

Abstract
There is a significant drive to identify alternative materials that exhibit room temperature phosphorescence for technologies including bio-imaging, photodynamic therapy and organic light-emitting diodes. Ideally, these materials should be non-toxic and cheap, and it will be possible to control their photoluminescent properties. This was achieved here by embedding carbon nanodots within crystalline particles of alkaline earth carbonates, sulphates and oxalates. The resultant nanocomposites are luminescent and exhibit a bright, sub-second lifetime afterglow. Importantly, the excited state lifetimes, and steady-state and afterglow colours can all be systematically controlled by varying the cations and anions in the host inorganic phase, due to the influence of the cation size and material density on emissive and non-emissive electronic transitions. This simple strategy provides a flexible route for generating materials with specific, phosphorescent properties and is an exciting alternative to approaches relying on the synthesis of custom-made luminescent organic molecules.
Materials exhibiting room temperature phosphorescence (RTP) with short afterglow are desirable for bio-medical applications. Here the authors synthesise a library of compounds with tunable RTP properties, embedding carbon nanodots in non-toxic alkaline-earth carbonate, sulphate and oxalate hosts.
Introduction
Room temperature phosphorescence (RTP) is traditionally associated with inorganic phases such as ZnS:Cu and SrAl2O4:Eu, Dy1, where these exhibit afterglow lifetimes of minutes to hours. However, although ideal for applications such as emergency signage, they are less-suited to areas such as anti-counterfeiting and bio-imaging materials, where bright, sub-second afterglow is required2. These compounds are also unstable in water and require large domain sizes ( > 1 μm) for good performance. Significant efforts have therefore been made to develop alternative materials. For decades, RTP was only observed from organic materials at very low temperatures. As a major advance, it has now been shown that RTP can be observed in some transition metal complexes and N-heterocyclic molecular crystals2–6, where intermolecular effects and enhanced structural rigidity facilitates RTP. Structural rigidity has also been achieved by embedding metal-free polyaromatic hydrocarbon (PAH) luminophores in porous media such as metal-organic frameworks, clays and cyclodextrans; or in amorphous matrices4,7–10. Molecular crystals and PAHs show promise in sub-second afterglow applications, but the need for specific molecular design to give desired photoluminescent (PL) properties, known or predicted toxicity11, and challenges associated with poor processability remain.
Carbon dots (CDs) provide an exciting alternative to these materials, and overcome many of the drawbacks, being water-soluble, cheap and easy to synthesise, and exhibiting excellent PL properties12. Additionally, RTP is activated when they are integrated within host matrices such as poly(vinyl alcohol)13–15, amorphous silica16,17, and polyurethane18. In this case, RTP originates from surface aromatic carbonyls19 and/or N-heterocycles20, to which the host material prevents RTP quenching by molecular oxygen and hydrogen bonding confers rigidity. A few studies have also explored KAl(SO4)2·x(H2O)19, layered double hydroxides21, and zeolites22 as inorganic host materials, where these demonstrated comparable RTP activation to polymer hosts. Calcite (CaCO3) has also been used as a host phase for CDs, yielding fluorescent nanocomposites, but no RTP was reported23.
That CDs must be incorporated into a solid host to activate RTP suggests an exciting prospect—that it may be possible to tune the PL properties according to selection of the host material. Although polymeric hosts offer limited scope for varying atomic composition, inorganic materials can be synthesised using the periodic table of metal cations. Heavy atoms are predicted to influence emissive and non-emissive electronic transitions, and in particular spin-inversion transitions as a result of enhanced spin-orbit coupling (SOC). This article demonstrates the power of this approach by integrating carbon nanodots (CNDs) into a range of alkaline earth carbonates, sulphates, and oxalates24–28. This is achieved using a simple, one-pot method in which the CNDs are used as crystallisation additives24,28–30. Although other reports describing inorganic and hydrogen bonding-rich hosts for CDs may describe longer lifetimes and higher quantum yields22,31, our method provides a systematic and easy approach to controlling PL properties through the selection of both the cation and anion, and the functionality of the CNDs, yielding specific fluorescence and phosphorescence colours and lifetimes. Importantly, lifetimes decrease systematically with increasing mass of the metal cation, providing a powerful demonstration of the effects of heavy atoms on SOC. That this can be achieved using biologically friendly materials such as CaCO3—where these can readily incorporate a host of guest species including proteins and drug molecules—opens the door to alternative simple-to-prepare theranostic agents2.
Results
Synthesis and properties of folic acid-derived CNDs
Folic acid-derived CNDs (F-CNDs) were synthesised by thermal degradation of folic acid using a hydrothermal method. A sodium folate solution was heated at 200 °C for 12 h in an autoclave20, and subsequent filtration of the resulting orange F-CND solution removed any large aggregates. Characterisation of the F-CNDs using Fourier transform infrared (FTIR) and Raman spectroscopy confirmed the presence of N-heterocycle motifs, which are retained from the folic acid precursor (Supplementary Fig. 1). These were also detected by X-ray photoelectron spectroscopy (XPS), along with ketones and carboxylate groups (Supplementary Note 1 and Supplementary Fig. 2). The latter contribute to a negative surface potential of 20 mV at pH 8. No graphitic carbon was detected by XPS (Supplementary Note 1 and Supplementary Fig. 2), or by transmission electron microscopy (TEM) (Supplementary Fig. 1), which confirmed that the nanoparticles were between 3 and 5 nm is size, and amorphous.
The photophysical properties of the F-CNDs were then investigated. Blue fluorescence was observed from aqueous solutions of F-CNDs on excitation at 365 nm (Supplementary Fig. 1). No phosphorescence was seen. F-CNDs are prone to self-quenching such that the fluorescence intensity was constant at concentrations above ≈ 1.5 × 10−4 g mL−1 (Supplementary Fig. 3). Steady-state photoluminescence (SS-PL) spectroscopy of these solutions revealed an excitation maximum at 320 nm for an emission maximum at 398 nm with a quantum yield (ΦF) of approximately 11% (Supplementary Fig. 1). Longer wavelength excitation (365 nm and 390 nm) yielded red-shifted emission maxima at 430 nm and 442 nm, respectively, but with significantly and progressively lower intensities (Supplementary Fig. 4).
Solutions of the F-CNDs were also analysed using fluorescence lifetime imaging microscopy (FLIM), yielding fluorescence lifetimes of = 4.81 ns (13.4% of the decay signal) and = 0.59 ns (86.6%). Dried particles exhibited shorter fluorescence lifetimes of = 1.46 ns (1.3%) and = 0.26 ns (98.7%), where the lifetime and proportion of the decay signal of the longer lifetime component were significantly reduced, and the fluorescence intensity also dropped dramatically, whereas the effect on the shorter lifetime component was proportionally weaker. Longer lifetime radiative processes are expected to be governed by electron-hole trapping by carbonyls, carboxylic acids, and N-heterocycles that are present on the surfaces of CDs and CNDs, and are therefore likely to be influenced by drying of these nanoparticles, or their integration into a solid host32,33.
Synthesis and characterisation of F-CND/host nanocomposites
F-CND/inorganic nanocomposites were prepared by precipitating alkaline earth (Ca, Sr, and Ba) carbonate, sulphate, and oxalate crystals in the presence of F-CNDs using a simple, direct-mixing protocol. Powder X-ray diffraction (pXRD) (Supplementary Fig. 5) and Raman spectroscopy (Supplementary Fig. 6) were used to determine the structures of the product crystals, where the results are summarised in Supplementary Table 1. Scanning electron microscopy (SEM) showed that SrCO3, BaCO3, and BaC2O4·0.5H2O particles were polycrystalline and took the form of bundles of particles or spherulites (Supplementary Fig. 7), whereas the other nanocomposites precipitated as single crystals, and exhibited unmodified morphologies as compared with control experiments (Supplementary Fig. 8). The quantities of CNDs in the nanocomposite crystals were determined by dissolution of the crystals, and measurement of the fluorescence intensity of the resulting solutions. All of the inorganic particles examined contained 0.008 to 0.002 wt% F-CNDs. It is noted that the F-CNDs may be occluded between crystalline units in the polycrystalline samples rather than within the crystal lattice as occurs with the single crystals.
The interiors of the calcite/F-CND nanocomposite crystals were analysed using TEM, where thin sections were prepared by focussed ion beam (FIB) milling (Supplementary Fig. 9). The images were identical to sections cut through pure calcite crystals, which demonstrated that the CNDs are not aggregated within the host crystal. It is not possible to image individual amorphous CNDs as the electron beam interacts much more strongly with the crystalline host. Confirmation of the incorporation and location of the F-CNDs within their inorganic hosts was therefore obtained using confocal fluorescence microscopy (CFM). As the host phases are not luminescent (Supplementary Fig. 10), any luminescence observed derives from the F-CNDs. All polycrystals contained a uniform distribution of F-CNDs (Supplementary Fig. 7), whereas all single crystals exhibited preferential occlusion in specific zones (Fig. 1); BaSO4 crystals were too thin (≈ 0.2–2 µm) to determine the nanoparticle location (Supplementary Fig. 7). Calcite crystals exhibited higher fluorescence intensity in one half of their volume, where this is indicative of preferential binding to the acute over the obtuse step edges (Fig. 1)24,34. CaSO4·2H2O, SrSO4, CaC2O4·H2O, and SrC2O4·H2O, in contrast, showed intra-sectoral zoning, where this is characterised by comparable levels of additive incorporation in all zones associated with symmetry-related faces. This is indicative of face-specific additive binding (Fig. 1). Preferential association between the F-CNDs and {011} faces in CaSO4·2H2O, SrSO4 and SrC2O4·H2O, resulted in hourglass motifs. Some association with the {001} faces is also observed for SrSO4 and with the {010} faces for CaC2O4·H2O26.
Fig. 1.

Visualising the integration of folic acid-derived carbon nanodots (F-CNDs) in inorganic single crystals. Optical microscopy images (a-e), confocal fluorescence microscopy (CFM) images (f-j), and distribution models (k-o) of CaCO3 (a,f, k), CaSO4·2H2O (b, g, l), SrSO4 (c, h, l), CaC2O4·H2O (d, i, n), and SrC2O4·H2O (e, j, o). All CFM images have accompanying look-up table (LUT) scales signifying PL intensity, from white (max) to cyan to blue to black (zero). Scale bars: 20 µm (a,c, f, h), 150 µm (b, g), 5 µm (d, i) and 10 µm (e, j)
Photophysical properties of F-CND/host nanocomposites
F-CND/inorganic nanocomposites exhibited SS-PL upon excitation with 365 nm UV light (Fig. 2a). The colour displayed varied between hosts, being cyan for CaCO3, blue for CaSO4·2H2O and green for BaSO4. While it was not possible to achieve precise control over the quantities of F-CNDs in the nanocomposites, the F-CND content could be readily controlled within a range where bright fluorescence was achieved, and where the effects of self-quenching, including reduced afterglow lifetimes and quantum yields caused by enhanced non-radiative energy loss, were minimised (Supplementary Fig. 11). As a distinct difference from F-CNDs alone, all of the nanocomposites exhibited green RTP (Fig. 2a), and SS-PL emission spectra comprised a primary peak at 398 nm, which corresponds to fluorescence, and a secondary peak at 518 nm, which corresponds to phosphorescence. Assignment of phosphorescence was made by (dark-state) spectroscopy of F-CND/SrSO4 (Supplementary Fig. 12). Notably, the relative intensity of phosphorescence to fluorescence (relative RTP intensity) increased with the cation atomic number (Z, where Z(Ca) < Z(Sr) < Z(Ba)) such that the peak appears as a shoulder to the fluorescence peak in CaCO3, but as a separate peak in SrCO3, BaCO3, SrSO4, and BaSO4 (Figs. 2b-d). This increase in relative RTP intensity is most prominent in sulphates, followed by carbonates, and is marginal in oxalates (Figs. 2a-d). Quantum yields were estimated by relating the PL intensity to the F-CND content of each nanocomposite. Fluorescence quantum yields (ΦF) decreased with increasing Z (CaCO3, SrCO3, and BaCO3 = 6.8, 3.2, and 0.5% respectively), whereas phosphorescence quantum yields (ΦP) generally increased with increasing Z (CaCO3, SrCO3, and BaCO3 = 0.3, 0.6, and 1.3%). However, the total quantum yield (Φtot) decreased with increasing Z, suggesting enhanced quenching or non-radiative elimination of excited electronic states (Table 1).
Fig. 2.

Characterisation of the photoluminescent (PL) behaviour of folic acid-derived carbon nanodot (FF-CND)/host composites. Photographs of the nine F-CND/host nanocomposites with, and immediately after the removal of, UV (365 nm) excitation (a). Steady-state photoluminescence (SS-PL) emission spectra (λex = 320 nm) of Ca (black) Sr (red) and Ba (blue) carbonates (b, solid line), sulphates (c, dotted line), and oxalates (d, dashed line) F-CND/host nanocomposites. Images obtained during video stroboscopy experiments of afterglow from F-CND/CaCO3 nanocomposite (e). UV ON: Nanocomposite under UV (365 nm) excitation showing steady-state luminescence. UV OFF: Photographs of the afterglow at different times (160 ms intervals) showing a decay in afterglow intensity with time. Decay curves obtained from video stroboscopy for Ca (black square), Sr (red circle), and Ba (blue triangle) carbonates (f, solid line), sulphates (g, dotted line), and oxalates (h, dashed line) F-CND/host nanocomposites
Table 1.
Physical and PL properties of F-CND/host nanocomposites
| Mineral host | Density (g cm–3) | Relative RTP intensitya | ΦF (%) | ΦP (%) | Φtot(%) | (ns) | (ns) | (%) | (%) | τP b (ms) | c (ms) | c (ms) | c (ms) | c (%) | c (%) | c (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mg5(CO3)4(OH)2·4H2O | 2.25 | 0.03 | 8.6 | <0.1 | 8.6 | 3.45 | 0.47 | 5.6 | 94.4 | ND | 114.9 | 23.4 | 3.6 | 22.9 | 35.1 | 42.0 |
| CaCO3 (Calcite) | 2.71 | 0.13 | 6.8 | 0.3 | 7.1 | 3.27 | 0.56 | 11.0 | 89.0 | 127 | 102.7 | 34.0 | 2.9 | 50.2 | 40.7 | 9.1 |
| SrCO3 | 3.78 | 0.22 | 3.2 | 0.6 | 3.8 | 2.76 | 0.46 | 6.6 | 93.4 | 110 | 82.3 | 26.1 | 5.3 | 42.8 | 23.2 | 34.9 |
| BaCO3 | 4.3 | 0.84 | 0.5 | 1.3 | 1.8 | 2.26 | 0.39 | 3.2 | 96.8 | 72 | 54.7 | 26.7 | 7.9 | 28.9 | 61.1 | 10.0 |
| Pb3(CO3)2(OH)2 | 6.5 | >0.99 | 0.0 | <0.1 | <0.1 | 1.42 | 0.21 | 0.3 | 99.7 | ND | 3.6 | 0.4d | NA | 100 | NA | NA |
| CaCO3 (amorphous) | 1.62 | 0.03 | NC | NC | NC | 2.90 | 0.46 | 7.5 | 92.5 | 130 | 104.0 | 35.6 | 6.7 | 41.3 | 48.9 | 9.8 |
| CaCO3 (vaterite) | 2.65 | 0.08e | NC | NC | NC | ND | ND | ND | ND | 125 | 111.5 | 33.3 | 3.9 | 50.1 | 38.6 | 11.3 |
| CaSO4·2H2O | 2.31 | 0.13 | 7.2 | 0.6 | 7.8 | 2.14 | 0.31 | 1.0 | 99.0 | 121 | 95.7 | 30.4 | 2.3 | 43.7 | 32.5 | 23.8 |
| SrSO4 | 3.95 | 0.72 | 1.7 | 2.9 | 4.6 | 2.13 | 0.37 | 3.6 | 96.4 | 90 | 82.4 | 32.8 | 7.1 | 38.1 | 48.7 | 13.2 |
| BaSO4 | 4.48 | 0.98 | 0.0 | 2.2 | 2.2 | 1.38 | 0.25 | 0.9 | 99.1 | 73 | 48.7 | 19.9 | 3.9 | 38.9 | 53.6 | 7.5 |
| CaC2O4·H2O | 2.21 | 0.09 | 2.7 | 0.1 | 2.8 | 3.33 | 0.52 | 7.6 | 92.4 | 128 | ND | ND | ND | ND | ND | ND |
| SrC2O4·H2O | 2.85 | 0.18 | 0.3 | <0.1 | 0.3 | 2.66 | 0.58 | 7.2 | 92.8 | 105 | ND | ND | ND | ND | ND | ND |
| BaC2O4·0.5H2O | 3.45 | 0.28 | 0.6 | 0.4 | 1.0 | 1.99 | 0.25 | 10.3 | 89.7 | 82 | 57.6 | 20.8 | 6.2 | 28.1 | 57.9 | 14.0 |
| FolicCD (aqueous solution) | NA | 0 | 11 | 0.0 | 11 | 4.81 | 0.59 | 13.4 | 86.6 | ND | ND | ND | ND | ND | ND | ND |
| FolicCD (dried from solution) | NA | 0 | NC | NC | NC | 1.46 | 0.26 | 1.3 | 98.7 | ND | ND | ND | ND | ND | ND | ND |
Tabulated data of density, relative RTP intensity; and fluorescence and phosphorescence quantum yields (Φ), lifetimes (τ), and pre-exponential factors (a)
ND not detected, NA not applicable, NC Not calculated, F-CND folic acid-derived carbon nanodot, PL photoluminescent, RTP room temperature phosphorescence, SS-PL steady-state photoluminescence, TRPM time-resolved phosphorescence microscopy, PLIM phosphorescence lifetime imaging microscopy
aRelative RTP intensity is estimated from SS-PL spectroscopy
bFrom video stroboscopy studies
cFrom TRPM studies
dTaken from PLIM data
eEstimated from ‘polycrystalline’ sample
The PL lifetimes of the F-CNDs also varied according to the composition of the inorganic host. The fluorescence lifetimes (both and ) of the F-CNDs embedded in all inorganic hosts were shorter than in aqueous solutions. Increasing the cation Z resulted in a decrease in the value and proportion of the longer () lifetime (Table 1 and Figs. 3c, d). The shorter () lifetime, however, did not change proportionally with Z, suggesting a different trend (Supplementary Fig. 13). RTP decay curves were obtained from video stroboscopic studies (excitation at 365 nm at 40 ms intervals) (Figs. 2e-h), and these were fitted with a single exponential function, yielding RTP lifetimes (τP) of 127 ms, 110 ms, and 72 ms for CaCO3, SrCO3, and BaCO3, respectively (Table 1 and Figs. 3e, f). This indicates that τP also decreases with increasing cation Z. The sulphates and oxalates also followed this trend (Fig. 3e).
Fig. 3.

The relationship between host structure and photoluminescent (PL) behaviour in folic acid-derived carbon nanodot (FF-CND)/host nanocomposites. Plots of relative room temperature phosphorescence (RTP) intensity values (a, b), fluorescence () lifetimes derived from fluorescence lifetime imaging microscopy (FLIM) decay curves (c, d) and phosphorescence (τP) lifetimes derived from video stroboscopy decay curves (e, f) of F-CND-rich carbonates (black square/solid line), sulphates (red circle/dotted line), and oxalates (blue triangle/dashed line) against cation Z (a, c, e) and density (b, d, f) of the host mineral phase. The background colour in a and b represents approximately the steady-state PL (SS-PL) observed from photographs obtained from composites under UV (365 nm) excitation. Values for for F-CND aqueous solution and dried F-CND are given on c and d
Time-resolved phosphorescence microscopy (TRPM; 40 μW excitation at 340 nm at 5 ms intervals) was additionally used to examine the afterglow, and to confirm that the RTP originated from individual particles. RTP decay plots were fitted with a triple exponential function, yielding three lifetimes of = 102.7 ms (50.7%), = 34.0 ms (40.7%), and = 2.9 ms (9.1%) for calcite (Table 1 and Supplementary Fig. 13). This shows that the RTP behaviour is complex and originates from various relaxation events in the F-CNDs. These lifetimes are shorter than those determined from video stroboscopy due to differences in excitation intensities and wavelengths, but the same trend with respect to the host cation Z is observed (Supplementary Fig. 13).
Influence of CND synthesis on nanocomposite photoluminescence
RTP in CDs has been attributed to carbonyls and N-heterocycles on CD surfaces, where these form during hydrothermal treatment of precursors such as ethanolamine19 and 1,3-diaminobenzene15,16. However, precursors such as folic acid retain N-heterocycle and carboxylic acid moieties during CD formation13,20, which suggests that the PL behaviour can be tailored through selection of the precursor. This strategy was investigated by preparing CDs from riboflavin rather than folic acid as a precursor, where this comprises an N-heterocycle flavin and ribose. Structurally, the N-heterocycle flavin moiety is comparable to the pterin moiety found in folic acid, but it has more extensive conjugation (Supplementary Fig. 14). The excitation and emission maxima of the flavin moiety are therefore at lower energies than those of pterin35,36. Preservation of this moiety during CD synthesis was therefore anticipated to deliver nanocomposites with longer wavelength fluorescence and phosphorescence.
Riboflavin-derived CNDs (R-CNDs) were comparable to F-CNDs in that they were amorphous, as observed by TEM (Supplementary Fig. 15), and contained similar chemical functionalities, including N-heterocycles and C=O bonds, as determined by Raman (Supplementary Fig. 15) and XPS spectroscopy (Supplementary Note 1 and Supplementary Fig. 2). SS-PL data of R-CND solutions revealed an excitation maximum at 380 nm for an emission maximum at 442 nm (Supplementary Fig. 15). The asymmetric emission peak has a broad shoulder at longer wavelengths such that the solution appears blue-green under UV (365 nm) excitation. Taking the example of sulphate hosts, incorporation of R-CNDs (Fig. 4) yielded nanocomposites with a white luminescence under UV (365 nm) excitation, and a yellow afterglow (572 nm cf. 54 nm longer than for F-CND under the same excitation wavelength) (Supplementary Fig. 16). SS-PL demonstrated an emission maximum at 465 nm (cf. 23 nm longer than in aqueous solution under the same excitation), with a very broad shoulder at longer wavelengths corresponding to RTP. As the Z-value of the cation increased, the relative strength of the RTP peak also increased, although the effect was far less pronounced than with F-CNDs (Fig. 4). RTP lifetimes obtained from TRPM studies also followed the same trend as the F-CNDs, becoming shorter with increasing cation Z. However, R-CND/SrSO4 crystals reproducibly exhibited a much longer lifetime than expected, which requires further examination. This study therefore demonstrates that the composition of the CND surface can be varied according to the synthesis method, providing another route to control the PL behaviour of the nanocomposites.
Fig. 4.

Exploring the impact of host phase synthesis and carbon nanodot (CND) precursors on photoluminescent (PL) behaviour. Photographs with, and immediately after the removal of, UV (365 nm) excitation of folic acid-derived CND (F-CND) nanocomposites with a amorphous CaCO3 (amorphous calcium carbonate (ACC)) and b calcite (CaCO3) nanoparticles, and c riboflavin–derived CND (R-CND) nanocomposites with SrSO4. Powder X-ray diffraction (pXRD) patterns of d ACC and e CaCO3 nanoparticles, where the peaks are indexed to calcite. Scanning electron microscopy (SEM) micrographs of nanocomposites are given as insets. (f) Steady-state photoluminescence (SS-PL) emission spectra (λex = 320 nm) for ACC (purple) and CaCO3 nanoparticles (cyan) (Calcite (λex = 320 nm, black), where the SS-PL spectrum is given for comparison. g Video stroboscopy decay plots for F-CND/ACC (purple circle) and F-CND/CaCO3 nanoparticles (cyan inverted triangle)(F-CND/calcite (black square) given for comparison). h SS-PL emission spectra (λex = 380 nm) of R-CND/CaSO4 2H2O (dotted black), R-CND/SrSO4 (dotted red) and R-CND/BaSO4 (dotted blue). i Time-resolved phosphorescence microscopy (TRPM) lifetime measurements for R-CND/inorganic host nanocomposites compared with host density (carbonates (black squares) and sulphates (red circles)). The outlier is R-CND/SrSO4. Scale bars: 500 nm (d) and 300 nm (e)
RTP nanoparticles
We also extended our strategy to the synthesis of water-stable RTP nanoparticles. Although CDs are advantageous in bio-imaging due to excellent PL properties and biocompatibility37–39, they typically require high excitation powers and continuous stimulation, which causes extensive background autofluorescence. This problem can be overcome by integrating them into organic or inorganic matrices. RTP calcite nanoparticles with an average size of 65 ± 7 nm were therefore synthesised by adding CO2 to a F-CND/Ca(OH)2 slurry24 (Fig. 4) where they were characterised using SEM and pXRD. RTP activation confirmed the incorporation of the F-CNDs, and the PL spectrum was comparable to that of the bulk F-CND/CaCO3 nanocomposites (cf. Fig. 2). Our synthetic strategy therefore provides a simple method for preparing water-stable, biocompatible afterglow nanoparticles, whose surfaces can be readily modified.
Discussion
These experiments show that PL behaviour in RTP F-CND/host nanocomposites can be tuned through judicious selection of the inorganic host. Our data show that all fluorescence and phosphorescence quantum yields, lifetimes and relative intensities are related to host cation Z (Table 1). The condensed host phase forces cations close to the F-CND surface, and heavier atoms interfere with electronic transitions (internal conversion (IC), intersystem crossing (ISC), fluorescence, and phosphorescence) within the photoactive carbonyl and N-heterocycles F-CND surface groups (Fig. 5)20. Each transition has an associated rate constant (i.e., , kISC, kF, and kP), which increases with Z, ultimately leading to fluorescence quenching and RTP activation.
Fig. 5.

The mechanism for PL behaviour modification in carbon nanodot (CND)/host nanocomposites. Modified Jablonski diagram indicating electronic states (S0 (black), S1 (blue), and T1 (green) and their relative energy levels; electronic transitions (non-emissive absorption (dotted purple), intersystem crossed (ISC, dotted black), internal conversion (IC, dotted black), and thermal relaxations (jagged black); and emissive fluorescence (solid blue) and phosphorescence (solid green)) and associated rate constants; and electronic spin states (a). The effect of increasing Z on rate constants is summarised in a. The implications of increasing Z on fluorescence and phosphorescence intensities (b) and lifetimes (c) are summarised with photoluminescent (PL) spectra (b) and decay curves (c). PL spectra and decay curves are given in greyscale, from low Z (white) to high Z (black)
Fluorescence quenching is caused by increased and kISC, which depopulate the excited singlet state non-radiatively, and increased kF leads to shortened fluorescence lifetimes. This is known as the Kasha effect, where heavy atoms interfere with electron-hole recombination in luminophores32. RTP activation is due to enhanced ISC as kISC increases, where this populates the first excited triplet state, as is necessary for phosphorescence. ISC is a forbidden spin-inversion process, which is facilitated through SOC, a phenomenon that effectively mixes singlet and triplet character. This is enhanced with both increasing Z and decreasing distance between the heavy atom and the electron undergoing spin inversion40–43, in the well-known heavy atom effect. Phosphorescence, which is also a spin-inversion process, is therefore similarly enhanced, leading to a higher rate of relaxation (kP) to the ground singlet state and a shorter observed lifetime as observed in our study with increasing Z. Also, although ΦP increased with Z, Φtot decreased significantly with Z, which is a result of enhanced radiationless IC (). This competes against other electronic transitions, and effectively quenches the net luminescence.
Further insight into the relationship between Z and RTP was gained by examining hosts with lighter (Mg, Z = 12) and heavier (Pb, Z = 82) metals. Hydromagnesite (Mg5(CO3)4(OH)2·4H2O) and hydrocerrusite (Pb3(CO3)2(OH)2) were precipitated from aqueous solution in the presence of F-CNDs (Supplementary Fig. 17). Mg5(CO3)4(OH)2·4H2O fluoresced blue under UV excitation, and produced a very weak green afterglow (Table 1). The SS-PL emission spectrum featured a significantly less intense shoulder at longer wavelengths than F-CND/CaCO3, signifying a lower relative RTP intensity (Fig. 6) and a much smaller kISC than for calcite. Similarly, TRPM indicates a longer RTP lifetime (Table 1). At the opposite end of the scale, the F-CND/Pb3(CO3)2(OH)2 nanocomposites exhibited a weak yellow SS-PL with a short RTP lifetime of 0.4 ms (Table 1). The SS-PL spectrum featured a single phosphorescence peak, which was red-shifted as compared with F-CND/BaCO3 nanocomposites (Supplementary Fig. 17). A much lower quantum yield (Table 1), and a significantly shorter lifetime suggests that the very high Z of Pb causes a disproportionate increase in compared with kISC.
Fig. 6.
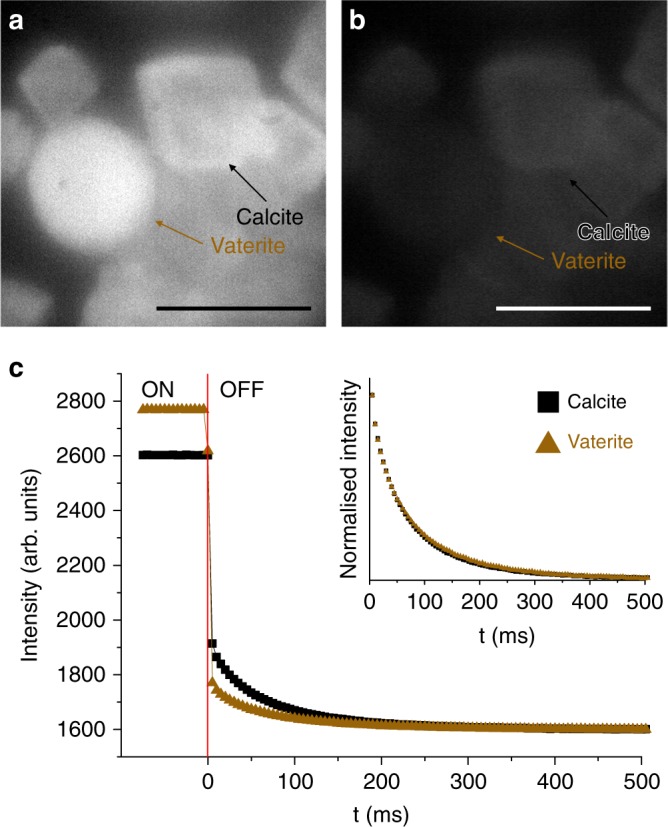
Dependence of room temperature phosphorescence activation on the crystal phase. Time-resolved phosphorescence microscopy images with UV excitation (a) on and (b) 5 ms after the removal of UV excitation, and (c) intensity vs time plots for calcite (black square) and vaterite (brown triangle)-based RTP nanocomposites. The plot in (c) shows the intensity with UV light on and off, and the point at which the light is removed is shown with a vertical red line at t = 0. The normalised decay curves for calcite and vaterite overlap, indicating identical phosphorescent lifetimes (inset; scale bar 50 μm)
The quantum yields, relative RTP intensity, and lifetimes are also influenced by the anion present. This can be related to two effects. First, F-CND/oxalate nanocomposites have much lower quantum yields than the corresponding carbonates and sulphates, which suggests that the oxalate anion may enhance —and therefore quenching—more efficiently than the other anions. Oxalate host phases also contain structural water, which may influence RTP activation. However, as bright luminescence and RTP activation occurs in water-containing CaSO4·2H2O and ACC (CaCO3·H2O) nanocomposites (Figs. 2 and 4), as well as the water-rich KAl(SO4)2·12H2O19 and cyanuric acid-based hosts31, the large reduction in Φtot in oxalate-based hosts is attributed to the oxalate ion itself. It is proposed that the structural water has a passive role, neither actively promoting nor quenching RTP, but causing a decrease in the density of the host crystal when present. This change in the host’s physical properties then influences the RTP behaviour. Second, hosts containing different anions have different densities (ρ). The magnitude of SOC is roughly proportional to the fourth power of Z, and also the inverse cube of distance (r) between the perturbed electron and perturbing nucleus. A denser phase with a cation of the same Z should therefore result in a shorter r, and therefore greater RTP activation44. For a given cation, exchange of the anions yields different crystal structures with different densities, e.g., SrC2O4·H2O, SrCO3, and SrSO4 have densities of 2.85, 3.78, and 3.95 g cm−3, respectively. Sulphates (with the exception of gypsum), generally have higher densities for their respective cations (CaSO4·2H2O, SrSO4, and BaSO4 ρ = 2.32, 3.95, and 4.48 g cm−3), followed by carbonates (CaCO3, SrCO3, and BaCO3 ρ = 2.71, 3.78, and 4.3 g cm−3) and finally oxalates (CaC2O4·H2O, SrC2O4·H2O, and BaC2O4·0.5H2O ρ = 2.21, 2.8545, and 3.4546 g cm−3). The relative RTP intensities follow this pattern, where they increase exponentially with density until a threshold (100%) relative RTP intensity is attained (Fig. 3).
These arguments suggest that single crystals—with their high densities and ability to stabilise and interact with functional groups on CD surfaces—should provide excellent hosts for the RTP activation from CDs28. Supporting this, nanocomposites comprising F-CND nanoparticles within amorphous calcium carbonate (ACC), where ρ = 1.62 g cm−347, exhibited a much lower relative RTP intensity than F-CND/calcite (ρ = 2.71 g cm−3) (Fig. 4), but a comparable RTP lifetime. Similarly, F-CND/vaterite particles also possessed a lower relative RTP intensity than F-CND/calcite, and a brighter fluorescence (Fig. 6). The decay curves are identical, however, which suggests that although calcite is more effective at activating RTP, the crystal structure has no effect on the lifetime. Both fluorescence and phosphorescence lifetimes are therefore more related to Z than to density (Fig. 3).
Finally, our synthetic strategy provides a unique opportunity to create materials with specific PL properties. By simply creating mixed cation hosts—where it is possible to tune the elemental composition of the simple inorganics employed here over a wide range—one can systematically control the average atomic mass and density of the host environment. This strategy was demonstrated here with Sr/BaCO3 and Sr/CaCO3 solid solutions as hosts for F-CNDs (Fig. 7). Mixed metal carbonates were precipitated from solutions containing different ratios of CaCl2, SrCl2, and BaCl2, and defined concentrations of Na2CO3 and F-CND (Supplementary Table 2), and their compositions were determined using XRD (Fig. 7b). All of the product crystals produced RTP after UV excitation (Fig. 7a). Normalised fluorescence spectra (Supplementary Fig. 18) revealed the anticipated progressive increase in relative RTP intensity with increasing average Z (Fig. 7c) while RTP lifetimes decreased with increasing Z (Fig. 7d). Quantum yields determined from absolute PL intensities exhibited a progressive decrease in ΦF, an increase in ΦP, and a significant decrease in Φtot with increasing Z (Fig. 7e). This demonstrates that subtle, controllable changes in the host composition can be used to make predictable changes in the PL behaviour of incorporated F-CNDs.
Fig. 7.

Fine control over folic acid-derived carbon nanodot (F-CND) photoluminescent (PL) properties by changing the composition of the host crystal. a Photographs with, and immediately after the removal of, UV (365 nm) excitation of F-CND nanocomposites with different Ca/Sr/BaCO3 solid solutions as labelled. b X-ray diffraction (XRD) patterns in the range 2θ = 23–27° of samples, where the peak centre for [111] reflection is shown to move to a smaller angle with an increasing average Z of the host. c Relative room temperature phosphorescence (RTP) intensity values obtained from background-subtracted normalised SS-PL spectra showing the progressive activation of RTP with increasing Z. d Lifetime measurements obtained by video stroboscopy showing shorter lifetimes as Z increases. e Fluorescence quantum yields decrease with Z, whereas phosphorescence/RTP quantum yields increase with Z. Overall, the total quantum yield falls.
Materials that exhibit RTP are required for many applications that often demand low toxicity and the ability to tune the PL properties. Although materials based on small organic molecules have shown considerable promise, they can be difficult to process into structures such as nanoparticles; and variation in luminescent behaviour relies on the de novo synthesis of structurally distinct molecules. The work presented here—in which we synthesise luminescent, non-toxic CND/inorganic nanocomposites—offers a powerful strategy for achieving these goals. Rather than varying the luminophore, we show that the PL behaviour of the nanocomposites can be controlled by systematically varying the composition of the crystalline host material. Indeed, by simply selecting the cations and anions, we can not only activate RTP, but also tune the luminescence colour and lifetime. Our approach is therefore ideally suited to the preparation of theranostic agents for backgroundless bio-imaging2 and pH-responsive controlled-release materials48. It also profits from a wealth of existing knowledge about controlling crystallisation processes, such that it is straightforward to optimise the size, shape and surface chemistry of the nanocomposite particles49. This one-pot synthetic protocol therefore offers many advantages over multi-step processes similar Ca-based theranostic agents48. Finally, it is envisaged that our simple and powerful approach can be extended to alternative luminophores, and more widely to other functional materials, such as electroluminescent, catalytic and light-harvesting materials, where a high degree of control over electronic properties is essential.
Methods
Materials
Folic acid, calcium carbonate, calcium chloride dihydrate, strontium chloride hexahydrate, barium chloride, magnesium chloride hexahydrate, sodium carbonate, sodium sulphate, riboflavin, fluorescein sodium salt, dextran (Sigma-Aldrich, UK); lead nitrate, 13% sodium hypochlorite solution, and sodium hydroxide (Fisher, UK) were used as purchased, without further purification. Deionised (DI) water was obtained from an in-house Millipore Reference A+ water purification system (MilliQ, 1–2 ppm OC, 18.2 MΩ).
CND synthesis
In all, 0.1 g folic acid was added to 20 mL DI water and stirred for 20 min yielding a yellow suspension. Under constant stirring, 3 M aqueous NaOH solution was added dropwise to the suspension, resulting in dissolution of folic acid to sodium folate in a yellow solution. The pH of the final solution was 6.0–6.1. The solution was added to a 40 mL stainless steel hydrothermal autoclave with a Teflon insert and sealed. The autoclave was placed in an oven preheated to 200 °C and heated for 12 h. After the oven cooled down to room temperature naturally, the orange/yellow product solution was filtered using a syringe-driven 0.22 µm pore size Millipore polycarbonate filter to remove large particles/aggregates. F-CND powders were obtained by lyophilisation and stored in a desiccator, or used from solution as prepared. For R-CNDs, the same protocol was repeated except folic acid was replaced with riboflavin. All other aspects were identical.
Cation/anion stock solution preparation
All stocks were prepared by adding 20 mL DI water to a calculated amount of solid in a clean 28 mL vial capped with a plastic lid as shown in Supplementary Table 3, and stirred until all powder was fully dissolved. Solutions were filtered before use through a syringe-driven 0.22 µm pore size Millipore polycarbonate filter. Although most of these stock solutions were stable for a few weeks if correctly stored and sealed, Na2CO3 and NaHCO3 stock solutions were made fresh before each experiment.
Crystal growth
In all experiments, 20 mL Petri dish and 400 mL glass beaker reaction vessels were cleaned in a base bath (1 M KOH in iPrOH), rinsed in an acid bath (1 M HCl in H2O), then rinsed with DI water and dried in a 60 °C oven for at least 2 h before use. In total, 25 mL glass vials were used as received. Each reaction vessel was charged with a Piranha solution-cleaned glass substrate before crystallisation liquor was added.
All crystalline phases were precipitated from an aqueous solution, but a range of physical conditions, crystallisers, concentrations, and final volumes were used due to the distinct differences between the phases. For instance, differences in solubility require a consideration for the initial concentrations of reagents; and some materials are distinctly sensitive to pH (e.g., hydromagnesite and hydrocerrusite). Full experimental parameters are provided in Supplementary Table 4, and an example experiment is outlined below.
Calcite was precipitated from a 10 mL final volume of aqueous solution containing [Ca2+] = [CO32−] = 25 mM in a clean, 20 mL glass Petri dish charged with a clean, glass substrate. 200 µL CD solution, 1.25 mL 200 mM CaCl2 aqueous solution and 7.3 mL DI water were added to the clean Petri dish and gently swirled to ensure mixing. Then, the reaction was initiated by adding 1.25 mL 200 mM Na2CO3 aqueous solution and the Petri dish swirled again, yielding a homogeneous suspension. The Petri dish was then covered with parafilm and allowed to react for 2 days at room temperature. When the reaction was complete, the glass substrate, now supporting CND/calcite composite single crystals, was removed, rinsed with DI water and then submerged for 5 min in a 13% sodium hypochlorite solution to remove any loosely bound surface-associated organic components. After bleaching, the glass slide was removed and rinsed with excess DI water, followed by ethanol, and allowed to dry.
The remaining CND/calcite composite single crystals on the surface of the Petri dish were obtained by careful disposal of the crystallisation liquor, followed by rinsing with excess DI water and ethanol. Then, a small amount ( ≤ 5 mL) of ethanol was added into the Petri dish, and the crystals were agitated from the glass surfacing using a spatula. When liberated from the surface and free-flowing in suspension, the crystals were collected from the ethanol by vacuum-driven filtration using a Millipore filtration system and a 0.45 µm polycarbonate membrane. The dried powder was then added to 2 mL 13% sodium hypochlorite solution in a 15 mL glass vial and left for 5 min. After bleaching, the bleach was carefully removed from the vial using a pipette, and carefully disposed. The bleached crystals were then carefully rinsed with excess water and filtered using the Millipore filtration system. Extra washing with DI water and ethanol took place in the filtration system itself. This protocol can be easily adapted for any of the target phases making appropriate adjustments to conditions, reaction vessels, concentrations, volumes, and salts by following the instruction in Supplementary Table 4.
ACC synthesis
A Millipore vacuum-driven filtration system with a 0.45 µm pore size polycarbonate membrane was prepared before the reaction by running approx. 20 mL ethanol through the filter. The vacuum was kept on, while the ACC was prepared. In all cases, 200 µL F-CND solution was mixed with 2.5 mL 200 mM CaCl2 aqueous solution in a 5 mL centrifuge tube. Then, 2.5 mL 200 mM Na2CO3 solution was added rapidly, before the centrifuge was closed and shaken vigorously for 5 s. The gel-like suspension was then poured into the filtration system, and the water drawn away immediately. The centrifuge tube and the retained F-CND/ACC composite was washed with ethanol. The composite was allowed to dry, and was then stored over dried silica desiccant. Due to the sensitivity to moisture, samples were only removed immediately before analysis.
Calcite nanoparticle synthesis
In all experiments, 50 mL DI water was degassed/decarbonated by refluxing at 80 °C in a three-necked round bottom flask under N2 flow for 6 h. In total, 0.44 g CaO, formed by calcination of pure CaCO3 at 900 °C for 8 h, was added, and stirring, heating, and N2 bubbling was continued for 15 min to facilitate Ca(OH)2 formation. The solution was aged in the closed, sealed vessel at room temperature for 16 h. In all, 2 mL F-CND solution was then mixed with aged Ca(OH)2 for 10 min. The solution was carbonated by bubbling a 3:1 mixture N2:CO2 through the solution at an overall flow rate of 1 L min−1 under stirring at room temperature. The reaction progress was monitored by pH, such that the reaction was stopped when pH = 7 by removing gas supply and closing the system. Dry, clean nanoparticles were obtained by centrifugation and twice washing with ethanol, before suspension in sodium hypochlorite (5%) solution for 2 h and further centrifugation and drying steps.
Raman microscopy and spectroscopy
Raman microscopy was conducted on inorganic CND/host nanocomposites grown on glass substrates using a Renishaw inVia Raman Microscope (785 nm laser) with a 50× objective using MS20 encoded sample stage control through rollerball XYZ peripheral. Data acquisition was undertaken with Renishaw WiRE 3.4 with a laser intensity of 0.1% under three accumulated acquisitions (3 × scan time 30 s) between 1200 and 100 cm−1.
Raman spectroscopy of CNDs was conducted using a Renishaw inVia Raman confocal inverted microscope integrated with a Leica DMi8/SP8 laser scanning confocal microscope system, with a 785 nm diode laser (laser power of 4.5 mW on the sample, intensity of ~ 5.7 × 105 W cm−2) and a 1200 l mm−1 grating. Light was collected using a near infrared enhanced CCD array detector (1024 × 256 pixels). Prior to every experiment, a spectrum of a silicon sample was collected and the microscope was calibrated to the peak position (520.5 cm−1). The sample was drop cast onto a quartz slide from an aqueous suspension and dried under nitrogen. Spectra were collected with a 40× objective (NA 0.85 HCX PL APO CORR CS) acquiring for 200 s. Baseline subtraction was done using the Matlab function f_baseline_corr with bandwidth of 350, smoothwidth of 30 and 20 iterations50. Spectra were analysed with reference to Raman spectra of folic acid51 and riboflavin52.
pXRD
pXRD was conducted using a Bruker D2 Phaser with a LYNXEYE detector for phase confirmation studies. Dry powdered samples were deposited onto a silicon substrate as dried powder or from ethanolic suspension. Data were obtained between 2θ = 5 and 95° over 20 min, with the smallest possible step size, on spinning samples. All data acquisition, instrument control, and data conversion were conducted in DIFFRAC.SUITE software package.
SEM
SEM was conducted using an FEI NanoSEM Nova 450 of samples grown directly on clean glass substrates. Samples were mounted on aluminium stubs with double sided Cu tape, with tape folded to a portion of the top surface of the substrate to minimise charging. All samples were coated with 2 nm Ir conductive layer prior to analysis.
Analysis of the CND content of the nanocomposites
A known mass of each nanocomposite was dissolved in a known volume of HCl/EDTA (approx. 2% HCl, 150 mM 2Na.EDTA). The concentration of the CNDs in solution was then determined by comparing the fluorescence intensity (obtained on a Perkin-Elmer Envision 2103, λex = 320 nm, λem = 405 nm) against a calibration curve prepared from known masses of CNDs in the same solvent. These data were then employed to calculate the wt% of CNDs in the nanocomposites.
CFM
CFM was conducted using a Zeiss LSM510 Upright Confocal Microscope of samples grown directly on clean glass substrates under oil immersion where required. Laser and imaging settings were controlled with Zeiss ZEN software (excitation laser at 405 nm, emission low pass filter from 440 nm).
Image analysis
For all confocal fluorescence and SEM micrographs, rendering and analysis was conducted in ImageJ or Fiji applet. For CFM z-stacks, optical images were taken from the central most plane. Fluorescence confocal micrographs were obtained by forming a z-projection for z-stacks. Surface images, as viewed from various angles, were obtained by rendering confocal fluorescence z-stacks into 3D, and rotating the rendered image manually.
SS-PL spectroscopy
SS-PL spectra (excitation and emission) were obtained from diluted CND solution and solid powders using a Jobin Yvon Horiba FluoroMax-3 fluorescence spectrometer operated by FluorEssence (v3.5) software. Powdered samples were prepared for fluorescence spectroscopy in two methods, depending on the required analysis. For relative RTP intensity calculations, nanocomposite samples were mixed with grease and smeared onto a glass substrate. PL spectra of grease and glass slide were used as background signals. For absolute intensity measurements and quantum yield estimates, nanocomposite samples were pulverised with a pestle and mortar, then pelletised with a piston-driven IR press at 7 tons. Pellets were then mounted onto glass slides with grease. PL spectra of a pellet of pure CaCO3 was used as a background signal. In both cases, background subtraction was essential due to high amounts of scattered light. For SS-PL of CND solutions, samples were diluted until optimal signal was obtained at emission maxima, and a quartz fluorescence cuvette were used for obtaining data.
Phosphorescence spectroscopy
Phosphorescence (dark-state) spectra were obtained using Ocean Optics 2000+ interfacing fibre optic signal inlet to a Sony ILX511B CCD detector with Overture (v1.0.1) software control. Powdered samples were prepared as above, and excited with an Applied Photophysics 150 W shuttered xenon arc lamp. Excitation light (360 nm) was filtered with a Comar Optics 360–50 band pass filter (bandwidth between 300 and 400 nm, asymmetric transmittance peak centred on 360 nm).
UV-Visible (UV-Vis) absorbance spectroscopy
Absorption spectra of aqueous CND solutions were obtained on a Perkin-Elmer Lambda 35 spectrometer in a quartz cuvette, against a water blank. Spectra were obtained between 200 and 700 nm at 240 nm min−1, with 1 nm intervals. Data acquisition, instrument control, and automatic background deletion were handled by the UV WinLab software.
FTIR spectroscopy
FTIR spectra were obtained with Perkin-Elmer Spectrum 100 with ATR accessory and sample mounting. Spectra were obtained across 650 to 4000 cm−1 at 1 cm−1 intervals, accumulated over four runs. Data acquisition, instrument control, and automatic background deletion were handled by the Spectrum software.
Zeta potential determination
Zeta potential measurements were obtained using a Malvern Instruments Zetasizer Nano from samples prepared in a Zetasizer Nano disposable capilliary cell. All data were obtained on diluted samples, typically over 20 accumulations, and run six times to ensure agreement of data. Data acquisition, instrument control, data analysis, and conversion were conducted with the Zetasizer family software package (v7). Note that fluorescent, coloured samples are not ideal for zeta potential/dynamic light scattering studies.
Video stroboscopy
Videos of RTP composites were obtained with Canon EOS 7D SLR camera in video mode (25 fps) with manual focus and manual exposure settings; with a Canon EF 100 mm f/2.8 Macro USM lens. Photoluminescence was stimulated with a Spectroline UV lamp (6 W longwave 365 nm), and shuttered manually. Video processing was conducted in VirtualDub 1.10.4 (64-bit) for frame isolation and image sequence generation, followed by image analysis and recomposition in ImageJ 1.46r. These videos were used for low-resolution stroboscopy. Stroboscopic data were obtained by plotting mean grey values against time with a 0.04 s interval, with lifetimes (τ) obtained using Origin Pro 8.
TRPM
High-resolution stroboscopic studies, at a time interval of 5 ms, were conducted using a Nikon Eclipse Ti optical microscope (40× quartz objective) with a sCMOS detector (Hamamatsu ORCA-Flash4.0) controlled by the MicroManager 1.4 software suite. Powders for afterglow nanocomposites were deposited onto a glass coverslip, and illuminated with an LED (40 µW illumination at sample, 340 nm LED with condenser from ThorLabs, powered by either ThorLabs LED driver LEDd1b, set at 0.7 A maximum for continuous illumination; or Thandar TG501 function generator set with a 0.5 or 0.25 Hz squarewave for light/dark measurements). Image analysis was conducted in ImageJ, where stroboscopic data were extracted from individual images as absolute intensity values. Lifetimes and pre-exponential factors (a) were obtained using Origin Pro 8. Pre-exponential factors indicate the proportion of the decay curve, which is calculated to have the corresponding lifetime in a multiexponential plot, and are expressed as a percentage. Here, it is referred to as “x % of the decay signal” or “proportion of the decay curve”.
Fluorescence/phosphorescence lifetime imaging microscopy
Fluorescence/phosphorescence lifetime imaging microscopy (FLIM/PLIM) was performed using a Nikon Eclipse Ti optical microscope with a Becker and Hickl DSC-120 dual channel confocal scanning head and an SPC-150 time correlated-single photon counting controller, using Becker and Hickl in-house software (v 9.77 in 64-bit). All data were obtained in Fifo mode, with both picosecond and microsecond decay traces per pixel. Afterglow nanocomposites were grown directly onto glass slides from conditions as stated above, and imaged with a 60× water immersion objective. Nanocomposites were excited with a two-photon process via a Coherent Mira 700 F, (pumped by a 10 W Verdi CW laser at 532 nm) at 730 nm with variable power and monitored using a Thorlabs silica diode power metre. A Tektronix TDS 3052B Oscilloscope was used to monitor the output of the Ti:Sapphire laser. All data were analysed using a Becker and Hickl image analysis software suite, SPCimage, version 6, or using FlimFit 5.0.3. All lifetime and pre-exponential factors values were taken from the brightest regions of each crystal.
Crystal morphology modelling
XPS
XPS spectra (Supplementary Figure 2) were collected using a Thermo Escalab 250 XPS instrument equipped with a monochromatic Al Kα X-ray source (150 W). These were used to identify elemental composition (Supplementary Table 5) and functional groups (Supplementary Figure 2). Calibration of the binding energies was performed using the carbon 1-s peak at 285 eV. Survey scans were collected between 0 and 1250 eV with a pass energy of 150 eV. The spot size was 500 μm and the analyses were done with a power of 150 W. High-resolution spectra were collected with a pass energy of 20 eV and a step size of 0.1 eV. The data were processed with the CasaXPS software using relative sensitivity factors for the individual elements based upon the scheme where C = 1.
TEM
TEM was conducted using an FEI Tecnai TF20 FEG-TEM fitted with an Oxford Instruments INCA 350 EDX system/80 mm X-Max SDD detector and a Gatan Orius SC600A CCD camera operating at 200 kV. Samples were loaded onto carbon-coated Cu grids.
FIB lithography
Glass substrates loaded with calcite grown in the presence of CNDs were adhered to an aluminium SEM sample holder using silver paint. Milling was performed using a FEI Helio G4 CX FEG SEM instrument equipped with a FIB. The operating voltage was 30 kV and the beam current was varied between 0.1 and 5 nA. All samples were coated in a thick layer of platinum prior to milling to protect the sample where milling was not required.
Fluorescence and phosphorescence quantum yield calculations
The quantum yield of an aqueous solution of F-CNDs was estimated by comparing the luminescence intensity of known particle/molecular concentrations against fluorescein sodium salt solutions. SS-PL data were obtained at λexc = 365 nm with slits at 1 nm, where the quantum yield of fluorescein was calculated as 11.3%. The relative absorbance/excitation for F-CND at 365 nm was ≈ 0.07 of that at the excitation maximum at 320 nm, and this was factored into quantum yield estimate (Supplementary Fig. 1).
Fluorescein solid standards were prepared by mixing a known number of molecules of fluorescein in solution with a known mass (100 mg, which equates to 0.037 cm3 when compacted) of solid and a trace (≈ 0.025 μg) of dextran to prevent aggregation. Samples were then finely ground, and then pelletized with a piston-driven press at 7 tons. Pellets were then analysed by SS-PL, and fluorescence intensities obtained from background-subtracted spectra (background spectrum obtained from pure CaCO3, finely ground and pelletized). Where PL intensity was in a linear relationship with the number of molecules per unit volume, there was no aggregation/quenching, and this region was used in quantum yield estimates.
Quantum yields of F-CND/host nanocomposite solids were estimated by comparing their luminescence (fluorescence and phosphorescence) intensity with fluorescein/CaCO3 mixtures. Particle per unit volume was calculated for all samples. Samples were finely ground, and then pelletized as above. Background-subtracted SS-PL for each sample were used to obtain fluorescence intensities and phosphorescence intensities (and therefore total PL intensities). Phosphorescence intensities were calculated by subtracting a fitted fluorescence peak from the spectrum. These values were compared with luminescence intensities of fluorescein at equivalent concentrations to obtain a quantum yield estimate. Quantum yields were estimated assuming a single absorption/emission event occurred per particle.
Supplementary information
Acknowledgements
This project was supported by Engineering and Physical Sciences Research Council (EPSRC) grants [EP/P005241/1] (D.C.G. and F.C.M.), [EP/M003027/1] and [EP/N002423/1] (M.A.H. and F.C.M.). J.Gd.P. thanks the EPRSC Award (1654179). We would like to thank the Faculty of Biological Sciences Bio-Imaging Suite at the University of Leeds for assistance with confocal fluorescence microscopy; and Dr. Mark Blitz and Mr. David Fogerty at the School of Chemistry, University of Leeds for advice on PL spectroscopy. We are grateful to Dr. Alex Kulak for preparation of FIB samples, and Dr. Yi-Yeoun Kim for assistance with TEM analysis. We also extend our thanks to the UKRI-STFC-Central laser facility at RCaH for access to the Octopus facility under proposal 17330015.
Author contributions
F.C.M. ran the project. D.C.G. designed and conducted all experiments and analysis unless otherwise stated. A.W. and S.W.B. designed and supported FLIM/PLIM and TRPM experiments. D.C.G., M.A.H., M.A.L., and S.Z. ran FLIM/PLIM and TRPM studies. B.R.G.J., J.Gd.P., and D.C.G. supported and ran XPS and Raman studies on CNDs, and analysed the data. D.C.G. and F.C.M. wrote the paper with contributions from all authors.
Data availability
Data (PL, FTIR, Raman, and UV-Vis spectra; FLIM/PLIM, confocal and SEM images; pXRD patterns; and TRPM and stroboscopy videos) that supports the findings of this study are available in the Research Data Leeds Repository with the identifier [http://doi.org/10.5518/371]55.
Competing interests
The authors declare no competing interests.
Footnotes
Journal peer review information: Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
David C. Green, Email: D.C.Green@leeds.ac.uk
Fiona C. Meldrum, Email: F.Meldrum@leeds.ac.uk
Supplementary information
Supplementary Information accompanies this paper at 10.1038/s41467-018-08214-6.
References
- 1.Matsuzawa T, Aoki Y, Takeuchi N, Murayama Y. New long phosphorescent phosphor with high brightness, SrAl2O4:Eu2+,Dy3+ J. Electrochem. Soc. 1996;143:2670–2673. doi: 10.1149/1.1837067. [DOI] [Google Scholar]
- 2.Hirata S. Recent advances in materials with room temperature phosphorescence: photophysics for triplet exciton stabilization. Adv. Opt. Mater. 2017;5:1700116. doi: 10.1002/adom.201700116. [DOI] [Google Scholar]
- 3.Mukherjee S, Thilagar P. Recent advances in purely organic phosphorescent materials. Chem. Commun. 2015;51:10988–11003. doi: 10.1039/C5CC03114A. [DOI] [PubMed] [Google Scholar]
- 4.Bolton O, Lee K, Kim HJ, Lin KY, Kim J. Activating efficient phosphorescence from purely organic materials by crystal design. Nat. Chem. 2011;3:205–210. doi: 10.1038/nchem.984. [DOI] [PubMed] [Google Scholar]
- 5.An ZF, et al. Stabilizing triplet excited states for ultralong organic phosphorescence. Nat. Mater. 2015;14:685–690. doi: 10.1038/nmat4259. [DOI] [PubMed] [Google Scholar]
- 6.Zhen X, et al. Ultralong phosphorescence of water-soluble organic nanoparticles for in vivo afterglow imaging. Adv. Mater. 2017;29:1606665. doi: 10.1002/adma.201606665. [DOI] [PubMed] [Google Scholar]
- 7.Hirata S, et al. Efficient persistent room temperature phosphorescence in organic amorphous materials under ambient conditions. Adv. Funct. Mater. 2013;23:3386–3397. doi: 10.1002/adfm.201203706. [DOI] [Google Scholar]
- 8.Wei JB, et al. Induction of strong long-lived room-temperature phosphorescence of N-phenyl-2-naphthylamine molecules by confinement in a crystalline dibromobiphenyl matrix. Angew. Chem. Int. Ed. 2016;55:15589–15593. doi: 10.1002/anie.201607653. [DOI] [PubMed] [Google Scholar]
- 9.Yang XG, Yan DP. Long-afterglow metal-organic frameworks: reversible guest-induced phosphorescence tunability. Chem. Sci. 2016;7:4519–4526. doi: 10.1039/C6SC00563B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hirata S, Vacha M. White afterglow room-temperature emission from an isolated single aromatic unit under ambient condition. Adv. Opt. Mater. 2017;5:1600996. doi: 10.1002/adom.201600996. [DOI] [Google Scholar]
- 11.Nisbet ICT, Lagoy PK. Toxic equivalency factors (Tefs) for polycyclic aromatic-hydrocarbons (Pahs) Regul. Toxicol. Pharm. 1992;16:290–300. doi: 10.1016/0273-2300(92)90009-X. [DOI] [PubMed] [Google Scholar]
- 12.Lim SY, Shen W, Gao ZQ. Carbon quantum dots and their applications. Chem. Soc. Rev. 2015;44:362–381. doi: 10.1039/C4CS00269E. [DOI] [PubMed] [Google Scholar]
- 13.Deng YH, et al. Long lifetime pure organic phosphorescence based on water soluble carbon dots. Chem. Commun. 2013;49:5751–5753. doi: 10.1039/c3cc42600a. [DOI] [PubMed] [Google Scholar]
- 14.Chen YH, et al. Room temperature phosphorescence from moisture-resistant and oxygen-barred carbon dot aggregates. J. Mater. Chem. C. 2017;5:6243–6250. doi: 10.1039/C7TC01615H. [DOI] [Google Scholar]
- 15.Jiang K, et al. Triple-mode emission of carbon dots: applications for advanced anti-counterfeiting. Angew. Chem. Int. Ed. 2016;55:7231–7235. doi: 10.1002/anie.201602445. [DOI] [PubMed] [Google Scholar]
- 16.Jiang K, Wang YH, Cai CZ, Lin HW. Activating room temperature long afterglow of carbon dots via covalent fixation. Chem. Mater. 2017;29:4866–4873. doi: 10.1021/acs.chemmater.7b00831. [DOI] [Google Scholar]
- 17.Joseph J, Anappara AA. Cool white, persistent room-temperature phosphorescence in carbon dots embedded in a silica gel matrix. Phys. Chem. Chem. Phys. 2017;19:15137–15144. doi: 10.1039/C7CP02731A. [DOI] [PubMed] [Google Scholar]
- 18.Tan J, et al. Large-scale synthesis of N-doped carbon quantum dots and their phosphorescence properties in a polyurethane matrix. Nanoscale. 2016;8:4742–4747. doi: 10.1039/C5NR08516K. [DOI] [PubMed] [Google Scholar]
- 19.Dong XW, et al. Efficient long lifetime room temperature phosphorescence of carbon dots in a potash alum matrix. J. Mater. Chem. C. 2015;3:2798–2801. doi: 10.1039/C5TC00126A. [DOI] [Google Scholar]
- 20.Li QJ, et al. Efficient room-temperature phosphorescence from nitrogen-doped carbon dots in composite matrices. Chem. Mater. 2016;28:8221–8227. doi: 10.1021/acs.chemmater.6b03049. [DOI] [Google Scholar]
- 21.Bai LQ, Xue N, Wang XR, Shi WY, Lu C. Activating efficient room temperature phosphorescence of carbon dots by synergism of orderly non-noble metals and dual structural confinements. Nanoscale. 2017;9:6658–6664. doi: 10.1039/C6NR09648D. [DOI] [PubMed] [Google Scholar]
- 22.Liu J, et al. Carbon dots in zeolites: a new class of thermally activated delayed fluorescence materials with ultralong lifetimes. Sci. Adv. 2017;3:e1603171. doi: 10.1126/sciadv.1603171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo SS, et al. Bioinspired synthesis of fluorescent calcium carbonate/carbon dot hybrid composites. Dalton. T. 2015;44:8232–8237. doi: 10.1039/C5DT00837A. [DOI] [PubMed] [Google Scholar]
- 24.Green DC, et al. 3D visualization of additive occlusion and tunable full-spectrum fluorescence in calcite. Nat. Commun. 2016;7:13524. doi: 10.1038/ncomms13524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kahr B, Gurney RW. Dyeing crystals. Chem. Rev. 2001;101:893–951. doi: 10.1021/cr980088n. [DOI] [PubMed] [Google Scholar]
- 26.Cook AF, Grover PK, Ryall RL. Face-specific binding of prothrombin fragment 1 and human serum albumin to inorganic and urinary calcium oxalate monohydrate crystals. BJU Int. 2009;103:826–835. doi: 10.1111/j.1464-410X.2008.08195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Green DC, et al. Rapid screening of calcium carbonate precipitation in the presence of amino acids: kinetics, structure, and composition. Cryst. Growth Des. 2016;16:5174–5183. doi: 10.1021/acs.cgd.6b00741. [DOI] [Google Scholar]
- 28.Kim YY, et al. Tuning hardness in calcite by incorporation of amino acids. Nat. Mater. 2016;15:903–910. doi: 10.1038/nmat4631. [DOI] [PubMed] [Google Scholar]
- 29.Kim YY, et al. An artificial biomineral formed by incorporation of copolymer micelles in calcite crystals. Nat. Mater. 2011;10:890–896. doi: 10.1038/nmat3103. [DOI] [PubMed] [Google Scholar]
- 30.Kim YY, et al. Structure and properties of nanocomposites formed by the occlusion of block copolymer worms and vesicles within calcite crystals. Adv. Funct. Mater. 2016;26:1382–1392. doi: 10.1002/adfm.201504292. [DOI] [Google Scholar]
- 31.Li Q, et al. Induction of long-lived room temperature phosphorescence of carbon dots by water in hydrogen bond matrices. Nat. Commun. 2018;9:734. doi: 10.1038/s41467-018-03144-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao HX, et al. Highly selective detection of phosphate in very complicated matrixes with an off-on fluorescent probe of europium-adjusted carbon dots. Chem. Commun. 2011;47:2604–2606. doi: 10.1039/c0cc04399k. [DOI] [PubMed] [Google Scholar]
- 33.Bao L, et al. Electrochemical tuning of luminescent carbon nanodots: from preparation to luminescence mechanism. Adv. Mater. 2011;23:5801–5806. doi: 10.1002/adma.201102866. [DOI] [PubMed] [Google Scholar]
- 34.Paquette J, Reeder RJ. Relationship between surface-structure, growth-mechanism, and trace-element incorporation in calcite. Geochim. Cosmochim. Acta. 1995;59:735–749. doi: 10.1016/0016-7037(95)00004-J. [DOI] [Google Scholar]
- 35.Thomas AH, et al. Fluorescence of pterin, 6-formylpterin, 6-carboxypterin and folic acid in aqueous solution: pH effects. Photoch. Photobio. Sci. 2002;1:421–426. doi: 10.1039/b202114e. [DOI] [PubMed] [Google Scholar]
- 36.Kotaki A, Yagi K. Fluorescence properties of flavins in various solvents. J. Biochem. 1970;68:509–516. doi: 10.1093/oxfordjournals.jbchem.a129381. [DOI] [PubMed] [Google Scholar]
- 37.Wang L, et al. Gram-scale synthesis of single-crystalline graphene quantum dots with superior optical properties. Nat. Commun. 2014;5:5357. doi: 10.1038/ncomms6357. [DOI] [PubMed] [Google Scholar]
- 38.Zheng M, et al. Integrating oxaliplatin with highly luminescent carbon dots: an unprecedented theranostic agent for personalized medicine. Adv. Mater. 2014;26:3554–3560. doi: 10.1002/adma.201306192. [DOI] [PubMed] [Google Scholar]
- 39.Ge JC, et al. Red-emissive carbon dots for fluorescent, photoacoustic, and thermal theranostics in living mice. Adv. Mater. 2015;27:4169–4177. doi: 10.1002/adma.201500323. [DOI] [PubMed] [Google Scholar]
- 40.Becker RS. Theory and Interpretation of Fluorescence and Phosphorescence. New York, United States of America: John Wiley & Sons, Inc.; 1969. [Google Scholar]
- 41.Turro, N. J., Ramamurthy, V. & Scaiano, J. C. Modern Molecular Photochemistry of Organic Molecules (University Science Books, Sausalito, CA, 2010).
- 42.Chen ZQ, Bian ZQ, Huang CH. Functional Ir-III complexes and their applications. Adv. Mater. 2010;22:1534–1539. doi: 10.1002/adma.200903233. [DOI] [PubMed] [Google Scholar]
- 43.Ulbricht C, Beyer B, Friebe C, Winter A, Schubert US. Recent developments in the application of phosphorescent iridium(III) complex systems. Adv. Mater. 2009;21:4418–4441. doi: 10.1002/adma.200803537. [DOI] [Google Scholar]
- 44.Marian CM. Spin-orbit coupling and intersystem crossing in molecules. Wires Comput. Mol. Sci. 2012;2:187–203. doi: 10.1002/wcms.83. [DOI] [Google Scholar]
- 45.Christensen AN, Hazell RG. Thermal analysis and crystal structure of tetragonal strontium oxalate dihydrate and of triclinic strontium oxalate hydrate. Acta Chem. Scand. 1998;52:508–512. doi: 10.3891/acta.chem.scand.52-0508. [DOI] [Google Scholar]
- 46.Christensen AN, Hazell RG, Madsen IC. Synthesis and characterization of the barium oxalates BaC2O4·0.5H2O, alpha-BaC2O4 and beta-BaC2O4. Acta Crystallogr. B. 2002;58:808–814. doi: 10.1107/S0108768101020717. [DOI] [PubMed] [Google Scholar]
- 47.Liu J, et al. Evaluation of the particle growth of amorphous calcium carbonate in water by means of the Porod invariant from SAXS. Langmuir. 2010;26:17405–17412. doi: 10.1021/la101888c. [DOI] [PubMed] [Google Scholar]
- 48.Qi C, et al. Calcium-based biomaterials for diagnosis, treatment, and theranostics. Chem. Soc. Rev. 2018;47:357–403. doi: 10.1039/C6CS00746E. [DOI] [PubMed] [Google Scholar]
- 49.Mitragotri S, Lahann J. Physical approaches to biomaterial design. Nat. Mater. 2009;8:15–23. doi: 10.1038/nmat2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koch M, Suhr C, Roth B, Meinhardt-Wollweber M. Iterative morphological and mollifier-based baseline correction for Raman spectra. J. Raman Spectrosc. 2017;48:336–342. doi: 10.1002/jrs.5010. [DOI] [Google Scholar]
- 51.Sun ZJ, Jiang ZW, Li YF. Poly(dopamine) assisted in situ fabrication of silver nanoparticles/metal–organic framework hybrids as SERS substrates for folic acid detection. RSC Adv. 2016;6:79805–79810. doi: 10.1039/C6RA16042E. [DOI] [Google Scholar]
- 52.Liu F, et al. Surface-enhanced Raman scattering study of riboflavin on borohydride-reduced silver colloids: Dependence of concentration, halide anions and pH values. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2012;85:111–119. doi: 10.1016/j.saa.2011.09.043. [DOI] [PubMed] [Google Scholar]
- 53.Kaminsky W. WinXMorph: a computer program to draw crystal morphology, growth sectors and cross sections with export files in VRML V2.0 utf8-virtual reality format. J. Appl. Crystallogr. 2005;38:566–567. doi: 10.1107/S0021889805012148. [DOI] [Google Scholar]
- 54.Kaminsky W. From CIF to virtual morphology using the WinXMorph program. J. Appl. Crystallogr. 2007;40:382–385. doi: 10.1107/S0021889807003986. [DOI] [Google Scholar]
- 55.Green. D. C. et al. Dataset for ‘Controlling the fluorescence and phosphorescence behaviour of carbon nanodots with inorganic crystalline nanocomposites’. Research Data Leeds Repository http://doi.org/10.5518/371 (2018). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data (PL, FTIR, Raman, and UV-Vis spectra; FLIM/PLIM, confocal and SEM images; pXRD patterns; and TRPM and stroboscopy videos) that supports the findings of this study are available in the Research Data Leeds Repository with the identifier [http://doi.org/10.5518/371]55.