

Abstract
The transcription factor Blimp1 is not only an essential regulator of plasma cells, but also a risk factor for the development of autoimmune disease in humans. Here, we demonstrate in the mouse that the Prdm1 (Blimp1) gene was partially activated at the chromatin and transcription level in early B cell development, although mature Prdm1 mRNA did not accumulate due to posttranscriptional regulation. By analyzing a mouse model that facilitated ectopic Blimp1 protein expression throughout B lymphopoiesis, we could demonstrate that Blimp1 impaired B cell development by interfering with the B cell gene expression program, while leading to an increased abundance of plasma cells by promoting premature plasmablast differentiation of immature and mature B cells. With progressing age, these mice developed an autoimmune disease characterized by the presence of autoantibodies and glomerulonephritis. Hence, these data identified ectopic Blimp1 expression as a novel mechanism, through which Blimp1 can act as a risk factor in the development of autoimmune disease.
Keywords: autoimmune disease, Blimp1‐mediated loss of B cells, ectopic expression throughout the B cell lineage, increased plasma cells differentiation, Prdm1 (Blimp1) transcription in developing B cells
Subject Categories: Immunology
Introduction
Plasma cells serve an important role in the acute response to infection and in long‐term protection of the host by providing humoral immunity through continuous secretion of antibodies. Moreover, plasma cells often contribute also to the pathogenesis of autoimmune disease by secreting self‐reactive antibodies (Suurmond & Diamond, 2015; Tsokos et al, 2016). The zinc finger transcription factor Blimp1 (encoded by the Prdm1 gene) is a key regulator of plasma cells (Nutt et al, 2007), which was initially discovered by its ability to induce plasmacytic differentiation upon ectopic expression in mature B cells (Turner et al, 1994). Within the B‐lymphoid lineage, Blimp1 is predominantly expressed in antibody‐secreting cells, where its highest expression is observed in quiescent long‐lived plasma cells (Kallies et al, 2004). Consistent with this expression pattern, antibody‐secreting cells are lost in mice with a B cell‐specific deletion of the Prdm1 gene, demonstrating that Blimp1 is essential for the generation of plasmablasts and plasma cells (Shapiro‐Shelef et al, 2003; Kallies et al, 2007). Blimp1 expression is furthermore required in long‐lived bone marrow plasma cells to maintain their secretory function (Tellier et al, 2016). Interestingly, the human PRDM1 gene is frequently mutated on both alleles in activated B cell‐like diffuse large B cell lymphoma (ABC‐DLBCL), demonstrating that loss of the tumor‐suppressor gene PRDM1 contributes to lymphomagenesis by preventing plasma cell differentiation (Pasqualucci et al, 2006; Tam et al, 2006). In addition, genome‐wide association studies (GWAS) have identified PRDM1 as a susceptibility gene for the autoimmune diseases systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA), as two informative single nucleotide polymorphisms (SNPs) have been specifically mapped in the intergenic region between the PRDM1 and ATG5 loci in SLE and RA patients (Gateva et al, 2009; Raychaudhuri et al, 2009; Zhou et al, 2011; Appendix Fig S1A). To date, there are, however, no functional data available that would causally implicate PRDM1 in the pathogenesis of SLE or RA.
At the molecular level, Blimp1 functions as a transcriptional repressor and activator by recruiting chromatin‐remodeling and histone‐modifying complexes to its target genes in plasmablasts (Minnich et al, 2016). Systematic analysis of regulated target genes identified multiple roles of Blimp1 in coordinating plasma cell differentiation (Minnich et al, 2016; Tellier et al, 2016). For instance, Blimp1 promotes the migration and adhesion of plasmablasts. It directly represses several key genes including those coding for the transcription factor Pax5, the co‐activator CIITA, and the cytidine deaminase AID, which leads to the silencing of B cell‐specific gene expression, antigen presentation and class switch recombination in plasmablasts, respectively (Shaffer et al, 2002; Minnich et al, 2016). Blimp1 furthermore directly activates genes, leading to increased expression of the plasma cell regulator IRF4 and proteins involved in immunoglobulin secretion (Minnich et al, 2016). Importantly, Blimp1 strongly induces the transcription of the immunoglobulin heavy‐chain (Igh) and κ light‐chain (Igk) genes and also regulates the posttranscriptional expression switch from the membrane‐bound to secreted Ig heavy‐chain protein in plasmablasts (Minnich et al, 2016; Tellier et al, 2016).
Little is so far known about how Blimp1 expression is regulated in plasmablasts and plasma cells. MicroRNAs (miR‐30b,d,e and miR125b) and the RNA‐binding protein ZFP36L1 have been implicated in the posttranscriptional regulation of Blimp1 expression by controlling mRNA decay and translation through binding to Prdm1 3′ UTR sequences in plasma cell lines (Gururajan et al, 2010; Nasir et al, 2012; Kassambara et al, 2017). At the transcriptional level, the Prdm1 gene is known to be activated by the transcription factors IRF4, E2A, and E2‐2 in plasma cells (Sciammas et al, 2006; Kwon et al, 2009; Gloury et al, 2016; Wöhner et al, 2016). Moreover, the regulatory landscape of the Prdm1 locus is rather complex, as it consists of eight open chromatin regions that interact with the Prdm1 promoter in plasmablasts and are located up to 272 kb upstream of the Prdm1 gene (Wöhner et al, 2016).
Here, we demonstrate that the Prdm1 locus is partially activated at the chromatin level already in early B cell development and is transcribed in the nucleus, although mRNA does not accumulate in the cytoplasm of B cells due to posttranscriptional regulation. To study the effect of ectopic Blimp1 expression in the B cell lineage, we generated a mouse model that resulted in early Blimp1 expression due to insertion of the Moloney murine leukemia virus (MoMLV) enhancer together with the IRES‐hCd2 (ihCd2) reporter gene between the stop codon and the 3′ UTR of the Prdm1 ihCd2 allele (Minnich et al, 2016). Consequently, Prdm1 transcription was strongly activated, and the Blimp1 protein was expressed already from lymphoid progenitors throughout B cell development with highest expression being observed in pro‐B, pre‐B, and immature B cells in the bone marrow of Prdm1 ihCd2/+ mice. Blimp1 interfered with the normal B cell gene expression program by activating and repressing many genes, which led to decreased B cell development in Prdm1 ihCd2/+ mice. In contrast, plasma cell development was strongly increased in Prdm1 ihCd2/+ mice, consistent with the finding that immature and mature B cells had an enhanced potential to undergo in vitro plasmablast differentiation. With progressing age, Prdm1 ihCd2/+ mice developed an autoimmune phenotype characterized by the generation of anti‐nuclear antibodies, immune complex deposition, and kidney pathology. Together, these data demonstrate that precocious Blimp1 expression can cause autoimmune disease.
Results
The Prdm1 locus is transcriptionally active already in early B cell development
We previously characterized open chromatin regions (sites A‐H) upstream of the Prdm1 (Blimp1) gene in plasmablasts, which highly express Blimp1 (Wöhner et al, 2016; Fig 1A). To investigate the epigenetic status of the Prdm1 locus in early B cell development, we mapped open chromatin regions and different histone modifications by ATAC‐seq and ChIP‐seq analyses in pro‐B cells, which do not express Blimp1. Unexpectedly, the Prdm1 promoter was partially open and contained bivalent chromatin as shown by the presence of active (H3K4me2, H3K4me3, H3K9ac) and repressive (H3K27me3) histone marks. The upstream regions A, B, and C were also partially activated in pro‐B cells, as shown by the presence of bivalent chromatin at region A and a subset of open chromatin sites in regions B and C compared to plasmablasts (Fig 1A). Notably, the far upstream regions D, E, F, G, and H, which also interact with the Prdm1 promoter in plasmablasts (Wöhner et al, 2016), were present in closed chromatin in pro‐B cells in marked contrast to plasmablasts (Fig 1A). Together, these data indicate that the Prdm1 locus has undergone partial epigenetic activation already in pro‐B cells.
Figure 1. Partial epigenetic and transcriptional activation of the Prdm1 locus in B cells.

- Mapping of open chromatin as well as active and repressive histone modifications at upstream regulatory regions of the Prdm1 locus in pro‐B cells and plasmablasts. Open chromatin was determined by ATAC‐seq (Buenrostro et al, 2013), and active (H3K4me2, H3K4me3, H3K9ac) and repressive (H3K27me3) histone marks were mapped by ChIP‐seq analysis in ex vivo sorted pro‐B cells (ATAC‐seq and H3K27me3, this study), short‐term in vitro cultured pro‐B cells (H3K4me2, H3K4me3, H3K9ac; Revilla‐i‐Domingo et al, 2012), and in vitro LPS‐induced plasmablasts (Minnich et al, 2016; Table EV4). The indicated upstream regions were previously shown by 3C analysis to interact with the Prdm1 promoter (Wöhner et al, 2016). The mm9 genomic coordinates of mouse chromosome 10 and the respective positions of two human SNPs (rs658431 and rs548234) are indicated. RPM, reads per million mapped sequence reads.
- Mapping of nascent transcripts or mature mRNA at the Prdm1 and Atg5 loci by GRO‐seq or RNA‐seq analyses of ex vivo sorted pro‐B and FO B cells, respectively (Table EV4). Strand‐specific GRO‐seq reads are indicated in blue and red, respectively.
- Quantification of nascent transcript and mRNA levels. The nascent transcription or mRNA expression of the Atg5 and Prdm1 genes is shown as mean expression value (TPM) with SEM based on two different GRO‐seq or RNA‐seq experiments for each B cell type, respectively. TPM, transcripts per million (Wagner et al, 2012).
The observed activation at the chromatin level may indicate that the Prdm1 gene is already transcribed during B cell development. To test this hypothesis, we measured the nascent transcript levels in bone marrow pro‐B cells and splenic follicular B cells by global run‐on sequencing (GRO‐seq; Core et al, 2008). As shown in Fig 1B and C, Prdm1 and its neighboring gene Atg5 were similarly transcribed in bone marrow pro‐B cells and splenic follicular (FO) B cells. Despite similar transcription rates, only a low amount of Prdm1 mRNA could be detected by RNA‐seq analysis in both cell types in contrast to the relatively high abundance of Atg5 mRNA (Fig 1B and C). Hence, these data indicate that posttranscriptional regulation prevents the accumulation of Prdm1 mRNA during B cell development.
Posttranscriptional control can be mediated by microRNAs that act principally through the control of mRNA decay and translation by binding to the 3′ untranslated region (3′ UTR) of mRNA (Pasquinelli, 2012). Alternatively, RNA‐binding proteins interact with AU‐rich elements (AREs) in the 3′ UTR, which promotes mRNA deadenylation and decay (Turner et al, 2014). Both mechanisms have been implicated in the posttranscriptional control of Prdm1 mRNA (Gururajan et al, 2010; Nasir et al, 2012; Parlato et al, 2013; Kassambara et al, 2017). To investigate the role of the 3′ UTR in the posttranscriptional regulation of Prdm1, we deleted most (2,253 bp; 90%) of the 2,490‐bp long 3′ UTR by CRISPR/Cas9‐mediated mutagenesis, which left only one consensus ARE motif and one microRNA‐binding site in the truncated 3′ UTR of the Prdm1 ∆3′U(90) allele (Appendix Fig S1B and C). Contrary to expectation, this large deletion did not lead to increased Prdm1 mRNA accumulation (Appendix Fig S1D) or elevated Blimp1 protein expression (Appendix Fig S1E) in early B cells of Prdm1 ∆3′U(90)/∆3′U(90) mice and had no effect on B cell development in these mice (Appendix Fig S1F). We conclude therefore that a large part (90%) of the 3′ UTR is dispensable for the posttranscriptional regulation of Prdm1 expression.
Premature expression of Blimp1 in lymphoid lineages of the Prdm1 ihCd2/+ mouse
Given the observed transcriptional activity of Prdm1 in early B lymphopoiesis, we asked the question whether B cell development would be affected by precocious expression of the Blimp1 protein in the B cell lineage. To this end, we generated a mouse model facilitating ectopic Blimp1 expression throughout B lymphopoiesis by inserting a 258‐bp long terminal repeat (LTR) sequence of the Moloney murine leukemia virus (MoMLV) together with the IRES‐hCd2 (ihCd2) reporter gene between the Prdm1 stop codon and the 3′ UTR (Fig 2A) in the Prdm1 ihCd2 allele (Minnich et al, 2016). The insertion contains both copies of the 75‐bp repeat of the MoMLV enhancer, which binds multiple transcription factors and is active in the lymphoid system (Speck et al, 1990; Fig 2A). The inserted MoMLV enhancer induced active chromatin at the 3′ end of the Prdm1 gene in Prdm1 ihCd2/+ pro‐B cells (Appendix Fig S2A) and led to a 86‐ and 58‐fold increase of Prdm1 mRNA expression in Prdm1 ihCd2/+ pro‐B and pre‐B cells compared to wild‐type counterpart cells, as determined by RNA‐seq (Fig 2B). RT–qPCR analysis revealed that the nascent Prdm1 transcripts were increased 26‐ to 28‐fold in Prdm1 ihCd2/+ pre‐B cells relative to wild‐type pre‐B cells (Fig 2C). Moreover, the level of nascent Prdm1 transcripts in Prdm1 ihCd2/+ pre‐B cells was only 3.4‐ to 4‐fold below that of LPS‐induced plasmablasts of both the Prdm1 ihCd2/+ and wild‐type genotype, which furthermore revealed that the MoMLV enhancer did not contribute to the high transcription rate of Prdm1 in plasmablasts (Fig 2C). Notably, the inserted MoMLV enhancer did not affect Atg5 transcription and mRNA expression (Fig 2B and C) and was part of the Prdm1‐ihCd2 transcript that still contained the 3′ UTR sequence of the Prdm1 gene (Appendix Fig S2B). Moreover, Flpe‐mediated deletion of the frt‐flanked sequences containing the MoMLV enhancer and ihCd2 reporter gene (Fig 2A) restored normal physiological expression of Prdm1 in the B cell lineage (Minnich et al, 2016). In summary, these data demonstrate that the inserted MoMLV enhancer strongly activated Prdm1 transcription and mRNA expression in Prdm1 ihCd2/+ pro‐B and pre‐B cells.
Figure 2. Premature expression of the Blimp1 protein in Prdm1 ihCd2/+ lymphocytes.

-
ASchematic diagram of the 3′ end of the Prdm1 ihCd2 allele (Minnich et al, 2016). The frt‐flanked IRES‐hCd2 (ihCD2) reporter gene, which was linked to a 258‐bp long terminal repeat (LTR) sequence of the Moloney murine leukemia virus (MoMLV, green), was inserted between the stop codon and 3′ UTR of the Prdm1 gene. C‐terminal sequences of exon 8 contained in‐frame tag sequences encoding the Flag and V5 epitopes, two TEV protease cleavage sites, and a biotin acceptor sequence. The inserted MoMLV enhancer sequence is shown below together with its regulatory elements (Speck et al, 1990). GRE, glucocorticoid response element; LV, leukemia virus factor‐binding site; NF1, nuclear factor 1; polyadenylation site, pA.
-
BExpression of Prdm1 and Atg5 mRNA in ex vivo sorted pro‐B and pre‐B cells from the bone marrow of Prdm1 ihCd2/+ or wild‐type (WT) mice. mRNA expression is shown as mean expression value (TPM) with SEM based on two independent RNA‐seq experiments for each cell type and genotype.
-
CRT–qPCR analysis of nascent Prdm1 and Atg5 transcripts in ex vivo sorted pre‐B cells and in in vitro differentiated plasmablasts (PB) of the indicated genotypes. Plasmablasts were generated by stimulation of splenic FO B cells for 4 days with LPS. The data were normalized to those of the ubiquitously expressed control gene Tbp and are presented relative to those of the wild‐type pre‐B cells (set as 1). Nascent transcripts were PCR‐amplified with primers located in the indicated introns (Table EV3). Statistical data are shown as mean value with SEM and were analyzed by the Student's t‐test; *P < 0.05, **P < 0.01. Each dot corresponds to one mouse.
-
D, EFlow cytometric analysis of hCD2 cell surface expression and intracellular Blimp1 expression of the indicated cell types. Bone marrow of Prdm1 ihCd2/+ (red) or wild‐type (WT, gray) mice was used to analyze ALPs, BLPs, pro‐B, pre‐B, immature B cells, NK cells, and granulocytes, whereas the spleen was used for flow cytometric analysis of FO B, MZ B, and plasma cells as well as naïve CD4 T and naïve CD8 T cells of both genotypes. The histograms show hCD2 (top row) and Blimp1 (bottom row) expression for the different cell types, which were defined as described in the Appendix Supplementary Methods. Wild‐type FO B cells (dashed line) were used as negative control for the Blimp1 staining in plasma cells. The difference in mean fluorescence intensity (ΔMFI) between the two genotypes is shown for each cell type.
-
FImmunoblot analysis of Blimp1 and the TATA‐binding protein (TBP) in nuclear extracts prepared from short‐term cultured wild‐type or Prdm1 ihCd2/+ pro‐B cells. Marker proteins of the indicated size (in kilodaltons) are shown to the right.
We next analyzed the expression of the hCD2 protein, reporting Prdm1 mRNA expression, at different B cell developmental stages in Prdm1 ihCd2/+ mice by flow cytometric analysis. hCD2 expression was already detected in uncommitted lymphoid progenitors (LMPPs, ALPs, BLPs); was increased in pro‐B, pre‐B, and immature B cells of the bone marrow; was reduced in marginal zone (MZ) and follicular (FO) B cells of the spleen as well as in B‐1 cells of the peritoneal cavity; and, as expected, was most highly activated in splenic plasma cells (Fig 2D and Appendix Fig S2C). hCD2 expression was low in double‐negative (DN) thymocytes, increased in double‐positive (DP) thymocytes, and was still observed in splenic CD4 and CD8 T cells as well as in natural killer (NK) cells, whereas hCD2 expression was absent in granulocytes and macrophages (Fig 2E, Appendix Fig S2D and data not shown). Next, we directly investigated expression of the Blimp1 protein by intracellular staining (Fig 2D and E, and Appendix Fig S2C and D), which was, however, less sensitive compared to the hCD2 analysis, but closely correlated with the hCD2 expression pattern (Appendix Fig S2E). Blimp1 expression was readily detectable in BLPs, pro‐B, pre‐B, immature B cells, and plasma cells of the bone marrow as well as in DP thymocytes of Prdm1 ihCd2/+ mice (Fig 2D and Appendix Fig S2D) and was confirmed for pro‐B cells by immunoblotting (Fig 2F). We next generated Prdm1 Gfp/ihCd2 mice to examine whether ectopic Blimp1 expression from the Prdm1 ihCd2 allele may auto‐regulate and thus activate expression of the second Prdm1 Gfp allele (Kallies et al, 2004). Despite hCD2 expression, the pro‐B, pre‐B, and immature B cells of Prdm1 Gfp/ihCd2 mice did not express GFP (Appendix Fig S2F), demonstrating that Blimp1‐mediated auto‐regulation did not occur in these B cell subsets. Collectively, these data demonstrate that the MoMLV enhancer insertion in the Prdm1 locus resulted in premature Blimp1 expression during B cell development and, to a lower degree, in mature T cells.
Impaired B cell development in Prdm1 ihCd2/+ mice
Ectopic expression of Blimp1 resulted in a 2.4‐fold decrease in total B cells (CD19+B220+) in the bone marrow of Prdm1 ihCd2/+ mice compared to wild‐type mice, as shown by flow cytometry (Fig 3A). Surprisingly however, B cells were almost completely lost in homozygous Prdm1 ihCd2/ihCd2 mice (Fig 3A), indicating that a further increase of ectopic Blimp1 expression effectively disrupted B cell development. While pro‐B cells (KithiCD19+CD25−IgM−IgD−) were present at similar numbers in the bone marrow of Prdm1 ihCd2/+ mice, pre‐B (Kit−CD19+CD25+IgM−IgD−), immature B (CD19+IgM+IgD−), and recirculating B (CD19+IgMloIgD+) cells were reduced 3.2‐, 2.5‐, and 8‐fold, respectively (Fig 3B). Moreover, splenic MZ (CD19+CD21hiCD23lo) and FO (CD19+CD21intCD23hi) B cells as well as peritoneal B‐1a cells (CD5+CD19+CD23−) were decreased 2.6‐, 6.9‐, and 21‐fold, respectively, in Prdm1 ihCd2/+ mice compared to wild‐type mice (Fig 3C and Appendix Fig S3A). We next investigated whether the observed developmental defects were caused by apoptosis. Notably, the analysis of ex vivo Prdm1 ihCd2/+ pro‐B cells by two different apoptosis assays did not reveal increased cell death compared to wild‐type pro‐B cells (Fig 3D and Appendix Fig S3B), consistent with the observed similar abundance of Prdm1 ihCd2/+ and wild‐type pro‐B cells in the bone marrow (Fig 3B). In contrast, Prdm1 ihCd2/+ pre‐B cells exhibited a 2‐fold increase in apoptosis relative to wild‐type pre‐B cells (Fig 3D and Appendix Fig S3B). Interestingly, B cell development in the bone marrow and, to a lower degree, in the spleen was rescued in Vav‐Bcl2 Prdm1 ihCd2/+ mice (Appendix Fig S3C), which constitutively expressed the pro‐survival protein Bcl2 from the Vav‐Bcl2 transgene in all hematopoietic cell types (Ogilvy et al, 1999). Hence, apoptosis contributes to the B cell development defects observed in Prdm1 ihCd2/+ mice.
Figure 3. Impaired B cell development in Prdm1 ihCd2/+ mice.
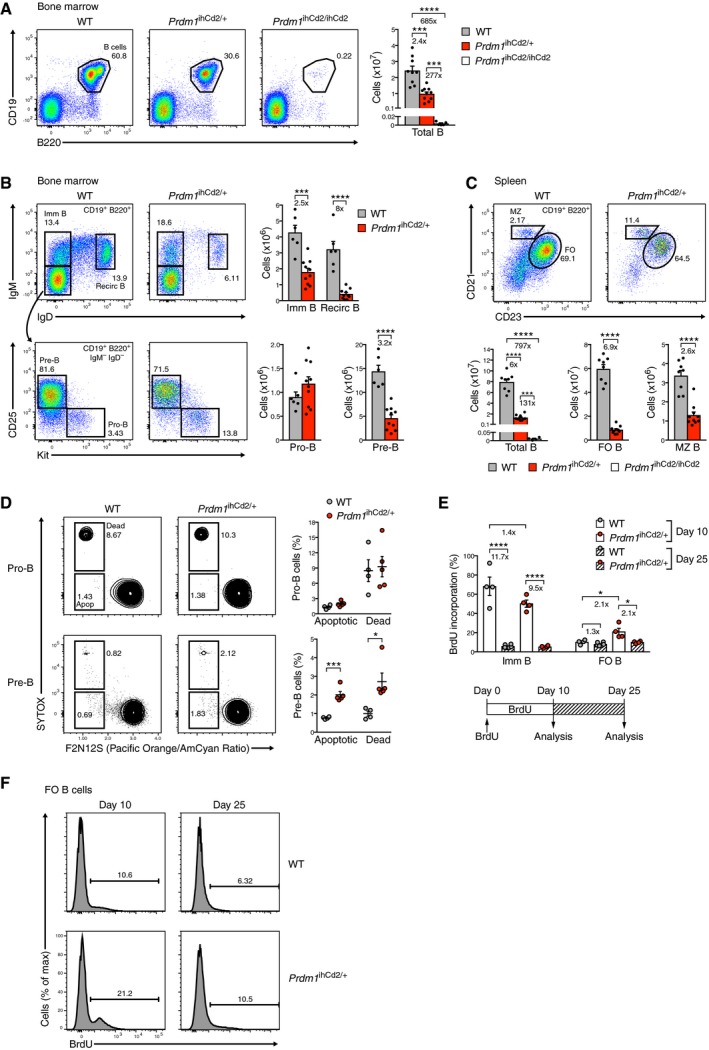
-
A, BFlow cytometric analysis of total B cells (A) as well as pro‐B, pre‐B, immature (imm) B, and recirculating (recirc) B cells (B) from the bone marrow of wild‐type (WT, gray), Prdm1 ihCd2/+ (red), and Prdm1 ihCd2/ihCd2 (white) mice at the age of 6‐8 weeks.
-
CFlow cytometric analysis of total B, FO B, and MZ B cells from the spleens of wild‐type, Prdm1 ihCd2/+, and Prdm1 ihCd2/ihCd2 mice. Bar graphs show absolute cell numbers for each cell type and indicated genotype. The different cell types were defined as described in detail in the Appendix Supplementary Methods. The total numbers of bone marrow cells and splenocytes are shown in Appendix Fig S5A.
-
DDetermination of cell death of ex vivo pro‐B and pre‐B cells from the bone marrow of Prdm1 ihCd2/+ (red) and wild‐type (gray) mice by flow cytometric analysis of the loss of plasma membrane asymmetry (F2N12S ratiometric dye staining) and the loss of cell membrane integrity (SYTOX staining). Representative flow cytometry plots (left) and the quantification of dead and apoptotic (apop) cells (right) are shown.
-
E, FBrdU labeling of immature (imm) and FO B cells in the spleen of 3‐month‐old Prdm1 ihCd2/+ mice (red dots) and control wild‐type littermates (gray dots). The percentage of BrdU+ B cells was determined for each B cell type by flow cytometric analysis after 10 days of continuous BrdU labeling (day 10, white bar) or after a subsequent 15‐day chase period (day 25; hatched bar) without BrdU in the drinking water (E). A diagram (below) indicates the design of the BrdU labeling and chase experiments. Flow cytometric data obtained with FO B cells from one mouse of each genotype are shown in (F). The percentage of BrdU+ FO B cells is shown above the indicated gates (F).
To measure the in vivo lifespan of Blimp1‐expressing FO B cells, we continuously labeled Prdm1 ihCd2/+ and wild‐type mice with the thymidine analogue bromodeoxyuridine (BrdU) for 10 days prior to flow cytometric analysis of BrdU incorporation in FO B cells (Fig 3E and F). As previously published (Rolink et al, 1998), BrdU was incorporated in only 10% of all FO B cells in contrast to 68% of the immature B cells (CD21−CD23−B220+CD19+) in the spleen of control wild‐type mice (Fig 3E and F), indicating that few immature B cells were recruited into the quiescent FO B cell pool during the 10‐day labeling period. In contrast, 21% (2.1‐fold increase) of the splenic FO B cells in Prdm1 ihCd2/+ mice incorporated BrdU during the first 10 days, but half of them were then replaced by unlabeled Prdm1 ihCd2/+ FO B cells during the subsequent 15‐day chase period in contrast to the wild‐type FO B cells (Fig 3E and F). These data therefore revealed a shortened lifespan and thus more rapid turnover of Prdm1 ihCd2/+ FO B cells compared to control FO B cells in the spleen.
The development of T and NK cells was only moderately affected even in homozygous Prdm1 ihCd2/ihCd2 mice (Appendix Fig S3D–F). We conclude therefore that ectopic Blimp1 expression from the Prdm1 ihCd2 allele preferentially impaired B lymphopoiesis.
Blimp1‐dependent deregulation of the B cell gene expression program
As an important role of Blimp1 in plasma cells is to suppress the B cell gene expression program (Shaffer et al, 2002; Minnich et al, 2016), we investigated the degree to which the ectopically expressed Blimp1 protein could interfere with the transcriptional program of developing B cells in Prdm1 ihCd2/+ mice. By comparing the gene expression changes between Prdm1 ihCd2/+ and wild‐type pro‐B cells, we identified 208 Blimp1‐activated and 113 Blimp1‐repressed genes, based on an expression difference of > 3‐fold, an adjusted P‐value of < 0.05, and an expression value of > 5 TPM (transcripts per million) in one of the two pro‐B cell types (Fig 4A and Table EV1). The same expression analysis identified 280 Blimp1‐activated and 125 Blimp1‐repressed genes in Prdm1 ihCd2/+ pre‐B cells (Fig 4B and Table EV1) with an overlap of 67 activated and 42 repressed genes between pro‐B and pre‐B cells (Fig 4C and Appendix Fig S4A). Notably, the comparison of Blimp1‐regulated genes between Prdm1 ihCd2/+ pro‐B or pre‐B cells and wild‐type pre‐plasmablasts (Minnich et al, 2016) revealed an overlap of 12 or 38 Blimp1‐activated and 17 or 36 Blimp1‐repressed genes (Appendix Fig S4A), respectively, indicating that some genes were similarly regulated by Blimp1 in early B cells and plasmablasts.
Figure 4. Blimp1‐dependent deregulation of the B cell gene expression program.

-
A, BScatter plot of gene expression differences in ex vivo sorted Prdm1 ihCd2/+ and wild‐type (WT) pro‐B (A) and pre‐B (B) cells, based on two RNA‐seq experiments for each cell type and genotype. The expression data of individual genes (indicated by dots) were plotted as normalized (norm) rlog values. Genes with an expression difference of > 3‐fold, an adjusted P‐value of < 0.05, and a TPM value of > 5 (in one of the two pro‐B or pre‐B cell types, respectively) are colored in blue or red, corresponding to activation or repression by Blimp1.
-
CExpression of commonly activated (blue) and commonly repressed (red) genes in pro‐B and pre‐B cells. The log2‐fold expression change observed between Prdm1 ihCd2/+ and wild‐type pro‐B cells (horizontal axis) as well as between Prdm1 ihCd2/+ and wild‐type pre‐B cells (vertical axis) is plotted for each gene. Lines indicated 2‐fold (2×) and 3‐fold (3×) expression differences.
-
DIdentification of 762 Blimp1 peaks in short‐term cultured Prdm1 ihCd2/+ pro‐B cells, as detected by ChIP‐seq with an anti‐V5 antibody and MACS peak calling with a P‐value of < 10−10. The 9,320 Blimp1 peaks identified in plasmablasts (PB; Minnich et al, 2016) are shown for comparison. Common (black) and unique (white) peaks are shown for both cell types.
-
EDensities of Blimp1 binding. Average read density profiles aligned at the center of the Blimp1 peak are shown for the common Blimp peaks of both cell types.
-
FBlimp1 binding at activated and repressed genes in Prdm1 ihCd2/+ pro‐B and pre‐B cells. Regulated genes, which are bound by Blimp1 in Prdm1 ihCd2/+ pro‐B cells, are shown in black.
-
GBlimp1 binding and regulation of the commonly repressed target genes Sell (CD62L) and B3gnt5. The ChIP‐seq data (left) were obtained with Prdm1 ihCd2/+ pro‐B cells (this study) and Prdm1 Bio/Bio Rosa26 BirA/BirA plasmablasts (PB; Minnich et al, 2016). The RNA‐seq data (right) were determined in pro‐B and pre‐B cells of the Prdm1 ihCd2/+ (red) and wild‐type (gray) genotype and in Blimp1‐deficient (Prdm1 Gfp/∆, blue) and wild‐type (gray) pre‐plasmablasts (Pre‐PB; Minnich et al, 2016). Cell types expressing Blimp1 (+) are indicated.
-
H, IExpression of selected Blimp1‐activated and Blimp1‐repressed genes coding for transcriptional regulators (H) and intracellular signal transducers (I) in Prdm1 ihCd2/+ (red) and wild‐type (gray) pre‐B cells. Blimp1‐bound genes are underlined. The mRNA expression of the indicated genes is shown as mean expression value (TPM) with SEM, based on two different RNA‐seq experiments for pre‐B cells of each genotype.
As the ectopically expressed Blimp1 protein contained a V5 epitope sequence (Fig 2A), we determined the genome‐wide Blimp1‐binding pattern in Prdm1 ihCd2/+ pro‐B cells by ChIP‐seq analysis with an anti‐V5 antibody. Peak calling with a stringent P‐value of < 10−10 identified 762 Blimp1 peaks with a consensus Blimp1‐binding motif in Prdm1 ihCd2/+ pro‐B cells (Fig 4D and Appendix Fig S4B). Although the Blimp1 peaks in Prdm1 ihCd2/+ pro‐B cells were 12‐fold reduced in number compared to the Blimp1‐bound sites (9,320) in plasmablasts (Minnich et al, 2016), the majority (88%) of them were also present in plasmablasts, but exhibited a 2.5‐fold lower Blimp1‐binding density compared to the corresponding Blimp1 peaks in plasmablasts (Fig 4D and E). Blimp1 binding was observed at one‐third of all repressed genes in Prdm1 ihCd2/+ pro‐B or pre‐B cells (Fig 4F and G), which resulted in nine commonly repressed Blimp1 target genes in Blimp1‐expressing pro‐B cells, pre‐B cells, and plasmablasts (Fig 4G and Appendix Fig S4C and D). In contrast, Blimp1 binding was detected at only 4.3% of all activated genes in Prdm1 ihCd2/+ pro‐B or pre‐B cells (Fig 4F), which resulted in the identification of only one commonly activated target gene in Blimp1‐expressing pre‐B cells and plasmablasts (Appendix Fig S4C). We conclude therefore that Blimp1 regulated gene expression in pro‐B and pre‐B cells primarily in an indirect manner, which is in marked contrast to the high proportion of directly regulated Blimp1 target genes identified in plasmablasts (Minnich et al, 2016).
Annotation of the Blimp1‐regulated genes in Prdm1 ihCd2/+ pre‐B cells revealed that half of these genes coded for proteins of the following functional classes: 45 activated and 23 repressed cell surface proteins, 51 activated and 18 repressed signal transducers, 24 activated and 10 repressed transcriptional regulators, and 21 activated and 13 repressed metabolic enzymes (Appendix Fig S4E and Table EV1). The deregulated transcriptional regulators likely explain the indirect control of gene expression by Blimp1 in pro‐B and pre‐B cells (Fig 4H and Table EV1). Notably, the B cell commitment gene Pax5 was 2.8‐fold repressed in Prdm1 ihCd2/+ pre‐B cells (Fig 4H), which was observed only at the pre‐B and immature B cell stages of B lymphopoiesis (Appendix Fig S4F). The deregulation of multiple cell surface receptors and intracellular signal transducers suggested that Blimp1 affected the normal signaling responses of B cells (Fig 4I and Appendix Fig S4G). In summary, these data indicate that premature Blimp1 expression in Prdm1 ihCd2/+ mice strongly interfered with the B cell gene expression program controlling B cell development.
Increased plasma cell development in the absence of GC B cells in Prdm1 ihCd2/+ mice
As ectopic Blimp1 expression interfered with the B‐lymphoid gene expression program in B cells in a manner similar to Blimp1's physiological role in plasma cells (Minnich et al, 2016), we next investigated whether premature Blimp1 expression could lead to increased plasma cell differentiation in Prdm1 ihCd2/+ mice. Indeed, plasma cells (Lin−B220int/−CD138+CD28+) were significantly increased in the spleen and bone marrow of non‐immunized Prdm1 ihCd2/+ mice compared to wild‐type mice at the age of 2–12 months (Fig 5A and B, and Appendix Fig S5A). Accordingly, the serum titers of IgM and IgG antibodies (measured by ELISA) as well as the number of plasma cells secreting different IgG isotype antibodies (determined by ELISPOT assay) were also increased in Prdm1 ihCd2/+ mice compared to wild‐type littermates (Appendix Fig S5B and C). Interestingly, FO B cells in the spleen of non‐immunized Prdm1 ihCd2/+ mice already expressed higher levels of the activation markers CD40, CD80, CD86, and MHCII (Appendix Fig S5D), suggesting that the altered threshold for B cell activation may contribute to enhanced plasma cell differentiation in Prdm1 ihCd2/+ mice. We next immunized experimental and control mice with 4‐hydroxy‐3‐nitrophenylacetyl‐conjugated keyhole limpet hemocyanin (NP‐KLH). At day 14 after immunization, anti‐NP‐IgM antibody‐secreting cells (ASCs) were 4.6‐fold increased in the spleen of Prdm1 ihCd2/+ mice, as determined by ELISPOT assay (Fig 5C), and the titers of anti‐NP‐IgM were 2.4‐fold higher in the serum of Prdm1 ihCd2/+ mice compared to wild‐type littermates, as measured by ELISA (Fig 5D). Notably, the increase of plasma cells was observed despite a 6‐fold decrease of B cells in the presence of a similar number of total splenocytes in Prdm1 ihCd2/+ mice relative to wild‐type littermates (Fig 3C and Appendix Fig S5E). We conclude therefore that plasma cell development was strongly enhanced also in immunized Prdm1 ihCd2/+ mice.
Figure 5. Increased plasma cell development and decreased TFH cell differentiation in the absence of GC B cell formation in Prdm1 ihCd2/+ mice.
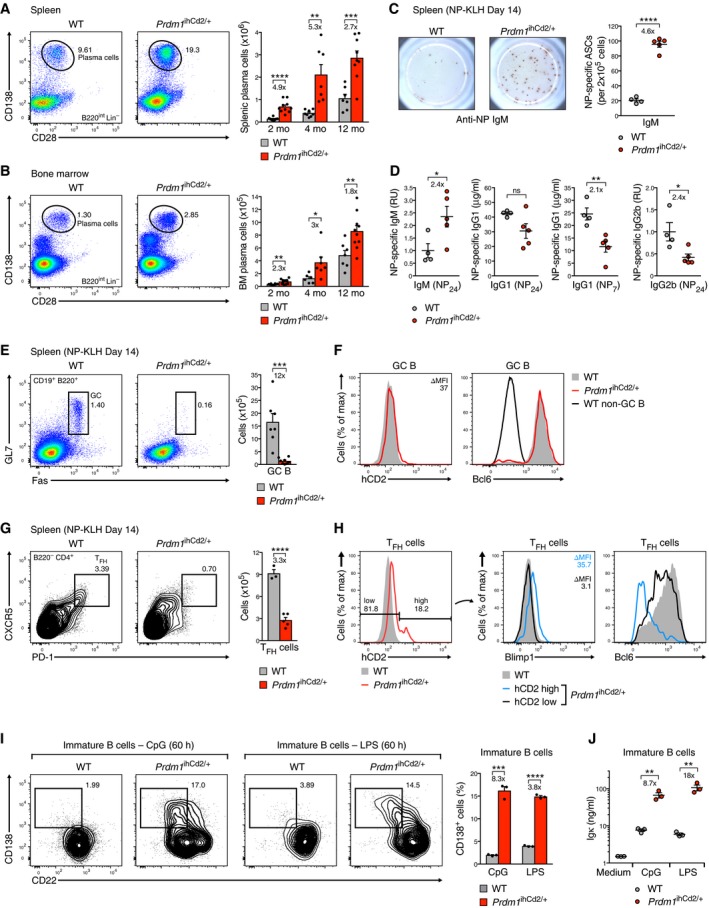
-
A, BFlow cytometric analysis of plasma cells from the spleen (A) and bone marrow (B) of Prdm1 ihCd2/+ and wild‐type (WT) mice under steady‐state conditions. Representative flow cytometric data are shown for 12‐month‐old mice (left), while bar graphs present absolute numbers of plasma cells for Prdm1 ihCd2/+ (red) and wild‐type (gray) mice at the age of 2, 4, and 12 months (right). The total number of cells in the spleen and bone marrow of the analyzed mice is shown in Appendix Fig S5A.
-
C, DT cell‐dependent immune responses. Prdm1 ihCd2/+ (red dots) and wild‐type (gray dots) mice at the age of 2 months were immunized with NP‐KLH (in alum) and analyzed at day 14 after immunization by ELISPOT assay (C) and ELISA (D). The number of anti‐NP‐IgM antibody‐secreting cells (ASCs) in the spleen was determined by ELISPOT assay (C) using NP24‐BSA‐coated plates for detecting cells secreting total anti‐NP‐IgM antibodies. ELISPOT results are shown as representative pictures (left) or absolute ASC numbers (right) (C). The serum titers of anti‐NP‐specific IgM, IgG1, and IgG2b antibodies were analyzed by ELISA using NP7‐BSA‐ or NP24‐BSA‐coated plates for detecting high‐affinity IgG1 or total IgM, IgG1, and IgG2b antibodies, respectively (D). Dot plots display the serum immunoglobulin titers of Prdm1 ihCd2/+ (red dots) and wild‐type (gray dots) mice. NP‐specific IgG1 concentrations (μg/ml) were determined relative to a standard NP‐binding IgG1 antibody, whereas IgM and IgG2b amounts are indicated as relative units (RU) by setting the WT data to 1. The total number of splenocytes in the immunized mice is shown in Appendix Fig S5E.
-
E, FFlow cytometric analysis of splenic GC B cells in Prdm1 ihCd2/+ (red) and wild‐type (gray) mice (at the age of 2 months) at day 14 after NP‐KLH immunization. Bar graphs display absolute numbers of GC B cells detected in the spleen of both genotypes (E). Cell surface expression of hCD2 and intracellular staining of Bcl6 are shown for Prdm1 ihCd2/+ (red) and wild‐type (gray) GC B cells (F). The absence of Bcl6 expression in non‐GC B cells (black) is shown as reference.
-
G, HFlow cytometric analysis of splenic TFH cells from the same mice analyzed in (E, F). Bar graphs indicate absolute numbers of TFH cells (G), and histograms display cell surface expression of hCD2 and intracellular staining of Bcl6 (H) in hCD2lo (black) and hCD2hi (blue) Prdm1 ihCd2/+ as well as wild‐type (gray) TFH cells.
-
IEfficient in vitro plasmablast differentiation of immature B cells. Immature B cells sorted from the bone marrow of Prdm1 ihCd2/+ (red) and wild‐type (gray) mice were stimulated with CpG oligodeoxynucleotides or LPS for 60 h followed by flow cytometric analysis of CD138+CD22lo plasmablasts (PB). Bar graphs (right) summarize the plasmablast data of the CpG and LPS stimulation experiments.
-
JELISA detection of Igκ‐containing antibodies in the supernatants of immature B cells at 60 h of CpG or LPS stimulation.
In contrast to the increased anti‐NP‐IgM levels, the serum titers of anti‐NP‐IgG1 and anti‐NP‐IgG2b were decreased in immunized Prdm1 ihCd2/+ mice compared to control littermates (Fig 5D). Moreover, the frequency of somatic hypermutation (SHM) at the rearranged Igh gene was reduced in splenic plasma cells of Prdm1 ihCd2/+ mice compared to wild‐type littermates (Appendix Fig S5F). As suggested by the observed reduction of SHM, germinal center (GC) B cells (CD19+B220+GL7+Fas+) were strongly decreased in the spleen of Prdm1 ihCd2/+ mice relative to control mice at day 14 after NP‐KLH immunization (Fig 5E) as well as in non‐immunized mice (Appendix Fig S5G). Moreover, the few residual Prdm1 ihCd2/+ GC B cells did not express hCD2 (Blimp1) and revealed normal expression of the transcription factor Bcl6 (Fig 5F), which is an essential regulator of GC B cell differentiation (Dent et al, 1997; Ye et al, 1997). Notably, follicular helper T (TFH) cells (CXCR5+PD‐1+CD4+B220−), which provide T cell help to support GC B cell differentiation (Vinuesa et al, 2016), were 3.3‐fold decreased in the spleen of immunized Prdm1 ihCd2/+ mice relative to wild‐type mice and expressed hCD2 (Blimp1), albeit at different levels (Fig 5G and H). The hCD2lo population of the Prdm1 ihCd2/+ TFH cells expressed little Blimp1 protein and minimally reduced levels of Bcl6 (Fig 5H), which is also an essential regulator of TFH cell differentiation (Johnston et al, 2009; Nurieva et al, 2009; Yu et al, 2009). In contrast, the hCD2hi subset of Prdm1 ihCd2/+ TFH cells expressed Blimp1, leading to strongly decreased Bcl6 expression (Fig 5H), which suggests that the ectopically expressed Blimp1 protein repressed Bcl6 in this subset, consistent with an antagonistic role of these transcription factors in TFH cells (Johnston et al, 2009). Hence, these data imply that ectopic Blimp1 expression suppresses TFH cell differentiation by interfering with the Bcl6‐regulated gene expression program, which likely contributes to the loss of GC B cells in Prdm1 ihCd2/+ mice (Johnston et al, 2009; Nurieva et al, 2009; Yu et al, 2009).
Blimp1 strongly enhances plasmablast differentiation of immature and mature B cells
We next directly investigated whether precocious Blimp1 expression may endow immature and mature B cells with an enhanced potential to differentiate to plasmablasts. To this end, we stimulated immature B cells (CD19+B220+IgM+IgD−) from the bone marrow of Prdm1 ihCd2/+ or control wild‐type mice with CpG oligodeoxynucleotides for 60 h, as activation of the Toll‐like receptor 9 (TLR9) is known to promote differentiation of immature B cells to IgM‐secreting plasmablasts (Azulay‐Debby et al, 2007). Interestingly, the immature B cells of Prdm1 ihCd2/+ mice underwent differentiation to plasmablasts (CD138+CD22lo) at a 8.3‐fold higher frequency than wild‐type immature B cells (Fig 5I). Likewise, lipopolysaccharide (LPS)‐mediated TLR4 activation of Prdm1 ihCd2/+ immature B cells resulted in a 3.8‐fold increase of plasmablast formation compared to that of wild‐type immature B cells (Fig 5I). These findings were confirmed by the presence of Igκ‐containing antibodies at 8.7‐ and 18‐fold higher levels in the supernatant of the CpG‐ and LPS‐stimulated Prdm1 ihCd2/+ plasmablasts compared to identically treated wild‐type plasmablasts (Fig 5J). Finally, mature FO B cells from the spleen of Prdm1 ihCd2/+ mice also differentiated more efficiently to plasmablasts than wild‐type FO B cells after 4 days of stimulation with CpG, LPS or a combination of IL‐4, IL‐5, and anti‐CD40 (Appendix Fig S5H–J). We conclude therefore that precocious Blimp1 expression strongly enhanced the plasmablast differentiation potential of the first IgM+ immature B cells in the bone marrow as well as of mature FO B cells in the spleen.
Blimp1 expression in TFH cells causes the loss of GC B cells in Prdm1 ihCd2/+ mice
As CD8 and CD4 T cells including TFH cells ectopically expressed Blimp1 in Prdm1 ihCd2/+ mice (Figs 2E and 5H), we next examined whether the B cell phenotype of these mice is B cell‐intrinsic or depends on Blimp1‐expressing T cells. To this end, we generated mixed bone marrow chimeras by reconstituting lethally irradiated Rag2 −/− mice with a mixture of Eβ−/− Prdm1 ihCd2/+ and JHT Prdm1 +/+ bone marrow at a ratio of 15:1 (Fig 6A). A higher amount of the Prdm1 ihCd2/+ bone marrow was required as ectopic Blimp1 expression in uncommitted lymphoid progenitors (LMPPs, ALPs, BLPs; Fig 2D and Appendix Fig S2C) conferred a competitive disadvantage to the Prdm1 ihCd2/+ progenitors relative to the JHT Prdm1 +/+ progenitors (data not shown). All B cells in the chimeric mice originated from the Eβ−/− Prdm1 ihCd2/+ stem cells, as the homozygous JHT mutation, eliminating the DHQ52, JH, and Eμ elements of the Igh locus (Gu et al, 1993), prevented B cell development of the JHT Prdm1 +/+ progenitors. In contrast, all T cells were derived from JHT Prdm1 +/+ stem cells, as the Eβ−/− Prdm1 ihCd2/+ progenitors could not contribute to αβ T cell development due to deletion of the Eβ enhancer of the Tcrb locus (Bouvier et al, 1996). For comparison, we generated bone marrow chimeras with a mixture of Eβ−/− Prdm1 +/+ and JHT Prdm1 +/+ bone marrow and analyzed all chimeric mice 4 months after transplantation (Fig 6A). As the lower competitive fitness of the Eβ−/− Prdm1 ihCd2/+ progenitors compared to the control Eβ−/− Prdm1 +/+ progenitors could also contribute to the observed difference in B cell development, we analyzed the trend, but not the absolute fold difference of B cell numbers in these chimeras. Whereas all B cell subsets were reduced in the bone marrow and spleen of Eβ−/− Prdm1 ihCd2/+ chimeric mice compared to the control Eβ−/− Prdm1 +/+ chimeric mice (Fig 6B and C), plasma cells were slightly increased in both lymphoid organs of the Eβ−/− Prdm1 ihCd2/+ chimeras (Fig 6D). A similar B cell phenotype was observed by comparing non‐chimeric Eβ−/− Prdm1 ihCd2/+ mice with control Eβ−/− Prdm1 +/+ mice (Appendix Fig S6A–D). Together, these data indicate that decreased B cell development and increased plasma cell differentiation are a B cell‐intrinsic property of the Prdm1 ihCd2/+ mouse. However, GC B cells were the exception, as they were present at similar numbers in the spleen of Eβ−/− Prdm1 ihCd2/+ and control Eβ−/− Prdm1 +/+ chimeric mice (Fig 6E) in marked contrast to the loss of GC B cells in Prdm1 ihCd2/+ mice (Fig 5E). Since the splenic CD4 T cells in Eβ−/− Prdm1 ihCd2/+ chimeric mice did not express hCD2 as expected (Fig 6F), we conclude that the ectopic expression of Blimp1 in T cells interfered with the development of GC B cells possibly due to the observed decrease of functional TFH cells in Prdm1 ihCd2/+ mice.
Figure 6. Cell‐autonomous B cell phenotype except for the T cell‐dependent loss of GC B cells in Prdm1 ihCd2/+ mice.
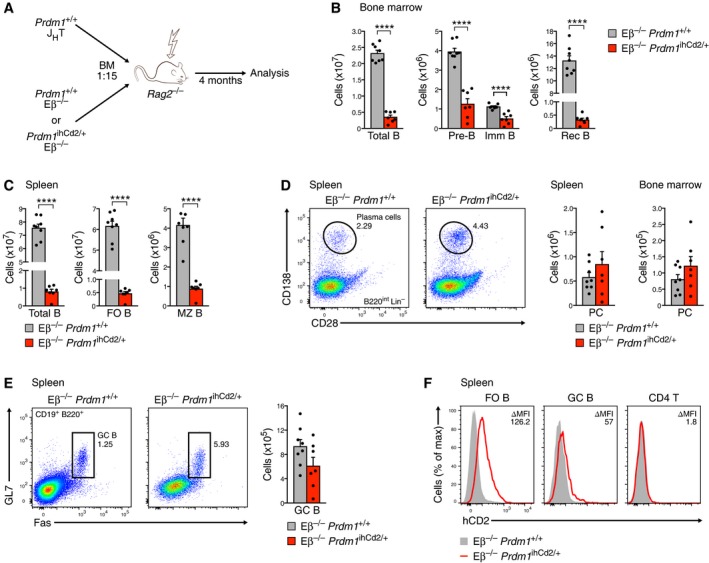
-
ASchematic diagram describing the generation of mixed bone marrow chimeras. Lineage‐depleted bone marrow of B cell‐deficient JHT Prdm1 +/+ mice was mixed at ratio of 1:15 with lineage‐depleted bone marrow of T cell‐deficient Eβ−/− Prdm1 ihCd2/+ or control Eβ−/− Prdm1 +/+ mice prior to injection into lethally irradiated Rag2 −/− recipient mice and subsequent analysis 4 months after transplantation.
-
B–FFlow cytometric analysis of total B cells and different B cell subsets in the bone marrow (B, D) and spleen (C–F) of non‐immunized Eβ−/− Prdm1 ihCd2/+ (red) and Eβ−/− Prdm1 +/+ (gray) chimeric mice. Bar graphs indicate absolute numbers of the different B cell types in these chimeric mice. (F) Histograms display the expression of hCD2 (Blimp1) in splenic FO B, GC B, and naïve CD4 T cells from the chimeric mice of the indicated genotypes. Statistical data (B–E) are shown as mean value with SEM and were analyzed by the Student's t‐test; ****P < 0.0001. Each dot corresponds to one mouse.
Precocious Blimp1 expression generates an autoimmune disease with progressing age
Given the identification of PRDM1 as a susceptibility gene for human SLE and RA (Gateva et al, 2009; Raychaudhuri et al, 2009; Zhou et al, 2011), we next investigated whether Prdm1 ihCd2/+ mice develop an autoimmune phenotype with progressing age. Autoimmune diseases, such as SLE, are characterized by circulating autoantibodies that recognize double‐stranded (ds) DNA, nuclear proteins [such as SSA (Ro‐52) and SSB (La)], and mitochondrial cardiolipin (Suurmond & Diamond, 2015). Hence, we compared, by ELISA, the titers of IgM and IgG recognizing dsDNA, cardiolipin, SSA (Ro‐52), and SSB (La) in the serum of Prdm1 ihCd2/+ mice with those of the Fasl gld/gld mouse, a known model of severe systemic autoimmune disease (Takahashi et al, 1994). The levels of IgM antibodies recognizing dsDNA, cardiolipin, SSA (Ro‐52), and SSB (La) were already increased at 2 and 4 months in Prdm1 ihCd2/+ mice compared to wild‐type mice (Appendix Fig S7A), and elevated titers of the corresponding IgG antibodies were observed at 4 and 12 months in Prdm1 ihCd2/+ mice, although they did not reach the high level of the respective IgG antibodies measured in the serum of Fasl gld/gld mice (Fig 7A). Moreover, anti‐nuclear antibodies (ANA) of the IgG isotype could readily be detected in the serum of eight out of 22 Prdm1 ihCd2/+ mice at the age of 12 months, whereas these autoantibodies were rarely found in the serum of wild‐type mice at this age (Fig 7B). Finally, histological analysis of the kidney revealed a significant increase of pathologically altered glomeruli in 12‐month‐old Prdm1 ihCd2/+ mice relative to wild‐type mice (Fig 7C and Table EV2). Consistent with these pathological changes, the deposition of IgG immune complexes was detected in glomeruli of the kidneys of some Prdm1 ihCd2/+ females (Appendix Fig S7B), further demonstrating that Prdm1 ihCd2/+ mice could develop a moderate form of glomerulonephritis.
Figure 7. Precocious Blimp1 expression causes autoimmunity in Prdm1 ihCd2/+ mice with progressing age.

- Presence of IgG antibodies detecting dsDNA, cardiolipin, SSA (Ro‐52), and SSB (La) in the serum of Prdm1 ihCd2/+ (red dots) and wild‐type (gray dots) mice at the age of 2, 4, and 12 months. The titers of the different IgG antibodies were determined in the serum of male and female mice by ELISA and are displayed as arbitrary units (AU). The serum of 6‐month‐old Fasl gld/gld mice was used as positive control.
- Representative images of anti‐nuclear antibody (ANA) staining obtained with serum from 12‐month‐old Prdm1 ihCd2/+ or wild‐type mice (right), as detected by indirect immunofluorescence assay using HEp‐2 cells and an Alexa488‐conjugated anti‐mouse IgG detection antibody. The serum of Fasl gld/gld mice was used as positive control. The presence of ANA‐IgG antibodies was analyzed for 22 Prdm1 ihCd2/+ and 12 wild‐type mice, as summarized in the pie chart (right).
- Periodic acid‐Schiff (PAS) staining of paraffin‐embedded kidney sections of Prdm1 ihCd2/+ and wild‐type mice at 12 months of age. The mean score of kidney pathology was determined for each mouse by evaluating 40 individual glomeruli to assess the severity of glomerulonephritis as shown in Table EV2 and described in the Appendix Supplementary Methods. Representative pictures of glomeruli of Prdm1 ihCd2/+ kidneys with different pathology scores are shown to the left, and dot plots with average scores for male and female mice (Table EV2) are shown to the right. Arrows denote intracapillary hyaline “thrombi,” asterisks indicate mesangial sclerosis with obscured capillary loops, and the arrowhead points to an obsolescent glomerulus with collapse of the glomerular tuft architecture. Statistical data (A, C) are shown as mean value with SEM and were analyzed by the Mann–Whitney U‐test (A) or the Student's t‐test (C); *P < 0.05, **P < 0.01, ***P < 0.001. Each dot corresponds to one mouse.
- Proposed model explaining the autoimmune phenotype of Prdm1 ihCd2/+ mice. Precocious Blimp1 expression in Prdm1 ihCd2/+ mice results in decreased numbers of developing B cells (red arrows) and increased differentiation of immature and mature B cells to plasmablasts (blue arrows) and plasma cells (green arrow). As a consequence, autoreactive immature B cells may differentiate to plasmablasts prior to their elimination by central tolerance mechanisms (receptor editing or clonal deletion; Nemazee, 2017), and autoreactive mature B cells may escape their anergic state, imposed by anergy‐induced BCR desensitization (peripheral tolerance; Theofilopoulos et al, 2017), by premature differentiation to plasmablasts, which leads to an increase of autoantibody‐secreting plasma cells in Prdm1 ihCd2/+ mice.
As defective clearance of apoptotic debris has been causally associated with SLE development (Tsokos et al, 2016), we next sought to accelerate the development of the autoimmune phenotype in Prdm1 ihCd2/+ mice by five sequential intravenous injections of syngeneic apoptotic thymocytes at weekly intervals (Appendix Fig S8A), as described (Duhlin et al, 2016). Ten days after the last injection, Prdm1 ihCd2/+ and wild‐type mice were analyzed together with non‐injected littermates. The serum titers of anti‐dsDNA, anti‐cardiolipin, anti‐SSA (Ro‐52), and anti‐SSB (La) IgM antibodies were already higher in non‐injected Prdm1 ihCd2/+ mice compared to wild‐type mice (Appendix Fig S8B), as expected for 2‐month‐old mice (Appendix Fig S7A), but these titers were even further increased in the injected Prdm1 ihCd2/+ mice (Appendix Fig S8B). By contrast, the anti‐dsDNA and anti‐cardiolipin IgG antibodies were only increased in the injected Prdm1 ihCd2/+ mice (Appendix Fig S8B). Finally, while anti‐nuclear antibodies of the IgM or IgG isotype were absent in the non‐injected Prdm1 ihCd2/+ or injected wild‐type mice, these antibodies could be readily detected in the serum of three out of four injected Prdm1 ihCd2/+ mice (Appendix Fig S8C and data not shown). Together, these data demonstrate that precocious expression of Blimp1 in the B cell lineage predisposes Prdm1 ihCd2/+ mice to develop an autoimmune disease with progressing age.
Discussion
The human PRDM1 (Blimp1) locus has been identified as a susceptibility gene for development of the autoimmune diseases systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA; Gateva et al, 2009; Raychaudhuri et al, 2009; Zhou et al, 2011). In the B cell lineage, Blimp1 normally acts as an essential regulator of plasma cell development and function (Shapiro‐Shelef et al, 2003; Kallies et al, 2007; Tellier et al, 2016). Here, we discovered that the mouse Prdm1 gene was partially activated at the chromatin and transcription level already in the earliest committed pro‐B cells, although mature Prdm1 mRNA did not accumulate during B cell development due to posttranscriptional regulation. By analyzing a mouse model that facilitated ectopic Blimp1 protein expression throughout B lymphopoiesis, we could demonstrate that Blimp1 impaired B cell development by interfering with the B cell gene expression program, while leading to increased plasma cell differentiation. With progressive aging, these mice developed an autoimmune disease characterized by the appearance of autoantibodies and a moderate form of glomerulonephritis. These data therefore suggest that mutations in the PRDM1 locus that lead to premature expression of the Blimp1 protein in developing B cells may cause autoimmune diseases such as SLE and RA.
While the expression of key lineage‐specific regulators, such as the B cell commitment factor Pax5, is controlled at the transcriptional level (Decker et al, 2009), we made the surprising observation that the Prdm1 locus was already accessible at the chromatin level and was transcriptionally active in early B cell development long before the onset of plasmablast differentiation. Nascent Prdm1 transcripts were, however, present at a 100‐fold lower level in wild‐type pre‐B cells compared to the fully activated state in wild‐type plasmablasts (Fig 2C). Despite transcriptional activity, mature Prdm1 mRNA did not accumulate in early B cell development possibly due to stringent posttranscriptional control. Low expression of Blimp1 mRNA was reported only for peritoneal B‐1 and splenic MZ B cells (Fairfax et al, 2007), suggesting that the posttranscriptional control mechanism may operate less stringently in these two B cell types. Posttranscriptional regulation is often mediated by microRNAs or RNA‐binding proteins that interact with microRNA targets or AU‐rich elements in the 3′ UTR of mRNAs, thereby inhibiting translation and/or inducing mRNA decay (Pasquinelli, 2012; Turner et al, 2014). Although two microRNAs (miR‐30b,d,e and miR‐125b) and the RNA‐binding protein ZFP36L1 have been implicated in the posttranscriptional control of Prdm1 mRNA (Gururajan et al, 2010; Nasir et al, 2012; Parlato et al, 2013; Kassambara et al, 2017), Prdm1 mRNA and Blimp1 protein expression did not increase upon deletion of 90% of the Prdm1 3′ UTR sequences in developing B cells of Prdm1 ∆3′U(90)/∆3′U(90) mice. These data suggest that microRNAs and RNA‐binding proteins are either not involved in the posttranscriptional control of Prdm1 mRNA or mediate their effect through the residual consensus AU‐rich element and microRNA‐binding site, which are still present in the Prdm1 ∆3′U(90) allele.
By inserting the MoMLV enhancer between the stop codon and 3′ UTR of the Prdm1 ihCd2 allele, we created an ideal mouse model for studying the effect of ectopic Blimp1 expression in B cells, as Blimp1 expression was high enough to observe a B cell developmental defect in heterozygous Prdm1 ihCd2/+ mice, while a 2‐fold higher level of Blimp1 protein already eliminated all B cells in homozygous Prdm1 ihCd2/ihCd2 mice. B cell subsets from the pre‐B cell stage onwards were reduced in Prdm1 ihCd2/+ mice largely due to increased apoptosis. This finding is consistent with a previous report demonstrating that ectopic Blimp1 expression in established immature and mature B cell lines activates a strong apoptotic response (Messika et al, 1998). The observed cell death is likely caused by the strong interference of Blimp1 with the normal B cell gene expression program by activating and repressing many genes in pro‐B and pre‐B cells of Prdm1 ihCd2/+ mice. Unexpectedly, Blimp1 bound only to a small fraction of the Blimp1‐regulated genes in pro‐B and pre‐B cells, suggesting that Blimp1 predominantly regulated gene expression in early B cells in an indirect manner in marked contrast to its significantly higher occupancy at regulated genes in terminally differentiated plasmablasts (Minnich et al, 2016). Blimp1 deregulated the expression of many transcription factors including those encoded by the directly repressed target genes Spib, Hhex, Aff3, Irf2bp2, and Pax5, which together likely mediate the indirect effects of Blimp1 in Prdm1 ihCd2/+ B cells. In addition to transcription factors, Blimp1 deregulated the expression of multiple cell surface receptors and intracellular signal transducers, suggesting that ectopic Blimp1 interfered with normal signaling in B cells.
GC B cells were strongly reduced in Prdm1 ihCd2/+ mice, although the few residual GC B cells did not express hCD2 (Blimp1) and showed normal expression of the essential regulator Bcl6. TFH cells, which provide T cell help for GC B cell development (Vinuesa et al, 2016), were also reduced in immunized Prdm1 ihCd2/+ mice. However, these TFH cells expressed hCD2 (Blimp1) and exhibited low Bcl6 expression, as Blimp1 is known to repress Bcl6 in TFH cells (Johnston et al, 2009). Consequently, the loss of GC B cells in Prdm1 ihCd2/+ mice was not a B cell‐intrinsic phenotype, as the presence of “wild‐type” TFH cells efficiently supported the differentiation of Prdm1 ihCd2/+ GC B cells in mixed bone marrow chimeras.
A prominent B cell‐intrinsic feature of ectopic Blimp1 expression was the strong increase in plasma cells of extrafollicular origin, which was in stark contrast to the reduced B cell development and almost complete absence of GC B cells in Prdm1 ihCd2/+ mice. Notably, immature and mature B cells of the Prdm1 ihCd2/+ genotype rapidly differentiated in vitro to plasmablasts in response to TLR signaling, which strongly suggests that precocious Blimp1 expression in Prdm1 ihCd2/+ B cells led to increased plasma cell numbers by promoting premature plasmablast differentiation of B cells (Fig 7D). In this context, it is interesting to note that the lowly Blimp1‐expressing B‐1 and MZ B cells of wild‐type mice also undergo efficient plasmablast differentiation in response to TLR signaling (Fairfax et al, 2007; Genestier et al, 2007). Hence, the low Blimp1 expression may also contribute to the enhanced plasmablast differentiation of these wild‐type B cell subsets (Fairfax et al, 2007). As a likely consequence of increased plasma cell generation, Prdm1 ihCd2/+ mice developed, with progressing age, an autoimmune disease, which was characterized by the appearance of autoantibodies and a moderate form of glomerulonephritis. Based on the autoimmune phenotype of this mouse model, we hypothesize that human PRDM1 mutations, which may result in premature Blimp1 expression in immature and mature B cells, could cause a predisposition for the development of autoimmune diseases such as SLE and RA. This predisposition could be achieved by increasing the PRDM1 transcription rate in B cells through mutation of its regulatory elements, by stabilizing the PRDM1 mRNA through inactivation of its posttranscriptional regulation or by stabilizing the Blimp1 protein by mutations that prevent polyubiquitination and subsequent degradation of Blimp1 (Yang et al, 2014).
The majority (55–75%) of early immature B cells express autoreactive BCRs as a consequence of the vast antibody diversity generated by V(D)J recombination (Wardemann et al, 2003). A large fraction of the immature B cells with autoreactive BCRs are eliminated in the bone marrow by central tolerance mechanisms involving receptor editing, apoptotic deletion, or AID‐mediated elimination (Nemazee, 2017; Cantaert et al, 2015; Fig 7D). Although Blimp1 represses Aicda (AID) transcription in plasma cells (Minnich et al, 2016), ectopically expressed Blimp1 in immature B cells did not repress Aicda expression (data not shown), suggesting that the AID‐mediated elimination of autoreactive immature B cells (Cantaert et al, 2015) was not affected in Prdm1 ihCd2/+ mice. Self‐reactive immature B cells in the bone marrow are, however, known to be responsive to CpG stimulation and thus have the potential to circumvent negative selection by prematurely differentiating to plasmablasts that secrete autoantibodies (Azulay‐Debby et al, 2007). While peripheral tolerance silences mature B cells with autoreactive BCRs through anergy induction (Theofilopoulos et al, 2017), it is conceivable that precocious Blimp1 expression may promote TLR‐mediated differentiation of anergic mature B cells to autoreactive plasma cells in Prdm1 ihCd2/+ mice (Fig 7D). Ectopic Blimp1 expression in immature and mature B cells can enhance premature plasma cell differentiation in two ways (Fig 7D). First, the enhanced apoptosis of Blimp1‐expressing B cells likely results in an increase of cellular debris, apoptotic blebs, and extruded nuclei, which exposes self‐antigens to polyreactive BCRs that, upon endocytosis, present these self‐antigens to endosomal TLRs (TLR3,7,8,9), thus resulting in TLR activation. Second, ectopic Blimp1 expression partially activates the plasma cell program in developing B cells by interfering with the intrinsic B cell gene expression pattern and by regulating plasmablast‐specific genes, which may further accelerate plasmablast differentiation, thus allowing autoreactive immature B cells to evade central tolerance and anergic B cells to escape anergy control by differentiating to plasma cells. In summary, the Prdm1 ihCd2/+ mouse model of ectopic Blimp1 expression has identified a novel mechanism that can explain how Blimp1 as a risk factor contributes to the development of autoimmune disease.
Materials and Methods
Detailed methods can be found in the Appendix Supplementary Methods available online.
Mice
The following mice were maintained on the C57BL/6 genetic background: Prdm1 ihCd2/ihCd2 (Minnich et al, 2016), Prdm1 Gfp/+ (Kallies et al, 2004), Eβ−/− (Bouvier et al, 1996), JHT (Gu et al, 1993), Rosa26 BirA/BirA (Driegen et al, 2005), Pax5 iGfp/iGfp (Fuxa & Busslinger, 2007), Rag2 −/− (Shinkai et al, 1992), Fasl gld/gld (Takahashi et al, 1994), and transgenic Vav‐Bcl2 (Ogilvy et al, 1999) mice. All animal experiments were carried out according to valid project licenses, which were approved and regularly controlled by the Austrian Veterinary Authorities.
Generation of Prdm1 ∆3′U(90)/∆3′U(90) mice
The 3′ UTR in the endogenous Prdm1 locus was deleted by CRISPR/Cas9‐mediated genome editing by co‐injecting mouse zygotes with Cas9 mRNA and two specific sgRNAs (Table EV3). PCR genotyping with the primers shown in Table EV3 yielded a 399‐bp and 218‐bp PCR fragment for the wild‐type and the Prdm1 ∆3′U(90)/+ allele, respectively.
Antibodies
The following monoclonal antibodies were used for flow cytometry: B220/CD45R (RA3‐6B2), CD3ε (145‐2C11), CD4 (GK1.5), CD5 (53‐7.3), CD8α (53‐6.7), CD11b/Mac1 (M1/70), CD19 (1D3), CD21 (7G6), CD22 (Cy34.1), CD23 (B3B4), CD25 (PC61), CD28 (37.51), CD40 (3/23), CD44 (IM7), CD49b (DX5), CD62L (MEL‐14), CD69 (H1.2F3), CD80 (16‐10A1), CD86 (GL1), CD90.2/Thy1.2 (30‐H12), CD95/Fas (Jo2), CD117/c‐Kit (2B8), CD127/IL7Rα (A7R34), CD135/Flt3 (A2F10), CD138 (281‐2), CD279/PD‐1 (J43), CXCR5 (2G8), F4/80 (CI:A3‐1), GL7 (GL7), Gr1 (RB6‐8C5), IgD (11‐26c.2a), IgM (II/41), Ly6C (6C3), Ly6D (49‐H4), MHCII (M5‐114), NK1.1 (PK136), Sca‐1 (D7), TCRβ (H57‐597), TCRγδ (GL3), and human CD2 (RPA‐2.10).
Generation of bone marrow chimeras
For the generation of mixed bone marrow chimeras, donor‐derived bone marrow cells of the indicated genotypes (Fig 6A) were stained with PE‐conjugated, lineage‐specific antibodies (CD19, CD4, CD8α, TCRβ, TCRγδ, NK1.1, and CD49b) followed by magnetic depletion of the PE‐labeled cells using MACS cell separation (Miltenyi Biotec). The donor cells, mixed at a 1:15 ratio (JHT : Eβ−/− or Eβ−/− Prdm1 ihCd2/+), were intravenously injected into lethally irradiated (1,000 rads) Rag2 −/− recipients.
Injection of apoptotic thymocytes
To generate apoptotic cells, thymocytes of 6‐ to 8‐week‐old C56BL/6 mice were cultured for 6 h at 22°C in IMDM medium containing 10% FCS (GE Healthcare; A15‐101), as described (Duhlin et al, 2016). Approximately 1 × 107 apoptotic thymocytes were intravenously injected into each mouse.
Immunization, ELISPOT, and ELISA analyses
The immune response to a T cell‐dependent antigen was studied by intraperitoneal injection of 100 μg of NP‐KLH (Biosearch Technologies) in alum. The frequencies of NP‐specific IgM antibody‐secreting cells (ASCs) were determined in the spleen by enzyme‐linked immunospot (ELISPOT) assay, as described (Smith et al, 1997).
The serum titer of NP‐specific IgM, IgG1, and IgG2b antibodies was determined by enzyme‐linked immunosorbent assay (ELISA; Smith et al, 1997) by using ELISA plates (Sigma‐Aldrich), which were coated with 25 μg/ml of NP7‐BSA or NP24‐BSA to capture high‐affinity IgG1 or total NP‐specific IgM, IgG1, and IgG2b antibodies, respectively.
ELISA measurements of autoantibodies
ELISA plates coated with mouse liver DNA were incubated with mouse serum for 2 h at 22°C. Anti‐DNA‐specific antibodies were detected by incubation with horseradish peroxidase‐coupled goat anti‐mouse IgG or goat anti‐mouse IgM antibodies (SouthernBiotech) in the presence of the TMB substrate (Biolegend). The absorbance was measured at 650 nm in an Epoch Microplate Spectrophotometer (BioTek Instruments). For measuring anti‐cardiolipin antibodies, ELISA plates were coated with cardiolipin (Sigma‐Aldrich) overnight. Serum was added after blocking, and antigen‐reactive IgG and IgM were measured with alkaline phosphate‐ or horseradish peroxidase‐conjugated anti‐mouse antibodies (SouthernBiotech). Antibodies against SSA (Ro‐52) and SSB (La) were measured using commercial kits (all from Signosis Inc) following the manufacturer's instruction.
Indirect immunofluorescence assay using HEp‐2 slides
Diluted mouse serum (1:100 in PBS) was incubated on HEp‐2 slides (Orgentech) for 30 min at 22°C, before the slides were washed twice for 5 min with PBS. For detection of mouse IgM or IgG, the slides were incubated for 30 min at 22°C with an Alexa488‐conjugated goat anti‐mouse IgM antibody or an Alexa488‐conjugated goat anti‐mouse IgG (H + L) antibody (both from Thermo Fisher Scientific) as a secondary antibody. Following two washing steps, DAPI‐containing mounting medium (Life Technologies) was added together with a cover slip. Images were acquired with a Zeiss Axio Imager 2 microscope and were analyzed with the Fiji software.
ChIP‐seq analysis of Blimp1 binding
Chromatin of 1 × 108 in vitro cultured pro‐B cells from Prdm1 ihCd2/+ mice was prepared using a lysis buffer containing 0.25% SDS and was then subjected to ChIP with anti‐V5 agarose beads (Sigma‐Aldrich), as described (Wöhner et al, 2016). About 1–5 ng of ChIP‐precipitated DNA was used for library preparation and Illumina deep sequencing (Table EV4).
RNA‐sequencing
RNA from ex vivo sorted B cells was isolated with the RNeasy Plus Mini Kit (Qiagen). mRNA was obtained by two rounds of poly(A) selection and used for library preparation and Illumina deep sequencing as described (Minnich et al, 2016).
Bioinformatic analysis of RNA‐ and ChIP‐seq data
The bioinformatic analysis of RNA‐ and ChIP‐seq data was performed as described in detail (Minnich et al, 2016).
Statistical analysis
Statistical analysis was performed with the GraphPad Prism 7 software. The two‐tailed Student's t‐test analysis was used to assess the statistical significance of differences between two experimental groups except for ELISA data, which were analyzed using the Mann–Whitney U‐test.
Accession numbers
RNA‐seq, ChIP‐seq, and GRO‐seq data (Table EV4), which are first reported in this study, are available at the Gene Expression Omnibus (GEO) repository under the accession numbers GSE111692. Previously published ATAC‐seq, ChIP‐seq, and RNA‐seq datasets are available under the GEO accession numbers indicated in Table EV4.
Author contributions
PB performed most experiments; MW performed the ELISPOT, ELISA, BrdU incorporation, and cell injection experiments; MM generated the Prdm1 ihCd2 allele and discovered the Blimp1 overexpression phenotype of the Prdm1 ihCd2/+ mouse; HT generated the GRO‐seq data; MG measured the anti‐cardiolipin, anti‐SSA, and anti‐SSB antibody titers; MCIK provided advice for analysis of the autoimmune phenotype; AK evaluated the pathology of the kidneys; MF and MJ performed the bioinformatic analysis of all RNA‐seq and ChIP‐seq data, respectively; MB and PB planned the project and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Table EV1
Table EV2
Table EV3
Table EV4
Review Process File
Acknowledgements
We thank G. Schmauß and M. Weninger for FACS sorting, A. Sommer's team at the Vienna Biocenter Support Facilities GmbH (VBCF) for Illumina sequencing, and T. Engelmeier at VBCF for histological service. This research was supported by Boehringer Ingelheim, the European Community's Seventh Framework Program (European Research Council Advanced Grant 291740‐LymphoControl), and the Austrian Industrial Research Promotion Agency (Headquarter Grant FFG‐852936).
The EMBO Journal (2019) 38: e100010
References
- Azulay‐Debby H, Edry E, Melamed D (2007) CpG DNA stimulates autoreactive immature B cells in the bone marrow. Eur J Immunol 37: 1463–1475 [DOI] [PubMed] [Google Scholar]
- Bouvier G, Watrin F, Naspetti M, Verthuy C, Naquet P, Ferrier P (1996) Deletion of the mouse T‐cell receptor β gene enhancer blocks αβ T‐cell development. Proc Natl Acad Sci USA 93: 7877–7881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ (2013) Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA‐binding proteins and nucleosome position. Nat Methods 10: 1213–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantaert T, Schickel J‐N, Bannock JM, Ng Y‐S, Massad C, Oe T, Wu R, Lavoie A, Walter JE, Notarangelo LD, Al‐Herz W, Kilic SS, Ochs HD, Nonoyama S, Durandy A, Meffre E (2015) Activation‐induced cytidine deaminase expression in human B cell precursors is essential for central B cell tolerance. Immunity 43: 884–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Waterfall JJ, Lis JT (2008) Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 322: 1845–1848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker T, Pasca di Magliano M, McManus S, Sun Q, Bonifer C, Tagoh H, Busslinger M (2009) Stepwise activation of enhancer and promoter regions of the B cell commitment gene Pax5 in early lymphopoiesis. Immunity 30: 508–520 [DOI] [PubMed] [Google Scholar]
- Dent AL, Shaffer AL, Yu X, Allman D, Staudt LM (1997) Control of inflammation, cytokine expression, and germinal center formation by BCL‐6. Science 276: 589–592 [DOI] [PubMed] [Google Scholar]
- Driegen S, Ferreira R, van Zon A, Strouboulis J, Jaegle M, Grosveld F, Philipsen S, Meijer D (2005) A generic tool for biotinylation of tagged proteins in transgenic mice. Transgenic Res 14: 477–482 [DOI] [PubMed] [Google Scholar]
- Duhlin A, Chen Y, Wermeling F, Sedimbi SK, Lindh E, Shinde R, Halaby MJ, Kaiser Y, Winqvist O, McGaha TL, Karlsson MC (2016) Selective memory to apoptotic cell‐derived self‐antigens with implications for systemic lupus erythematosus development. J Immunol 197: 2618–2626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairfax KA, Corcoran LM, Pridans C, Huntington ND, Kallies A, Nutt SL, Tarlinton DM (2007) Different kinetics of blimp‐1 induction in B cell subsets revealed by reporter gene. J Immunol 178: 4104–4111 [DOI] [PubMed] [Google Scholar]
- Fuxa M, Busslinger M (2007) Reporter gene insertions reveal a strictly B lymphoid‐specific expression pattern of Pax5 in support of its B cell identity function. J Immunol 178: 8222–8228 [DOI] [PubMed] [Google Scholar]
- Gateva V, Sandling JK, Hom G, Taylor KE, Chung SA, Sun X, Ortmann W, Kosoy R, Ferreira RC, Nordmark G, Gunnarsson I, Svenungsson E, Padyukov L, Sturfelt G, Jonsen A, Bengtsson AA, Rantapaa‐Dahlqvist S, Baechler EC, Brown EE, Alarcon GS et al (2009) A large‐scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat Genet 41: 1228–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genestier L, Taillardet M, Mondiere P, Gheit H, Bella C, Defrance T (2007) TLR agonists selectively promote terminal plasma cell differentiation of B cell subsets specialized in thymus‐independent responses. J Immunol 178: 7779–7786 [DOI] [PubMed] [Google Scholar]
- Gloury R, Zotos D, Zuidscherwoude M, Masson F, Liao Y, Hasbold J, Corcoran LM, Hodgkin PD, Belz GT, Shi W, Nutt SL, Tarlinton DM, Kallies A (2016) Dynamic changes in Id3 and E‐protein activity orchestrate germinal center and plasma cell development. J Exp Med 213: 1095–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, Zou YR, Rajewsky K (1993) Independent control of immunoglobulin switch recombination at individual switch regions evidenced through Cre‐loxP‐mediated gene targeting. Cell 73: 1155–1164 [DOI] [PubMed] [Google Scholar]
- Gururajan M, Haga CL, Das S, Leu C‐M, Hodson D, Josson S, Turner M, Cooper MD (2010) MicroRNA 125b inhibition of B cell differentiation in germinal centers. Int Immunol 22: 583–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, Dent AL, Craft J, Crotty S (2009) Bcl6 and Blimp‐1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science 325: 1006–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallies A, Hasbold J, Tarlinton DM, Dietrich W, Corcoran LM, Hodgkin PD, Nutt SL (2004) Plasma cell ontogeny defined by quantitative changes in Blimp‐1 expression. J Exp Med 200: 967–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallies A, Hasbold J, Fairfax K, Pridans C, Emslie D, McKenzie BS, Lew AM, Corcoran LM, Hodgkin PD, Tarlinton DM, Nutt SL (2007) Initiation of plasma‐cell differentiation is independent of the transcription factor Blimp‐1. Immunity 26: 555–566 [DOI] [PubMed] [Google Scholar]
- Kassambara A, Jourdan M, Bruyer A, Robert N, Pantesco V, Elemento O, Klein B, Moreaux J (2017) Global miRNA expression analysis identifies novel key regulators of plasma cell differentiation and malignant plasma cell. Nucleic Acids Res 45: 5639–5652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon H, Thierry‐Mieg D, Thierry‐Mieg J, Kim HP, Oh J, Tunyaplin C, Carotta S, Donovan CE, Goldman ML, Tailor P, Ozato K, Levy DE, Nutt SL, Calame K, Leonard WJ (2009) Analysis of interleukin‐21‐induced Prdm1 gene regulation reveals functional cooperation of STAT3 and IRF4 transcription factors. Immunity 31: 941–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messika EJ, Lu PS, Sung YJ, Yao T, Chi JT, Chien YH, Davis MM (1998) Differential effect of B lymphocyte‐induced maturation protein (Blimp‐1) expression on cell fate during B cell development. J Exp Med 188: 515–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minnich M, Tagoh H, Bönelt P, Axelsson E, Fischer M, Cebolla B, Tarakhovsky A, Nutt SL, Jaritz M, Busslinger M (2016) Multifunctional role of the transcription factor Blimp‐1 in coordinating plasma cell differentiation. Nat Immunol 17: 331–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasir A, Norton JD, Baou M, Zekavati A, Bijlmakers M‐J, Thompson S, Murphy JJ (2012) ZFP36L1 negatively regulates plasmacytoid differentiation of BCL1 cells by targeting BLIMP1 mRNA. PLoS One 7: e52187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemazee D (2017) Mechanisms of central tolerance for B cells. Nat Rev Immunol 17: 281–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, Wang Y‐H, Dong C (2009) Bcl6 mediates the development of T follicular helper cells. Science 325: 1001–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutt SL, Fairfax KA, Kallies A (2007) BLIMP1 guides the fate of effector B and T cells. Nat Rev Immunol 7: 923–927 [DOI] [PubMed] [Google Scholar]
- Ogilvy S, Metcalf D, Print CG, Bath ML, Harris AW, Adams JM (1999) Constitutive Bcl‐2 expression throughout the hematopoietic compartment affects multiple lineages and enhances progenitor cell survival. Proc Natl Acad Sci USA 96: 14943–14948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parlato S, Bruni R, Fragapane P, Salerno D, Marcantonio C, Borghi P, Tataseo P, Ciccaglione AR, Presutti C, Romagnoli G, Bozzoni I, Belardelli F, Gabriele L (2013) IFN‐α regulates Blimp‐1 expression via miR‐23a and miR‐125b in both monocytes‐derived DC and pDC. PLoS One 8: e72833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualucci L, Compagno M, Houldsworth J, Monti S, Grunn A, Nandula SV, Aster JC, Murty VV, Shipp MA, Dalla‐Favera R (2006) Inactivation of the PRDM1/BLIMP1 gene in diffuse large B cell lymphoma. J Exp Med 203: 311–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquinelli AE (2012) MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat Rev Genet 13: 271–282 [DOI] [PubMed] [Google Scholar]
- Raychaudhuri S, Thomson BP, Remmers EF, Eyre S, Hinks A, Guiducci C, Catanese JJ, Xie G, Stahl EA, Chen R, Alfredsson L, Amos CI, Ardlie KG, BIRAC Consortium , Barton A, Bowes J, Burtt NP, Chang M, Coblyn J, Costenbader KH et al (2009) Genetic variants at CD28, PRDM1 and CD2/CD58 are associated with rheumatoid arthritis risk. Nat Genet 41: 1313–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revilla‐i‐Domingo R, Bilic I, Vilagos B, Tagoh H, Ebert A, Tamir IM, Smeenk L, Trupke J, Sommer A, Jaritz M, Busslinger M (2012) The B‐cell identity factor Pax5 regulates distinct transcriptional programmes in early and late B lymphopoiesis. EMBO J 31: 3130–3146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolink AG, Andersson J, Melchers F (1998) Characterization of immature B cells by a novel monoclonal antibody, by turnover and by mitogen reactivity. Eur J Immunol 28: 3738–3748 [DOI] [PubMed] [Google Scholar]
- Sciammas R, Shaffer AL, Schatz JH, Zhao H, Staudt LM, Singh H (2006) Graded expression of interferon regulatory factor‐4 coordinates isotype switching with plasma cell differentiation. Immunity 25: 225–236 [DOI] [PubMed] [Google Scholar]
- Shaffer AL, Lin K‐I, Kuo TC, Yu X, Hurt EM, Rosenwald A, Giltnane JM, Yang L, Zhao H, Calame K, Staudt LM (2002) Blimp‐1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity 17: 51–62 [DOI] [PubMed] [Google Scholar]
- Shapiro‐Shelef M, Lin K‐I, McHeyzer‐Williams LJ, Liao J, McHeyzer‐Williams MG, Calame K (2003) Blimp‐1 is required for the formation of immunoglobulin secreting plasma cells and pre‐plasma memory B cells. Immunity 19: 607–620 [DOI] [PubMed] [Google Scholar]
- Shinkai Y, Rathbun G, Lam K‐P, Oltz EM, Stewart V, Mendelsohn M, Charron J, Datta M, Young F, Stall AM, Alt FW (1992) RAG‐2‐deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell 68: 855–867 [DOI] [PubMed] [Google Scholar]
- Smith KG, Light A, Nossal GJ, Tarlinton DM (1997) The extent of affinity maturation differs between the memory and antibody‐forming cell compartments in the primary immune response. EMBO J 16: 2996–3006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speck NA, Renjifo B, Hopkins N (1990) Point mutations in the Moloney murine leukemia virus enhancer identify a lymphoid‐specific viral core motif and 1,3‐phorbol myristate acetate‐inducible element. J Virol 64: 543–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suurmond J, Diamond B (2015) Autoantibodies in systemic autoimmune diseases: specificity and pathogenicity. J Clin Invest 125: 2194–2202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Tanaka M, Brannan CI, Jenkins NA, Copeland NG, Suda T, Nagata S (1994) Generalized lymphoproliferative disease in mice, caused by a point mutation in the Fas ligand. Cell 76: 969–976 [DOI] [PubMed] [Google Scholar]
- Tam W, Gomez M, Chadburn A, Lee JW, Chan WC, Knowles DM (2006) Mutational analysis of PRDM1 indicates a tumor‐suppressor role in diffuse large B‐cell lymphomas. Blood 107: 4090–4100 [DOI] [PubMed] [Google Scholar]
- Tellier J, Shi W, Minnich M, Liao Y, Crawford S, Smyth GK, Kallies A, Busslinger M, Nutt SL (2016) Blimp‐1 controls plasma cell function through the regulation of immunoglobulin secretion and the unfolded protein response. Nat Immunol 17: 323–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theofilopoulos AN, Kono DH, Baccala R (2017) The multiple pathways to autoimmunity. Nat Immunol 18: 716–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsokos GC, Lo MS, Costa Reis P, Sullivan KE (2016) New insights into the immunopathogenesis of systemic lupus erythematosus. Nat Rev Rheumatol 12: 716–730 [DOI] [PubMed] [Google Scholar]
- Turner CAJ, Mack DH, Davis MM (1994) Blimp‐1, a novel zinc finger‐containing protein that can drive the maturation of B lymphocytes into immunoglobulin‐secreting cells. Cell 77: 297–306 [DOI] [PubMed] [Google Scholar]
- Turner M, Galloway A, Vigorito E (2014) Noncoding RNA and its associated proteins as regulatory elements of the immune system. Nat Immunol 15: 484–491 [DOI] [PubMed] [Google Scholar]
- Vinuesa CG, Linterman MA, Yu D, MacLennan IC (2016) Follicular helper T cells. Annu Rev Immunol 34: 335–368 [DOI] [PubMed] [Google Scholar]
- Wagner GP, Kin K, Lynch VJ (2012) Measurement of mRNA abundance using RNA‐seq data: RPKM measure is inconsistent among samples. Theory Biosci 131: 281–285 [DOI] [PubMed] [Google Scholar]
- Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC (2003) Predominant autoantibody production by early human B cell precursors. Science 301: 1374–1377 [DOI] [PubMed] [Google Scholar]
- Wöhner M, Tagoh H, Bilic I, Jaritz M, Kostanova Poliakova D, Fischer M, Busslinger M (2016) Molecular functions of the transcription factors E2A and E2‐2 in controlling germinal center B cell and plasma cell development. J Exp Med 213: 1201–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Qiu Q, Gao B, Kong S, Lin Z, Fang D (2014) Hrd1‐mediated BLIMP‐1 ubiquitination promotes dendritic cell MHCII expression for CD4 T cell priming during inflammation. J Exp Med 211: 2467–2479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye BH, Cattoretti G, Shen Q, Zhang J, Hawe N, de Waard R, Leung C, Nouri‐Shirazi M, Orazi A, Chaganti RSK, Rothman P, Stall AM, Pandolfi P‐P, Dalla‐Favera R (1997) The BCL‐6 proto‐oncogene controls germinal‐centre formation and Th2‐type inflammation. Nat Genet 16: 161–170 [DOI] [PubMed] [Google Scholar]
- Yu D, Rao S, Tsai LM, Lee SK, He Y, Sutcliffe EL, Srivastava M, Linterman M, Zheng L, Simpson N, Ellyard JI, Parish IA, Ma CS, Li Q‐J, Parish CR, Mackay CR, Vinuesa CG (2009) The transcriptional repressor Bcl‐6 directs T follicular helper cell lineage commitment. Immunity 31: 457–468 [DOI] [PubMed] [Google Scholar]
- Zhou X‐J, Lu X‐L, Lv J‐C, Yang H‐Z, Qin L‐X, Zhao M‐H, Su Y, Li Z‐G, Zhang H (2011) Genetic association of PRDM1‐ATG5 intergenic region and autophagy with systemic lupus erythematosus in a Chinese population. Ann Rheum Dis 70: 1330–1337 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Table EV1
Table EV2
Table EV3
Table EV4
Review Process File