dynamically affecting host-pathogen interactions. In a study by Masaki and colleagues, infection of adult Schwann cells with Mycobacterium leprae resulted in their differentiation into a pluripotent stem cell state, followed by promoter demethylation and upregulation of several genes controlling epithelial-to-mesenchymal transition programs. Reprogrammed cells then go on to differentiate into skeletal and smooth muscle cells and form granuloma-like structures that promote dissemination of infection [82]. Finally, pathogen-induced abnormal cellular reprogramming has been causally linked to oncogenesis. Helicobacter pylori infection induces aberrant DNA methylation in human gastric mucosa at promoter regions of methylated genes found in gastric carcinoma cells [83]. Similarly, promoter hypermethylation and subsequent down-regulation of the tumor-suppressor gene PTEN is associated with Epstein Barr virus-related gastric cancer [84]. Altogether, these studies demonstrate the potential for epigenetic therapeutic interventions in the management of infectious diseases. Antibiotic resistance is a global health problem that mandates development of novel drugs, and modulation of both pathogen-induced epigenetic modifications could represent viable therapeutic targets.
Epigenetic regulation of autoimmune diseases
There are over 80 different known autoimmune diseases collectively affecting 7% of the United States population [85]. Despite the high prevalence of autoimmune diseases, there are still significant gaps in our understanding of the pathobiology and causative factors of these diseases. Thus far, we know that autoimmunity ensues when a genetically predisposed host is challenged by distinct environmental factors resulting in a dysregulated immune response and loss of self-tolerance. The epigenome connects these environmental cues with gene expression patterns during immune cell development and maintenance and provides the necessary plasticity required to respond to cellular stressors throughout the lifespan. Therefore, researchers have recently shifted their focus onto the analysis of differential epigenetic patterns in autoimmune diseases, as epigenetic phenomena may explain the molecular mechanisms underpinning disease development, progression and phenotypic variability in patients with similar genetic backgrounds.
The study of monozygotic twins presents an opportunity to uncover the role played by environmental epigenetic modifications in the susceptibility to autoimmune diseases, as they likely explain how identical genotypes lead to distinct phenotypes. Javierre and colleagues performed a global DNA methylation analysis in identical twins discordant for systemic lupus erythematosus (SLE) and found a significant reduction in the overall DNA methylation content in the SLE-affected twin. Furthermore, gene ontology-based functional enrichment analysis demonstrated differential gene expression profiles related to immune function and molecular pathways linked to the pathogenesis of SLE [86].
The majority of autoimmune disease-affected patients are females, with female:male ratios ranging from 3:1 for rheumatoid arthritis (RA) to 9:1 for SLE, suggesting a potential contribution of X-chromosome-located genes to the pathogenesis of autoimmune diseases [87]. Since epigenetic mechanisms (prominently DNA methylation) control X-chromosome inactivation, it is unsurprising that new-onset epigenetic modifications can result in differential expression of genes encoded on the X-chromosome. For example, the X-chromosome-encoded gene CD40L is a B cell costimulatory transmembrane protein that shows increased expression on the surface of activated CD4+ T cells from patients with SLE [88]. Two independent studies found that DNA demethylation of CD40L regulatory elements leads to overexpression of this gene in the CD4+ T cells of SLE and systemic sclerosis patients [89, 90], thereby contributing to the overproduction of auto-antibodies that drive disease pathogenesis.
B cells and regulatory T cells: drivers and mitigators of autoimmunity
Epigenetic modifications modulate B cell maturation and production of autoreactive antibodies by B cell-derived plasma cells. Upon antigenic stimulation and T cell-mediated activation (i.e., CD40L costimulatory signal) mature naïve B cells migrate to secondary lymphoid organs and differentiate into germinal center B cells. Marked activation-induced demethylation and increased heterogeneity of methylation patterning of germinal center B cells accompanies this transition [91, 92]. Through initiation of somatic hypermutation and class switch recombination, activation-induced cytidine deaminase generates a diversified repertoire of high-affinity antibodies that modulate adaptive immune responses. After egressing germinal centers, B cells undergo further differentiation into either antibody-producing plasma cells or long-lived memory B cells capable of mounting a more efficient response to subsequent challenges. DNA methylation profiling comparing naïve versus memory B cells showed differentially methylated regions associated with key genes regulating their immune function (for example, RUNX3, RELA and PAX5), revealing that DNA methylation reprogramming poises memory B cells to exhibit a more sustained and enhanced recall response compared to naïve B cells [93].
Drug-induced lupus erythematosus is an autoimmune disorder triggered by the chronic use of certain medications with DNA methyltransferase inhibitor (DNMTI) characteristics, including hydralazine and procainamide. Mazari and colleagues showed that passive transfer of hydralazine-treated B cells into syngenic mice leads to increased detection of autoreactive antibodies through disruption of receptor editing and B cell tolerance, demonstrating a causal role for the loss of B cell DNA methylation and the development of autoimmunity [94]. The association of dysregulated B cell methylation and autoimmunity is not only limited to SLE. In a genome-wide DNA methylation study of patients with primary Sjögren’s Syndrome, investigators demonstrated that DNA methylation alterations were more prevalent in genetic risk loci of B cells when compared with T cells. Moreover, methylation alterations in B cells were enriched for pathways involved in inflammation, interferon signaling and positively correlated with disease activity [95]. These studies demonstrate that epigenetic regulation of B cell function is a key mechanism driving the development and severity of autoimmune disease (Fig 3).
Figure 3.
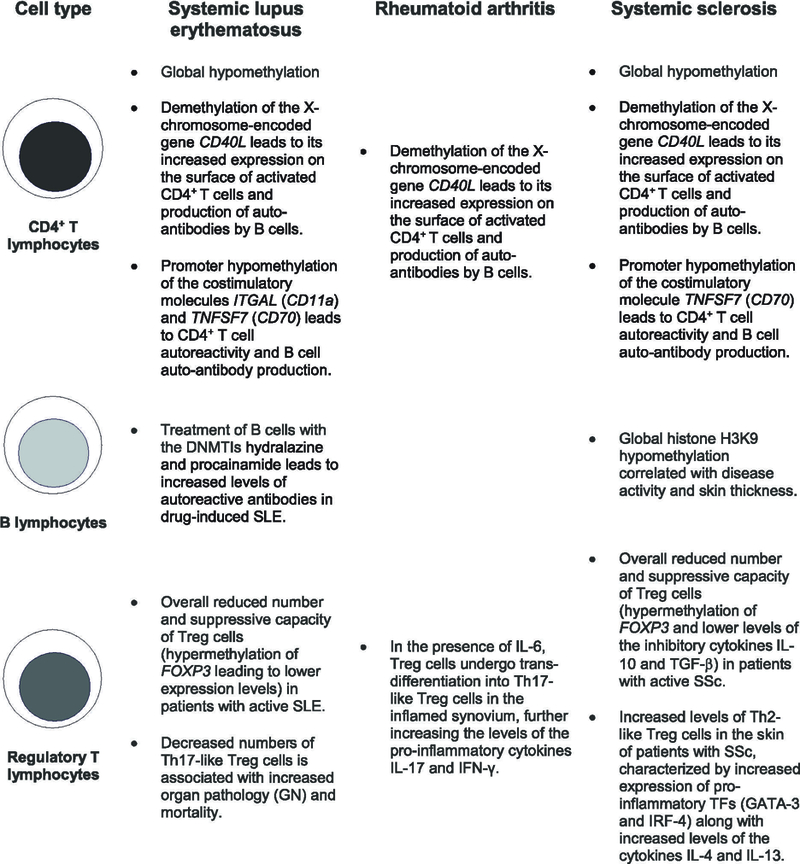
Epigenetic patterns of immune cell subsets in the pathogenesis of autoimmune diseases. Cell-specific methylation changes are linked to immune cell differentiation and function during the development of autoimmune processes. Epigenetic marks can identify subjects at increased risk for organ damage, disease progression and overall mortality in these human pathologies. Systemic lupus erythematosus (SLE), systemic sclerosis (SSc), regulatory T cells (Treg cells), glomerulonephritis (GN), TFs (transcription factors).
Immunomodulatory regulatory T (Treg) cells constitute a subset of CD4+ T cells with a key role in mitigation of autoimmunity and maintenance of immune homeostasis. The master transcription factor of Treg cells is Forkhead box P3 (FOXP3), a member of the fork-winged helix family exclusively expressed at stable levels in Treg cells and responsible for their differentiation and repressive function [96]. Early after its discovery, investigators demonstrated that FOXP3 mutations result in human neonatal onset of immune dysregulation, polyendocrinopathy, enteropathy X-linked (IPEX) syndrome, implicating Treg cell dysfunction as a cause of severe autoimmunity [97, 98]. Induction and stabilization of FOXP3 expression is under tight epigenetic regulation. Specifically, both the FOXP3 promoter and one of its supporting conserved noncoding DNA sequences (CNS), CNS2, display hypomethylation in Treg cells and relative hypermethylation in conventional T cells, linking DNA methylation to T cell lineage commitment and function [99–103].
Treg cells exert their repressive function on effector T cells through expression of inhibitory checkpoint molecules, such as cytotoxic T lymphocyte antigen 4 (CTLA-4), which is involved in cell contact-mediated suppression. Impairment of T cell proliferation through secretion of inhibitory cytokines, such as IL-10 and transforming growth factor-β (TGF-β) constitutes another immunosuppressive effect of Treg cells [104]. Compared with patients with inactive SLE, isolated Treg cells from patients with active SLE showed a significant decrease in their ability to suppress CD4+ effector T cell proliferation and cytokine secretion. FOXP3 message and protein from patients with active SLE also showed significant reductions [105, 106]. Moreover, several groups of researchers described a global reduction of Treg cells that inversely correlated with SLE activity [107, 108]. Interestingly, glucocorticoid therapy increased Treg cell numbers in these patients [109]. Similarly, in a mixed pool of patients with either limited or diffuse systemic sclerosis, the number of Treg cells along with levels of serum IL-10 and TGF-β were significantly lower when compared with healthy controls [110]. Moreover, Baraut and colleagues showed that in patients with diffuse systemic sclerosis, the decrease of both Treg cell overall numbers and Treg cell-mediated suppressive capacity were rescued after autologous hematopoietic stem cell transplantation [111]. Collectively, these studies showed that impaired suppressive function of Treg cells promotes the development of autoimmunity (Fig 3). Interventions that reconstitute the Treg cell pool (e.g., through adoptive transfer or IL-2 therapy) or modulate epigenetic modifications that translate into enhancement of Treg cell number and suppressive function could provide keys to preventing and managing these disorders.
Epigenome-wide association studies in autoimmune diseases
Genome-wide association studies (GWAS) have been fundamental in identifying single-nucleotide polymorphisms (SNPs) associated with human diseases, and over 300 loci have been associated with autoimmune diseases [112]. However, the vast majority of SNPs do not alter protein-coding sequences, limiting our ability to elucidate their molecular function in disease pathogenesis. Moreover, the effect of environmental factors on disease initiation, progression and phenotypic variation are not included in GWAS. In order to circumvent this limitation and integrate the effect of environmental exposures with known genetic variations to uncover inter-individual variation in common diseases, investigators have focused on the analysis of epigenome-wide association studies (EWAS) [113]. EWAS aim to quantify distinct epigenetic modifications, such as DNA methylation, in order to derive changes in the epigenetic landscape with causality of a specific pathology or trait. Moreover, the identification of SNPs with differentially methylated positions, known as methylation quantitative trait loci (meQTLs), permits the integration of GWAS and EWAS as a means to reveal how distinct genotypes can drive gene expression through epigenetic modifications [114, 115]. Recently, Imgenber-Kreuz and colleagues uncovered several SLE-associated meQTLs, which included PTPRC (encodes CD45), MHC-class II, UHRF1BP1, IRF5, IRF7, IKZF3 and UBE2L3. These findings suggested that some SLE-associated genetic variants exert their effect on disease phenotype through DNA methylation variance [116]. The combined study of genetic and epigenetic modifications offers a comprehensive tool to analyze the mechanisms underlying disease initiation, progression, severity and therapeutic response profile, which could ultimately lead to the development of novel preventive and therapeutic interventions.
Limitations of epidemiological epigenetic studies must be understood in order to enhance their clinical value. Whole blood has been the tissue of choice for most EWAS, and therefore cellular heterogeneity has been a known limitation hindering the interpretation of these studies. Another complicating factor for EWAS interpretation is whether epigenetic modifications drive causality or are the result of the disease (reverse causality). After cell-type proportion adjustment and use of mediation analysis in order to exclude associations likely related to reverse causality, investigators reported nine differentially methylated positions in the major histocompatibility cluster that strongly correlated with genetic risk for RA, providing a potential marker for disease development [117]. One of the key effector cells in the pathogenesis of RA are fibroblast-like synoviocytes, which contribute to joint destruction through the production of pro-inflammatory cytokines and proteases. DNA methylation analysis of isolated fibroblast-like synoviocytes from RA and osteoarthritis patients revealed significant hypomethylation and a simultaneous increase in the expression of genes involved in inflammation, chemotaxis and extracellular matrix regulation among RA patients, highlighting the potential of EWAS in accurately differentiating different pathologies [118]. Besides the potential for discovery of novel mechanisms involved in disease pathogenesis, identification of biomarkers, and characterization of phenotypic variation in disease, EWAS could also enable the recognition of selective therapies that benefit individual patients. Based on this premise, in a cohort of prospectively-followed RA patients, close to 20 differentially methylated positions were found to be associated with response to disease modifying anti-rheumatic agents with the strongest associations found for the ADAMTSL2 and BTN3A2 loci [119]. Altogether, these data validate the need to integrate EWAS findings with GWAS in order to identify high-risk patients based on their genetic background and cumulative environmental exposures. In patients with established disease, these studies could provide insight into the active determinants of therapeutic response, allowing clinicians to modify pharmacotherapy.
Targeting the epigenetic landscape in the treatment of human disease
With an understanding of how epigenetic modifications can drive immune system pathology, there is now a focus on translational applications to modify the epigenome for treatment of human disease. The most well established therapies alter DNA methylation and histone acetylation. The DNMTIs, 5-azacytadine and 5-aza-2’-deoxycytidine (decitabine), were originally developed in the 1960s as cytotoxic agents [120], but it was nearly 20 years before their effects on DNA methylation were discovered [121] (Table 3). Initial studies with these agents were unsuccessful, as conventional dose escalation strategies caused toxicities and poor tolerability [122]. With improved understanding of their mechanism-of-action, researchers discovered that nanomolar doses could achieve effective inhibition of DNA methylation while also improving tolerability [123].
Table 3. DNA methyltransferase inhibitors.
5-azacitadine is a ribonucleotide that can be incorporated into RNA as well as DNA. Guadacitabine is a dinucleotide followed by deoxyguanosine, which makes it less prone to breakdown by cytidine deaminase [136]. DNA methyltransferase (DNMT), myelodysplastic syndrome (MDS), acute myeloid leukemia (AML), non-small cell lung cancer (NCSLC).
| Therapeutic Agent | Mechanism Of Action | Clinical Indications (ref) |
|---|---|---|
| 5-azacytadine | Cytosine analogs that incorporate into DNA and |
- MDS [126] - AML [148] - NSCLC [129] - ovarian cancer [130] - AML [149] - MDS [127] - ovarian cancer [150] |
|
5-aza-2’-deoxycytidine (decitabine) |
• at low doses, covalently bind DNMT resulting in hypomethylation |
|
|
2’-deoxy 5-azacytidylyl-(3’→5’)- 2’deoxyguanosine (guadacitabine) |
• at high doses, halts DNA replication resulting in direct cytotoxicity |
- AML [138] |
As discussed previously, multiple hematologic malignancies are associated with dysregulation of DNA methylation and were therefore initial targets for epigenetic therapies. Myelodysplastic syndrome represents a heterogeneous group of hematologic disorders derived from abnormal progenitor cells that result in hypoproliferative bone marrow and place patients at risk for transformation into various forms of acute leukemia. 5-azacytidine was first studied in patients with MDS, as it was known in vitro to cause immature cell differentiation, especially of malignant cells such as promyelocytes [124, 125]. In the first clinical application of DNMTIs to treat MDS, study participants who received 5-azacytidine had improved response rates, less time to transformation to acute leukemia, and prolonged survival (18 months vs 11 months, p=0.03) when compared with supportive therapy alone [126]. In phase II trials, decitabine showed similar clinical outcomes [127]. DNMTI therapy relies on integration into DNA through multiple cell cycles, and thus studies have reported improvements in survival with longer treatment courses [128].
Following establishment of DNMTI-based therapies as effective with high tolerability in hematologic malignancies, investigators evaluated their efficacy in solid organ malignancies. Non-small cell lung carcinoma [129] and ovarian cancer [130, 131] have shown response to treatment with DNMTIs, although response is highly variable between individuals. The greatest potential of these therapies appears to be in combination with cytotoxic agents, with which DNMTIs appear to sensitize tumors and increase efficacy of conventional cytotoxic agents, even in those patients who have not responded to these therapies previously [132]. Recently, epigenetic therapies have shown promise when used synergistically with novel immunotherapies. Immune checkpoint inhibitors, such as CTLA-4 inhibitors and programmed death-1 (PD-1) inhibitors, have potential for increased efficacy when used with epigenetic modifiers [133]. In melanoma, 5-azacytidine induces specific double stranded RNA production, utilized in host viral defense mechanisms, which upregulates transcription of interferon-β and sensitizes malignant cells to CTLA-4 inhibitors [134]. A similar mechanism has also been shown in colon cancer [135], raising the possibility that this mechanism could be effective against multiple different malignancies. Clinical trial NCT01928576 is currently underway to investigate the efficacy of combined PD-1 inhibition with both 5-azacytidine and a histone deacetylase (HDAC) inhibitor (entinostat) in non-small-cell lung carcinoma.
The first next-generation DNMTI, guadecitabine, may increase the efficacy of hypomethylating agents in the treatment of cancer. Guadecitabine’s novel structure makes it resistant to degradation by the enzyme cytidine deaminase, which prolongs in vivo exposure [136, 137]. Early phase I studies demonstrated the efficacy and safety when used to treat AML [138], with a phase III study currently ongoing (NCT02920008). Outside of AML, there are also multiple preliminary trials evaluating the efficacy of this novel hypomethylating agent in solid tumors such as hepatocellular carcinoma and ovarian cancer.
Conclusions and future directions
A single genome gives rise to the myriad cellular phenotypes that constitute the progenitors and effectors of the immune system. The cellular transcriptional machinery is sensitive to DNA methylation and results in gene expression profiles that confer different cellular functions while maintaining cellular identity. These epigenetic modifications link a cell’s heritable and developmental history with their functional programs in the context of dynamic changes from environmental input, aging and stochasticity. The National Institutes of Health Roadmap Epigenomics Mapping Consortium and the International Human Epigenome Consortium have spearheaded recent efforts to map DNA methylation and other epigenetic marks at a genome-wide scale across different species and human tissues. Through development of novel molecular techniques and bioinformatic approaches allowing single-cell analysis, investigators have been able to uncover how cellular heterogeneity can drive immune system development and cancer progression. Equipped with well-annotated databases, these highly dimensional sequencing and machine-learning approaches promise to provide similar insights into the role of DNA methylation in regulating host defense and autoimmunity. Currently available hypomethylating agents lead to global (even if non-uniform) epigenetic changes. The use of CRISPR-based DNA methylation editing technologies offers the potential for locus-specific methylation editing [139], which could add specificity to epigenetic therapies. Integration of DNA information and DNA modifications (genome and epigenome) with RNA information and RNA modifications (transcriptome and epitranscriptome) will provide a better understanding of the regulatory mechanisms underpinning complex immune system disorders. These datasets could then inform the design of epigenetic pharmacotherapies that target blood malignancies, promote homeostasis in microbial host defense, and mitigate autoimmunity.
Acknowledgments
BDS is supported by NIH/NIAID grant U19AI135964, NIH/NHLBI grant K08HL128867, the Parker B. Francis Research Opportunity Award of the Francis Family Foundation, and the Eleanor Wood-Prince Grants Initiative of the Woman’s Board of Northwestern Memorial Hospital. This manuscript was prepared in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. All authors have read and agree to the journal’s authorship agreement and policy on disclosure of potential conflicts of interest.
Abbreviations:
- (5’ – cytosine – phosphate – guanine – 3’)
CpG dinucleotides
- (mCH)
non-CG methylation
- (DNMT)
DNA methyltransferase
- (5mC)
5-methylcytosine
- (TET)
ten-eleven translocation
- (5hmC)
5-hydroxymethylcytosine
- (5fC)
5-formylcytosine
- (5caC)
5-carboxylcytosine
- (TDG)
thymine DNA glycosylase
- (BER)
base excision repair
- (AID/APOBEC)
activation-induced deaminase/apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like; proteins
- (UHRF)
ubiquitin-like, containing PHD and RING finger domains
- (5mU)
5-methyluracil
- (5hmU)
5-hydroxymethyluracil
- (AP)
apyrimidinic
- (MeDIP)
methyl-DNA immunoprecipitation
- (ChIP)
chromatin immunoprecipitation
- (MBD)
methyl-CpG binding domain
- (RRBS)
reduced representation bisulfite sequencing
- (WGBS)
whole-genome bisulfite sequencing
- (MPP)
multipotent progenitors
- (CLP)
common lymphoid progenitors
- (CMP)
common myeloid progenitors
- (CTCF)
CCCTC-binding factor
- (CHIP)
clonal hematopoiesis of indeterminate potential
- (MDS)
myelodysplastic syndrome
- (AML)
acute myeloid leukemia
- (NMP1)
nucleophosmin 1
- (MLL)
mixed lineage leukemia
- (IDH 1 and IDH 2)
isocitrate dehydrogenase 1 and 2
- (α-KG)
α-ketoglutarate
- (2-HG)
2-hydroxyglutarate
- (CD4-CD8-, DN)
double-negative cells
- (CD4+CD8+, DP)
double-positive cells
- (Th)
helper T
- (LPS)
lipopolysaccharide
- (Dam)
DNA adenine methyltransferases
- (SLE)
systemic lupus erythematosus
- (RA)
rheumatoid arthritis
- (DNMTI)
DNA methyltransferase inhibitor
- (Treg cells)
regulatory T cells
- (FOXP3)
Forkhead box P3
- (IPEX)
immune dysregulation, polyendocrinopathy, enteropathy X-linked
- (CNS)
conserved noncoding DNA sequences
- (CTLA-4)
cytotoxic T lymphocyte antigen 4
- (TGF-β)
transforming growth factor-β
- (GWAS)
genome-wide association studies
- (SNPs)
single-nucleotide polymorphisms
- (EWAS)
epigenome-wide association studies
- (meQTLs)
methylation quantitative trait loci
- (PD-1)
programmed death-1
- (HDAC)
histone deacetylase
- (NSCLC)
non-small cell lung cancer
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Waddington CH. The epigenotype. 1942. Int J Epidemiol 2012;41:10–3. [DOI] [PubMed] [Google Scholar]
- 2.Goldberg AD, Allis CD, Bernstein E. Epigenetics: A landscape takes shape. Cell 2007;128:635–8. [DOI] [PubMed] [Google Scholar]
- 3.Bird A DNA methylation patterns and epigenetic memory. Genes Dev 2002;16:6–21. [DOI] [PubMed] [Google Scholar]
- 4.Schultz MD, He Y, Whitaker JW, et al. Human body epigenome maps reveal noncanonical DNA methylation variation. Nature 2015;523:212–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saxonov S, Berg P, Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci U S A 2006;103:1412–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lyko F The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat Rev Genet 2018;19:81–92. [DOI] [PubMed] [Google Scholar]
- 7.Hendrich B, Bird A. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol Cell Biol 1998;18:6538–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu H, Wang G, Qian J. Transcription factors as readers and effectors of DNA methylation. Nat Rev Genet 2016;17:551–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu X, Zhang Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat Rev Genet 2017;18:517–34. [DOI] [PubMed] [Google Scholar]
- 10.Laird PW. Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet 2010;11:191–203. [DOI] [PubMed] [Google Scholar]
- 11.Parkin J, Cohen B. An overview of the immune system. Lancet 2001;357:1777–89. [DOI] [PubMed] [Google Scholar]
- 12.Maximov A Der lymphozyt als gemeinsame stammzelle der verschiedenen blutelemente in der embryonalen entwicklung und im postfetalen leben der säugetiere. Fol Haematol 1909;8:125–34. [Google Scholar]
- 13.Farlik M, Halbritter F, Muller F, et al. DNA methylation dynamics of human hematopoietic stem cell differentiation. Cell Stem Cell 2016;19:808–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tadokoro Y, Ema H, Okano M, Li E, Nakauchi H. De novo DNA methyltransferase is essential for self-renewal, but not for differentiation, in hematopoietic stem cells. J Exp Med 2007;204:715–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Challen GA, Sun D, Mayle A, et al. Dnmt3a and Dnmt3b have overlapping and distinct functions in hematopoietic stem cells. Cell Stem Cell 2014;15:350–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Broske AM, Vockentanz L, Kharazi S, et al. DNA methylation protects hematopoietic stem cell multipotency from myeloerythroid restriction. Nat Genet 2009;41:1207–15. [DOI] [PubMed] [Google Scholar]
- 17.Trowbridge JJ, Snow JW, Kim J, Orkin SH. DNA methyltransferase 1 is essential for and uniquely regulates hematopoietic stem and progenitor cells. Cell Stem Cell 2009;5:442–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bock C, Beerman I, Lien WH, et al. DNA methylation dynamics during in vivo differentiation of blood and skin stem cells. Mol Cell 2012;47:633–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ji H, Ehrlich LI, Seita J, et al. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature 2010;467:338–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cabezas-Wallscheid N, Klimmeck D, Hansson J, et al. Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome, transcriptome, and DNA methylome analysis. Cell Stem Cell 2014;15:507–22. [DOI] [PubMed] [Google Scholar]
- 21.Ong CT, Corces VG. Ctcf: An architectural protein bridging genome topology and function. Nat Rev Genet 2014;15:234–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jimenez-Useche I, Ke J, Tian Y, et al. DNA methylation regulated nucleosome dynamics. Sci Rep 2013;3:2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schuyler RP, Merkel A, Raineri E, et al. Distinct trends of DNA methylation patterning in the innate and adaptive immune systems. Cell Rep 2016;17:2101–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee PP, Fitzpatrick DR, Beard C, et al. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity 2001;15:763–74. [DOI] [PubMed] [Google Scholar]
- 25.Rodriguez RM, Suarez-Alvarez B, Mosen-Ansorena D, et al. Regulation of the transcriptional program by DNA methylation during human alphabeta T-cell development. Nucleic Acids Res 2015;43:760–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sellars M, Huh JR, Day K, et al. Regulation of DNA methylation dictates cd4 gene expression during development of helper and cytotoxic T cell lineages. Nat Immunol 2015;16:746–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsagaratou A, Aijo T, Lio CW, et al. Dissecting the dynamic changes of 5-hydroxymethylcytosine in T-cell development and differentiation. Proc Natl Acad Sci U S A 2014;111:E3306–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ichiyama K, Chen T, Wang X, et al. The methylcytosine dioxygenase Tet2 promotes DNA demethylation and activation of cytokine gene expression in T cells. Immunity 2015;42:613–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scharer CD, Barwick BG, Youngblood BA, Ahmed R, Boss JM. Global DNA methylation remodeling accompanies CD8 T cell effector function. J Immunol 2013;191:3419–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kumar BV, Connors TJ, Farber DL. Human T cell development, localization, and function throughout life. Immunity 2018;48:202–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Youngblood B, Hale JS, Kissick HT, et al. Effector CD8 T cells dedifferentiate into long-lived memory cells. Nature 2017;552:404–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee JH, Park SJ, Nakai K. Differential landscape of non-CpG methylation in embryonic stem cells and neurons caused by dnmt3s. Sci Rep 2017;7:11295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ehrlich M, Gama-Sosa MA, Huang LH, et al. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res 1982;10:2709–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983;301:89–92. [DOI] [PubMed] [Google Scholar]
- 35.Esteller M CpG island hypermethylation and tumor suppressor genes: A booming present, a brighter future. Oncogene 2002;21:5427–40. [DOI] [PubMed] [Google Scholar]
- 36.Xie M, Lu C, Wang J, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med 2014;20:1472–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kyle RA, Therneau TM, Rajkumar SV, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med 2002;346:564–9. [DOI] [PubMed] [Google Scholar]
- 38.Rawstron AC, Bennett FL, O’Connor SJ, et al. Monoclonal B-cell lymphocytosis and chronic lymphocytic leukemia. N Engl J Med 2008;359:575–83. [DOI] [PubMed] [Google Scholar]
- 39.Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015;126:9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014;371:2488–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Genovese G, Kahler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014;371:2477–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stegelmann F, Bullinger L, Schlenk RF, et al. DNMT3A mutations in myeloproliferative neoplasms. Leukemia 2011;25:1217–9. [DOI] [PubMed] [Google Scholar]
- 43.Walter MJ, Ding L, Shen D, et al. Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia 2011;25:1153–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fried I, Bodner C, Pichler MM, et al. Frequency, onset and clinical impact of somatic DNMT3A mutations in therapy-related and secondary acute myeloid leukemia. Haematologica 2012;97:246–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ley TJ, Ding L, Walter MJ, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med 2010;363:2424–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thol F, Damm F, Ludeking A, et al. Incidence and prognostic influence of DNMT3A mutations in acute myeloid leukemia. J Clin Oncol 2011;29:2889–96. [DOI] [PubMed] [Google Scholar]
- 47.Figueroa ME, Lugthart S, Li Y, et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell 2010;17:13–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moran-Crusio K, Reavie L, Shih A, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 2011;20:11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. New England Journal of Medicine 2013;368:2059–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ko M, Huang Y, Jankowska AM, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 2010;468:839–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ko M, Bandukwala HS, An J, et al. Ten-eleven-translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc Natl Acad Sci U S A 2011;108:14566–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pronier E, Almire C, Mokrani H, et al. Inhibition of TET2-mediated conversion of 5-methylcytosine to 5-hydroxymethylcytosine disturbs erythroid and granulomonocytic differentiation of human hematopoietic progenitors. Blood. 2011;118:2551–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Losman JA, Looper RE, Koivunen P, et al. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 2013;339:1621–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Anso E, Weinberg SE, Diebold LP, et al. The mitochondrial respiratory chain is essential for haematopoietic stem cell function. Nat Cell Biol 2017;19:614–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ono R, Taki T, Taketani T, Taniwaki M, Kobayashi H, Hayashi Y. Lcx, leukemia-associated protein with a cxxc domain, is fused to MLL in acute myeloid leukemia with trilineage dysplasia having t(10;11)(q22;q23). Cancer Res 2002;62:4075. [PubMed] [Google Scholar]
- 56.Lorsbach RB, Moore J, Mathew S, Raimondi SC, Mukatira ST, Downing JR. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10;11)(q22;q23). Leukemia 2003;17:637. [DOI] [PubMed] [Google Scholar]
- 57.Tahiliani M, Koh KP, Shen Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009;324:930–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Delhommeau F, Dupont S, Della Valle V, et al. Mutation in TET2 in myeloid cancers. N Engl J Med 2009;360:2289–301. [DOI] [PubMed] [Google Scholar]
- 59.Langemeijer SM, Kuiper RP, Berends M, et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet 2009;41:838–42. [DOI] [PubMed] [Google Scholar]
- 60.Abdel-Wahab O, Mullally A, Hedvat C, et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood 2009;114:144–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gross S, Cairns RA, Minden MD, et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med 2010;207:339–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009;462:739–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mardis ER, Ding L, Dooling DJ, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 2009;361:1058–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Figueroa ME, Abdel-Wahab O, Lu C, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010;18:553–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xu W, Yang H, Liu Y, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011;19:17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.DiNardo CD, Stein EM, de Botton S, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med 2018;378:2386–98. [DOI] [PubMed] [Google Scholar]
- 67.Lavin Y, Winter D, Blecher-Gonen R, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 2014;159:1312–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Heinz S, Benner C, Spann N, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 2010;38:576–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Saeed S, Quintin J, Kerstens HH, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 2014;345:1251086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Okabe Y, Medzhitov R. Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell 2014;157:832–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ostuni R, Piccolo V, Barozzi I, et al. Latent enhancers activated by stimulation in differentiated cells. Cell 2013;152:157–71. [DOI] [PubMed] [Google Scholar]
- 72.Pacis A, Tailleux L, Morin AM, et al. Bacterial infection remodels the DNA methylation landscape of human dendritic cells. Genome Res 2015;25:1801–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wallner S, Schroder C, Leitao E, et al. Epigenetic dynamics of monocyte-to-macrophage differentiation. Epigenetics Chromatin 2016;9:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang X, Wang X, Liu D, Yu L, Xue B, Shi H. Epigenetic regulation of macrophage polarization by DNA methyltransferase 3b. Mol Endocrinol 2014;28:565–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yu J, Qiu Y, Yang J, et al. DNMT1-ppary pathway in macrophages regulates chronic inflammation and atherosclerosis development in mice. Sci Rep 2016;6:30053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pereira JM, Hamon MA, Cossart P. A lasting impression: Epigenetic memory of bacterial infections? Cell Host Microbe. 2016;19:579–82. [DOI] [PubMed] [Google Scholar]
- 77.Luca MD, Pels K, Moleirinho S, Curtale G. AIMS Molecular Science 4:110–39. [Google Scholar]
- 78.Medzhitov R Recognition of microorganisms and activation of the immune response. Nature 2007;449:819–26. [DOI] [PubMed] [Google Scholar]
- 79.Novakovic B, Habibi E, Wang S- Y, et al. B-glucan reverses the epigenetic state of LPS-induced immunological tolerance. Cell 2016;167:1354–68.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Arts RJW, Joosten LAB, Netea MG. The potential role of trained immunity in autoimmune and autoinflammatory disorders. Front Immunol 2018;9:298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.del Campo R, Martinez E, del Fresno C, et al. Translocated LPS might cause endotoxin tolerance in circulating monocytes of cystic fibrosis patients. PLoS One 2011;6:e29577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Masaki T, Qu J, Cholewa-Waclaw J, Burr K, Raaum R, Rambukkana A. Reprogramming adult schwann cells to stem cell-like cells by leprosy bacilli promotes dissemination of infection. Cell 2013;152:51–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Maekita T, Nakazawa K, Mihara M, et al. High levels of aberrant DNA methylation in helicobacter pylori-infected gastric mucosae and its possible association with gastric cancer risk. Clin Cancer Res 2006;12:989–95. [DOI] [PubMed] [Google Scholar]
- 84.Hino R, Uozaki H, Murakami N, et al. Activation of DNA methyltransferase 1 by EBV latent membrane protein 2A leads to promoter hypermethylation of PTEN gene in gastric carcinoma. Cancer Res 2009;69:2766–74. [DOI] [PubMed] [Google Scholar]
- 85.Miller FW, Alfredsson L, Costenbader KH, et al. Epidemiology of environmental exposures and human autoimmune diseases: Findings from a national institute of environmental health sciences expert panel workshop. J Autoimmun 2012;39:259–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Javierre BM, Fernandez AF, Richter J, et al. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res 2010;20:170–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Brooks WH, Renaudineau Y. Epigenetics and autoimmune diseases: The X chromosome-nucleolus nexus. Front Genet 2015;6:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ciferska H, Horak P, Hermanova Z, et al. The levels of sCD30 and of sCD40L in a group of patients with systemic lupus erythematodes and their diagnostic value. Clin Rheumatol 2007;26:723–8. [DOI] [PubMed] [Google Scholar]
- 89.Lu Q, Wu A, Tesmer L, Ray D, Yousif N, Richardson B. Demethylation of CD40LG on the inactive X in T cells from women with lupus. J Immunol 2007;179:6352–8. [DOI] [PubMed] [Google Scholar]
- 90.Lian X, Xiao R, Hu X, et al. DNA demethylation of cd40l in CD4+ T cells from women with systemic sclerosis: A possible explanation for female susceptibility. Arthritis Rheum 2012;64:2338–45. [DOI] [PubMed] [Google Scholar]
- 91.Shaknovich R, Cerchietti L, Tsikitas L, et al. DNA methyltransferase 1 and DNA methylation patterning contribute to germinal center B-cell differentiation. Blood 2011;118:3559–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dominguez PM, Teater M, Chambwe N, et al. DNA methylation dynamics of germinal center B cells are mediated by AID. Cell Rep 2015;12:2086–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lai AY, Mav D, Shah R, et al. DNA methylation profiling in human B cells reveals immune regulatory elements and epigenetic plasticity at alu elements during B-cell activation. Genome Res 2013;23:2030–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mazari L, Ouarzane M, Zouali M. Subversion of B lymphocyte tolerance by hydralazine, a potential mechanism for drug-induced lupus. Proc Natl Acad Sci U S A 2007;104:6317–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Miceli-Richard C, Wang-Renault S- F, Boudaoud S, et al. Overlap between differentially methylated DNA regions in blood B lymphocytes and genetic at-risk loci in primary Sjögren’s syndrome. Ann Rheum Dis. 2016;75:933–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell 2008;133:775–87. [DOI] [PubMed] [Google Scholar]
- 97.Wildin RS, Ramsdell F, Peake J, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet 2001;27:18–20. [DOI] [PubMed] [Google Scholar]
- 98.Bennett CL, Christie J, Ramsdell F, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet 2001;27:20–1. [DOI] [PubMed] [Google Scholar]
- 99.Baron U, Floess S, Wieczorek G, et al. DNA demethylation in the human FOXP3 locus discriminates regulatory T cells from activated FOXP3(+) conventional T cells. Eur J Immunol 2007;37:2378–89. [DOI] [PubMed] [Google Scholar]
- 100.Kim HP, Leonard WJ. Creb/atf-dependent T cell receptor-induced FoxP3 gene expression: A role for DNA methylation. J Exp Med 2007;204:1543–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Singer BD, King LS, D’Alessio FR. Regulatory T cells as immunotherapy. Front Immunol 2014;5:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Singer BD, Mock JR, Aggarwal NR, et al. Regulatory T cell DNA methyltransferase inhibition accelerates resolution of lung inflammation. Am J Respir Cell Mol Biol 2015;52:641–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.McGrath-Morrow SA, Ndeh R, Helmin KA, et al. DNA methylation regulates the neonatal CD4(+) T-cell response to pneumonia in mice. J Biol Chem. 2018;293:11772–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shu Y, Hu Q, Long H, Chang C, Lu Q, Xiao R. Epigenetic variability of CD4+CD25+ Tregs contributes to the pathogenesis of autoimmune diseases. Clin Rev Allergy Immunol. 2017;52:260–72. [DOI] [PubMed] [Google Scholar]
- 105.Valencia X, Yarboro C, Illei G, Lipsky PE. Deficient CD4+CD25high T regulatory cell function in patients with active systemic lupus erythematosus. J Immunol 2007;178:2579–88. [DOI] [PubMed] [Google Scholar]
- 106.Valencia X, Lipsky PE. CD4+CD25+FoxP3+ regulatory T cells in autoimmune diseases. Nat Clin Pract Rheumatol 2007;3:619–26. [DOI] [PubMed] [Google Scholar]
- 107.Crispin JC, Martinez A, Alcocer-Varela J. Quantification of regulatory T cells in patients with systemic lupus erythematosus. J Autoimmun 2003;21:273–6. [DOI] [PubMed] [Google Scholar]
- 108.Miyara M, Amoura Z, Parizot C, et al. Global natural regulatory T cell depletion in active systemic lupus erythematosus. J Immunol 2005;175:8392–400. [DOI] [PubMed] [Google Scholar]
- 109.Suarez A, Lopez P, Gomez J, Gutierrez C. Enrichment of CD4+ CD25high T cell population in patients with systemic lupus erythematosus treated with glucocorticoids. Ann Rheum Dis 2006;65:1512–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Antiga E, Quaglino P, Bellandi S, et al. Regulatory T cells in the skin lesions and blood of patients with systemic sclerosis and morphoea. Br J Dermatol 2010;162:1056–63. [DOI] [PubMed] [Google Scholar]
- 111.Baraut J, Grigore EI, Jean-Louis F, et al. Peripheral blood regulatory T cells in patients with diffuse systemic sclerosis (SSc) before and after autologous hematopoietic SCT: A pilot study. Bone Marrow Transplant 2013;49:349. [DOI] [PubMed] [Google Scholar]
- 112.MacArthur J, Bowler E, Cerezo M, et al. The new nhgri-ebi catalog of published genome- wide association studies (GWAS catalog). Nucleic Acids Res 2017;45:D896–D901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Paul DS, Beck S. Advances in epigenome-wide association studies for common diseases. Trends Mol Med 2014;20:541–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rakyan VK, Down TA, Balding DJ, Beck S. Epigenome-wide association studies for common human diseases. Nat Rev Genet 2011;12:529–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Feinberg AP. The key role of epigenetics in human disease prevention and mitigation. N Engl J Med 2018;378:1323–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Imgenberg-Kreuz J, Carlsson Almlof J, Leonard D, et al. DNA methylation mapping identifies gene regulatory effects in patients with systemic lupus erythematosus. Ann Rheum Dis 2018;77:736–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Liu Y, Aryee MJ, Padyukov L, et al. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat Biotechnol 2013;31:142–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nakano K, Boyle DL, Firestein GS. Regulation of DNA methylation in rheumatoid arthritis synoviocytes. J Immunol 2013;190:1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Glossop JR, Nixon NB, Emes RD, et al. DNA methylation at diagnosis is associated with response to disease-modifying drugs in early rheumatoid arthritis. Epigenomics 2016;9:419–28. [DOI] [PubMed] [Google Scholar]
- 120.Sorm F, Vesely J. Effect of 5-aza-2’-deoxycytidine against leukemic and hemopoietic tissues in AKR mice. Neoplasma 1968;15:339–43. [PubMed] [Google Scholar]
- 121.Jones PA, Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation. Cell 1980;20:85–93. [DOI] [PubMed] [Google Scholar]
- 122.Saiki JH, McCredie KB, Vietti TJ, et al. 5-azacytidine in acute leukemia. Cancer 1978;42:2111–4. [DOI] [PubMed] [Google Scholar]
- 123.Issa JP. Optimizing therapy with methylation inhibitors in myelodysplastic syndromes: Dose, duration, and patient selection. Nat Clin Pract Oncol 2005;2 Suppl 1:S24–9. [DOI] [PubMed] [Google Scholar]
- 124.Christma JK, Mendelsoh N, Herzog D, Schneiderman N. Effect of 5-azacytidine on differentiation and DNA methylation in human promyelocytic leukemia cells (HL-60). Cancer Res 1983;43:763–9. [PubMed] [Google Scholar]
- 125.Creusot F, Acs G, Christman JK. Inhibition of DNA methyltransferase and induction of friend erythroleukemia cell differentiation by 5-azacytidine and 5-aza-2’-deoxycytidine. J Biol Chem 1982;257:2041–8. [PubMed] [Google Scholar]
- 126.Silverman LR, Demakos EP, Peterson BL, et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: A study of the cancer and leukemia group B. J Clin Oncol 2002;20:2429–40. [DOI] [PubMed] [Google Scholar]
- 127.Kantarjian H, Issa JP, Rosenfeld CS, et al. Decitabine improves patient outcomes in myelodysplastic syndromes: Results of a phase iii randomized study. Cancer 2006;106:1794– 803. [DOI] [PubMed] [Google Scholar]
- 128.Silverman LR, Fenaux P, Mufti GJ, et al. Continued azacitidine therapy beyond time of first response improves quality of response in patients with higher-risk myelodysplastic syndromes. Cancer. 2011;117:2697–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Juergens RA, Wrangle J, Vendetti FP, et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov 2011;1:598–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Fu S, Hu W, Iyer R, et al. Phase 1b-2a study to reverse platinum resistance through use of a hypomethylating agent, azacitidine, in patients with platinum-resistant or platinum-refractory epithelial ovarian cancer. Cancer 2011;117:1661–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Meng F, Sun G, Zhong M, Yu Y, Brewer MA. Anticancer efficacy of cisplatin and trichostatin a or 5-aza-2’-deoxycytidine on ovarian cancer. Br J Cancer 2013;108:579–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Azad N, Zahnow CA, Rudin CM, Baylin SB. The future of epigenetic therapy in solid tumours--lessons from the past. Nat Rev Clin Oncol 2013;10:256–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dear AE. Epigenetic modulators and the new immunotherapies. N Engl J Med 2016;374:684–6. [DOI] [PubMed] [Google Scholar]
- 134.Chiappinelli KB, Strissel PL, Desrichard A, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsrna including endogenous retroviruses. Cell 2015;162:974– 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Roulois D, Loo Yau H, Singhania R, et al. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell 2015;162:961–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chuang JC, Warner SL, Vollmer D, et al. S110, a 5-aza-2’-deoxycytidine-containing dinucleotide, is an effective DNA methylation inhibitor in vivo and can reduce tumor growth. Mol Cancer Ther 2010;9:1443–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Griffiths EA, Choy G, Redkar S, Taverna P, Azab M, Karpf AR. Sgi-110: DNA methyltransferase inhibitor oncolytic. Drugs Future 2013;38:535–43. [PMC free article] [PubMed] [Google Scholar]
- 138.Roboz GJ, Kantarjian HM, Yee KWL, et al. Dose, schedule, safety, and efficacy of guadecitabine in relapsed or refractory acute myeloid leukemia. Cancer 2018;124:325–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Liu XS, Wu H, Ji X, et al. Editing DNA methylation in the mammalian genome. Cell 2016;167:233–47 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Nair SS, Coolen MW, Stirzaker C, et al. Comparison of methyl-DNA immunoprecipitation (MeDIP) and methyl-CpG binding domain (MBD) protein capture for genome-wide DNA methylation analysis reveal CpG sequence coverage bias. Epigenetics 2011;6:34–44. [DOI] [PubMed] [Google Scholar]
- 141.Aberg KA, Chan RF, Xie L, Shabalin AA, van den Oord E. Methyl-CpG-binding domain sequencing: MBD-seq. Methods Mol Biol 2018;1708:171–89. [DOI] [PubMed] [Google Scholar]
- 142.Cokus SJ, Feng S, Zhang X, et al. Shotgun bisulphite sequencing of the arabidopsis genome reveals DNA methylation patterning. Nature 2008;452:215–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Meissner A, Gnirke A, Bell GW, Ramsahoye B, Lander ES, Jaenisch R. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res 2005;33:5868–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lu R, Wang P, Parton T, et al. Epigenetic perturbations by arg882-mutated DNMT3A potentiate aberrant stem cell gene expression program and acute leukemia development. Cancer cell 2016;30:92–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Rasmussen KD, Jia G, Johansen JV, et al. Loss of TET2 in hematopoietic cells leads to DNA hypermethylation of active enhancers and induction of leukemogenesis. Genes & Development 2015;29:910–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Falini B, Nicoletti I, Martelli MF, Mecucci C. Acute myeloid leukemia carrying cytoplasmic/mutated nucleophosmin (npmc+ AML): Biologic and clinical features. Blood 2007;109:874. [DOI] [PubMed] [Google Scholar]
- 147.Ayton PM, Cleary ML. Transformation of myeloid progenitors by MLL oncoproteins is dependent on hoxa7 and hoxa9. Genes & Development 2003;17:2298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia. J Clin Oncol 2010;28:562–9. [DOI] [PubMed] [Google Scholar]
- 149.Kantarjian HM, Thomas XG, Dmoszynska A, et al. Multicenter, randomized, open-label, phase iii trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J Clin Oncol 2012;30:2670–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Matei D, Fang F, Shen C, et al. Epigenetic resensitization to platinum in ovarian cancer. Cancer Res 2012;72:2197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]