

Abstract
Intestinal handling of dietary proteins usually prevents local inflammatory and immune responses and promotes oral tolerance. However, in ~ 1% of the world population, gluten proteins from wheat and related cereals trigger an HLA DQ2/8‐restricted TH1 immune and antibody response leading to celiac disease. Prior epithelial stress and innate immune activation are essential for breaking oral tolerance to the gluten component gliadin. How gliadin subverts host intestinal mucosal defenses remains elusive. Here, we show that the α‐gliadin‐derived LGQQQPFPPQQPY peptide (P31–43) inhibits the function of cystic fibrosis transmembrane conductance regulator (CFTR), an anion channel pivotal for epithelial adaptation to cell‐autonomous or environmental stress. P31–43 binds to, and reduces ATPase activity of, the nucleotide‐binding domain‐1 (NBD1) of CFTR, thus impairing CFTR function. This generates epithelial stress, tissue transglutaminase and inflammasome activation, NF‐κB nuclear translocation and IL‐15 production, that all can be prevented by potentiators of CFTR channel gating. The CFTR potentiator VX‐770 attenuates gliadin‐induced inflammation and promotes a tolerogenic response in gluten‐sensitive mice and cells from celiac patients. Our results unveil a primordial role for CFTR as a central hub orchestrating gliadin activities and identify a novel therapeutic option for celiac disease.
Keywords: celiac disease, CFTR, gliadin, P31–43 peptide, mucosal immunology
Subject Categories: Immunology, Membrane & Intracellular Transport, Molecular Biology of Disease
Introduction
The intestinal immune system is confronted with the permanent challenge to distinguish between safe and potentially harmful luminal triggers. Under physiological conditions, a finely tuned system of cellular adaptation ensures tissue homeostasis and provides the gut mucosa with the unique capacity of suppressing inflammation and promoting oral tolerance to non‐self‐antigens from dietary origin or commensal microbes (Kim et al, 2016). This tolerogenic response can be subverted by environmental triggers, such as viral infections (Bouziat et al, 2017), or as‐yet‐undefined predisposing factors, leading to immune and inflammatory responses.
Celiac disease (CD), a permanent intolerance to dietary proteins from wheat, rye, and barley, occurring in ~ 1% of individuals worldwide, is a paradigm of antigen mishandling. In a subset of genetically susceptible individuals bearing the human leukocyte antigen (HLA) DQ2/DQ8, the ingestion of gluten proteins switches tolerance toward an adaptive immune response with an autoimmune component (Meresse et al, 2012; Sollid & Jabri, 2013). In the CD gut, DQ2/DQ8‐restricted gluten‐specific CD4+ T cells act in concert with intestinal B cells to promote the production of IgA antibodies against the self‐antigen tissue transglutaminase (TG2; Meresse et al, 2012; Sollid & Jabri, 2013). However, additional factors are required to ignite an epithelial stress response with cytotoxic activation of intraepithelial CD8+ T lymphocytes, which ultimately cause villous atrophy and disease pathology (Meresse et al, 2009, 2012; Cerf‐Bensussan & Meresse, 2015; Setty et al, 2015). Such stressors possibly include reovirus infections (Bouziat et al, 2017) or other yet‐to‐be‐defined factors (Cerf‐Bensussan & Meresse, 2015) that provide the danger signals for adaptive immune response to immunodominant gliadin epitopes (Maiuri et al, 2003; Meresse et al, 2009, 2012; DePaolo et al, 2011; Barone et al, 2014; Cerf‐Bensussan & Meresse, 2015; Jabri & Abadie, 2015; Setty et al, 2015). However, the exact mechanisms through which gliadin can ignite a stress response are still unclear.
An approximately threefold increase in the prevalence of fully diagnosed CD, as well as a ~ 4% prevalence of positive anti‐TG2‐IgA autoantibodies, a serological marker of CD, even in the absence of villous atrophy (Lionetti et al, 2015), has been reported in several cohorts of patients with cystic fibrosis (CF; Fluge et al, 2009; Walkowiak et al, 2010; De Lisle & Borowitz, 2013), including ours (Appendix Table S1). CF is the most frequent monogenic lethal disease worldwide (Cutting, 2015), caused by loss‐of‐function mutations of the gene coding for cystic fibrosis transmembrane conductance regulator (CFTR), a cyclic adenosine monophosphate (cAMP)‐regulated anion channel that mediates chloride/bicarbonate transport across epithelia (Gadsby et al, 2006; Cutting, 2015). CF is best known for its respiratory phenotype, yet also frequently leads to intestinal problems, as CFTR protein is strongly expressed all along the intestine (Gadsby et al, 2006; Ooi & Durie, 2016). CFTR is not only an anion channel, but also orchestrates proteostasis at respiratory and intestinal epithelial surfaces, meaning that it regulates adaptation to cell‐autonomous or environmental stress signals (Luciani et al, 2010b; Villella et al, 2013a,b; Ferrari et al, 2011). CFTR malfunction generates epithelial stress, early TG2 activation, inhibition of autophagy, and activation of innate immunity (Maiuri et al, 2008; Luciani et al, 2009; Luciani et al, 2010b; Villella et al, 2013a,b; Ferrari et al, 2011), features that are reminiscent of those triggered by gliadin in intestinal epithelial cells and celiac duodenal mucosa (Maiuri et al, 2003; Meresse et al, 2009; Barone et al, 2014). These considerations led us to hypothesize that CFTR might be involved in the pathogenesis of CD. Specifically, we tested the hypothesis that gliadin may induce a stress response and subvert host mucosal defenses by reducing CFTR function at the intestinal surface.
Results
CFTR malfunction favors gliadin responsiveness in vivo
To determine whether the constitutive activation of innate immunity in the CF intestine (De Lisle & Borowitz, 2013; Nichols & Chmiel, 2015; Ooi & Durie, 2016) may favor the inflammatory and immune response to the gluten component gliadin, we administered gliadin to constitutively CFTR‐deficient mice, either CFTR knock‐out (B6.129P2‐KOCftrtm1UNC, Cftr −/−) mice or knock‐in mice harboring the most common loss‐of‐function F508del‐CFTR mutation (Cftrtm1EUR, F508del, FVB/129, Cftr F508del/F508del; Cutting, 2015). Before gliadin challenge, the small intestine from CFTR‐defective mice exhibited increased TG2 protein levels (Fig 1A), high NLRP3 activation, and caspase‐1 cleavage (Fig 1B) and increased levels of the pro‐inflammatory cytokines IL‐1β, MIP‐2α, TNF‐α, and IL‐17A (Fig 1C; Appendix Fig S1A), but reduced IFN‐γ production (De Lisle & Borowitz, 2013; Nichols & Chmiel, 2015; Fig 1C), together with cytoskeletal disassembly and increased intestinal permeability (Appendix Fig S1B and C) as compared to their wild‐type (WT) controls. Consistent with the constitutive NF‐κB activation in CF tissues (De Lisle & Borowitz, 2013; Nichols & Chmiel, 2015) and with the presence of an active NF‐κB binding motif in the gene promoter of IL‐15 (Stone et al, 2011), a master pro‐inflammatory cytokine that critically contributes to breaking oral tolerance to gluten and hence to causing CD‐associated pathology (Meresse et al, 2009, 2012; DePaolo et al, 2011; Cerf‐Bensussan & Meresse, 2015; Jabri & Abadie, 2015; Setty et al, 2015), CFTR‐defective mice showed a constitutive increase in IL‐15 mRNA and protein levels in the small intestine, irrespective of differences in genetic background (P < 0.001 vs. WT mice; Fig 1D). Thus, CFTR malfunction suffices to activate the intestinal IL‐15 system. IL‐15 upregulation in CFTR‐deficient mouse intestine was mediated by the constitutive activation of TG2, which is known to induce NF‐κB activation through sequestering IK‐Bα protein (Luciani et al, 2009), as IL‐15 expression in the small intestine was largely reduced in Cftr F508del/F508del mice backcrossed into a TG2‐knock‐out background (TG2−/−/Cftr F508del/F508del; Fig 1E). Next, we administered gliadin to CFTR‐deficient mice or their WT littermates for four consecutive weeks (5 mg/daily for 1 week and then 5 mg/daily thrice a week for 3 weeks; Galipeau et al, 2011; Papista et al, 2012; Larsen et al, 2015; Moon et al, 2016). Both FVB/129 and B6.129P2 littermates bearing WT‐CFTR are not sensitive to gliadin, as oral gliadin administration failed to stimulate inflammatory response and IFN‐γ production. Conversely, CFTR‐mutated mice (De Stefano et al, 2014; Tosco et al, 2016) fed with gliadin greatly increased both IL‐15 and IL‐17A levels in their small intestine and exhibited a 3.5‐fold increase in IFN‐γ production (P < 0.01 vs. vehicle‐treated mice; Fig 1F; Appendix Fig S1D and E). These results indicate that the constitutive stress response and innate immunity activation in CFTR‐deficient intestines favor an immune response to gliadin.
Figure 1. CFTR malfunction favors gliadin responsiveness in vivo .

-
AImmunoblot with anti‐TG2 or anti‐β‐actin as loading control in whole lysates from small intestine homogenates of Cftr F508del/F508del and wild‐type (Cftr WT) mice (n = 5 per group) and densitometric analysis of immunoblots. Mean ± SD of triplicates of independent pooled samples. **P < 0.01 (Student's t‐test).
-
BDetection of NLRP3 expression (top) and caspase‐1 cleavage (bottom) by immunoblot of whole lysates from small intestine homogenates of Cftr WT and Cftr F508del/F508del mice (n = 5) and densitometric analysis of immunoblots. Mean ± SD of triplicates of independent pooled samples. **P < 0.01 Cftr WT vs. Cftr F508del/F508del (Student's t‐test).
-
CProtein levels of IL‐17A and IFN‐γ from small intestine homogenates of Cftr WT and Cftr F508del/F508del (n = 10). Mean ± SD of triplicates of independent pooled samples. ***P < 0.001 Cftr WT vs. Cftr F508del/F508del (Student's t‐test).
-
DIL‐15 mRNA (left) and protein (right) levels in small intestine homogenates from Cftr F508del/F508del, Cftr −/− or their Cftr WT littermates (n = 10 per group). Mean ± SD of triplicates of independent pooled samples. **P < 0.01 Cftr F508del/F508del vs. Cftr WT(FVB/129), or °°P < 0.01 Cftr −/− vs. Cftr WT(B6.129P2) (ANOVA, Bonferroni post hoc test).
-
EIL‐15 mRNA levels in small intestine homogenates from Cftr WT mice or Cftr F508del/F508del or TG −/−/Cftr F508del/F508del or TG −/− mice (n = 10 per group). Mean ± SD of triplicates of independent pooled samples. ***P < 0.001 vs. Cftr WT, °°P < 0.01, ## P < 0.01 vs. Cftr F508del/F508del (ANOVA, Bonferroni post hoc test).
-
FEffects of 4 weeks of oral administration of gliadin on IL‐15, IL‐17A, and IFN‐γ protein levels in small intestine homogenates from Cftr F508del/F508del and Cftr WT mice (n = 10 mice per group of treatment). Mean ± SD of triplicates of independent pooled samples. **P < 0.01, ***P < 0.001 (Cftr F508del/F508del vs. Cftr WT mice prior gliadin challenge), °°°P < 0.001 (Cftr F508del/F508del mice vs. Cftr F508del/F508del mice after gliadin challenge; ANOVA, Bonferroni post hoc test).
-
G–IBALB/c mice (G) fed with a gluten‐free diet for at least three generations, or (H) NOD or (I) NOD‐DQ8 mice orally challenged with vehicle or gliadin for 4 weeks (5 mg/daily for 1 week and then 5 mg/daily thrice a week for 3 weeks). Representative traces of CFTR‐dependent Cl− secretion measured by forskolin (Fsk)‐induced increase in chloride current [Isc (μA/cm2)] in small intestines mounted in Ussing chambers; quantification of the peak CFTR inhibitor 172 (CFTRinh172)‐sensitive Isc (∆Isc) in tissue samples (n = 3 independent experiments). Mean ± SD of samples assayed; **P < 0.01, ***P < 0.001 vs. challenged with gliadin (Student's t‐test).
Gliadin inhibits CFTR function in vivo in the small intestine of gliadin‐sensitive mice
To determine whether gliadin may reduce CFTR function in the small intestine in vivo, we took advantage of established mouse models of gliadin sensitivity. In the first model, 10‐week‐old BALB/c mice were fed for three generations with a gluten‐free diet and then challenged with gliadin for 4 weeks (5 mg/daily for 1 week and then 5 mg/daily thrice a week for 3 weeks), following established protocols (Galipeau et al, 2011; Papista et al, 2012; Larsen et al, 2015; Moon et al, 2016). In all tested mice, gliadin reduced CFTR function measured as the forskolin‐inducible chloride current in the small intestine mounted in Ussing chambers (Fig 1G). In the second model of gluten sensitivity, we resorted to non‐obese diabetic (NOD) female mice, which are prone to the development of autoimmune diseases (Maurano et al, 2005; Galipeau et al, 2011; Papista et al, 2012; Larsen et al, 2015; Moon et al, 2016). When orally administered to these mice, gliadin suppressed CFTR function (Fig 1H). Notably, we confirmed the CFTR inhibitory effects of gliadin in NOD mice transgenic for the CD‐predisposing HLA molecule DQ8 (NOD‐DQ8; Galipeau et al, 2011; Papista et al, 2012; Korneychuk et al, 2015; Larsen et al, 2015; Moon et al, 2016; Fig 1I). Altogether, the aforementioned results indicate that gliadin inhibits CFTR function in vivo in the small intestine of gliadin‐sensitive mice.
The α‐gliadin LGQQQPFPPQQPY peptide (P31–43) inhibits CFTR function in intestinal epithelial cells
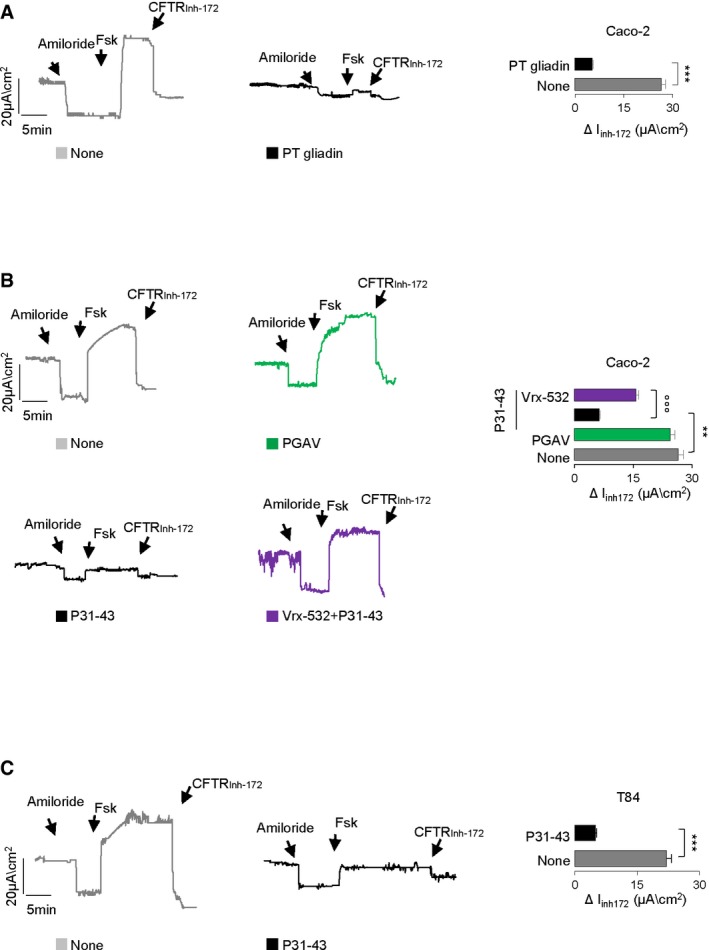
To determine whether gliadin may perturb CFTR channel activity at the intestinal epithelial surface, we resorted to human intestinal epithelial cell lines, either Caco‐2 or T84 cells, which are reportedly sensitive to gliadin or gliadin‐derived peptides (Barone et al, 2014). When confluent cells were cultured for 3 h with a peptic‐tryptic digest of gliadin from bread wheat (PT gliadin; 500 μg/ml; Maiuri et al, 2003; Barone et al, 2014), we noted a strong suppression of the forskolin‐inducible chloride current, as compared to controls kept in medium alone (Fig EV1A).
Figure EV1. Effects of gliadin and gliadin peptides on CFTR channel function in intestinal epithelial cells.
-
A–CTreatment of Caco‐2 cells (A, B) or T84 (C) cells with a PT gliadin (500 μg/ml) (A) or P57–68 or PGAV or P31–43 (20 μg/ml; 3 h) in the presence or absence of pre‐treatment with Vrx‐532. CFTR‐dependent Cl− secretion measured by means of forskolin‐induced (Fsk) increase in the chloride current [Isc (μA/cm2)] in cells mounted in Ussing chambers; quantification of the peak CFTR Inhibitor 172 (CFTRinh172)‐sensitive Isc (∆Isc; n = 3 independent experiments). Mean ± SD of samples assayed. ***P < 0.001 vs. PT gliadin (A) or vs. P31–43 (C) or **P < 0.01 vs. P31–43 (B), °°°P < 0.01 vs. P31–43 + Vrx‐532.
Source data are available online for this figure.
In vivo, two main α‐gliadin peptides remain undigested, the 25‐mer (P31–55) that is not recognized by T cells but damages the celiac intestine in vitro and in vivo (Maiuri et al, 2003; Meresse et al, 2009; Barone et al, 2014), and the 33‐mer (P55–87) that is deamidated by TG2, binds to HLA‐DQ2/DQ8 and induces an adaptive Th1 response (Barone et al, 2014). We challenged Caco‐2 or T84 cells for 3–24 h with gliadin‐derived LGQQQPFPPQQPY (P31–43) and QLQPFPQPQLPY (P57–68) peptides (20 μg/ml), which are fragments of the 25‐mer and 33‐mer, respectively (Maiuri et al, 2003; Meresse et al, 2009; Barone et al, 2014). These gliadin‐derived peptides are capable of inducing the enterocyte stress response (P31–43) or of activating T cells in the absence of any toxic effect on epithelial cells (P57–68; Maiuri et al, 2003; Meresse et al, 2009; Barone et al, 2014). The scrambled GAVAAVGVVAGA (PGAV) peptide was used as a control. As soon as after 3 h following incubation, P31–43 (but not P57–68, P33‐mer or PGAV) reduced the forskolin‐inducible chloride current in Caco‐2 (Figs 2A and EV1B) and T84 cell lines (Fig EV1C), although P31–43 did not affect cell viability at this time (Appendix Fig S2A), as reported (Rauhairta et al, 2011). This effect was prevented by a short (up to 20 min) pre‐incubation with the CFTR potentiators VX‐770 (10 μM), a Food and Drug Administration (FDA)‐ and European Medicines Agency (EMA)‐approved drug for the treatment of CF patients bearing plasma membrane (PM)‐resident CFTR mutants (Cutting, 2015) or Vrx‐532 (20 μM; Figs 2A and EV1B). These CFTR potentiators allow CFTR channels to stay longer in an open conformational state and hence increase the probability of CFTR channel opening (Eckford et al, 2012; Jih & Hwang, 2013).
Figure 2. P31–43 binds NBD1 and inhibits CFTR channel function.

-
ARepresentative traces of CFTR‐dependent Cl− secretion measured by forskolin (Fsk)‐inducible chloride current [Isc (μA/cm2)] in Caco‐2 cells mounted in Ussing chambers after 3 h of incubation with P57–68 or P33‐mer or P31–43 peptides (20 μg/ml) and of P31–43 in the presence of up to 30 min of pre‐treatment of VX‐770 (10 μM); quantification of the peak CFTR inhibitor 172 (CFTRinh172)‐sensitive Isc (∆Isc) in Caco‐2 cells (n = 3 independent experiments). Mean ± SD of samples assayed. ***P < 0.001 vs. P31–43 challenge, ## P < 0.01 vs. Caco‐2 challenged with P31–43 in the presence of VX‐770 (ANOVA, Bonferroni post hoc test).
-
BIncubation of Caco‐2 cells with P31–43 or control peptides (P57–68 or PGAV) for 1 h that were preceded or not by 20 min of pre‐treatment with VX‐770. Immunoprecipitation in non‐reducing and non‐denaturing conditions of CFTR protein and immunoblot with streptavidin‐HRP or CFTR antibody.
-
CProtein–protein docking and molecular dynamics of P31–43 (violet) bound to NBD1 (orange). Left side: general view of P31–43 and NBD1 interaction. Upper right: detailed interaction pattern, highlighting the most important amino acids. Lower right: in silico NBD1/P31–43 complex compared to the original crystallographic positions of Trp401 (red) and ATP (yellow).
-
DGraphical view of the in silico sampling percentage of P31–43 against NBD1/NBD2.
-
ESurface plasmon resonance (SPR) analysis of increasing concentrations of P31–43 and P57–68 peptides on rhNBD1 covalently bound to the CM5 sensor chip.
-
FSurface plasmon resonance (SPR) analysis of rhNBD1 binding to P31–43‐ and P57–68‐biotinylated peptides immobilized on SA sensor chip.
-
G, HBlue native polyacrylamide gel electrophoresis (PAGE) Western blotting of P31–43 and P57–68 biotinylated peptides in the presence of rhNBD1 (G) and of WT and double NBD1 mutants in the presence of biotinylated P31–43 (H). All the recombinant proteins and the indicated peptides were pre‐incubated in an appropriate buffer at 4°C for 30 min and then resolved in native conditions to preserve the formation of peptide/protein complexes.
-
IP31–43 induced modifications on NBD1 ATP binding site using the intrinsic W401 fluorescence.
-
JP31–43 effect on NBD1 ATPase activity. For further details, see Materials and Methods.
-
KP31–43 effect on NBD1 ATPase activity, applied at its IC50, compared to that obtained with 5 μM of the non‐hydrolyzable ATP analogue (P‐ATP) as a positive control of inhibition. Mean ± SD of triplicates of independent experiments.
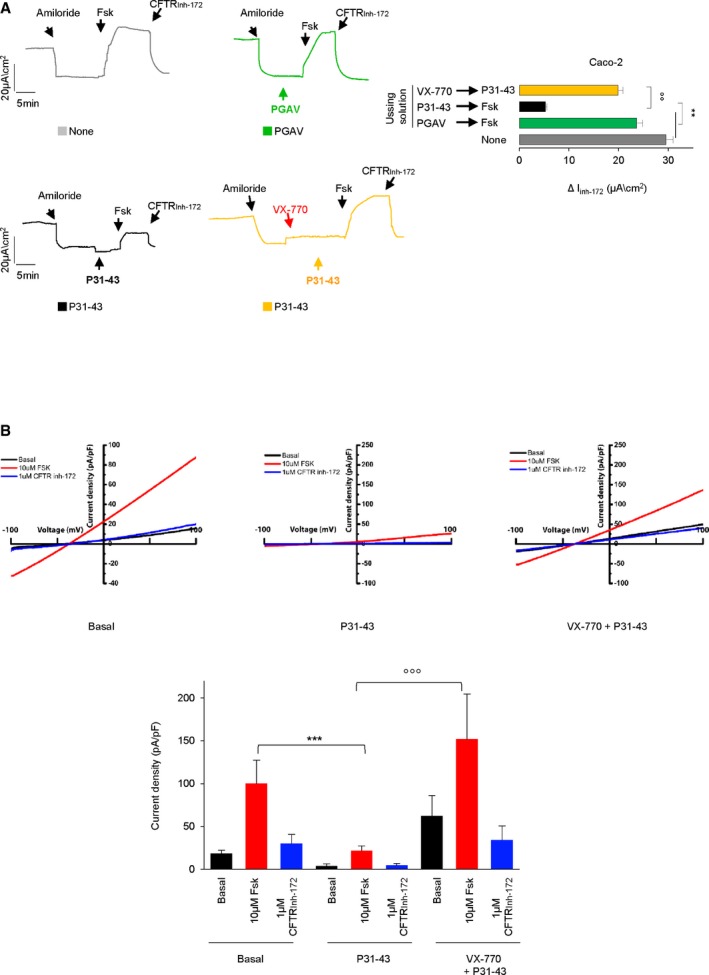
Notably, measurements of whole‐cell CFTR current of Caco‐2 cells revealed that P31–43 inhibits the forskolin‐inducible chloride current within a few minutes, unless the cells were pre‐treated with VX‐770 (Fig EV2A and B). Such a protective effect of VX‐770 suggests that P31–43 is likely not capable of inhibiting activated CFTR. Accordingly, we found that P31–43 failed to inhibit the forskolin‐inducible chloride current when forskolin was added to the cells before P31–43 (Appendix Fig S2B). Moreover, VX‐770 was poorly active if it was added after P31–43 addition (Appendix Fig S2B).
Figure EV2. Effects of P31–43 stimulation on forskolin‐activated CFTR channel.
-
ARepresentative traces of CFTR‐dependent Cl− secretion measured by forskolin (Fsk)‐inducible chloride current [Isc (μA/cm2)] in Caco‐2 cells mounted in Ussing chambers: effects of P31–43 (100 μM) and PGAV (100 μM) directly added to the bathing solution. After amiloride, the solution was directly supplied with buffer only (none, gray traces), with PGAV (green traces) or P31–43 (black traces) for 3–5 min (as detailed in Materials and Methods) and then pulsed with forskolin (Fsk; 20 μM) and finally with the CFTR inhibitor 172 (CFTRinh172). Yellow traces: after amiloride, the solution was first supplied with VX‐770 (10 μM) followed by P31–43 (100 μM) and then pulsed with forskolin (Fsk) followed by CFTRinh172. Quantification of the peak CFTR inhibitor 172 (CFTRinh172)‐sensitive Isc (∆Isc) in Caco‐2 cells (n = 3 independent experiments, mean ± SD). **P < 0.01 None or PGAV (gray or green traces) vs. P31–43 followed by Fsk (black traces), °°P < 0.01 P31–43 followed by Fsk (black trace) vs. VX‐770 followed by P31–43 and then by Fsk (yellow trace; ANOVA, Bonferroni post hoc test).
-
BWhole‐cell CFTR current densities induced by 10 μM forskolin (Fsk) at +100 mV in Caco‐2 cells w/o treatment (left), with 100 μM P31–43 peptide in the luminal solution during patch clamping (middle), or after treatment with 20 μM VX‐770 (20 min), followed by an application of 100 μM P31–43 peptide in the luminal solution while patch clamping (right). Current–voltage (I–V) relationships were elicited by ramps from −100 mV to +100 mV (holding potential, −40 mV). Statistical analysis of average CFTR current densities induced by forskolin and blocked by CFTR inh‐172 in cells treated as described. n ≥ 3; ***P < 0.001 basal vs. P31–43, °°°P < 0.001 P31–43 vs. VX770 + P31–43, two‐way ANOVA with Bonferroni post‐test. Data are presented as means ± SEM (bottom).
Source data are available online for this figure.
The effects of VX‐770 and Vrx‐532 in protecting intestinal epithelial cells against P31–43‐mediated CFTR inhibition were confirmed by assessing the rate of iodide efflux that was usually impaired by P31–43 (but not by P57–68 or PGAV; Appendix Fig S2C–H).
P31–43 inhibits the ATPase activity of the NBD1 domain of CFTR
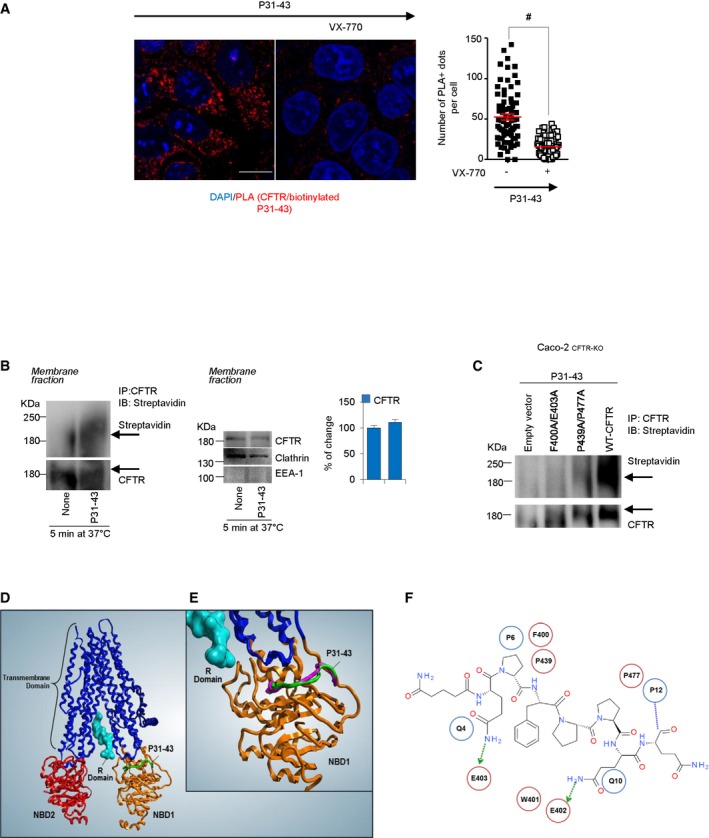
To explore the mechanism through which gliadin may interfere with the CFTR channel activity, we investigated whether P31–43 might interact with CFTR protein at the epithelial surface. When Caco‐2 cells were cultured for 1 h in the presence of biotinylated P31–43, subsequent immunoprecipitation of CFTR revealed the presence of the biotinylated peptide (Fig 2B; Appendix Fig S3A). In line with the protective effect of VX‐770 on P31–43‐mediated CFTR inhibition (Fig EV2A and B), a short pre‐incubation with VX‐770 abolished the interaction of P31–43 with CFTR, as demonstrated by two different methods, namely co‐immunoprecipitation and proximity ligation assay (Figs 2B and EV3A). Notably, CFTR and P31–43 co‐immunoprecipitated in the clathrin+ EEA1− plasma membrane (PM) protein fractions from Caco‐2 cells as soon as after 5 min following incubation with P31–43 (Fig EV3B), supporting the hypothesis that P31–43 may encounter and bind CFTR in clathrin+ PM fractions. Indeed, both CFTR and P31–43 enter the endosomal compartment through clathrin+ vesicles for either recycling or lysosomal degradation (Lukacs et al, 1997; Barone & Zimmer, 2016).
Figure EV3. P31–43 interaction with NBD1.
-
ACaco‐2 cells challenged with P31–43 peptide in the presence or absence of pretreatment of VX‐770. Confocal analysis of endogenous CFTR interaction with P31–43 by proximity ligation assay (PLA). Scale bar, 10 μm. Quantifications were obtained from five randomly selected fields per condition, with each containing ˜ 20–25 cells. Data are presented as means ± SEM, # P < 0.05 relative to VX‐770 + P31–43; two‐tailed unpaired Student's t‐test.
-
BCaco‐2 cells challenged with biotinylated P31–43 for 5 min at 37°C. Immunoprecipitation of purified protein from membrane fractions in non‐reducing and non‐denaturing conditions of CFTR protein and immunoblot with streptavidin‐HRP or CFTR antibody (left). Immunoblot of purified protein from membrane fractions with anti‐CFTR, anti‐clathrin, and anti‐EEA‐1 antibodies. EEA‐1 and clathrin were used as controls of purification of clathrin‐positive membrane fraction vesicles (middle). Densitometric analysis of protein levels (right). Mean ± SD of three independent experiments.
-
CCaco‐2CFTR‐KO cells were transfected with pcDNA3.1_F400A/E403A‐CFTR, pcDNA3.1_P439A/P477A‐CFTR, or pcDNA3.b WT‐CFTR plasmids (as positive control) or empty vector (as negative control). After 24 h, the cells were challenged with P31–43. Immunoprecipitation in non‐reducing and non‐denaturing conditions of CFTR protein and immunoblot with streptavidin‐HRP or CFTR antibody (n = 3 independent experiments).
-
DProtein–protein docking and molecular dynamics of P31–43 (green) bound to CFTR.
-
EComparison between the docking poses of P31–43 against CFTR structure (green) and against NBD1 alone (violet).
-
F2D interaction map, between P31–43 peptide retrieved from the molecular docking procedure (PDB: 2BBO). Most important amino acids are detailed (blue circle for P31–43, red circle for NBD1).
Next, we investigated whether P31–43 may target the nucleotide‐binding domains (NBDs) of CFTR, which are responsible for ATP binding and hydrolysis (Sheppard & Welsh, 1999; Gadsby et al, 2006). Protein–protein docking analysis suggested a high probability (78%) for a P31–43 interaction with NBD1 (between amino acids 400 and 477; Fig 2C) but a low probability (7%) for its interaction with NBD2 (Fig 2D). Molecular dynamics simulation indicated that P31–43 forces the movement of tryptophan at the position 401 (W401), which normally interacts with the adenine moiety of ATP via π‐π stacking (Sheppard & Welsh, 1999), from its original position (red) to a novel one (orange) that is not any more compatible with the interaction with ATP and hence with ATPase activity (Fig 2C). Surface plasmon resonance (SPR) confirmed the predicted interaction of P31–43 (but not P57–68) with recombinant human NBD1 (rhNBD1). This selective interaction was detected by immobilization of rhNBD1 on CM5 chips and both P31–43 (which yielded a dose‐dependent signal) and P57–68 (which failed to induce any signal) as analytes in the flow phase (Fig 2E and F). Molecular modeling indicated that, within NBD1, the amino acid couples F400/E403 and P439/P477 are critical for the interaction with P31–43 (Fig 2C). This model was confirmed by incubating native rhNBD1 or its two double mutants F400A/E403A and P439A/P477A with P31–43 and subjecting the complex to native polyacrylamide gel electrophoresis. P31–43 (but not P57–68) bound to rhNBD1 (Fig 2G), and this interaction was attenuated for both rhNBD1 mutants (Fig 2H). A higher molecular weight complex of biotinylated P31–43 bound to NBD1 was also detectable, in line with previous reports of higher molecular weight aggregates of NBD1 occurring in the presence of 2 mM ATP (Galeno et al, 2011; Fig 2G and H). To confirm the hypothesis that NBD1 interacts with P31–43, we replaced endogenous CFTR with mutant versions bearing either F400A/E403A or P439A/P477A double NBD1 mutations in Caco‐2 cells (that were first rendered CFTR‐null by CRISP/CAS9 technology and then transfected with mutant CFTR or WT‐CFTR as a control). Then, we measured the interaction of CFTR with P31–43 added to the cells. P31–43 co‐immunoprecipitated with WT but not with F400A/E403A or P439A/P477A mutant CFTR (Fig EV3C). These cell‐based experiments confirm the results obtained in the cell‐free system, suggesting that P31–43 indeed interacts with NBD1.
Next, we investigated whether the interaction between P31–43 and NBD1 would interfere with ATP binding as this was predicted from the altered spatial orientation of W401. When measuring the intrinsic tryptophan fluorescence of rhNBD1, we observed that the quenching effect of ATP on W401 fluorescence (Fig 2I, compare black and green curves) was reduced by pre‐incubation with P31–43 (Fig 2I, blue line). Noteworthy, the data shown in Fig 2I suggest that P31–43 does not enter the ATP binding pocket (compare black and red lines), thus confirming the in silico hypothesis that a conformational change occurs when P31–43 binds NBD1. Finally, we assessed the ATPase activity of NBD1 in the presence of P31–43 or of a non‐hydrolyzable ATP analogue (P‐ATP) as a positive control of inhibition. P31–43 inhibited NBD1 ATPase activity with an IC50 of 55.11 μM (Fig 2J and K). These experiments indicate that the direct binding of P31–43 to NBD1 negatively affects the ATPase activity of NBD1 via a conformational effect. The recently published electron microscopic structure of CFTR (in its dephosphorylated, ATP‐free form; Liu et al, 2017) allowed us to confirm that the theoretical P31–43 binding to the entire CFTR protein (Fig EV3D and E) is superimposable to the one calculated for P31–43 interacting with the isolated NBD1 domain (Fig 2C).
Mutual relationship between gliadin, CFTR, and TG2 in intestinal epithelial cells
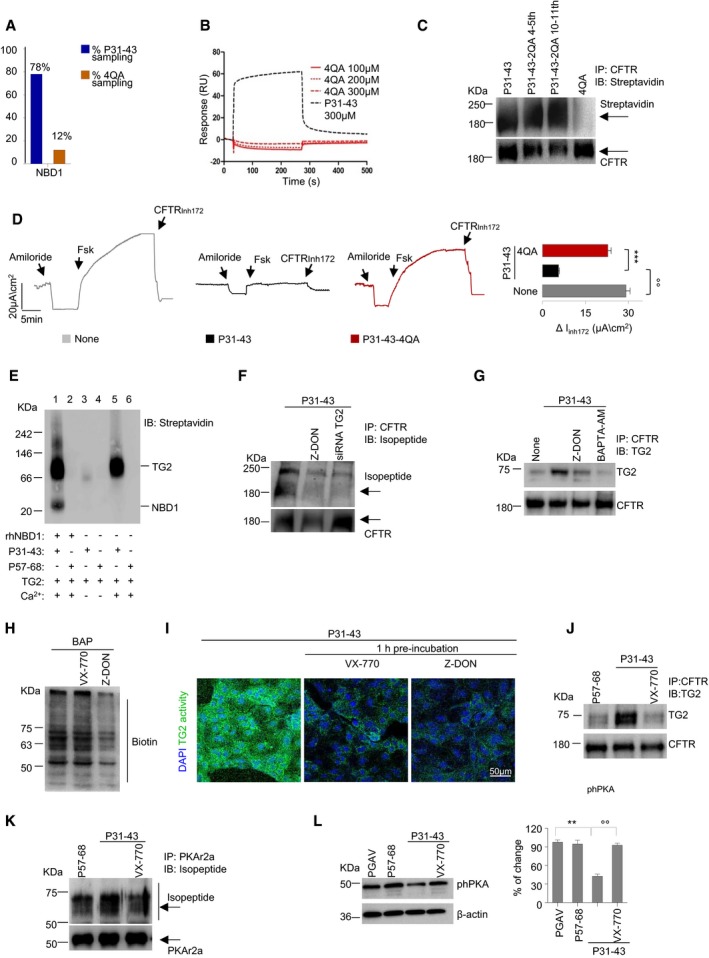
The in silico model (see Fig 2C) also led to the prediction that substitution of the glutamines at position 4, 5, 10, and 11 with alanine residues in the P31–43 sequence (LGQAAPFPPAAPY, below referred to as 4QA peptide) would attenuate binding to NBD1 (Fig 3A). SPR confirmed that the interaction of mutant peptide 4QA with hrNBD1 was strongly reduced as compared to unmutated P31–43 (Fig 3B). Indeed, the model predicts that Q4 and Q10 within the gliadin peptide P31–P43 directly interact with NBD1 residues E403 and E402, respectively (Fig EV3F), while Q5 and Q10 establish intrapeptide interactions with P6 and P12 that are likely essential for their correct orientation and binding to NBD1 residues P439/F400 and P477, respectively. Of interest, the interaction of P31–43 with E402 may account for the forced movement of W401 described above (see Fig 2C). Consistent with the in silico prediction and cell‐free experiments, the quadruple‐mutated P31–43 peptide 4QA showed marginal interaction with CFTR protein in Caco‐2 cells (Fig 3C) and was largely unable to inhibit CFTR function (Fig 3D), contrasting with single or double Q/A substitutions in P31–43 that did not affect the CFTR inhibitory effect of P31–43 (Fig 3C and Appendix Fig S3B).
Figure 3. Mutual relationship between gliadin, TG2, and CFTR in intestinal epithelial cells.
-
AGraphical view of the in silico sampling percentage of P31–43 and of P31–43/4QA (4QA) against NBD1.
-
BSPR analysis of 4QA peptide on rhNBD1 covalently bound to CM5 sensor chip. For further details, see Materials and Methods.
-
C, DIncubation of Caco‐2 cells with P31–43 or with modified P31–43 (P31–43‐2QA 4–5th or P31–43‐2QA 10–11th or P31–43/4QA mutant (4QA) peptides) for 1 h. (C) Immunoprecipitation in non‐reducing and non‐denaturing conditions of CFTR protein and immunoblot with streptavidin‐HRP or CFTR antibody (n = 3 independent experiments). (D) Representative traces of CFTR‐dependent Cl− secretion measured by forskolin (Fsk)‐induced increase in chloride current [Isc (μA/cm2)] in Caco‐2 cells mounted in Ussing chambers; quantification of the peak CFTR inhibitor 172 (CFTRinh172)‐sensitive Isc (∆Isc) in Caco‐2 cells (n = 3 independent experiments). Mean ± SD of samples assayed. °°P < 0.01 vs. P31–43 challenge, ***P < 0.001 vs. Caco‐2 challenged with 4QA (ANOVA, Bonferroni post hoc test).
-
EIn vitro Blue native PAGE Western blotting of enhanced binding of P31–43 to rhNBD1 in the presence of activated rTG2 that in turn promotes the formation of higher molecular weight complexes. As described in the text, only P31–43 (lanes 1, 3, 5), and not P57–68 (lanes 2, 4, 6), has the ability to form stable complexes with NBD1 and TG2 (n = 3 independent experiments).
-
F–LIncubation of Caco‐2 cells with P57–68 or P31–43, in the presence or absence of pre‐treatment with TG2 inhibitor Z‐DON, TG2‐siRNA, BAPTA‐AM, or VX‐770. (F) Immunoprecipitation in non‐reducing and non‐denaturing conditions of CFTR protein and immunoblot with anti‐isopeptide glutamime‐lysine and CFTR or (G) immunoprecipitation of CFTR and then immunoblot with anti‐TG2 or anti‐CFTR antibodies (n = 3 independent experiments). (H and I) Effects of treatment with VX‐770 and Z‐DON on TG2 transamidating activity in situ. (H) In situ detection of TG2 activity in Caco‐2 cells pulsed with Ca2+ by immunoblotting of the TG‐catalyzed incorporation of 5‐biotinamidopentylamine (BAP) and blotting with anti‐biotin antibody (n = 3 independent experiments). (I) Assay of TG2 activity by immunostaining of the TG‐catalyzed incorporation of monodansylcadaverine in Caco‐2 cells pre‐treated with VX‐770 or Z‐DON and then pulsed with P31–43. Scale bar, 50 μm. (J) Immunoprecipitation of CFTR and immunoblot with anti‐CFTR and anti‐TG2 antibodies. (K) Immunoprecipitation in non‐reducing and non‐denaturing conditions of PKAr2α protein and immunoblot with isopeptide glutamine‐lysine and PKAr2α antibodies (n = 3 independent experiments). (L) Immunoblot of phospho‐PKA (phPKA) protein; densitometric analysis of protein levels relative to β‐actin. Mean ± SD of triplicates of independent experiments. **P < 0.01 vs. P31–43, °°P < 0.001 vs. VX‐770 (ANOVA, Bonferroni post hoc test).
Cystic fibrosis transmembrane conductance regulator channel activity in vivo is the result of a dynamic sequence of opening and closing events initiated by cycles of ATP binding and hydrolysis (Sheppard & Welsh, 1999; Gadsby et al, 2006). Notably, CFTR works in concert with multiple neighboring proteins that can impact on, and can be influenced by, the dynamic events of CFTR channel activation. Knowing that CFTR inhibition can result in oxidative stress and Ca2+‐dependent TG2 activation (Maiuri et al, 2008; Luciani et al, 2009; Luciani et al, 2010b) and that TG2 can transamidate P31–43 (Barone et al, 2014), we determined whether the interaction of P31–43 with NBD1 would be influenced by TG2. For this, rhNBD1 was incubated in vitro in the absence or presence of biotinylated P31–43 (or biotinylated P57–68 as a negative control) and Ca2+ to activate TG2, followed by native polyacrylamide gel electrophoresis to measure protein complex formation (Fig 3E and Appendix Fig S3C). This procedure indicates that P31–43 (but not P57–68; compare lanes 1 and 2, as well as 5 and 6 in Fig 3E) binds to Ca2+‐activated (but not inactive, lanes 3 and 4) TG2 and actually favors the formation of a supramolecular complex involving TG2 and NBD1 (lane 1). Thus, TG2 may sustain the inhibitory effects of P31–43 on the NBD1‐dependent CFTR channel activity.
Notably, P31–43, which covalently interacted with TG2 in Caco‐2 cells shortly upon challenge (Appendix Fig S4A), induced the TG2‐catalyzed formation of glutamime‐lysine bonds that were detectable in CFTR immunoprecipitates and became undetectable after inhibition of the TG2 transamidating activity with Z‐DON or the depletion of TG2 with siRNAs (Fig 3F and Appendix Fig S4B). Moreover, the addition of P31–43 to cells stimulated the interaction between TG2 and CFTR, as detectable by co‐immunoprecipitation (Fig 3G). This effect was prevented when TG2 activity was suppressed by Z‐DON or the Ca2+chelator BAPTA‐AM (Fig 3G). Notably, pre‐incubation of Caco‐2 cells with the CFTR potentiator VX‐770, which itself has no impact on TG2‐mediated transamidation reactions (Fig 3H and Appendix Fig S4C), negated the capability of P31–43 to induce TG2 activation (Luciani et al, 2010a; Barone et al, 2014; Fig 3I) and abolished the P31–43‐induced interaction of TG2 and CFTR (Fig 3J). Altogether, these results indicate that the P31–43‐mediated inhibition of CFTR function drives TG2 activation that covalently links P31–43, CFTR, and TG2 in a trimolecular complex.
The early TG2 activation consequent to P31–43 driven CFTR inhibition also stimulated TG2‐mediated crosslinking of several TG2 substrate proteins, among which the regulatory subunit 2α of protein kinase A (PKAr2α; Fig 3K and Appendix Fig S4D). This leads to a subsequent decrease in the abundance of phosphorylated PKA protein after 24 h following incubation with P31–43 (Fig 3L). Since PKA is essential for CFTR phosphorylation and activity (Chin et al, 2017), this may enhance the detrimental effects of P31–43 on CFTR function. Importantly, VX‐770 counteracted these TG2‐mediated effects of P31–43 (Fig 3K and L).
CFTR potentiators protect intestinal epithelial cells from P31–43‐driven epithelial stress and innate immune activation
Prompted by the evidence that the inhibition of CFTR function suffices to generate epithelial stress (Maiuri et al, 2008; Luciani et al, 2009; Luciani et al, 2010b; Villella et al, 2013a,b; Cutting, 2015), we determined whether CFTR inhibition may account for the P31–43‐induced epithelial stress response, which is pivotal for CD pathogenesis (Meresse et al, 2009; Barone et al, 2014; Cerf‐Bensussan & Meresse, 2015; Jabri & Abadie, 2015). Pre‐incubation with the CFTR potentiator VX‐770 (which prevents the P31–43‐mediated inhibition of CFTR function, see above) protected Caco‐2 cells from signs of P31–43‐induced epithelial stress including the downregulation of PPARγ (Luciani et al, 2010a; Fig 4A, top), the phosphorylation of ERK1/2 (Luciani et al, 2010a; Barone et al, 2014; Fig 4A, bottom), and the upregulation of heat‐shock protein (HSP) 70 expression (Appendix Fig S5A). VX‐770 also prevented additional pro‐inflammatory P31–43 effects on Caco‐2 cells, namely NF‐κB p65 translocation into the nucleus (Fig 4B), NLRP3 expression (Fig 4C), and caspase‐1 cleavage (Fig 4D).
Figure 4. CFTR malfunction drives P31–43‐induced epithelial stress response.
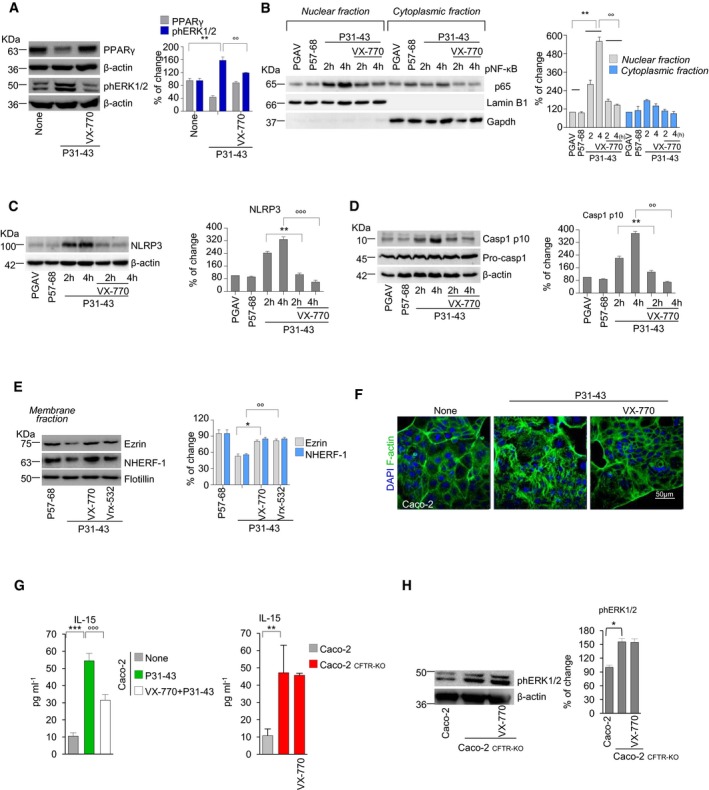
-
AImmunoblot of PPARγ or phospho‐ERK1/2 (phERK1/2) and densitometric analysis of protein levels relative to β‐actin. Mean ± SD of triplicates of independent experiments. **P < 0.01 vs. P31–43, °°P < 0.001 vs. VX‐770 (ANOVA, Bonferroni post hoc test).
-
B–DImmunoblotting with specific antibodies in Caco‐2 cells challenged for 2 or 4 h in the presence or absence of VX‐770. NF‐κB p65 in cytoplasmic and nuclear extracts (B), NLRP3 expression (C), and caspase‐1 cleavage (D). Densitometric analysis of protein levels relative to loading control. Mean ± SD of triplicates of independent experiments. **P < 0.01 and °°P < 0.001 vs. 2 h or 4 h of culture with P31–43 in the presence of VX‐770 (ANOVA, Bonferroni post hoc test).
-
EImmunoblot of membrane protein fractions with anti‐NHERF‐1, anti‐ezrin Abs, and anti‐flotillin as a control. Densitometric analysis of immunoblot (mean ± SD of triplicates of independent experiments); *P < 0.01 vs. treatment with VX‐770 or °°P < 0.01 vs. treatment with Vrx‐532 (ANOVA, Bonferroni post hoc test).
-
FConfocal image staining with anti‐F‐actin. DAPI (blue), nuclear counterstaining. Scale bar, 50 μm.
-
GIL‐15 production (quantified by specific ELISA) in CFTR‐WT Caco‐2 cells treated or not with P31–43 (left) or in Caco‐2CFTR‐KO cells (right), in the presence or absence of VX‐770. Mean ± SD of triplicates of independent experiments. ***P < 0.001 vs. untreated and °°°P < 0.001 vs. CFTR‐WT Caco‐2 cells treated with P31–43 + VX‐770 (ANOVA, Bonferroni post hoc test). **P < 0.01 vs. CFTR‐WT Caco‐2 (Student's t‐test).
-
HCaco‐2 or Caco‐2CFTR‐KO cells in the presence or absence of pre‐treatment with VX‐770. Immunoblot of phERK1/2 or β‐actin (left); densitometric analysis of protein levels (right). Mean ± SD of triplicates of independent experiments. *P < 0.05 (Student's t‐test).
In line with the evidence that CFTR inhibition may stimulate CFTR ubiquitination and its consequent depletion from the PM following an interaction with the ubiquitin‐binding protein SQSTM1/p62 (Villella et al, 2013a,b), we found that 24 h of challenge with P31–43 increased the susceptibility of CFTR to trypsin digestion, CFTR ubiquitination by carboxy terminal of hsp70‐interacting protein (CHIP), and SQSTM1/p62 accumulation at the epithelial surface (Appendix Fig S5B–D). Moreover, P31–43 resulted in the decrease in mature CFTR (band C) in PM fractions (Appendix Fig S5E) after 24 h of challenge. These effects of P31–43 were prevented by pre‐incubation with either VX‐770 or Vrx‐532 (Appendix Fig S5F), as well as by the enforced expression of a dominant‐negative SQSTM1/p62 mutant lacking the UBA domain (Villella et al, 2013a; Appendix Fig S5G). A 24‐h‐long incubation with P31–43 also led to the VX‐770 and Vrx‐532 inhibitable decrease in the abundance of the CFTR interactor proteins ezrin and Na+/H+ exchange regulatory factor isoform 1 (NHERF‐1; Fig 4E), a cytoskeletal scaffolding protein that promotes the organization of the ezrin–radixin–moesin (ERM) membrane complex and F‐actin assembly (Casaletto et al, 2011; Kawaguchi et al, 2017). Accordingly, VX‐770 negated the capacity of P31–43 to induce the formation of F‐actin stress fibers after 24 h of challenge (Barone et al, 2014; Fig 4F).
Importantly, pre‐treatment of Caco‐2 cells with VX‐770 prevented the P31–43‐induced production of IL‐15 (P < 0.001; Fig 4G, left). Stable CFTR deletion by CRISP/CAS9 in Caco‐2 cells (Caco‐2CFTR‐KO) led to a threefold increase in IL‐15 protein levels (P < 0.01; Fig 4G, right) as well as 1.6‐fold increase in ERK1/2 phosphorylation (Fig 4H). VX‐770 failed to modulate both IL‐15 expression and ERK1/2 phosphorylation in Caco‐2CFTR‐KO cells, thus confirming that VX‐770 controls IL‐15 production through on‐target (via CFTR) rather than off‐target effects (Fig 4G and H).
CFTR potentiators protect intestinal epithelial cells from the detrimental effects of P31–43 on endosomal trafficking
A major consequence of CFTR malfunction in epithelial cells is the impairment of endosomal maturation and trafficking consequent to TG2‐mediated sequestration of the phosphatidylinositol‐3‐kinase (PI3K) complex‐3 (Luciani et al, 2010b; Villella et al, 2013a) organized around the BECN1 protein, which is essential for autophagosome formation, and UV irradiation resistance‐associated gene (UVRAG), which is pivotal for endosomal maturation and trafficking (Liang et al, 2008; Marat & Haucke, 2016). P31–43, as well as the α‐gliadin fragment P31–49 (19‐mer; Zimmer et al, 2010), is known to stall in early endosomal vesicles, as well as in the late endosomal compartment (Luciani et al, 2010a; Zimmer et al, 2010; Barone et al, 2014), as they delay early‐to‐late endosomal maturation and vesicular trafficking of several cargos, including EGFR (Barone et al, 2014, 2010). This latter feature is recapitulated by CFTR inhibition in bronchial epithelial cells (Villella et al, 2013a). We observed that P31–43 reduced the total cellular abundance of BECN1, human vacuolar protein sorting (hVps)34, and UVRAG (Fig EV4A–C), with a subsequent decrease in the small GTPases Rab5 and Rab7 levels in endosomal protein fractions (Fig EV4D). These effects were counteracted by the pre‐incubation with the CFTR potentiators VX‐770 and Vrx‐532 (Fig EV4D and Appendix Fig S6A–C), unless the PI3K inhibitor 3‐methyladenine (3‐MA) was added to the system (Fig EV4C). Accordingly, VX‐770 restored the availability of phosphatidylinositol‐3‐phosphate (PI3P) at early endosomes (Villella et al, 2013a) that is compromised in P31–43‐treated cells (Appendix Fig S6D).
Figure EV4. Prevention by CFTR potentiators of P31–43 induced derangement of endosomal trafficking.
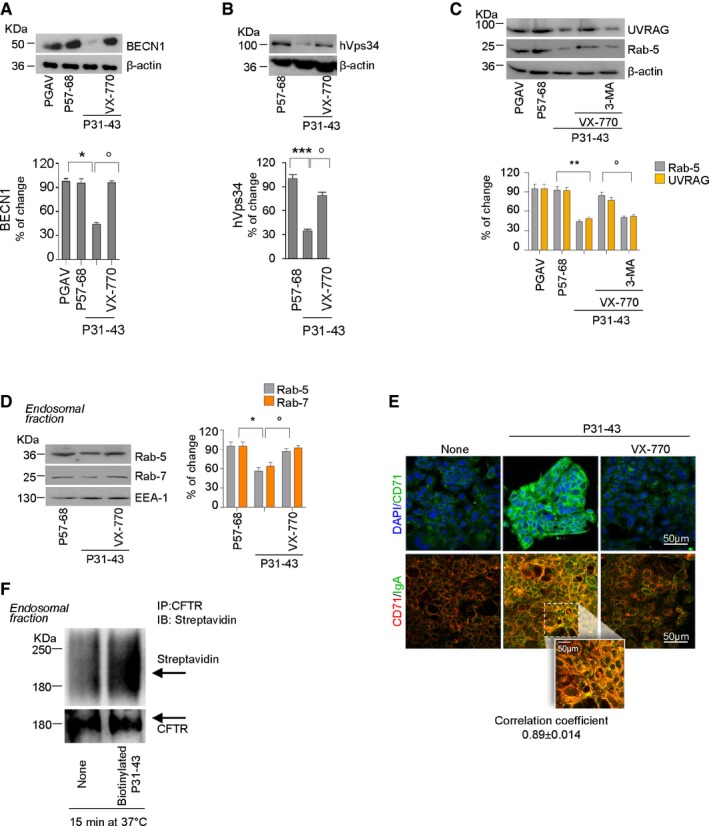
-
A–CImmunoblot of BECN‐1 (A), phosphatidylinositol‐3 (PI3)‐kinase (hVps34) (B), UVRAG and Rab5 (C), and β‐actin; densitometric analysis of protein levels (bottom). Mean ± SD of triplicates of independent experiments. *P < 0.05 or **P < 0.01 or ***P < 0.01 vs. P31–43, °P < 0.05 VX‐770+P31–43 vs. VX‐770 + 3‐MA+P31–43 (ANOVA, Bonferroni post hoc test).
-
DImmunoblot of Rab5 and Rab7 proteins in endosomal protein fractions from Caco‐2 cells challenged as described; endosomal antigen‐1 (EEA‐1) as loading control. Densitometric analysis of immunoblot is mean ± SD of triplicates of independent experiments. *P < 0.05 vs. P31–43, °P < 0.05 vs. P31–43 + VX‐770.
-
ECaco‐2 cells pre‐incubated for 1 h at 4°C with S‐IgA from hColostrum and then challenged with P31–43 in the presence or absence of VX‐770. Confocal images of apical localization of CD71. Staining with anti‐CD71 antibody and DAPI nuclear counterstaining (top) or anti‐CD71 (red) and S‐IgA (green) antibodies (bottom). Scale bar, 50 μm.
-
FImmunoprecipitation of CFTR protein in endosomal protein fractions in non‐reducing and non‐denaturing conditions, after 15 min of challenge with P31–43 and immunoblot with HRP‐streptavidin or CFTR.
Consistent with the effects of CFTR inhibition on endosomal trafficking (Villella et al, 2013a), VX‐770 blocked the P31–43‐mediated increase in the cell surface expression of transferrin receptor (CD71; Matysiak‐Budnik et al, 2008; Lebreton et al, 2012; Fig EV4E, top). When Caco‐2 cells were pre‐incubated for 1 h at 4°C with secretory IgA (SIgA), as described (Matysiak‐Budnik et al, 2008; Lebreton et al, 2012), VX‐770 abrogated the ability of P31–43 to increase the interaction of CD71 with SIgA (Fig EV4E, bottom), an interaction that is known to facilitate the transport of gliadin peptides across epithelia (Matysiak‐Budnik et al, 2008; Lebreton et al, 2012). In line with the ability of TG2 to control endocytosis of surface receptors (Davies et al, 1980) and to boost SIgA‐CD71 complex retrotranscytosis through the recycling pathway (Lebreton et al, 2012), TG2 was detected in the endosomal protein fraction from polarized Caco‐2 cells pulsed with biotinylated P31–43 as soon as after 15 min following internalization and co‐localized with P31–43 (Appendix Fig S6E and F). In addition, immunoprecipitation of CFTR from early endosomal protein fractions revealed the presence of biotinylated P31–43 (Fig EV4F). Notably, TG2 was detected within 5 min following challenge with P31–43 in clathrin+ PM fractions (Nurminskaya & Belkin, 2012; Barone & Zimmer, 2016; Appendix Fig S6G) that also contain CFTR and P31–43 (see Fig EV3A). Altogether, these results suggest that CFTR malfunction mediates the multifaceted effects of P31–43 on endosomal trafficking. Accordingly, CFTR potentiators prevented the reported (Luciani et al, 2010a) accumulation of biotinylated P31–43 within late endosomal vesicles (Appendix Fig S6H).
CFTR potentiators prevent gliadin‐induced immune dysregulation in vivo
To complete our demonstration, we investigated whether CFTR potentiators may counteract the pathogenic effects of gliadin on gliadin‐sensitive mice (Maurano et al, 2005; Galipeau et al, 2011; Papista et al, 2012; Korneychuk et al, 2015; Larsen et al, 2015; Moon et al, 2016). In this set of experiments, mice were orally challenged with gliadin for four consecutive weeks as above (see Fig 1), and the CFTR potentiator VX‐770 (2 mM) was administered intraperitoneally 15 min prior to gliadin challenge. Indeed, VX‐770 is known to potentiate the ion channel function of wild‐type mouse CFTR (Cui & McCarty, 2015; Cui et al, 2016; Zeng et al, 2017) which shares a high degree of sequence identity (~ 78%) with human CFTR (Cui & McCarty, 2015). In line with the data in Caco‐2 cell lines, VX‐770 prevented the gliadin‐induced decrease in CFTR function (Fig 5A) and protein expression (Fig 5B) in the small intestine from gliadin‐sensitive mice, as it abrogated the depletion of NHERF‐1 (Fig EV5A). VX‐770 also prevented the gliadin‐induced increase in intestinal permeability (Barone et al, 2014; Schumann et al, 2017; Figs 5C and EV5B). Notably, in short‐term experiments, in which intestinal permeability was not compromised, oral administration of gliadin (20 mg/mice in 100 μl saline) for two consecutive days led to a decrease in CFTR function (Appendix Fig S7A and B), meaning that gliadin can inhibit CFTR independently from gross alterations of the gut.
Figure 5. Effects of CFTR potentiators on gliadin‐induced immune response in mice and in celiac human PBMC .

-
A–HBALB/c mice fed with a gluten‐free diet for at least three generations, orally challenged with vehicle or gliadin for 4 weeks (5 mg/daily for 1 week and then 5 mg/daily thrice a week for 3 weeks) in the presence or absence of intraperitoneal VX‐770 administered 15 min prior gliadin challenge (n = 10 mice per group of treatment). (A) Representative traces of CFTR‐dependent Cl− secretion measured by forskolin (Fsk)‐induced increase in chloride current [Isc (μA/cm2)] in small intestines mounted in Ussing chambers; quantification of the peak CFTR inhibitor 172 (CFTRinh172)‐sensitive Isc (∆Isc) in tissue samples (n = 5). Mean ± SD of samples assayed. ***P < 0.001 gliadin vs. gliadin + VX‐770 (Student's t‐test). (B) Immunoblot with anti‐CFTR antibody and β‐actin loading control. Densitometric analysis is mean ± SD of triplicates of independent pooled samples. **P < 0.01 vehicle vs. gliadin; °P < 0.05, gliadin vs. VX‐770 + gliadin (ANOVA, Bonferroni post‐test). (C) Plasma markers of intestinal permeability in mice. Plasma concentration of FITC‐dextran 4000 (FITC‐D4000) measured 1 h after gavage of a single dose of 600 mg FITC‐D4000 per kg body weight. Quantification of plasma concentration from n = 10 mice per group of treatment expressed as mean ± SD of triplicates of independent pooled samples. ***P < 0.001 vehicle vs. gliadin; °°°P < 0.01, gliadin vs. VX‐770 + gliadin (ANOVA, Bonferroni post‐test). (D) NLRP3 expression by immunoblotting with specific antibodies in small intestines from three mice. Densitometric analysis is mean ± SD of triplicates of independent pooled samples. **P < 0.01 vehicle vs. gliadin; °P < 0.05, gliadin vs. VX‐770 + gliadin. (E, F) mRNA (E) or protein (by specific ELISA) (F) levels of IL‐15, IL‐21, IL‐17A, and IFN‐γ. Mean ± SD of triplicates of independent pooled samples. *P < 0.05, **P < 0.01, ***P < 0.001 vehicle vs. gliadin; °P < 0.05, °°P < 0.01, °°°P < 0.001 gliadin vs. VX‐770 + gliadin (ANOVA, Bonferroni post hoc test). (G, H) Transcript level of Rorc and Tbet (G) and protein levels (by specific ELISA) of IL‐10 and TGF‐β (H) from small intestine homogenates. Mean ± SD of triplicates of independent pooled samples. ***P < 0.001 or ****P < 0.0001 gliadin vs. VX‐770 + gliadin (ANOVA, Bonferroni post hoc test).
-
I, JIFN‐γ release (ELISA) in culture supernatants by PBMC from six celiac patients or four controls cultured in the lower compartment of a bidimensional co‐culture model upon 24 h challenge of confluent CaCo‐2 cells in the upper compartment with PT gliadin (I) or combination of P31–43 and P57–68 (J) in presence or absence of VX‐770. Mean ± SD of triplicates of independent pooled samples. ***P < 0.001, PT gliadin vs. medium; °°°P < 0.001, PT gliadin vs. PT gliadin + VX‐770 (n = 4); ***P < 0.001, P57–68 or P31–43 vs. P31–43/P57–68 combination (n = 6); °°°P < 0.001, P57–68/P31–43 combination vs. VX‐770 + P57–68/P31–43 (n = 3), (ANOVA, Bonferroni post hoc test).
-
KIL‐10 release (ELISA) in culture supernatants by PBMC from four celiac patients cultured as in (J). Mean ± SD of triplicates of independent pooled samples. ****P < 0.0001, P57–68/P31–43 combination vs. P57–68/P31–43+VX‐770 treatment (n = 3; ANOVA, Bonferroni post hoc test).
-
LIFN‐γ release (ELISA) into culture supernatants by PBMC from three celiac patients cultured in the lower compartment of a bidimensional co‐culture model upon a 24 h challenge of confluent Caco‐2CFTR‐KO cells in the upper compartment with a combination of P31–43 and P57–68 in the presence or absence of VX‐770, as in (J and K). *P < 0.05, medium vs. P57–68/P31–43 combination (n = 3; ANOVA, Bonferroni post hoc test).
Figure EV5. Prevention by VX‐770 of gliadin‐induced effects in gliadin‐sensitive mice.
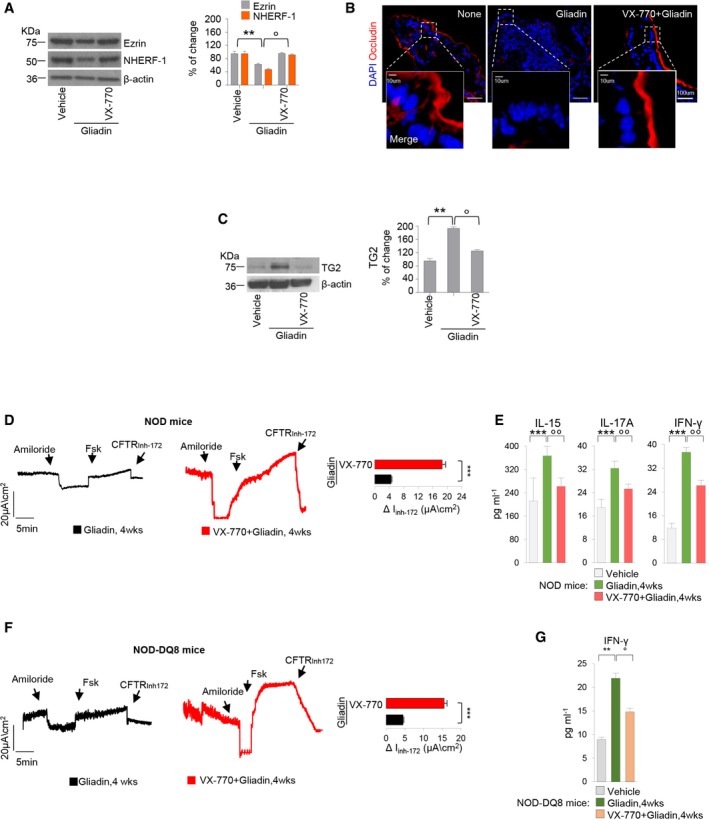
-
A–CBALB/c mice were fed for at least three generations with a gluten‐free diet, orally challenged with vehicle or gliadin for 4 weeks (5 mg/daily for 1 week and then 5 mg/daily thrice a week for 3 weeks) in the presence or absence of intraperitoneal VX‐770 administered 15 min prior gliadin challenge (n = 10 mice per group of treatment). (A and C) Immunoblot with antibodies against NHERF‐1 and ezrin (A) or TG2 (C) and β‐actin as loading control, in whole lysates from small intestine homogenates. Right panels, densitometric analysis of protein relative to β‐actin, mean ± SD of triplicates of independent pooled samples. **P < 0.01 vs. gliadin, °P < 0.05 vs. VX‐770, (ANOVA, Bonferroni post hoc test). (B) Confocal images of small intestine stained with antibodies against occludin (red) with nuclear counterstaining (blue). Scale bar, 100 μm.
-
D–GNOD mice (D and E) or NOD.scid AB0nullDQ8 mice (NOD‐DQ8 mice) (F and G) challenged with gliadin for consecutive 4 weeks in the presence or absence of intraperitoneal administration of VX‐770 15 min prior gliadin challenge (n = 10 mice per group of treatment). (D and F) Assessment in Ussing chambers of CFTR‐dependent Cl− secretion measured by forskolin‐induced (Fsk) increase in the chloride current [Isc (μA/cm2)]; quantification of the peak CFTR inhibitor 172 (CFTRinh172)‐sensitive Isc (∆Isc; n = 5). Mean ± SD of samples assayed. ***P < 0.001 vs. VX‐770 treatment (Student's t‐test). (E) IL‐15, IL‐17A, and IFN‐γ protein levels from small intestine homogenates of NOD mice. Mean ± SD of triplicates of independent pooled samples. ***P < 0.001 vs. gliadin, °°P < 0.01 vs. VX‐770 treatment (ANOVA, Bonferroni post hoc test). (G) IFN‐γ protein levels from small intestine homogenates of NOD‐DQ8 mice. Mean ± SD of triplicates of independent pooled samples. **P < 0.01 vs. gliadin, °P < 0.05 vs. VX‐770 treatment (ANOVA, Bonferroni post hoc test).
Next, we determined whether VX‐770 would be efficient in protecting gliadin‐sensitive mice from gliadin‐induced immunopathology. VX‐770 opposed the ability of gliadin to increase TG2 protein levels (Fig EV5C), counteracted gliadin‐induced NLRP3 expression (Fig 5D), and the (reportedly IL‐15‐dependent; Jabri & Abadie, 2015) upregulation of natural killer (NK) receptor NKG2D on intraepithelial lymphocytes (Appendix Fig S8A). VX‐770 also prevented the gliadin‐induced upregulation of IL‐15, IL‐17A, and IFN‐γ mRNA (Fig 5E) and protein (Fig 5F; P < 0.001) and inhibited the gliadin‐induced expression of IL‐21, which is known to positively correlate with Th1 and Th17 activity (Sarra et al, 2013; Fig 5E and F). Accordingly, both the Th17 (Rorc) and the Th1 (Tbet) transcription factors were downregulated upon VX‐770 treatment (Fig 5G). In contrast, VX‐770 improved IL‐10 and TGF‐β production in gliadin‐sensitive mice (Fig 5H). Notably, a short (2‐day) challenge with gliadin did not induce IFN‐γ production (Appendix Fig S8B) although it was effective in reducing CFTR function (see Appendix Fig S7B), further supporting that gliadin‐induced CFTR inhibition drives gliadin‐induced immunopathology. The protective effects of VX‐770 were confirmed in NOD mice challenged with gliadin after diabetes onset (Fig EV5D and E) and, importantly, in NOD‐DQ8 mice as well (Fig EV5F and G).
In conclusion, it appears that CFTR potentiators can reduce epithelial stress and local immune dysregulation induced by gliadin.
CFTR potentiators oppose the gliadin‐induced immune response ex vivo in celiac patients
To translate our findings into the relevant clinical setting, we determined whether CFTR potentiators would prevent the HLA‐restricted immune response to gliadin in two different experimental models of CD.
In the first model, we took advantage from a bidimensional co‐culture system in which confluent intestinal epithelial (Caco‐2) cells were placed in the upper compartment while peripheral blood mononuclear cells (PBMC) collected from six celiac patients and four healthy controls were cultured in the lower compartment (Vincentini et al, 2015a). Then, Caco‐2 cells were challenged with PT gliadin or with a combination of P31–43 and P57–68 (Maiuri et al, 2003; or P57–68 alone as a negative control) and IFN‐γ and IL‐10 were quantified in the supernatants from the lower compartment (Vincentini et al, 2015a). In this system, both PT gliadin (Fig 5I) and the combination of P31–43 and P57–68 (but not the P57–68 or P31–43 alone; Fig 5J) induced IFN‐γ production in the lower compartments in which celiac, but not control, PBMC were present (Fig 5I and J). VX‐770 prevented the production of IFN‐γ by PT gliadin or peptide combination (P < 0.01; Fig 5I and J) while it increased IL‐10 levels (P < 0.01; Fig 5K). Notably, the challenge of Caco‐2 cells with either PT gliadin or the peptide combination in the bidimensional culture model led to IL‐15 accumulation in the lower compartment (P < 0.001 vs. medium alone) regardless of the presence of PBMCs, an effect that was prevented by VX‐770 (P < 0.001 vs. peptide combination; Appendix Fig S9A). Notably, when normal Caco‐2 cells were replaced by Caco‐2CFTR‐KO cells, VX‐770 failed to prevent the production of IFN‐γ by celiac PBMC upon challenge with P31–43 and P57–68 (Fig 5L), confirming that VX‐770 controls gliadin‐induced immune response through on‐target (CFTR‐mediated) effects.
In the second model, we challenged duodenal biopsies from 5 CD patients with PT gliadin (500 μg/ml; Maiuri et al, 2003; Barone et al, 2014) or the combination of P31–43 and P57–68 (20 μg/ml each), as described (Maiuri et al, 2003) in the presence or absence of 3 h pre‐incubation with the CFTR potentiator Vrx‐532 (20 μM). Either PT gliadin or the combination of the two peptides (but neither of them alone) induced a significant increase in the number of CD3+CD25+ cells within the lamina propria, as described (Maiuri et al, 2003). Notably, Vrx‐532 was highly effective in abrogating these effects of gliadin peptides (Appendix Fig S9B).
These preclinical in vitro results suggest that CFTR potentiators might be efficient in reducing pathogenic inflammatory reactions in patients with CD.
Discussion
Our results shed light on the mechanisms through which the gluten component gliadin triggers epithelial stress in the intestine and ignites inflammatory reactions that are relevant to CD. We showed that one particular α‐gliadin fragment, P31–43, can directly interact with a functionally important domain of CFTR, NBD1, to cause a conformational change that interferes with the ATPase activity of NBD1 and ultimately reduces the cAMP‐stimulated chloride channel function of CFTR. Consistent with previous studies in CF, the functional inhibition of CFTR perturbs cellular physiology at multiple levels. Indeed, as a consequence of stressed cellular response, and much like in CF (Luciani et al, 2010b; Villella et al, 2013a,b), gliadin‐mediated CFTR inhibition resulted in TG2 activation that impaired endosomal trafficking through perturbing the activity of PI3K complex 3 (Fig 6). Notably, gliadin‐mediated CFTR inhibition stimulated pro‐inflammatory reactions including the secretion of IL‐15. Thus, CFTR inhibition may favor the emergence of a stressed local context that contributes to break gluten tolerance.
Figure 6. Schematic view of celiac disease pathogenesis.

The P31–43 peptide from the gluten‐associated protein gliadin is generated in the gut lumen and interacts with the NBD1 domain of CFTR, if the domain is in its inactive conformation. P31–43 binds to specific residues of NBD1 of inactive CFTR, competing with ATP binding, thus blocking CFTR function as an anion channel. The inhibition of CFTR disrupts cellular proteostasis through two effects (i) TG2 activation and (ii) BECN1 complex inhibition. This accessorily recruits TG2 to a tripartite complex that stabilizes P31–43 binding to CFTR, thus worsening CFTR inhibition. P31–43‐mediated CFTR inhibition leads to impaired endosomal trafficking, cytoskeleton disassembly, inflammasome activation resulting in IL‐1β secretion, NF‐κB activation, and consequent IL‐15 production. Stressed enterocytes stimulate local inflammation and immune responses against immunodominant gliadin peptides, in particular peptide P56–88, in the context of HLA‐DQ2/DQ8, thus triggering celiac disease. This pathogenic cascade may be theoretically interrupted at two levels, at the apex by CFTR potentiators that prevent P31–43 binding to CFTR (but are poorly active if they are added after P31–43 addition) or by reconstitution of cellular proteostasis by inhibiting TGase or activating the BECN1 complex.
The broad contribution of CFTR to normal epithelial function may explain how a constitutive or acquired CFTR malfunction may contribute to the pathogenesis of a number of diseases (Thiagarajah et al, 2014; Raju et al, 2016) beyond CF. The hitherto unsuspected involvement of CFTR in CD pathogenesis has potential clinical implications. Several distinct pharmacological agents that are considered to act as CFTR potentiators oppose the negative effects of gliadin on CFTR function. Thus, the CFTR potentiator VX‐770 (Ivacaftor), an FDA‐ and EMA‐approved drug for the treatment of CF patients bearing PM‐resident CFTR mutants, prevented gliadin‐induced pathology in several models, namely in human intestinal epithelial cells cultured in vitro, preclinical mouse models of gliadin sensitivity in vivo, as well as ex vivo models of CD patient‐derived cells and tissues. VX‐770 allows CFTR channel to maintain an open conformational state either via ATP‐independent mechanisms or by favoring multiple rounds of ATP hydrolysis (Jih & Hwang, 2013; Eckford et al, 2012). We speculate that VX‐770 opposes the effects of P31–43 on NBD1, thereby blocking the negative effects of gliadin‐derived peptides on CFTR function. Although other mechanisms could act in concert with CFTR inhibition in driving the pathogenic cascade of CD, our results highlight a primordial pathogenic role for CFTR in gluten sensitivity.
It remains to be determined whether measures designed to intercept pathogenic pathways that act downstream of CFTR inhibition (such as TG2 activation, PI3K complex 3 inhibition, inflammasome activation, or stimulation of the IL‐15 system) might be used in combination with CFTR potentiators to suppress CD pathogenesis. Yet, another possibility of therapeutic intervention might consist in the generation of competitive inhibitors of the physical interaction between pathogenic gliadin peptides and CFTR. Thus, agents that block the binding of the α‐gliadin fragment P31–43 to the NBD1 from CFTR and hence avoid P31–43‐mediated CFTR inhibition might constitute alternative options for preventing CD manifestations in genetically susceptible individuals.
In conclusion, this study identifies CFTR as a molecular target of gluten that contributes to CD pathogenesis, providing the scientific rationale for repurposing CFTR potentiators for the prevention or treatment of CD. Future clinical trials must explore whether oral administration of CFTR potentiators, possibly in galenic formulations that render them bioavailable after gastric passage, may be capable to interfere with CD pathogenesis and to allow celiac individuals to avoid autoimmune comorbidities without changing their diet.
Materials and Methods
Peptides
The following peptides were synthesized by Inbios (Napoli, Italy): α‐gliadin peptide LGQQQPFPPQQPY (P31–43) or QLQPFPQPQLPY (P57–68) or LQLQPFPQPQLPY PQPQLPYPQPQLPYPQPQPF (P56–88) or scrambled GAVAAVGVVAGA (PGAV) or modified P31–43 (different Q‐A or P‐A substitutions, single position or double positions, 4QA). All peptides were obtained with or without Biotin‐NH2‐tag.
Molecular modeling
NBD1 and NBD2 crystal structures were retrieved from Protein Data Bank (PDB: 2BBO and 3GD7). All the ligands and cofactors were removed; hydrogen atoms were added to the protein structure using Autodock 4.2 (Morris et al, 2009). The missing part of the structure 2BBO (aa 405–435) was added, even if it does not influence the following in silico procedures. Same can be applied for the amino acid mutations (F409L, F429S, F433L, G550E, R553Q, R555K, H667R). To minimize contacts between hydrogens, the structures were subjected to Amber force field keeping all the heavy atoms fixed. Peptides P31–43 (LGQQQPFPPQQPY) and 4QA (LGQAAPFPPAAPY) were built using PEP‐FOLD3, by generating five clusters sorted using sOPEP energy value (Morris et al, 2009).
Protein–protein docking analysis was performed using two FFT‐based docking software packages PIPER (Shen et al, 2014) and Zdock (Kozakov et al, 2006). The procedures were performed using NBD1 or NBD2 crystal structures as the target proteins while the peptides were considered as probes. 1,000 complexes were obtained from both docking algorithms and clusterized using the pairwise RMSD (root mean square deviation) into six clusters. The final complex was chosen according to the energy scoring function. In particular, among the five clusters generated by PEP‐FOLD3 one single P31–43 peptide conformation resulted to be selected efficiently by both protein–protein docking algorithms (75% of total conformations). Protein–protein docking analysis was not able to retrieve significant P31–43 peptide conformations able to interact with NBD2; indeed, the best solution selected presented a very low sample percentage (7%). P31–43 conformation bound to NBD1 was optimized by a remodeling of the peptide folding in close contact with NBD1 (PEP‐FOLD3), starting from the patch of interaction retrieved from the protein–protein docking procedure.
Molecular dynamics (MD) simulations of the final complex (parameterized with AMBER14SB/ff14SB) were performed with ACEMD (Accellera MC4‐node, 4× GeForce GTX980 GPUs; Chen et al, 2003; Harvey et al, 2009) in order to verify the complex stability over time; in particular, a 500 ns of NPT (isothermal–isobaric ensemble, 1 atm, 300 K) MD simulation was performed after an equilibration phase of 10 ns (positional restraints were applied on carbon atoms to equilibrate the solvent around the protein).
The interaction pattern between P31–43 and NBD1 obtained from the computational approach described above was used to design a peptide unable to efficiently interact with NBD1. In particular, an in silico Ala‐scanning was performed targeting the P31–43 residues mostly involved in the interaction with NBD1. All the peptides produced by the combination of different amino acid substitutions were subjected to PEP‐FOLD3 protocol and docked against NBD1 through the combined PIPER/Zdock approach. The peptide presenting an Ala substitution in 4th, 5th, 10th, and 11th positions (4QA) was selected as the best solution as control peptide unable to interact with NBD1 (sampling percentage of only 12% compared to the 75% of P31–43).
The same protein–protein docking and molecular dynamics approaches were used to investigate the binding of P31–43 to CFTR structure obtained via electron microscopy (PDB code: 5UAK) and parameterized as described before. In this particular case and during molecular dynamics processes, the transmembrane domains and the R‐Domain were kept fixed, the first being immersed in a membrane system, the latter because its position is not completely determined in the structure.
In vitro studies
Proteins cloning, expression, and purification
All the in vitro experiments were performed with recombinant human NBD1 domain corresponding to region 389–678 of full‐length protein NP_000483.3. Three different sources of recombinant NBD1 were used: (i) already purified wild‐type human NBD1[389–678], with the so‐called “solubilizing mutations” (F429S/F494N/L636E/Q637R), was generously provided by Dr. C.G. Brouillette (University of Alabama at Birmingham, AL, USA); (ii) already purified wild‐type human NBD1[389–673], without any solubilizing mutations, was obtained from CFTR Folding Consortium consortium in the distribution program supported by Cystic Fibrosis Foundation; (iii) plasmid encoding human recombinant NBD1 domain [389–678] with the solubilizing mutations F429S/F494N/Q637R was purchased from DNASU plasmid repository (clone id HsCD00287336).
From this construct, we obtained the two double mutants F400A/E403A and P439A/P477A (Primm, Milan, Italy). Wild‐type, F400A/E403A, and P439A/P477A contained six histidines in the N‐terminus to facilitate the in‐house purification following the protocol reported in http://www.cftrfolding.org. All the wild‐type hNBD1, listed above, was assayed in the same conditions and gave the same results.
Surface Plasmon Resonance
Peptide–protein interactions were studied using a Biacore™ T100 (GE Healthcare) instrument. hrNBD1 domain was immobilized by amino coupling on a CM5 (series S) sensor chip to a final density of 2000 resonance units (RU). A flow cell with no immobilized protein was used as control. Binding analysis was carried out at 20°C in a running buffer consisting of 10 mM HEPES, pH 7.4, 150 mM NaCl, 0.05% (v/v) Tween‐20, 2 mM ATP, 5 mM MgCl2, 1 mM DTT applying a flow rate of 20 μl/min. For each experiment, a Biacore™ method program was used. It included a series of three start‐up injections (running buffer), zero control (running buffer), and different concentrations of analyte. In all cases, the chip surface was regenerated with two 20 s injections of 0.03% (w/v) SDS; this treatment restored the baseline to the initial RU value. Each sensorgram (time‐course of the surface plasmon resonance signal) was corrected for the response obtained in the control flow cell and normalized to baseline. Data were analyzed using the 2.0.3 BIAevaluation software (GE Healthcare). At least three independent experiments for each analysis were performed.
Peptide–protein interactions were further studied exploiting a different biochemical strategy: recombinant NBD1, both wild type and/or its mutants, was incubated at 4°C for 30′ in 40 mM Tris pH 7.5 in the presence of the biotinylated P31–43 peptide unless otherwise indicated and resolved in native conditions by BN‐PAGE (Novex, Invitrogen). The complexes were then blotted onto PVDF membrane in accordance with the manufacturer's instructions, and the interacting partners (biotinylated peptide/binding proteins) were detected with streptavidin‐HRP antibody (PerkinElmer).
In vitro characterization of NBD1 ATPase activity
To assess the capability of P31–43 peptide to bind NBD1 domain and modify its ATP binding site, the intrinsic tryptophan fluorescence of NBD1 was measured. Experiments were performed in a 0.2‐ml fluorimeter cuvette at 25°C using Varian Cary Eclipse fluorescence spectrophotometer. NBD1 protein was diluted to 1 μM in a buffer containing 50 mM Tris/HCl (pH 7.6), 150 mM NaCl, and 5 mM MgCl2, supplemented with 2 mM ATP and P31–43 5 μM when indicated. All spectra were corrected for buffer fluorescence. Excitation wavelength was adjusted to 292 nm, and emission was scanned over the range 300–500 nm.
The measurement of NBD1 ATPase activity was performed with a malachite green‐based kit supplied by Sigma (cod. MAK113) in accordance with the manufacturer's instructions. The enzyme activity was measured at 620 nm at room temperature using a Multiskan 96‐well plate reader. In graph of Fig 2J is reported the NBD1 ATPase activity inhibition obtained with IC50 (50 μM) of P31–43 and 5 μM of P‐ATP as control.
Cells and treatments
Human colon adenocarcinoma‐derived Caco‐2 and T84 cells were obtained from the ATCC. Cells were maintained in T25 flask in modified Eagle's medium (MEM) for Caco‐2, or Ham's F12+ DMEM (1:1) for T84, supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine +1% non‐essential amino acids (NEAA), and the antibiotics penicillin/streptomycin (100 units/ml; all reagents from Lonza; Luciani et al, 2010a). Cells were grown in Transwells (Corning, 3,470 or 3,460) under the normal condition. Briefly, 8 × 104 or 5 × 105 cells were seeded in 6.5 mm diameter or 12 mm diameter, respectively, and grown until the RT reached 800–1,200 Ω cm2. Transwells with a pore size of 0.4 μm were used. Medium in both the apical and basolateral chambers was changed every other day (Luciani et al, 2012). Cells were treated with 20 μg/ml of either α‐gliadin peptide P31–43 or P57–68 or P56–88 or scrambled PGAV or modified P31–43 either biotin‐tagged or not, for different time point (from 1 h short challenge up to 24 h). Cells were also treated with 50 or 100 μg/ml of P57–68 or PGAV peptides for 3 h. Cells were also treated with pepsin‐trypsin‐gliadin (PT gliadin; 500 μg/ml; Maiuri et al, 2003; Barone et al, 2014, 2007) prepared as described (Drago et al, 2006) with minor modifications. Breifly, each 50 g Sigma gliadin (G‐3375; Sigma, St. Louis, MO) or 50 g wheat flour was dissolved in 500 ml 0.2 N HCl for 2 h at 37°C with 1 g pepsin (P‐7000; 800–2,500 units/mg protein, Sigma, St. Louis, MO). The resultant peptic digest was further digested by addition of 1 g trypsin (P‐8096, activity, 4× USP specifications; Sigma, St. Louis, MO), after pH adjusted to 7.4 using 2 M NaOH. The solution was stirred vigorously at 37°C for 4 h, boiled to inactivate enzymes for 30 min, lyophilized, and then stored at −20°C until used. PT gliadin was freshly suspended in a sterile phosphate‐buffered saline (PBS, 0.15 M NaCl, 0.0027 M KCl, 0.0081 M disodium phosphate, and 0.0015 M monopotassium phosphate, pH 7.2). Caco‐2 or T84 cells were also treated with CFTR potentiators VX‐770 (10 μM) or Vrx‐532 (20 μM; Selleck chemicals) or in presence or absence of CFTR inhibitor172 (CFTRinh172, 20 μM) or 3‐methyladenine (3‐MA, 20 μM; Calbiochem), Z‐DON (20 nM, Zedira), or BAPTA‐AM (10 μM, Calbiochem).
NBD1 mutant transfection
Caco‐2CFTR‐KO cells were transfected with NBD1 mutant CFTR plasmids pcDNA3.1_F400A/E403A‐CFTR and pcDNA3.1_P439A/P477A‐CFTR (GenScript, New Jersey, USA), or with pcDNA3b_WT‐CFTR (as positive control), by means of Lipofectamine 2000 (Life‐Technology), in accordance with the manufacturer's instructions. After 24 h of transfection, the cells were challenged with P31–43 for 3 h, and then, immunoprecipitations of transfected proteins were performed on 500 μg of cell lysates.
Mice and treatments
BALB/c mice (background BALB/cAnNCrl; Papista et al, 2012) were purchased from Charles River (Varese, Italy). Three‐generation gluten‐free diet (Mucedola srl, Milan), male and female, were challenged with gliadin for 4 weeks (Galipeau et al, 2011; Papista et al, 2012; Larsen et al, 2015; Moon et al, 2016). To assess the effects of VX‐770 into a controlled environment, mice were challenged via gavage for 4 weeks with (i) vehicle alone or (ii) gliadin (Sigma‐Aldrich, G3375; 5 mg/daily for 1 week and then 5 mg/daily thrice a week for 3 weeks; Galipeau et al, 2011; Papista et al, 2012; Larsen et al, 2015; Moon et al, 2016) in the presence or absence of intraperitoneal VX‐770 (0.075 mg/mice in 100 μl DMSO, 2 mM, Selleckchem S1144), administered 15 min prior each gliadin challenge (n = 10 mice per group of treatment).
CF mice homozygous for the F508del‐CFTR in the FVB/129 outbred background (Cftrtm1EUR, F508del, FVB/129, abbreviated Cftr F508del/F508del) and wild‐type littermates, male and female, were obtained from Bob Scholte, Erasmus Medical Center Rotterdam, The Netherlands; CF coordinated action program EU FP6 LSHMCT‐2005‐018932 (van Doorninck et al, 1995; De Stefano et al, 2014).
Transgenic KO Cftr mice (B6.129P2‐KOCftrtm1UNC, abbreviated Cftr −/−), and wild‐type littermates, were purchased from The Jackson Laboratory (Bar Harbor, ME, USA).
In order to obtain TG2−/− mice carrying F508del‐CFTR mutation, C57BL/6 mice KO for TG2 (obtained from Gerry Melino, Department of Experimental Medicine and Biochemical Sciences, University of Rome “Tor Vergata”, Rome, Italy) were crossed with 129/FVB Cftr F508del/F508del mice (abbreviated Cftr F508del/F508del/TG2 −/−), as described (Rossin et al, 2018). The newly generated mice were housed at Department of Experimental Medicine and Biochemical Sciences, University of Rome “Tor Vergata” (Rome, Italy).
All above‐described mice for the study were 10 weeks old. At least 10 mice per group per experiment were used.
Prediabetic NOD (Non‐obese diabetic) mice were purchased from Charles River (Varese, Italy). Diabetes incidence was followed weekly measuring of blood glucose levels with a Contour glucose meter (Bayer; US). At time 12–13 weeks, female mice with manifested diabetes (> 250 mg/dl) were challenged as described in (i) or (ii).
NOD.scid AB0nullDQ8 mice (NOD DQ8tg, transgenic mice that express HLA‐DQ8 in an endogenous MHC class II‐deficient background were backcrossed to NOD mice for 10 generations and intercrossed to produce congenic NOD AB° DQ8 mice; Galipeau et al, 2011; Moon et al, 2016) were purchased from The Jackson Laboratory (Bar Harbor, ME, USA) and were weaned and maintained on a low‐fat (4.4%), gluten‐free diet (Mucedola srl, Milan), and bred in a conventional, specific pathogen‐free colony at the San Raffaele Scientific Institute SOPF animal house (Milan, Italy). Mice were challenged as described in (i) or (ii).
At the end of the last daily treatment, mice were anesthetized with avertin (tribromoethanol, 250 mg/kg, Sigma‐Aldrich, T48402) and then killed; the intestines were collected for CFTR function analysis or stored for all described techniques.
These studies and procedures were approved by the local Ethics Committee for Animal Welfare (IACUC No 583, 849, 713, 661, 628) and conformed to the European Community regulations for animal use in research (2010/63 UE).
Purification of PBMC and transwell co‐culture model
Five milliliters of peripheral blood has been withdrawn from six untreated celiac patients (females and males, age range 8–25 years) and from four not CD‐affected controls. The Ethics Committee of the Istituto Superiore di Sanità (ISS) approved the protocol (#CE/12/341), and patients or patients’ parents signed the informed consent. Peripheral blood mononuclear cells were isolated using lympholite (Cederlane, UK) density gradient overlaid by heparin blood diluted 1:1 in PBS and centrifuged (20 min at 900 rpm). After being washed three times, PBMCs were resuspended in complete RPMI 1640 supplemented with 25 mM HEPES, 10% (v/v) heat‐inactivated FBS, 100 U/ml penicillin, 100 mg/ml streptomycin, and 1% 2 mM l‐glutamine.
For transwell experiments using polarized Caco‐2 cells, 3 weeks prior to the experiment, Caco‐2 cells were seeded at a density of 80 × 103 cells × cm2 on 0.4‐μm, 1‐cm2 tissue culture inserts (Costar, Corning Incorporated). Transwell cultures (12‐well) with confluent Caco‐2 monolayers were used for co‐culture with 1 ml PBMC (1.5 × 106 cells/ml) using PBMC medium and kept in an incubator at 37°C and 5% CO2. Cells were allowed to settle for 1 h before the starting of the experiment. The permeability of the epithelial monolayer was assessed just before the experiments, measuring the transwell electrical resistance between the upper and lower compartments. A value of transwell resistance > 800 Ω × cm2 has been considered index of a fully formed epithelial monolayer, not allowing the paracellular passage of molecules (Vincentini et al, 2015a).
Caco‐2 or Caco‐2CFTR‐KO cells were apically exposed for 3 h with P31–43 peptide (20 μg/ml) and then treated with P57–68 (20 μg/ml) in presence or absence of CFTR potentiators VX‐770. As negative control, cells were treated with medium alone and with P57–68 alone. After the treatments, supernatants from the basolateral compartment were collected, centrifuged, and stored at −20°C until cytokine measurement. At the same time, the cells from the apical compartment were harvested, lysated, and stored at −80°C.
Ussing chamber
Chambers for mounting either transwell cell cultures or mouse tissue biopsies were obtained from Physiologic Instruments (model P2300, San Diego, CA, USA). Chamber solution was buffered by bubbling with identical Ringer solution on both sides and were maintained at 37°C, vigorously stirred, and gassed with 95% O2/5% CO2. Cells or tissues were short circuited using Ag/AgCl agar electrodes. A basolateral‐to‐apical chloride gradient was established by replacing NaCl with Na‐gluconate in the apical (luminal) compartment to create a driving force for CFTR‐dependent Cl− secretion. To measure stimulated Isc, the changed sodium gluconate solution, after stabilization, was supplied with 100 μM amiloride. Agonists (forskolin) were added to the bathing solutions as indicated (for a minimum 5 min of observation under each condition) to activate CFTR channels present at the apical surface of the epithelium (either cell surface or lumen side of the tissue) and CFTRInh‐172 (10 μM) was added to the mucosal bathing solution to block CFTR‐dependent Isc. Short‐circuit current [expressed as Isc (μA/cm2)] and resistance were acquired or calculated using the VCC‐600 transepithelial clamp from Physiologic Instruments and the Acquire &Analyze2·3 software for data acquisition (Physiologic Instruments), as previously described (Gondzik & Awayda, 2011; Marchelletta et al, 2013; Tosco et al, 2016; Romani et al, 2017).
To evaluate the effect of the direct addition of P31–43 (100 μM each) to the bathing solution, the solution was supplied with 100 μM amiloride after stabilization and then: (i) P31–43 or the control peptide (100 μM each) was directly added to the bathing solution for 3–5 min before adding forskolin (20 μM; for a minimum of 5 min under each condition) or (ii) P31–43 (100 μM) was directly supplied, as before, 3–5 min after forskolin stimulation. In addition, the bathing solution was first supplemented with (i) VX‐770 (10 μM) for 5 min, followed by addition of P31–43 for 3–5 min first, and then forskolin or (iv) P31–43 followed by VX‐770 and then forskolin.
Finally, in all experimental conditions, CFTRInh‐172 (10 μM) was directly added to the bathing solution to block CFTR‐dependent Isc.
Whole‐cell patch‐clamp of Caco‐2 cells
Whole‐cell patch‐clamp recordings were performed by using an EPC 10‐patch‐clamp amplifier (HEKA Elektronik). Pipettes with a resistance of 5–10 MΩ were pulled from borosilicate glass capillary tubing (borosilicate glass with filament, fire polished, o.d. 1.5 mm, i.d. 0.75 mm, length 10 cm; Sutter Instrument, cat. no. BF150‐75‐10) using a Flaming/Brown‐type micropipette puller (Sutter Instrument, cat. no. P‐97). Seal resistances ranging from 3 to 10 GΩ were obtained. Current–voltage (I–V) relationships were built by clamping the membrane potential of Caco‐2 cells at −40 mV and by delivering ramps from −100 mV to +100 mV, each lasting 5 s. The pipette solution contained 113 mM l‐aspartic acid, 113 mM CsOH, 27 mM CsCl, 1 mM NaCl, 1 mM MgCl2, 1 mM EGTA and 10 mM TES (pH 7.2). MgATP (3 mM) was added just before the patch‐clamp experiments were started. The external solution contained 145 mM NaCl, 4 mM CsCl, 1 mM CaCl2, 10 mM glucose, and 10 mM TES (pH 7.4). Results were analyzed by means of PATCHMASTER software (HEKA Elektronik).
Permeability assay: Fluorescein Isothiocyanate‐Dextran 4000 (FITC‐D4000) Test
The FITC‐D4000 test in treated BALB/c mice (as described in mice and treatments section) was performed as previously described (Volynets et al, 2016). The 4,000‐Da test substance can be easily detected in blood plasma by a photometric approach. Briefly, FITC‐D4000 (Sigma‐Aldrich) was administered to the mice by gavage at a concentration of 600 mg/kg body weight and a volume of 200–300 μl, using a stock solution at 50 mg/ml. After gavage, the mice remained in the metabolic cages until the experiments were completed and the mice were killed. One hour after gavage, the animals were anesthetized and blood was taken by cardiocentesis, heparinized, and then centrifuged (10 min, 12,000 g, 4°C). Plasma was light protected and stored at −80°C for photometric analysis of FITC‐D4000. For use as a measurement, the plasma was diluted in an equal volume of phosphate‐buffered saline (PBS, pH 7.4). Standards (range 50–0.312 μg/ml) were obtained by diluting the FITC‐D4000 gavage stock solution in PBS. An amount of 100 μl of both diluted animal samples and standards, as well as blanks (PBS and diluted plasma from untreated animals), were transferred to black 96‐well microplates. Analysis of the FITC‐D4000 concentration was carried out with a fluorescence spectrophotometer (Multi‐Detection Microplate Reader) at an excitation wavelength of 485 nm and an emission wavelength of 528 nm.
CRISP/CAS9 CFTR knock‐out
CFTR CRISP/CAS9 KO plasmids were purchased from Santa Cruz Biotechnology and transfected in Caco‐2 cells by UltraCruz transfection reagent according to the manufacturer's instructions (Santa Cruz Biotech.). Successful transfection of CRISPR/Cas9 KO Plasmid was visually confirmed by detection of the green fluorescent protein (GFP) by immunofluorescence. The cells were then sorted by replacing selective media with puromycin antibiotic approximately every 2–3 days for a minimum of 3–5 days. The knock‐out was then confirmed by western blot with specific CFTR antibody and by functional assay (Ussing chamber or SPQ assay).
Real‐Time and reverse transcription analysis
The analysis was performed as previously described (Maiuri et al, 2008; Luciani et al, 2009; Luciani et al, 2010b; De Stefano et al, 2014; Tosco et al, 2016). Total RNA was extracted with the RNeasy Mini Kit (Qiagen, 74104) from mouse intestine homogenates. The mRNA was reverse transcribed with OneTranscript Plus cDNA Synthesis Kit (abm good). The sequences of mouse primers were as follows (Cavalieri et al, 2005; Cezário et al, 2011; Pallotta et al, 2011; Simard et al, 2011; De Stefano et al, 2014):
IL‐15: forward 5′‐ACCAGCCTACAGGAGGCC‐3′
reverse 5′‐TGAGCAGCAGGTGGAGGTAC‐3′
IL‐17: forward 5′‐ACCGCAATGAAGACCCTGAT‐3′
reverse 5′‐TCCCTCCGCATTGACACA‐3′
NF‐ϒ: forward 5′‐AGAGGATGGTTTGCATCTGGGTCA‐3′
reverse 5′‐ACAACGCTATGCAGCTTGTTCGTG‐3′
IL‐21: forward 5′‐CGCCTCCTGATTAGACTTCCG‐3′
reverse 5′‐TGGGTGTTCCTTTTCTCATACG‐3′
IL‐10: forward 5′‐CAGAGCCACATGCTCCTAGA‐3′
reverse 5′‐TGTCCAGCTGGTCCTTTGTT‐3′
Rorc: forward, 5′‐ACAACAGCAGCAAGTGATGG‐3′
reverse 5′‐CCTGGATTTATCCCTGCTGA‐3′
Tbet: forward, 5′‐GGACGATCATCTGGGTCACATTGT‐3′
reverse 5′‐GCCAGGGAACCGCTTATATG‐3′
Expression levels of genes were normalized to β‐actin (primer design HK‐sy‐mo600) levels in the same samples.
Immunoblot and immunoprecipitation
The whole lysate or membrane fraction proteins of cell lines and mice intestine homogenates were obtained from treated and untreated cells or mice as described (Barone et al, 2007; Maiuri et al, 2008; Luciani et al, 2009; Luciani et al, 2010b; Luciani et al, 2012; Villella et al, 2013a; De Stefano et al, 2014; Tosco et al, 2016). The equal amount of protein were resolved by SDS–PAGE gel and blotted with antibodies against: SQSTM1 (Sigma‐Aldrich, 108k4767) 1:1,000, PPARγ (Santa Cruz Biotechnology, sc7273) 1:500, BECN1 (Abcam, ab58878) 1:1,000, CFTR clone M3A7 (Abcam, ab4067) 1:500, phospho‐ERK1/2 (php42/44, Cell Signaling Technology, #91101) 1:1,000, UVRAG (Santa Cruz Biotechnology sc8215) 1:1,000, NHERF‐1 (BD, 611161) 1:1,000, ezrin (BD, 610603) 1:1,000, hVps34 (Sigma V9764) 1:300, biotin (Abcam, ab1227) 1:2,000, PKAr2α (BD, 610625) 1:500, ubiquitin (Cell Signaling clone P4D1, #3936) 1:1,000, CHIP (Calbiochem PC711) 1:500, Caspase‐1 p10 (M‐20; Santa Cruz Biotechnology, sc‐514) 1:200, NLRP3 (Abcam, ab4207), 1:500, used as primary antibodies. Normalization was performed by probing the membrane with anti‐β‐actin (Cell Signaling, #4970) 1:1,000, anti‐GAPDH (Sigma‐Aldrich, G8795) 1:1,000, and anti‐flotillin (Abcam ab15148) 1:1,000 antibodies. Immunoprecipitations of endogenous proteins were performed on 500 μg of lysates from Caco‐2 cells. The proteins were incubated for 12–18 h at 4°C with 10 μg/ml of specific antibody followed by the addition of Protein A/G‐agarose beads for 4 h at 4°C. The beads were then washed three times with lysis buffer, and Laemmli's sample buffer was added to the samples. The membranes were then blotted with: CFTR clone CF3 (Abcam, ab2784) 1:1,000 or anti‐CFTR antibody 660 (provided J.R. Riordan through a program of the CFFT, University of North Carolina, Chapel Hill, NC) 1:1,000, TG2 (clone CUB7402; NeoMarkers) 1:1,000, CHIP (Calbiochem PC711) 1:500, ubiquitin (Cell Signaling clone P4D1 3936), isopeptide (Abcam 422) 1:1,000, PKAr2α (BD 610625) 1:500, and anti‐Clathrin (Abcam ab21679) 1:1,000.
For detection of complex with biotinylated‐P31–43 or immune complex of glutamine‐lysine bonds, the beads were resolved in non‐reducing and non‐denaturing conditions, and then blotted with streptavidin‐HRP (Sigma S2438) 1:3,000 or anti‐biotin (Abcam ab2103) 1:2,500 and anti‐isopeptide (Abcam 422) 1:1,000.
Nuclear and cytoplasmic extraction
For cytoplasmic and nuclear extracts, Caco‐2 cells were harvested, washed in cold PBS twice, and centrifuged at 3,300 g for 5 min in cold room to collect pellets. Pellets were resuspended in double cell volume of cytoplasmic extract (CE) buffer [10 mM HEPES, pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.3% NP‐40, and 1× protease inhibitor cocktail (Thermo Scientific™)], incubated on ice for 10 min and centrifuged at 800 g for 5 min to obtain the supernatants as cytoplasmic fraction. The pellets (containing the nuclei) were resuspended in equal volume of nuclear extract (NE) buffer (20 mM HEPES, pH 7.9, 0.4 M NaCl, 1 mM EDTA, 25% glycerol, and 1× protease inhibitor cocktail), incubated on ice for 10 min, and centrifuged at 16,100 g for 5 min to obtain the supernatants as nuclear fraction. The protein concentrations were measured by Bradford protein assay (Protein Reagent, Bio‐Rad). For Western blots, cytoplasmic and nuclear proteins were fractionated on 10% SDS–PAGE and transferred to nitrocellulose blotting membranes (Amersham™Protran™ 0.45 μm NC, GE Healthcare, Life science). Membranes were incubated with anti‐phospho‐NF‐κB p65 (Ser536; (93H1), Cell Signaling, #3033), 1:1,000, Lamin B1‐Nuclear Envelope Marker (Abcam, ab16048) 1:5,000, and anti‐β‐actin (Cell Signaling, #4970) 1:1,000.
Membrane fractionation
Protein from membrane fractionation was obtained as described (Maiuri et al, 2008; Luciani et al, 2009; Luciani et al, 2010b; Villella et al, 2013a; De Stefano et al, 2014; Tosco et al, 2016). Cells were homogenized with a Potter‐Elvehjem pestle and centrifuged at 2,300 × g for 15 min at 4°C. Supernatants that contain the cytoplasmic and PM fractions were centrifuged 1 h at 16,000 × g at 4°C; the pellet was the intact membrane and was solubilized in BUFFER A (20 mM Tris–HCl pH 7.4, 2 mM EDTA, 20 mM 2‐ME, 1× PMSF, 1 μg/ml inhibitor protease cocktail) +1% Triton X‐100 and centrifuged 1 h at 60,000 × g in the ultracentrifuge. The supernatants were collected as PM fraction. Equivalent amounts of proteins (500 mg) were used for Immunoprecipitation assay. Proteins of PM fraction were used for IP or WB and immunoblotted against CFTR, Ezrin, NHERF‐1, SQSTM1/p62, CHIP, ubiquitin, clathrin, and flotillin antibodies.
Differential fractionation and separation of endosomes
The separation of endosomal fractions was performed as described (Villella et al, 2013a) from treated and untreated cells. Protein of endosomal fraction was resolved by SDS–PAGE and blotted with primary antibodies against Rab‐5 (Abcam 18211) 1:500, Rab‐7 (Abcam 77993) 1:500, EEA‐1 (Abcam 2900) 1:500, TG2, or CFTR. EEA‐1 was used as early endosome marker. For detection of complex with biotinylated‐P31–43, the beads were resolved in non‐reducing and non‐denaturing conditions and then blotted with streptavidin‐HRP (Sigma S2438) 1:3,000 or anti‐biotin (Abcam ab2103) 1:2,500.
ELISA
ELISA was performed on tissue samples using standard ELISA kits (R&D Systems) for IL‐1β, TNF‐α, Mip2α, IL‐15, IL‐17A, IFN‐γ, IL‐21, IL‐10, and TGF‐β. According to the manufacturer's instructions, samples were read in triplicate at 450 nm in a microplate Reader (Bio‐Rad, Milan, Italy) using Microplate Manager 5.2.1 software. Values were normalized to protein concentration evaluated by Bradford analysis.
TGM2 enzyme activity detection
TG2 enzymatic activity in Caco‐2 cells, treated as described above, was detected: (i) by incubating unfixed sections with biotin‐mono‐dansylcadaverine (MDC, 10 mM, Molecular Probe) for 1 h at room temperature and then stained with Alexa Fluor 488 Streptavidin, as described previously (Maiuri et al, 2008; Luciani et al, 2010b) or (ii) or by incorporation of 5‐(biotinamido)pentylamines (BAP) into protein substrates. For BAP‐incorporation, 2 mM BAP (Soltec Ventures, B110) was directly added into the medium together with the indicated treatments. In the presence of TG2 transamidating activity, BAP is incorporated into the substrates. To measure this activity, cells were lysed and proteins were resolved by SDS‐PAGE. The blots were incubated with anti‐biotin antibody (Altuntas et al, 2015).
Immunofluorescence assay
Tissue sections and cells were processed as previously described (Maiuri et al, 2008; Luciani et al, 2009, 2010a, 2010b; Villella et al, 2013a). Mouse tissues: briefly, tissue sections were fixed and then incubated with the following primary antibodies: occludin (Invitrogen, 71‐1500) 1:150, NKG2D (Abcam 2560) 1:300. Cell lines: the cells were fixed after challenge with P31–43 (i) for 3 h at 37°C and then incubated with Lamp1 (Abcam, 24270) 1:300 and Alexa Fluor 546 Streptavidin 1:300 or (ii) for 15 min at 37°C and then incubated with UVRAG (Santa Cruz, sc8215) 1:100 and EEA‐1 (Abcam, 2900) 1:300 or TG2 (clone CUB7402) 1:500 and Phalloidin‐Alexa Fluor 488 conjugated (Thermo‐Fischer, A12379) 1:500 or EEA‐1. All primary antibodies were incubated overnight at 4°C. After washing, the slides were incubated with Alexa Fluor 488 or 546 secondary antibodies (Molecular Probe) 1:300. Moreover, IgA staining in Caco‐2 cells was performed as described (Lebreton et al, 2012): P57–68 or P31–43 peptides were added for 2 h in the apical compartment of polarized Caco‐2 cell after 1 h at 4°C of pre‐incubation with colostrum‐derived S‐IgA (250 μg/ml, Sigma‐Aldrich). Cells were washed, fixed, and stained with anti‐human IgA‐FITC (Sigma‐Aldrich, F5259) 1:50 and CD71. Slides were washed, mounted on glass coverslips using ProLong Gold (Invitrogen), and imaged on a Zeiss LSM 510 confocal laser‐scanning microscope (Carl Zeiss MicroImaging) equipped with ×63 oil‐immersion or ×40 or ×20 objective. All of the experiments were repeated at least three times, and representative images are shown. To quantify the levels of co‐localization, confocal serial sections were acquired from 8 to 10 cells per experimental condition, exported in TIFF format, and processed as previously described (Villella et al, 2013a).
Proximity ligation assay
The proximity ligation assay was performed using Duolink® in situ reagents (Olink Biosciences) following the manufacturer's specifications. The cells were fixed with 4% PFA in PBS for 15 min at room temperature. After blocking with 0.1% Triton X‐100 and 0.5% BSA dissolved in PBS, the cells were incubated overnight at 4°C with primary antibodies. After incubation with Duolink PLA anti‐rabbit Plus and anti‐mouse Minus probes (1:5 dilution, Olink Bioscience) at 37°C for 1 h, ligation, rolling circle amplification, and detection were performed using the Duolink In Situ Detection Reagents Red (Olink Bioscience). Nuclei were counterstained with DAPI. PLA signals were documented by means of a Leica SP8 confocal microscope and quantified with CellProfiler software for image analysis (Festa et al, 2018).
Statistical analysis
GraphPad Prism software 6.01 (GraphPad Software) was used for analysis. Data are expressed as means ± SD. Statistical significance was calculated by ANOVA (Bonferroni's post hoc test) for multiple comparisons and by Student's t‐test for single comparisons. We considered all P‐values < 0.05 to be significant. The in vivo groups consisted of 10 mice/group. The data reported are either representative of at least three experiments.
Author contributions
LM conceived the study and designed the experimental plan together with VR and GK, supervised the experiments, analyzed the data, and wrote the paper; LR and MP provided the scientific knowledge on mucosal immune response and on TG2, respectively, and contributed to the analysis of data and writing of the manuscript; VRV contributed to the experimental design, to the analysis of data and editing of the manuscript and together with SE performed most cell cultures, transfection, immunoblotting experiments, confocal microscopy, and Ussing chamber experiments on cell lines; AV and GC designed and performed all in vitro and in silico experiments, respectively; VO and GR performed cytokine assessment and some immunofluorescence experiments; AT contributed to experiments on human PBMC and data analysis. EF performed in vivo experiments on mice and together with RM and MCS performed Ussing chamber experiments and data analysis on mouse intestines. FR performed some immunoblotting experiments. AL contributed to immunofluorescence, on cell lines and to data analysis; Y‐KC and CG performed patch‐clamp experiments on cell lines and contributed to data analysis; SG and EG contributed to data analysis. MS performed experiments on patient's cells and contributed to the analysis of data.
Conflict of interest
The authors declare that they have no conflict of interest. A patent application by LM is pending (filing date, July 26, 2017. No 102017000085714).
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
We thank B. Scholte (Erasmus Medical Center Rotterdam), who provided Cftrtm1EUR mice (F508del mice, European Economic Community European Coordination Action for Research in Cystic Fibrosis program EU FP6 SHMCT‐2005‐018932). This project was supported by the Italian Institute of Health, under the frame of E‐Rare‐2, the ERA‐Net for Research on Rare Diseases “RescueCFTRpreclinic” (to LM and GK), by the Specific Targeted Research Project FunMeta (ERC‐2011‐AdG‐293714 to LR). GK is supported by the Ligue contre le Cancer Comité de Charente‐Maritime (équipe labelisée); Agence Nationale de la Recherche (ANR)—Projets blancs; ANR under the frame of E‐Rare‐2; Association pour la Recherche sur le Cancer (ARC); Cancéropôle Ile‐de‐France; Chancelerie des universités de Paris (Legs Poix), Fondation pour la Recherche Médicale (FRM); a donation by Elior; the European Commission (ArtForce); the European Research Council (ERC); Fondation Carrefour; Institut National du Cancer (INCa); Inserm (HTE); Institut Universitaire de France; LeDucq Foundation; the LabEx Immuno‐Oncology; the RHU Torino Lumière; the Searave Foundation; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); the SIRIC Cancer Research and Personalized Medicine (CARPEM); and the Paris Alliance of Cancer Research Institutes (PACRI).
The EMBO Journal (2019) 38: e100101
See also: L Vachel & S Muallem (January 2019)
Contributor Information
Guido Kroemer, Email: kroemer@orange.fr.
Luigi Maiuri, Email: luigi.maiuri@gmail.com.
References
- Altuntas S, Rossin F, Marsella C, D'Eletto M, Diaz‐Hidalgo L, Farrace MG, Campanella M, Antonioli M, Fimia GM, Piacentini M (2015) The transglutaminase type 2 and pyruvate kinase isoenzyme M2 interplay in autophagy regulation. Oncotarget 6: 44941–44954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barone MV, Gimigliano A, Castoria G, Paolella G, Maurano F, Paparo F, Maglio M, Mineo A, Miele E, Nanayakkara M, Troncone R, Auricchio S (2007) Growth factor‐like activity of gliadin, an alimentary protein implication for coeliac disease. Gut 56: 480–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barone MV, Nanayakkara M, Paolella G, Maglio M, Vitale V, Troiano R, Ribecco MT, Lania G, Zanzi D, Santagata S, Auricchio R, Troncone R, Auricchio S (2010) Gliadin peptide P31–43 localises to endocytic vesicles and interferes with their maturation. PLoS One 5: e122469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barone MV, Troncone R, Auricchio S (2014) Gliadin peptides as triggers of the proliferative and stress/innate immune response of the celiac small intestinal mucosa. Int J Mol Sci 15: 20518–20537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barone MV, Zimmer KP (2016) Endocytosis and transcytosis of gliadin peptides. Mol Cell Pediatr 3: 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouziat R, Hinterleitner R, Brown JJ, Stencel‐Baerenwald JE, Ikizler M, Mayassi T, Meisel M, Kim SM, Discepolo V, Pruijssers AJ, Ernest JD, Iskarpatyoti JA, Costes LM, Lawrence I, Palanski BA, Varma M, Zurenski MA, Khomandiak S, McAllister N, Aravamudhan P et al (2017) Reovirus infection triggers inflammatory responses to dietary antigens and development of celiac disease. Science 356: 44–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casaletto JB, Saotome I, Curto M, McClatchey AI (2011) Ezrin‐mediated apical integrity is required for intestinal homeostasis. Proc Natl Acad Sci USA 108: 11924–11929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalieri H, Gamba G, Courreges MC, Massouh EJ, Benencia F (2005) Expression of IL‐15, IL‐18 and NOS‐II in contralateral eyes of BALB/c mice during the development of HSV‐induced keratitis. Immunol Lett 96: 295–298 [DOI] [PubMed] [Google Scholar]
- Cerf‐Bensussan N, Meresse B (2015) Coeliac disease & gluten sensitivity: epithelial stress enters the dance in coeliac disease. Nat Rev Gastroenterol Hepatol 12: 491–497 [DOI] [PubMed] [Google Scholar]
- Cezário GA, de Oliveira LR, Peresi E, Nicolete VC, Polettini J, de Lima CR, Gatto M, Calvi SA (2011) Analysis of the expression of toll‐like receptors 2 and 4 and cytokine production during experimental Leishmania chagasi infection. Mem Inst Oswaldo Cruz 106: 573–583 [DOI] [PubMed] [Google Scholar]
- Chen R, Li L, Weng Z (2003) ZDOCK: an initial‐stage protein docking algorithm. Proteins 52: 80–87 [DOI] [PubMed] [Google Scholar]
- Chin S, Hung M, Bear CE (2017) Current insights into the role of PKA phosphorylation in CFTR channel activity and the pharmacological rescue of cystic fibrosis disease‐causing mutants. Cell Mol Life Sci 74: 57–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui G, McCarty NA (2015) Murine and human CFTR exhibit different sensitivities to CFTR potentiators. Am J Physiol Lung Cell Mol Physiol 7: L687–L699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui G, Khazanov N, Brandon B, Stauffer Infield D (2016) Ion channels and transporters in lung function and disease potentiators exert distinct effects on human, murine, and Xenopus CFTR. Am J Physiol Lung Cell Mol Physiol 311: L192–L207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutting GR (2015) Cystic fibrosis genetics: from molecular understanding to clinical application. Nat Rev Genet 16: 45–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies PJ, Davies DR, Levitzki A, Maxfield FR, Milhaud P, Willingham MC, Pastan IH (1980) Transglutaminase is essential in receptor‐mediated endocytosis of alpha 2‐macroglobulin and polypeptide hormones. Nature 283: 162–167 [DOI] [PubMed] [Google Scholar]
- De Lisle RC, Borowitz D (2013) The cystic fibrosis intestine. Cold Spring Harb Perspect Med 3: a009753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stefano D, Villella VR, Esposito S, Tosco A, Sepe A, De Gregorio F, Salvadori L, Grassia R, Leone CA, De Rosa G, Maiuri MC, Pettoello‐Mantovani M, Guido S, Bossi A, Zolin A, Venerando A, Pinna LA, Mehta A, Bona G, Kroemer G et al (2014) Restoration of CFTR function in patients with cystic fibrosis carrying the F508del‐CFTR mutation. Autophagy 10: 2053–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePaolo RW, Abadie V, Tang F, Fehlner‐Peach H, Hall JA, Wang W, Marietta EV, Kasarda DD, Waldmann TA, Murray JA, Semrad C, Kupfer SS, Belkaid Y, Guandalini S, Jabri B (2011) Co‐adjuvant effects of retinoic acid and IL‐15 induce inflammatory immunity to dietary antigens. Nature 471: 220–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Doorninck JH, French PJ, Verbeek E, Peters RH, Morreau H, Bijman J, Scholte BJ (1995) A mouse model for the cystic fibrosis delta F508 mutation. EMBO J 14: 4403–4411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drago S, El Asmar R, Di Pierro M, Grazia Clemente M, Tripathi A, Sapone A, Thakar M, Iacono G, Carroccio A, D'Agate C, Not T, Zampini L, Catassi C, Fasano A (2006) Gliadin, zonulin and gut permeability: effects on celiac and non‐celiac intestinal mucosa and intestinal cell lines. Scand J Gastroenterol 41: 408–419 [DOI] [PubMed] [Google Scholar]
- Eckford PD, Li C, Ramjeesingh M, Bear CE (2012) Cystic fibrosis transmembrane conductance regulator (CFTR) potentiator VX‐770 (ivacaftor) opens the defective channel gate of mutant CFTR in a phosphorylation‐dependent but ATP‐independent manner. J Biol Chem 287: 36639–36649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari E, Monzani R, Villella VR, Esposito S, Saluzzo F, Rossin F, D'Eletto M, Tosco A, De Gregorio F, Izzo V, Maiuri MC, Kroemer G, Raia V, Maiuri L (2017) Cysteamine re‐establishes the clearance of Pseudomonas aeruginosa by macrophages bearing the cystic fibrosis‐relevant F508del‐CFTR mutation. Cell Death Dis 8: e2544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Festa BP, Chen Z, Berquez M, Debaix H, Tokonami N, Prange JA, Hoek GV, Alessio C, Raimondi A, Nevo N, Giles RH, Devuyst O, Luciani A (2018) Impaired autophagy bridges lysosomal storage disease and epithelial dysfunction in the kidney. Nat Commun 9: 161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fluge G, Olesen HV, Gilljam M, Meyer P, Pressler T, Storrösten OT, Karpati F, Hjelte L (2009) Co‐morbidity of cystic fibrosis and celiac disease in Scandinavian cystic fibrosis patients. J Cyst Fibros 8: 198–202 [DOI] [PubMed] [Google Scholar]
- Gadsby DC, Vergani P, Csanady L (2006) The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature 440: 477–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galeno L, Galfrè E, Moran O (2011) Small‐angle X‐ray scattering study of the ATP modulation of the structural features of the nucleotide binding domains of the CFTR in solution. Eur Biophys J 40: 811–824 [DOI] [PubMed] [Google Scholar]
- Galipeau HJ, Rulli NE, Jury J, Huang X, Araya R, Murray JA, David CS, Chirdo FG, McCoy KD, Verdu EF (2011) Sensitization to gliadin induces moderate enteropathy and insulitis in nonobese diabetic‐DQ8 mice. J Immunol 187: 4338–4346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gondzik V, Awayda MS (2011) Methods for stable recording of short‐circuit current in a Na+‐transporting epithelium. Am J Physiol Cell Physiol 301: C162–C170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey M, Giupponi G, De Fabritiis G (2009) ACEMD: accelerated molecular dynamics simulations in the microseconds timescale. J Chem Theory Comput 5: 1632 [DOI] [PubMed] [Google Scholar]
- Jabri B, Abadie V (2015) IL‐15 functions as a danger signal to regulate tissue‐resident T cells and tissue destruction. Nat Rev Immunol 15: 771–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jih KY, Hwang TC (2013) Vx‐770 potentiates CFTR function by promoting decoupling between the gating cycle and ATP hydrolysis cycle. Proc Natl Acad Sci USA 110: 4404–4409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi K, Yoshida S, Hatano R, Asano S (2017) Pathophysiological roles of ezrin/radixin/moesin proteins. Biol Pharm Bull 40: 381–390 [DOI] [PubMed] [Google Scholar]
- Kim KS, Hong SW, Han D, Yi J, Jung J, Yang BG, Lee JY, Lee M, Surh CD (2016) Dietary antigens limit mucosal immunity by inducing regulatory T cells in the small intestine. Science 351: 858–863 [DOI] [PubMed] [Google Scholar]
- Korneychuk N, Meresse B, Cerf‐Bensussan N (2015) Lessons from rodent models in celiac disease. Mucosal Immunol 8: 18–28 [DOI] [PubMed] [Google Scholar]
- Kozakov D, Brenke R, Comeau SR, Vajda S (2006) PIPER: an FFT‐based protein docking program with pairwise potentials. Proteins 65: 392–406 [DOI] [PubMed] [Google Scholar]
- Larsen J, Weile C, Antvorskov JC, Engkilde K, Nielsen SM, Josefsen K, Buschard K (2015) Effect of dietary gluten on dendritic cells and innate immune subsets in BALB/c and NOD mice. PLoS One 10: e0118618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebreton C, Ménard S, Abed J, Moura IC, Coppo R, Dugave C, Monteiro RC, Fricot A, Traore MG, Griffin M, Cellier C, Malamut G, Cerf‐Bensussan N, Heyman M (2012) Interactions among secretory immunoglobulin A, CD71, and transglutaminase‐2 affect permeability of intestinal epithelial cells to gliadin peptides. Gastroenterology 143: 698–707 [DOI] [PubMed] [Google Scholar]
- Liang C, Lee JS, Inn KS, Gack MU, Li Q, Roberts EA, Vergne I, Deretic V, Feng P, Akazawa C, Jung JU (2008) Beclin1‐binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol 10: 776–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lionetti E, Gatti S, Pulvirenti A, Catassi C (2015) Celiac disease from a global perspective. Best Pract Res Clin Gastroenterol 29: 365–379 [DOI] [PubMed] [Google Scholar]
- Liu F, Zhang Z, Csanády L, Gadsby DC, Chen J (2017) Molecular Structure of the Human CFTR Ion Channel. Cell 169: 85‐95 [DOI] [PubMed] [Google Scholar]
- Luciani A, Villella VR, Vasaturo A, Giardino I, Raia V, Pettoello‐Mantovani M, D'Apolito M, Guido S, Leal T, Quaratino S, Maiuri L (2009) SUMOylation of tissue transglutaminase as link between oxidative stress and inflammation. J Immunol 183: 2775–2778 [DOI] [PubMed] [Google Scholar]
- Luciani A, Villella VR, Vasaturo A, Giardino I, Pettoello‐Mantovani M, Guido S, Cexus ON, Peake N, Londei M, Quaratino S, Maiuri L (2010a) Lysosomal accumulation of gliadin p31–43 peptide induces oxidative stress and tissue transglutaminase‐mediated PPARgamma downregulation in intestinal epithelial cells and coeliac mucosa. Gut 59: 311–319 [DOI] [PubMed] [Google Scholar]
- Luciani A, Villella VR, Esposito S, Brunetti‐Pierri N, Medina D, Settembre C, Gavina M, Pulze L, Giardino I, Pettoello‐Mantovani M, D'Apolito M, Guido S, Masliah E, Spencer B, Quaratino S, Raia V, Ballabio A, Maiuri L (2010b) Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS‐mediated autophagy inhibition. Nat Cell Biol 12: 863–875 [DOI] [PubMed] [Google Scholar]
- Luciani A, Villella VR, Esposito S, Gavina M, Russo I, Silano M, Guido S, Pettoello‐Mantovani M, Carnuccio R, Scholte B, De Matteis A, Maiuri MC, Raia V, Luini A, Kroemer G, Maiuri L (2012) Targeting autophagy as a novel strategy for facilitating the therapeutic action of potentiators on ΔF508 cystic fibrosis transmembrane conductance regulator. Autophagy 8: 1657–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacs GL, Segal G, Kartner N, Grinstein S, Zhang F (1997) Constitutive internalization of cystic fibrosis transmembrane conductance regulator occurs via clathrin‐dependent endocytosis and is regulated by protein phosphorylation. Biochem J 328: 353–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiuri L, Ciacci C, Ricciardelli I, Vacca L, Raia V, Auricchio S, Picard J, Osman M, Quaratino S, Londei M (2003) Association between innate response to gliadin and activation of pathogenic T cells in coeliac disease. Lancet 362: 30–37 [DOI] [PubMed] [Google Scholar]
- Maiuri L, Luciani A, Giardino I, Raia V, Villella VR, D'Apolito M, Pettoello‐Mantovani M, Guido S, Ciacci C, Cimmino M, Cexus ON, Londei M, Quaratino S (2008) Tissue transglutaminase activation modulates inflammation in cystic fibrosis via PPARgamma down‐regulation. J Immunol 180: 7697–7705 [DOI] [PubMed] [Google Scholar]
- Marat AL, Haucke V (2016) Phosphatidylinositol 3‐phosphates‐at the interface between cell signalling and membrane traffic. EMBO J 35: 561–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchelletta RR, Gareau MG, McCole DF, Okamoto S, Roel E, Klinkenberg R, Guiney DG, Fierer J, Barrett KE (2013) Altered expression and localization of ion transporters contribute to diarrhea in mice with Salmonella induced enteritis. Gastroenterology 145: 1358–1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matysiak‐Budnik T, Moura IC, Arcos‐Fajardo M, Lebreton C, Ménard S, Candalh C, Ben‐Khalifa K, Dugave C, Tamouza H, van Niel G, Bouhnik Y, Lamarque D, Chaussade S, Malamut G, Cellier C, Cerf‐Bensussan N, Monteiro RC, Heyman M (2008) Secretory IgA mediates retrotranscytosis of intact gliadin peptides via the transferrin receptor in celiac disease. J Exp Med 205: 143–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano F, Mazzarella G, Luongo D, Stefanile R, D'Arienzo R, Rossi M, Auricchio S, Troncone R (2005) Small intestinal enteropathy in non‐obese diabetic mice fed a diet containing wheat. Diabetologia 48: 931–937 [DOI] [PubMed] [Google Scholar]
- Meresse B, Ripoche J, Heyman M, Cerf‐Bensussan N (2009) Celiac disease: from oral tolerance to intestinal inflammation, autoimmunity and lymphomagenesis. Mucosal Immunol 2: 8–23 [DOI] [PubMed] [Google Scholar]
- Meresse B, Malamut G, Cerf‐Bensussan N (2012) Celiac disease: an immunological jigsaw. Immunity 36: 907–919 [DOI] [PubMed] [Google Scholar]
- Moon SH, Kim J, Kim MY, Parkdo H, Song TJ, Kim SA, Lee SS, Seo DW, Lee SK, Kim MH (2016) Sensitization to and challenge with gliadin induce pancreatitis and extrapancreatic inflammation in HLA‐DQ8 Mice: an animal model of type 1 autoimmune pancreatitis. Gut Liv 10: 842–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) Autodock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 16: 2785–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols DP, Chmiel JF (2015) Inflammation and its genesis in cystic fibrosis. Pediatr Pulmonol 50(Suppl 40): S39–S56 [DOI] [PubMed] [Google Scholar]
- Nurminskaya MV, Belkin AM (2012) Cellular functions of tissue transglutaminase. Int Rev Cell Mol Biol 294: 1–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi CY, Durie PR (2016) Cystic fibrosis from the gastroenterologist's perspective. Nat Rev Gastroenterol Hepatol 13: 175–185 [DOI] [PubMed] [Google Scholar]
- Pallotta MT, Orabona C, Volpi C, Vacca C, Belladonna ML, Bianchi R, Servillo G, Brunacci C, Calvitti M, Bicciato S, Mazza EM, Boon L, Grassi F, Fioretti MC, Fallarino F, Puccetti P, Grohmann U (2011) Indoleamine 2,3‐dioxygenase is a signaling protein in long‐term tolerance by dendritic cells. Nat Immunol 12: 870–878 [DOI] [PubMed] [Google Scholar]
- Papista C, Gerakopoulos V, Kourelis A, Sounidaki M, Kontana A, Berthelot L, Moura IC, Monteiro RC, Yiangou M (2012) Gluten induces coeliac‐like disease in sensitised mice involving IgA, CD71 and transglutaminase 2 interactions that are prevented by probiotics. Lab Invest 92: 625–635 [DOI] [PubMed] [Google Scholar]
- Raju SV, Solomon GM, Dransfield MT, Rowe SM (2016) Acquired cystic fibrosis transmembrane conductance regulator dysfunction in chronic bronchitis and other diseases of mucus clearance. Clin Chest Med 37: 147–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauhairta T, Qiao SW, Jiang Z, Myrsky E, Loponen J, Korponay‐Szabó IR, Salovaara H, Garcia‐Horsman JA, Venäläinen J, Männistö PT, Collighan R, Mongeot A, Griffin M, Mäki M, Kaukinen K, Lindfors K (2011) Epithelial transport and deamidation of gliadin peptides: a role for coeliac disease patient immunoglobulin A. Clin Exp Immunol 164: 127–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romani L, Oikonomou V, Moretti S, Iannitti RG, D'Adamo MC, Villella VR, Pariano M, Sforna L, Borghi M, Bellet MM, Fallarino F, Pallotta MT, Servillo G, Ferrari E, Puccetti P, Kroemer G, Pessia M, Maiuri L, Goldstein AL, Garaci E (2017) Thymosin α1 represents a potential potent single‐molecule‐based therapy for cystic fibrosis. Nat Med 23: 590–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossin F, Villella VR, D'Eletto M, Farrace MG, Esposito S, Ferrari E, Monzani R, Occhigrossi L, Pagliarini V, Sette C, Cozza G, Barlev NA, Falasca L, Fimia GM, Kroemer G, Raia V, Maiuri L, Piacentini M (2018) TG2 regulates the heat‐shock response by the post‐translational modification of HSF1. EMBO Rep 19: e45067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarra M, Cupi ML, Monteleone I, Franzè E, Ronchetti G, Di Sabatino A, Gentileschi P, Franceschilli L, Sileri P, Sica G, Del Vecchio Blanco G, Cretella M, Paoluzi OA, Corazza GR, Pallone F, Monteleone G (2013) IL‐15 positively regulates IL‐21 production in celiac disease mucosa. Mucosal Immunol 6: 244–255 [DOI] [PubMed] [Google Scholar]
- Schumann M, Siegmund B, Schulzke JD, Fromm M (2017) Celiac disease: role of the epithelial barrier. Cell Mol Gastroenterol Hepatol 3: 150–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setty M, Discepolo V, Abadie V, Kamhawi S, Mayassi T, Kent A, Ciszewski C, Maglio M, Kistner E, Bhagat G, Semrad C, Kupfer SS, Green PH, Guandalini S, Troncone R, Murray JA, Turner JR, Jabri B (2015) Distinct and synergistic contributions of epithelial stress and adaptive immunity to functions of intraepithelial killer cells and active celiac disease. Gastroenterology 149: 681–691.e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Maupetit JD, Derreumaux P, Tufféry P (2014) Improved PEP‐FOLD approach for peptide and miniprotein structure prediction. J Chem Theor Comput 10: 4745–4758 [DOI] [PubMed] [Google Scholar]
- Sheppard DN, Welsh MJ (1999) Structure and function of the CFTR chloride channel. Physiol Rev 79(1 Suppl): S23–S45 [DOI] [PubMed] [Google Scholar]
- Simard N, Konforte D, Tran AH, Esufali J, Leonard WJ, Paige CJ (2011) Analysis of the role of IL‐21 in development of murine B cell progenitors in the bone marrow. J Immunol 186: 5244–5253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollid LM, Jabri B (2013) Triggers and drivers of autoimmunity: lessons from coeliac disease. Nat Rev Immunol 13: 294–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone KP, Kastin AJ, Pan W (2011) NFĸB is an unexpected major mediator of interleukin‐15 signaling in cerebral endothelia. Cell Physiol Biochem 28: 115–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiagarajah JR, Ko EA, Tradtrantip L, Donowitz M, Verkman AS (2014) Discovery and development of antisecretory drugs for treating diarrheal diseases. Clin Gastroenterol Hepatol 12: 204–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosco A, De Gregorio F, Esposito S, De Stefano D, Sana I, Ferrari E, Sepe A, Salvadori L, Buonpensiero P, Di Pasqua A, Grassia R, Leone CA, Guido S, De Rosa G, Lusa S, Bona G, Stoll G, Maiuri MC, Mehta A, Kroemer G et al (2016) A novel treatment of cystic fibrosis acting on‐target: cysteamine plus epigallocatechin gallate for the autophagy‐dependent rescue of class II‐mutated CFTR. Cell Death Differ 23: 1380–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villella VR, Esposito S, Bruscia EM, Vicinanza M, Cenci S, Guido S, Pettoello‐Mantovani M, Carnuccio R, De Matteis MA, Luini A, Maiuri MC, Raia V, Kroemer G, Maiuri L (2013a) Disease‐relevant proteostasis regulation of cystic fibrosis transmembrane conductance regulator. Cell Death Differ 20: 1101–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villella VR, Esposito S, Bruscia EM, Maiuri MC, Raia V, Kroemer G, Maiuri L (2013b) Targeting the intracellular environment in cystic fibrosis: restoring autophagy as a novel strategy to circumvent the CFTR defect. Front Pharmacol 4: 1‐9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincentini O, Maialetti F, Gonnelli E, Silano M (2015a) Gliadin‐dependent cytokine production in a bidimensional cellular model of celiac intestinal mucosa. Clin Exp Med 15: 447–454 [DOI] [PubMed] [Google Scholar]
- Volynets V, Reichold A, Bárdos G, Rings A, Bleich A, Bischoff SC (2016) Assessment of the intestinal barrier with five different permeability tests in healthy C57BL/6J and BALB/cJ mice. Dig Dis Sci 61: 737–746 [DOI] [PubMed] [Google Scholar]
- Walkowiak J, Blask‐Osipa A, Lisowska A, Oralewska B, Pogorzelski A, Cichy W, Sapiejka E, Kowalska M, Korzon M, Szaflarska‐Popławska A (2010) Cystic fibrosis is a risk factor for celiac disease. Acta Biochim Pol 57: 115–118 [PubMed] [Google Scholar]
- Zeng M, Szymczak M, Ahuja M, Zheng C, Yin H, Swaim W, Chiorini JA, Bridges RJ, Muallem S (2017) Restoration of CFTR activity in ducts rescues acinar cell function and reduces inflammation in pancreatic and salivary glands of mice. Gastroenterology 153: 1148–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer KP, Fischer I, Mothes T, Weissen‐Plenz G, Schmitz M, Wieser H, Büning J, Lerch MM, Ciclitira PC, Weber P, Naim HY (2010) Endocytotic segregation of gliadin peptide 31‐49 in enterocytes. Gut 59: 300–310 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5