

Abstract
Despite relevant residua and sequels, follow-up care of adults with congenital heart disease (ACHD) is too often not performed by/in specialized and/or certified physicians or centers although major problems in the long-term course may develop. The most relevant encompass heart failure, cardiac arrhythmias, heart valve disorders, pulmonary vascular disease, infective endocarditis (IE), aortopathy and non-cardiac comorbidities. The present publication emphasizes current data on IE, pulmonary and pulmonary arterial hypertension and aortopathy in ACHD and underlines the deep need of an experienced follow-up care by specialized and/or certified physicians or centers, as treatment regimens from acquired heart disease can not be necessarily transmitted to CHD. Moreover, the need of primary and secondary medical prevention becomes increasingly important in order to reduce the burden of disease as well as the socioeconomic burden and costs in this particular patient group.
Keywords: Congenital heart defect (CHD), endocarditis, heart failure, primary health care, pulmonary hypertension, aortopathy, prevention
Infective endocarditis (IE) in congenital heart disease
The exact percentage of IE in the general population is unknown. A scientific statement for healthcare professionals from the American Heart Association quotes an annual incidence ranging from 3–7 per 100,000 person-years in the most contemporary population surveys (1-4). The wide range of incidence rates, reported from 1–15 cases per 100,000 per year, vary due to different inclusion data. They result from inclusion of different populations at risk, diverse diagnostic criteria and inclusion of cases with “possible” IE and referral bias (5-8). Epidemiological studies on IE from hospital case series suffer from selection bias, while well conducted prospective studies from population-based investigations are scarce (6). Amongst adults with CHD the incidence of IE remains according to contemporary reports between 0.91 and 1.32 cases/1,000 patient-years (9,10).
The risk is particularly high in adults, in patients with complex congenital heart anomalies, ventricular septal defects, prosthetic valves, and with left sided heart disease (10,11).
Male gender appears to be an independent predictor for developing endocarditis. Right-sided IE is more frequently seen in CHD compared to acquired heart disease. The impact of cyanosis, of types of valves and valved conduits, or whether a valve has been surgically or percutaneously implanted, on the risk of endocarditis is not clearly defined (11-13).
If IE is suspected, all diagnostic measures should be initiated at an early stage, especially in patients with CHD and corresponding symptoms, e.g., fever, night sweats, unclear loss of body weight, and/or newly occurring heart failure. These tests include the modified Duke criteria, laboratory tests, blood cultures, transthoracic and transesophageal echocardiography, and sometimes computed tomography (CT) and positron emission computed tomography (PET-CT) (14).
Before the introduction of antibiotics, IE was almost always lethal. Despite the modern antibiotics and aggressive surgical procedures, this still applies to up to 20% of cases (7).
In any case, particularly in adult CHD a multidisciplinary approach to IE is strongly recommended, including congenital cardiologists, microbiologists, CHD surgeons, and specialists in infectious disease and other disciplines (9). Given the high mortality, IE-prophylaxis should be given to patients at risk. This hypothesis, based on the assumption that bacteremia during medical procedures can lead to IE in patients at risk, is not confirmed by randomized, placebo controlled studies. Since about 1955, national and international societies have published guidelines for IE-prophylaxis, with subsequent adjustments in accordance with the latest findings. Since about 2007, a substantial liberalization of IE-prophylaxis recommendations has taken place (15-18). At present, IE-prophylaxis is only recommended for high-risk patients and an expected severe course and fewer medical procedures have been classified as requiring IE-prophylaxis (1,16,19,20) (Figure 1).
Figure 1.
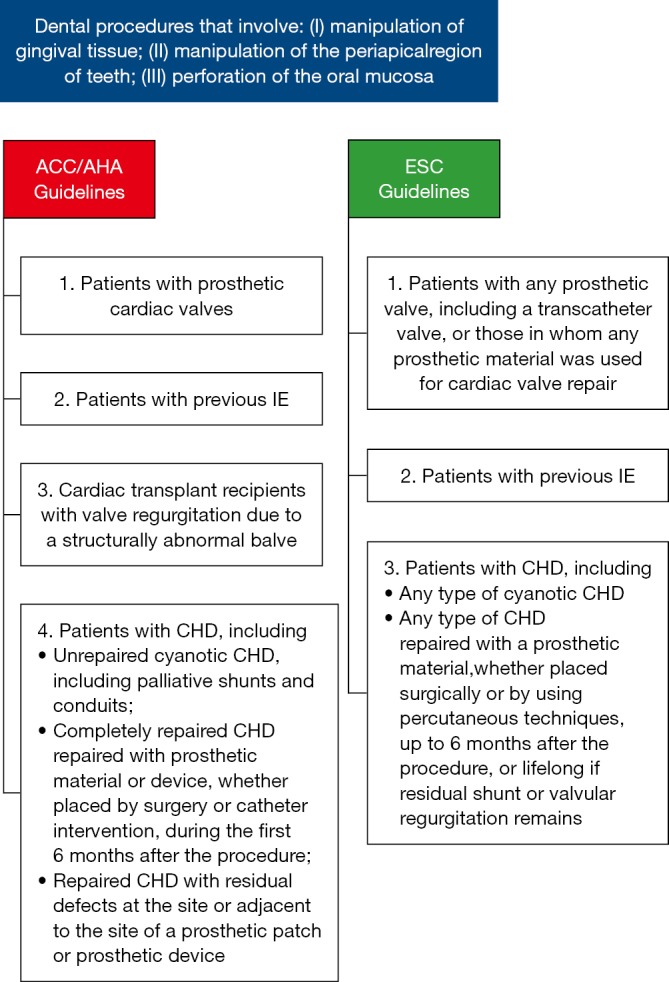
Use of antibiotic prophylaxis for the prevention of IE due to dental procedures, according to the ACC/AHA and ESC guidelines (20,21). IE, infective endocarditis; ACC, American College of Cardiology; AHA, American Heart Association; ESC, European Society of Cardiology.
In international comparison, however, the new recommendations and strategy changes are not uniform (22). It must be emphasized that the new recommendations are viewed controversial in the area of CHD and that at least some experienced clinicians see a wider spectrum of indications.
Before performing IE-prophylaxis, it has to be clarified, whether there is an increased risk of IE, which procedures are planned for diagnostic or therapeutic interventions, how IE-prophylaxis is optimally performed and the risks associated with antibiotic application.
In addition to antibiotic prophylaxis, good body and oral hygiene is of great importance. Tattoos and body piercing are generally not recommended. It is indispensable that not only physicians and dentists, but also all patients at risk, are given these information.
Unfortunately, the knowledge of the patients about the importance and the performance of IE-prophylaxis are often insufficient, despite intensive information from their treating physicians (23). In our experience, it is very successful to hand leaflets with precise recommendations on the IE-prophylaxis to the patients.
Pulmonary hypertension and pulmonary arterial hypertension in congenital heart disease [P(A)H-CHD]
Pulmonary hypertension and pulmonary arterial hypertension in children and adults with CHD [P(A)H-CHD] is a continuum from treatable CHD to severe pulmonary vascular disease (24).
Current estimates suggest that up to 10% of patients with CHD develop a P(A)H, which has a decisive impact on the ability and prognosis of those affected (25-27).
Primary left-right-shunt lesions, congenital obstructions of the left heart, cyanotic heart defects with increased pulmonary flow, and anomalies of the pulmonary artery are the most frequent lesions which can be complicated by a P(A)H, and in which P(A)H may considerably impair the quality and duration of life. Another group includes patients with univentricular hearts after Fontan operation that can develop pulmonary vascular disease (28-30).
Of clinical and prognostic importance is the assignment of the P(A)H-CHD to different classes of PH, including Eisenmenger syndrome, left-right shunt lesions with increased lung flow without cyanosis, PAH randomly associated with a small left-right shunt, or PAH persistent or developing after repair (Table 1, Figure 2) (25).
Table 1. Clinical classification of pulmonary arterial hypertension in congenital shunt lesions; modified from (28,31).
| Group | Class | Definition |
|---|---|---|
| A | Eisenmenger syndrome | ● All major intra- and extra-cardiac cardiovascular defects with initial systemic to pulmonary blood flow (shunt), in which PVR increases greatly over the course of the disease and in which there is a consecutive bidirectional shunt or a complete shunt reversal (blood flow from the lungs to the systemic circulation) ● In clinical terms, there is cyanosis, secondary erythrocytosis and cyanosis-remultiple organ involvement |
| B | Left-to-right shunts correctable (by intervention or surgery) | ● Medium- to large-size defects with mild to moderate systemic-pulmonary blood flow but no cyanosis at rest |
| C | PAH, coincidentally associated with a CHD | ● PAH is coincidentally associated with a congenital heart defect ● Markedly elevated PVR in the presence of small congenital defects that are not primarily responsible for the development of elevated PVR (in adults this is typically a ventricular septal defect with an effective diameter <1 cm or an atrial septal defect with an effective diameter <2 cm as measured by echocardiography) ● The clinical picture strongly resembles idiopathic PAH ● Defect closure is contraindicated ● The diameter measured does not always indicate the haemodynamic relevance of the defect! ● For a more precise assessment of the shunt haemodynamics, pressure gradients, shunt size and direction and the ratio of pulmonary to systemic flow (Qp/Qs) must be taken into consideration |
| D | PAH after previous repair | ● Persistent PAH that reoccurs within months or years after repair of the CHD, without haemodynamically relevant re- or residual shunts |
| E | Others | ● Segmental PAH ● Pulmonary vascular disease after previous Fontan-Operation |
PVR, pulmonary vascular resistance; PAH, pulmonary arterial hypertension; CHD, congenital heart defect.
Figure 2.

Differential diagnostic aspects in pulmonary hypertension due to congenital heart disease (32). PAH, pulmonary arterial hypertension.
Early and correct diagnosis of P(A)H is of outstanding importance for the care of these patients, as quite often a timely surgical or interventional treatment can improve the clinical course and the prognosis considerably.
The complexity of the underlying diseases makes it advisable to guide all ACHD jointly in cooperation between congenital cardiologists and P(A)H-experts (32). This is particularly important when decisions are made on the initiation of a PAH-targeted therapy, the options for an operative or interventional treatment (e.g., interventional Pott-shunt, atrioseptostomy) or decisions on intensive care measures and/or lung- or heart-lung transplantation (25,33).
Adults with P(A)H-CHD differ fundamentally from other P(A)H forms regarding diagnostics, risk stratification, and therapy decisions. This is especially true for cyanotic patients with and without Eisenmenger syndrome (34). Referring to this, certain special features, such as life style, IE prophylaxis, vaccination (influenza and pneumococcae), psychological support, physical activity and physical training, behavior on flights and recommendations for pregnancy, contraception or elective operations have to be taken into consideration (25,34).
It is important to recognize that some treatment forms may have negative systemic site-effects in these vulnerable patients. Therefore, the respective individual disorder has to be taken into account meticulously, particularly concerning of supportive therapy with vasodilators (AT-blockers, ACE-inhibitors), diuretics, oxygen, oral anticoagulation, phlebotomy as well as replacement for anemia and/or iron deficiency (35,36).
For specific pharmacotherapy in PAH-CHD several drug classes are available, including endothelin antagonists, PDE 5 inhibitors, prostanoids or IP prostacyclin receptor agonists, as well as stimulants of the soluble guanylyl cyclase (sGC). Their application can lead to a significant improvement in quality of life and prognosis (25,32).
Current trial data on P(A)H-CHD leave many questions unanswered, particularly which drugs are primarily used and whether mono- or combination therapy is preferable. Currently, treatment decisions for ACHD are based largely on clinical expertise and only to a limited extent on clinical trials; they must hence remain limited to experienced specialists (Figure 3) (25,32,36).
Figure 3.

Suspected diagnosis of PAH associated with congenital heart disease/Eisenmenger syndrome. Modified from (31). PAH, pulmonary arterial hypertension.
Aortopathy in ACHD
The association of aortic pathophysiological degeneration, aortic dilation and aortic-ventricular interaction can be recognized as a new clinical entity: “Aortopathy” (37,38).
Aortic ectasia is well recognized in Marfan syndrome, Turner syndrome, bicuspid aortic valve or aortic coarctation, and consistently associated with ascending aortic and/or para-coarctation medial degeneration (39-41). CHD, such as univentricular hearts, truncus arteriosus communis, transposition of the great arteries, hypoplastic left heart syndrome and tetralogy of Fallot, may also be associated with aortic medial degeneration and aortic dilatation (39,42,43).
Aortic medial degeneration, inaccurately called “cystic medial necrosis”, reach their severest form in Marfan syndrome and annuloaortic ectasia. In other CHD they are qualitatively similar but seldom quantitatively so pronounced. Aortic medial degeneration possibly reflect also in CHD a common developmental fault that weakens the aortic wall, causing aortic dilatation with decreased aorta elasticity and increased stiffness (44,45). A subset of adults with CHD exhibit ongoing ectasia of the aortic root and reduced aortic elasticity that may relate to intrinsic properties of the aortic root. This new concept of aortic dilatation is shifting in CHD-patients the paradigm of aortic dilatation from so called “poststenotic dilatation” to primary intrinsic aortopathy.
Aortic dilatation and increased aortic stiffness can induce aortic aneurysm, rupture and aortic valve regurgitation, but also left ventricular hypertrophy, reduced coronary artery flow and left ventricular failure (45).
For prevention of aortic dilatation beta-blockers and angiotensin-converting enzyme (ACE) inhibitor are frequently prescribed in Marfan syndrome. Since the histopathological changes are similar in CHD, it might be logical to administer beta-blockers and/or angiotensin receptor blocker also to the CHD patients with aortopathy, but scientific data are not robust and definite medication is not yet established.
Monogenetic aortic syndromes
Monogenetic aortic syndromes are a subgroup of CHD, frequently complicated by aortic root aneurysms. These syndromes are rare, and are usually inherited as an autosomal dominant trait. Marfan-, Loeys-Dietz- (46), vascular Ehlers-Danlos syndrome (47), or familial non-syndromic thoracic aortic aneurysm or dissection (TAAD) are the most frequent entities (48). In untreated Marfan patients, life expectancy is reduced by 30–40% of the normal population (49), while the other mentioned syndromes have a similar or even worse prognosis (48-50). If left untreated, most patients die from aortic dissection or rupture, but heart failure, ventricular arrhythmia, and IE may also cause mortality and morbidity.
Especially in Marfan syndrome a primary cardiomyopathy may exist, but it is rarely the exclusive cause of severe heart failure. Heart failure results typically from severe aortic valve regurgitation due to aortic root dilatation, from mitral valve prolapse, or after heart surgery. Myocardial dysfunction is probably the major cause of ventricular arrhythmias and sudden death in Marfan syndrome.
Optimal treatment of monogenetic aortic syndromes is possible only in multidisciplinary expert centres (50). Mainstay of treatment is medication with beta blockers, ACEI, or ARBs, and prophylactic surgery with replacement of the aortic root with aortic valve-sparing root replacement or composite graft-valve replacement. Such treatment has been shown to significantly improve life expectancy at least in Marfan patients. Unfortunately, under-diagnosis of Marfan syndrome remains a substantial problem, where misdiagnosis and diagnostic delay were shown to result in delay of treatment with fatal consequences in many patients (48).
In conclusion, although today most patients with CHD survive into adulthood, many of them have relevant residua and sequels, and deeply need an experienced follow-up care by specialized and/or certified physicians or centres. Thereby, it is important that treatment regimens from acquired heart disease are not necessarily transmitted to CHD. Even simple, postoperative heart defects that were until recently considered to be harmless can lead to problems with age, a fact that had not been expected so far. Moreover, the awareness of ACHD problems must be increased to close actual supply gaps.
The field of CHD, primary and secondary medical prevention will henceforth become increasingly important in order to reduce the burden of disease as well as the socioeconomic burden and costs.
Acknowledgements
The paper is dedicated to the memory of Dr. Joseph Kayle Perloff, Los Angeles, founder of the new specialty of Cardiology—‘Adults with congenital heart disease’—who passed away on August 18, 2014.
Funding: This work was supported by the German Heart Foundation (“Deutsche Herzstiftung e.V.) (grant number F-30-15), the patient organization “Herzkind e. V.”, Actelion Pharmaceuticals Germany GmbH (grant number MED-2015-495) and the German health care insurance AOK-Bayern.
Footnotes
Conflicts of Interest: R Neidenbach received research grants (“Unrestricted educational grant”) from Actelion Pharmaceuticals Deutschland GmbH and from the German Heart Foundation (“Deutsche Herzstiftung e.V.”) and the patient organization “Herzkind e. V.”; H Kaemmerer received fees and/or travel expenses for consulting activities and/or lectures from the following companies within the last 3 years: Actelion, Pfizer, Bayer-Healthcare, Bristol-Myers Squibb; D Pittrow has received speaker fees or honoraria for consultations from Actelion, Bayer, Genzyme, Boehringer Ingelheim, Novartis, MSD. and Dr. Erwin Oechslin currently holds the “Bitove Family Professorship for Adult Congenital Heart Disease”. Other author have no conflicts of interest to declare.
References
- 1.Baddour LM, Wilson WR, Bayer AS, et al. Infective endocarditis in adults: diagnosis, antimicrobial therapy, and management of complications: a scientific statement for healthcare professionals from the American Heart Association. Circulation 2015;132:1435-86. 10.1161/CIR.0000000000000296 [DOI] [PubMed] [Google Scholar]
- 2.Correa de Sa DD, Tleyjeh IM, Anavekar NS, et al. Epidemiological trends of infective endocarditis: a population-based study in Olmsted County, Minnesota. Mayo Clin Proc 2010;85:422-6. 10.4065/mcp.2009.0585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Duval X, Delahaye F, Alla F, et al. Temporal trends in infective endocarditis in the context of prophylaxis guideline modifications: three successive population-based surveys. J Am Coll Cardiol 2012;59:1968-76. 10.1016/j.jacc.2012.02.029 [DOI] [PubMed] [Google Scholar]
- 4.Federspiel JJ, Stearns SC, Peppercorn AF, et al. Increasing US rates of endocarditis with Staphylococcus aureus: 1999-2008. Arch Intern Med 2012;172:363-5. 10.1001/archinternmed.2011.1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abdulhak AA, Baddour LM, Erwin PJ, et al. Global and regional burden of infective endocarditis, 1990–2010: a systematic review of the literature. Global Heart 2014;9:131-43. 10.1016/j.gheart.2014.01.002 [DOI] [PubMed] [Google Scholar]
- 6.Cresti A, Chiavarelli M, Scalese M, et al. Epidemiological and mortality trends in infective endocarditis, a 17-year population-based prospective study. Cardiovasc Diagn Ther 2017;7:27-35. 10.21037/cdt.2016.08.09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murdoch DR, Corey GR, Hoen B, et al. Clinical presentation, etiology, and outcome of infective endocarditis in the 21st century: the International Collaboration on Endocarditis–Prospective Cohort Study. Arch Intern Med 2009;169:463-73. 10.1001/archinternmed.2008.603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tleyjeh IM, Abdel-Latif A, Rahbi H, et al. A systematic review of population-based studies of infective endocarditis. Chest 2007;132:1025-35. 10.1378/chest.06-2048 [DOI] [PubMed] [Google Scholar]
- 9.Montanaro C, Dimopoulos K, Shore DF. Infective endocarditis in patients with congenital heart disease: When, where and how. Int J Cardiol 2017;249:171-2. 10.1016/j.ijcard.2017.09.186 [DOI] [PubMed] [Google Scholar]
- 10.Moore B, Cao J, Kotchetkova I, et al. Incidence, predictors and outcomes of infective endocarditis in a contemporary adult congenital heart disease population. Int J Cardiol 2017;249:161-5. 10.1016/j.ijcard.2017.08.035 [DOI] [PubMed] [Google Scholar]
- 11.Kuijpers JM, Koolbergen DR, Groenink M, et al. Incidence, risk factors, and predictors of infective endocarditis in adult congenital heart disease: focus on the use of prosthetic material. Eur Heart J 2017;38:2048-56. [DOI] [PubMed] [Google Scholar]
- 12.Lluri G, Levi DS, Miller E, et al. Incidence and outcome of infective endocarditis following percutaneous versus surgical pulmonary valve replacement. Catheter Cardiovasc Interv 2018;91:277-84. 10.1002/ccd.27312 [DOI] [PubMed] [Google Scholar]
- 13.Diller GP, Baumgartner H. Endocarditis in adults with congenital heart disease: new answers–new questions. Eur Heart J 2017;38:2057-9. 10.1093/eurheartj/ehx044 [DOI] [PubMed] [Google Scholar]
- 14.Pizzi MN, Dos-Subirà L, Roque A, et al. 18F-FDG-PET/CT angiography in the diagnosis of infective endocarditis and cardiac device infection in adult patients with congenital heart disease and prosthetic material. Int J Cardiol 2017;248:396-402. 10.1016/j.ijcard.2017.08.008 [DOI] [PubMed] [Google Scholar]
- 15.Centre for Clinical Practice at NICE. Prophylaxis Against Infective Endocarditis: Antimicrobial Prophylaxis Against Infective Endocarditis in Adults and Children Undergoing Interventional Procedures. London: National Institute for Health and Clinical Excellence (UK), 2008. [PubMed] [Google Scholar]
- 16.Wilson W, Taubert KA, Gewitz M, et al. Prevention of infective endocarditis: guidelines from the American heart association: a guideline from the American heart association rheumatic fever, endocarditis, and Kawasaki disease committee, council on cardiovascular disease in the young, and the council on clinical cardiology, council on cardiovascular surgery and anesthesia, and the quality of care and outcomes research interdisciplinary working group. Circulation 2007;116:1736-54. 10.1161/CIRCULATIONAHA.106.183095 [DOI] [PubMed] [Google Scholar]
- 17.Taubert KA, Wilson W. Is endocarditis prophylaxis for dental procedures necessary? Heart Asia 2017;9:63-7. 10.1136/heartasia-2016-010810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Habib G, Hoen B, Tornos P, et al. Guidelines on the prevention, diagnosis, and treatment of infective endocarditis (new version 2009) The Task Force on the Prevention, Diagnosis, and Treatment of Infective Endocarditis of the European Society of Cardiology (ESC). Eur Heart J 2009;30:2369-413. 10.1093/eurheartj/ehp285 [DOI] [PubMed] [Google Scholar]
- 19.Naber CK, Al-Nawas B, Baumgartner H, et al. Prophylaxe der infektiösen Endokarditis. Der Kardiologe 2007;1:243-50. 10.1007/s12181-007-0037-x [DOI] [Google Scholar]
- 20.Habib G, Lancellotti P, Antunes MJ, et al. 2015 ESC Guidelines for the management of infective endocarditis: The Task Force for the Management of Infective Endocarditis of the European Society of Cardiology (ESC). Endorsed by: European Association for Cardio-Thoracic Surgery (EACTS), the European Association of Nuclear Medicine (EANM). Eur Heart J 2015;36:3075-128. 10.1093/eurheartj/ehv319 [DOI] [PubMed] [Google Scholar]
- 21.Regitz-Zagrosek V, Blomstrom Lundqvist C, Borghi C, et al. ESC Guidelines on the management of cardiovascular diseases during pregnancy: the Task Force on the Management of Cardiovascular Diseases during Pregnancy of the European Society of Cardiology (ESC). Eur Heart J 2011;32:3147-97. 10.1093/eurheartj/ehr218 [DOI] [PubMed] [Google Scholar]
- 22.Khan O, Shafi AM, Timmis A. International guideline changes and the incidence of infective endocarditis: a systematic review. Open Heart 2016;3:e000498. 10.1136/openhrt-2016-000498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bauer UM, Helm PC, Diller GP, et al. Are adults with congenital heart disease informed about their risk for infective endocarditis and treated in accordance to current guidelines? Int J Cardiol 2017;245:105-8. 10.1016/j.ijcard.2017.07.040 [DOI] [PubMed] [Google Scholar]
- 24.Zimmermann R, Schranz D, Ewert P, et al. Pulmonary arterial hypertension in congenital heart defects with shunt: a heterogeneous and complex constellation. Dtsch Med Wochenschr 2013;138:1244-6. [DOI] [PubMed] [Google Scholar]
- 25.Diller GP, Gatzoulis MA. Pulmonary vascular disease in adults with congenital heart disease. Circulation 2007;115:1039-50. 10.1161/CIRCULATIONAHA.105.592386 [DOI] [PubMed] [Google Scholar]
- 26.Duffels MG, Engelfriet PM, Berger RM, et al. Pulmonary arterial hypertension in congenital heart disease: an epidemiologic perspective from a Dutch registry. Int J Cardiol 2007;120:198-204. 10.1016/j.ijcard.2006.09.017 [DOI] [PubMed] [Google Scholar]
- 27.Hjortshøj CS, Jensen AS, Sørensen K, et al. Epidemiological changes in Eisenmenger syndrome in the Nordic region in 1977–2012. Heart 2017;103:1353-8. 10.1136/heartjnl-2016-310979 [DOI] [PubMed] [Google Scholar]
- 28.Kaemmerer H, Apitz C, Brockmeier K, et al. Pulmonary hypertension in grown-ups with congenital heart disease: recommendations of the cologne consensus conference 2016. Dtsch Med Wochenschr 2016;141:S70-S79. [DOI] [PubMed] [Google Scholar]
- 29.Derk G, Houser L, Miner P, et al. Efficacy of endothelin blockade in adults with Fontan physiology. Congenit Heart Dis 2015;10. [DOI] [PubMed] [Google Scholar]
- 30.Hays BS, Baker M, Laib A, et al. Histopathological abnormalities in the central arteries and veins of Fontan subjects. Heart 2018;104:324-31. [DOI] [PubMed] [Google Scholar]
- 31.Kaemmerer H, Apitz C, Brocmeier K, et al. Pulmonary hypertension in adults with congenital heart disease: Updated recommendations from the Cologne Consensus Conference 2018. Int J Cardiol 2018. [Epub ahead of print]. 10.1016/j.ijcard.2018.08.078 [DOI] [PubMed] [Google Scholar]
- 32.Galiè N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: the Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J 2016;37:67-119. 10.1093/eurheartj/ehv317 [DOI] [PubMed] [Google Scholar]
- 33.Brida M, Gatzoulis MA. Pulmonary arterial hypertension in adult congenital heart disease. Heart 2018;104:1568-74. 10.1136/heartjnl-2017-312106 [DOI] [PubMed] [Google Scholar]
- 34.D'alto M, Diller GP. Pulmonary hypertension in adults with congenital heart disease and Eisenmenger syndrome: current advanced management strategies. Heart 2014;100:1322-8. 10.1136/heartjnl-2014-305574 [DOI] [PubMed] [Google Scholar]
- 35.Kaemmerer H, Mebus S, Schulze-Neick I, et al. The adult patient with Eisenmenger syndrome: a medical update after dana point part I: epidemiology, clinical aspects and diagnostic options. Curr Cardiol Rev 2010;6:343-55. 10.2174/157340310793566154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaemmerer H, Niwa K, Oechslin E, et al. Pulmonary Arterial Hypertension in Congenital Heart Disease: Eisenmenger's Syndrome–A Global Perspective. Available online: http://www.uni-med.de/media/uploads/products/Downloads/Beschreibungen/le-kaemmerer-engl1s.pdf
- 37.Niwa K, Kaemmerer H. Aortopathy. 1st edition. Berlin: Springer Verlag, 2017. [Google Scholar]
- 38.Kuijpers JM, Mulder BJ. Aortopathies in adult congenital heart disease and genetic aortopathy syndromes: management strategies and indications for surgery. Heart 2017;103:952-66. 10.1136/heartjnl-2015-308626 [DOI] [PubMed] [Google Scholar]
- 39.Niwa K, Perloff JK, Bhuta SM, et al. Structural abnormalities of great arterial walls in congenital heart disease: light and electron microscopic analyses. Circulation 2001;103:393-400. 10.1161/01.CIR.103.3.393 [DOI] [PubMed] [Google Scholar]
- 40.Roberts CS, Roberts WC. Dissection of the aorta associated with congenital malformation of the aortic valve. J Am Coll Cardiol 1991;17:712-6. 10.1016/S0735-1097(10)80188-3 [DOI] [PubMed] [Google Scholar]
- 41.McKusick VA. Association of congenital bicuspid aortic valve and Erdheim's cystic medial necrosis. Lancet 1972;1:1026-7. 10.1016/S0140-6736(72)91211-1 [DOI] [PubMed] [Google Scholar]
- 42.Niwa K, Siu SC, Webb GD, et al. Progressive aortic root dilatation in adults late after repair of tetralogy of Fallot. Circulation 2002;106:1374-8. 10.1161/01.CIR.0000028462.88597.AD [DOI] [PubMed] [Google Scholar]
- 43.Yokoi S, Yasui K, Hasegawa Y, et al. Pathological background of subcortical hyperintensities on diffusion-weighted images in a case of neuronal intranuclear inclusion disease. Clin Neuropathol 2016;35:375. 10.5414/NP300961 [DOI] [PubMed] [Google Scholar]
- 44.Seki M, Kurishima C, Kawasaki H, et al. Aortic stiffness and aortic dilation in infants and children with tetralogy of Fallot before corrective surgery: evidence for intrinsically abnormal aortic mechanical property. Eur J Cardiothorac Surg 2012;41:277-82. 10.1016/j.ejcts.2011.05.024 [DOI] [PubMed] [Google Scholar]
- 45.Senzaki H, Iwamoto Y, Ishido H, et al. Arterial haemodynamics in patients after repair of tetralogy of Fallot: influence on left ventricular after load and aortic dilatation. Heart 2008;94:70-4. 10.1136/hrt.2006.114306 [DOI] [PubMed] [Google Scholar]
- 46.Loeys BL, Schwarze U, Holm T, et al. Aneurysm syndromes caused by mutations in the TGF-β receptor. N Engl J Med 2006;355:788-98. 10.1056/NEJMoa055695 [DOI] [PubMed] [Google Scholar]
- 47.Pepin MG, Schwarze U, Rice KM, et al. Survival is affected by mutation type and molecular mechanism in vascular Ehlers–Danlos syndrome (EDS type IV). Genet Med 2014;16:881-8. 10.1038/gim.2014.72 [DOI] [PubMed] [Google Scholar]
- 48.Von Kodolitsch Y, De Backer J, Schüler H, et al. Perspectives on the revised Ghent criteria for the diagnosis of Marfan syndrome. Appl Clin Genet 2015;8:137-55. 10.2147/TACG.S60472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murdoch JL, Walker BA, Halpern BL, et al. Life expectancy and causes of death in the Marfan syndrome. N Engl J Med 1972;286:804-8. 10.1056/NEJM197204132861502 [DOI] [PubMed] [Google Scholar]
- 50.von Kodolitsch Y, Rybczynski M, Vogler M, et al. The role of the multidisciplinary health care team in the management of patients with Marfan syndrome. J Multidiscip Healthc 2016;9:587-614. 10.2147/JMDH.S93680 [DOI] [PMC free article] [PubMed] [Google Scholar]