

Abstract
Although mitochondria play a multifunctional role in cancer progression and Ca2+ signaling is remodeled in a wide variety of tumors, the underlying mechanisms that link mitochondrial Ca2+ homeostasis with malignant tumor formation and growth remain elusive. Here, we show that phosphorylation at the N‐terminal region of the mitochondrial calcium uniporter (MCU) regulatory subunit MICU1 leads to a notable increase in the basal mitochondrial Ca2+ levels. A pool of active Akt in the mitochondria is responsible for MICU1 phosphorylation, and mitochondrion‐targeted Akt strongly regulates the mitochondrial Ca2+ content. The Akt‐mediated phosphorylation impairs MICU1 processing and stability, culminating in reactive oxygen species (ROS) production and tumor progression. Thus, our data reveal the crucial role of the Akt‐MICU1 axis in cancer and underscore the strategic importance of the association between aberrant mitochondrial Ca2+ levels and tumor development.
Keywords: Akt, calcium, cancer, MICU1, mitochondria
Subject Categories: Cancer, Membrane & Intracellular Transport
Introduction
Mitochondrial Ca2+ accumulation controls cellular energetics and cell metabolism by favoring ATP production, and mitochondrial Ca2+ also operates as a key regulator of cell fate (Giorgi et al, 2018). Multiple pathological contexts, including tumor formation and development, are strictly linked to mitochondrial deregulation, and a hallmark feature of cancer cells is the re‐programming of their mitochondrial metabolism (Hanahan & Weinberg, 2011; Boroughs & DeBerardinis, 2015). The connection between mitochondrial dysfunction and cancer is not only limited to the metabolic transformation of cancer cells but also triggers tumor‐promoting epigenetic changes (Gottlieb & Tomlinson, 2005; Gaude & Frezza, 2014). Therefore, it is not surprising that several oncogenes and tumor suppressors exert their activities by regulating mitochondrial function (Galluzzi et al, 2012; Frezza, 2014) and that many of them act on key Ca2+‐transport molecules to provoke deep mitochondrial Ca2+ homeostasis remodeling and promote certain malignant phenotypes (Prevarskaya et al, 2011; Danese et al, 2017).
The mitochondrial calcium uniporter (MCU), which is the channel that is responsible for Ca2+ accumulation inside the mitochondrial matrix, has been recently characterized at the molecular level (Baughman et al, 2011; De Stefani et al, 2011). Moreover, a series of proteins that contribute to the formation of the so‐called MCU complex have also been identified (Giorgi et al, 2018). Such proteins include the dominant‐negative form MCUb (Raffaello et al, 2013), the essential regulator EMRE (Sancak et al, 2013), and others that regulate MCU channel activity, such as MICU1 (Perocchi et al, 2010) and its paralog MICU2 (Plovanich et al, 2013). Among the different components of the mitochondrial Ca2+ uptake machinery, MICU1 functions have been the subject of extensive studies that revealed that MICU1 acts as a gatekeeper for the MCU complex, setting the threshold for mitochondrial Ca2+ uptake (Mallilankaraman et al, 2012; Csordas et al, 2013). Although genetic MICU1 ablations in both cells and tissues and loss‐of‐function mutations in the MICU1 gene have been associated with different pathological scenarios (Logan et al, 2014; Antony et al, 2016), little is known about the role of MICU1‐related mitochondrial Ca2+ homeostasis in tumor progression. Recent evidence indicates that both prostate and colon cancer cells overexpress cancer‐related microRNAs that target the channel pore‐forming subunit MCU, thus conferring resistance to apoptotic stimuli (Marchi et al, 2013). Moreover, MCU expression correlates with breast cancer progression, and the deletion of MCU reduces tumor growth and metastasis formation (Tosatto et al, 2016). No correlation has been observed between the expression of MCU regulators and tumor size, raising the possibility that instead of quantitative changes, qualitative alterations determined by post‐translational modifications could account for the altered MCU function in oncogenesis (Tosatto et al, 2016).
Here, we show that phosphorylation at the N‐terminal domain of the MICU1 protein robustly alters the basal mitochondrial Ca2+ content under resting conditions. The target amino acid residue is contained in an Akt consensus phosphorylation motif, and MICU1 is readily phosphorylated upon Akt activation inside the mitochondrial compartment. Akt‐mediated phosphorylation affects MICU1 proteolytic maturation and stability, thereby explaining the altered mitochondrial Ca2+ homeostasis. Importantly, the expression of a nonphosphorylatable MICU1 mutant significantly reduces the in vivo growth rate of tumors, even in the presence of activated Akt, suggesting a key role for the mitochondrial Akt‐MICU1 axis in cancer progression.
Results
N‐terminal MICU1 phosphorylation increases the basal mitochondrial Ca2+ levels
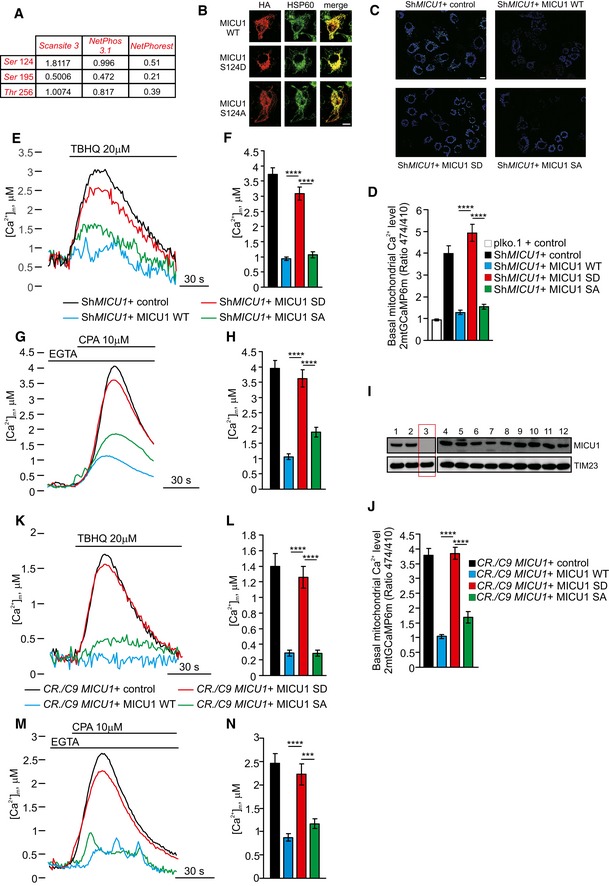
We investigated the potentially phosphorylated residues in the MICU1 sequence. Using the Scansite 3 software program (http://scansite3.mit.edu), we searched for motifs within the wild‐type (WT) MICU1 protein (NM_144822) that are likely to be phosphorylated by specific protein kinases. The following three candidates were identified: Ser124, Ser195, and Thr256 (Fig 1A). Among them, Ser124 displayed the highest value of surface accessibility, as well as a high phosphorylation prediction score (Fig 1A). Ser124 is localized in the N‐terminal region of MICU1, which has been proposed to extend into the intermembrane space (Csordas et al, 2013). As a result, we generated two MICU1 mutants, a nonphosphorylatable S124A and a phosphomimetic S124D MICU1 mutant. When transfected into cells, both mutated MICU1 proteins localized correctly to mitochondria (Fig 1B). It is now widely accepted that MICU1 functions as a gatekeeper of the MCU channel, meaning that loss of MICU1 facilitates Ca2+ accumulation inside the mitochondrial matrix even at low cytosolic Ca2+ levels (Mallilankaraman et al, 2012). Thus, we transfected the mitochondrial matrix‐targeted Ca2+ biosensor GCaMP6m into cells expressing the MICU1 WT or SD and SA mutants. Due to its characteristics as a ratiometric sensor, GCaMP6m is reputed to quantitatively measure even small differences in the resting mitochondrial Ca2+ concentration ([Ca2+]m) (Hill et al, 2014). We expressed WT or mutant forms of MICU1 in cells from which endogenous MICU1 was constitutively depleted using a short hairpin RNA (shRNA) (Fig EV1A). As expected, the resting mitochondrial Ca2+ levels were largely increased when MICU1 was stably downregulated, and the expression of both the WT MICU1 and SA mutant reduced the baseline [Ca2+]m levels in ShMICU1 cells. In contrast, the MICU1 SD variant failed to restore [Ca2+]m in ShMICU1 cells (Fig 1C and D). To verify the role of Ser124 phosphorylation in the regulation of MICU1 functionality, we analyzed the mitochondrial Ca2+ uptake following treatment with the sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA) inhibitor 2,5‐di‐tert‐butylhydroquinone (TBHQ), which induces slow and weak ER Ca2+ depletion (Waldeck‐Weiermair et al, 2015). The co‐transfection of the MICU1 WT, SD mutant, or SA mutant with an aequorin‐based mitochondrial Ca2+ probe revealed that only mock and MICU1 SD‐expressing cells were able to accumulate [Ca2+]m under these conditions, whereas the MICU1 WT and SA mutant failed to do so (Fig 1E and F). We obtained similar results when TBHQ was replaced with another SERCA antagonist, cyclopiazonic acid (CPA) (Fig 1G and H). The higher Ca2+ affinity of the MICU1 SD mutant‐expressing mitochondria was also investigated in permeabilized cells. After permeabilization in an EGTA‐containing Ca2+‐free buffer (IB/EGTA) that mimics the physiological ion milieu, Ca2+ uptake was generated by switching the perfusion buffer to IB, containing EGTA‐buffered Ca2+. Using different [Ca2+]s (500, 800 nM, and 1.5 μM), the rates of mitochondrial Ca2+ uptake were substantially increased in both MICU1‐depleted and SD‐expressing cells (Fig EV1B). Interestingly, MICU1 SA mutant slightly lowered the Ca2+ threshold for channel activation, showing reduced Ca2+ uptake rate compared to MICU1 WT (Fig EV1B). Overall, these data suggest that upon phosphorylation of serine 124, MICU1 loses its inhibitory function on the MCU complex.
Figure 1. MICU1 phosphorylation at the Ser124 position increases the mitochondrial basal Ca2+ levels.
-
APotentially phosphorylated residues in the MICU1 (NM_144822) sequence were detected using the Scansite 3 software (http://scansite3.mit.edu). The different values refer to the surface accessibility scores (Scansite) or the phosphorylation scores, which were obtained with both NetPhos 3.1 (http://www.cbs.dtu.dk/services/NetPhos/) and NetPhorest (http://www.netphorest.info) software.
-
BHeLa cells overexpressing the HA‐tagged MICU1 WT, MICU1 S124D, or MICU1 S124A mutants were stained for HA or HSP60 (mitochondrial marker). Merged images are indicated (merge). Scale bar 10 μm.
-
CRepresentative images of the 2mt‐GCaMP6m 474/410 ratio of ShMICU1 HeLa stable cells expressing an empty vector (ctrl) or the MICU1 WT, MICU1 SD, and MICU1 SA. Scale bar 10 μm.
-
DResting mitochondrial calcium levels, evaluated through ratiometric imaging of the mitochondrial‐targeted GCaMP6m, in ShRNA control (plko) or ShRNA MICU1 HeLa stable clone cells transfected with the indicated constructs (n = 5 independent experiments; 55–67 cells).
-
E, FRepresentative kinetics (E) and analysis (F) of aequorin‐based [Ca2+]m measurements in ShRNA MICU1 HeLa stable clone cells transfected with the indicated constructs and challenged with 20 μM 2,5‐di‐tert‐butylhydroquinone (TBHQ) in the absence of extracellular Ca2+ (n = 3 independent experiments).
-
G, HRepresentative kinetics (G) and analysis (H) of aequorin‐based [Ca2+]m measurements in intact ShRNA MICU1 HeLa stable clone cells transfected with the indicated constructs and challenged with 10 μM cyclopiazonic acid (CPA) in the presence of 100 μM EGTA (n = 3 independent experiments).
-
IWestern blot analysis for the presence of MICU1 in clones arising from single cells generated by CRISPR/Cas9‐mediated genome editing. The results for 12 of the 36 clones that were examined are shown.
-
JResting mitochondrial calcium levels, evaluated through ratiometric imaging of the mitochondrial‐targeted GCaMP6m, in MICU1 KO cells generated using the CRISPR/Cas9 technique and transfected with the indicated constructs (n = 3 independent experiments; 30–56 cells).
-
K, LRepresentative kinetics (K) and analysis (L) of aequorin‐based [Ca2+]m measurements in intact MICU1 KO cells generated using the CRISPR/Cas9 technique, transfected with the indicated constructs, and challenged with 20 μM TBHQ in the absence of extracellular Ca2+ (n = 3 independent experiments).
-
M, NRepresentative kinetics (M) and analysis (N) of aequorin‐based [Ca2+]m measurements in intact MICU1 KO cells generated using the CRISPR/Cas9 technique, transfected with the indicated constructs, and challenged with 10 μM CPA in the presence of 100 μM EGTA (n = 3 independent experiments).
Figure EV1. Akt‐mediated MICU1 phosphorylation increases mitochondrial basal Ca2+ levels.
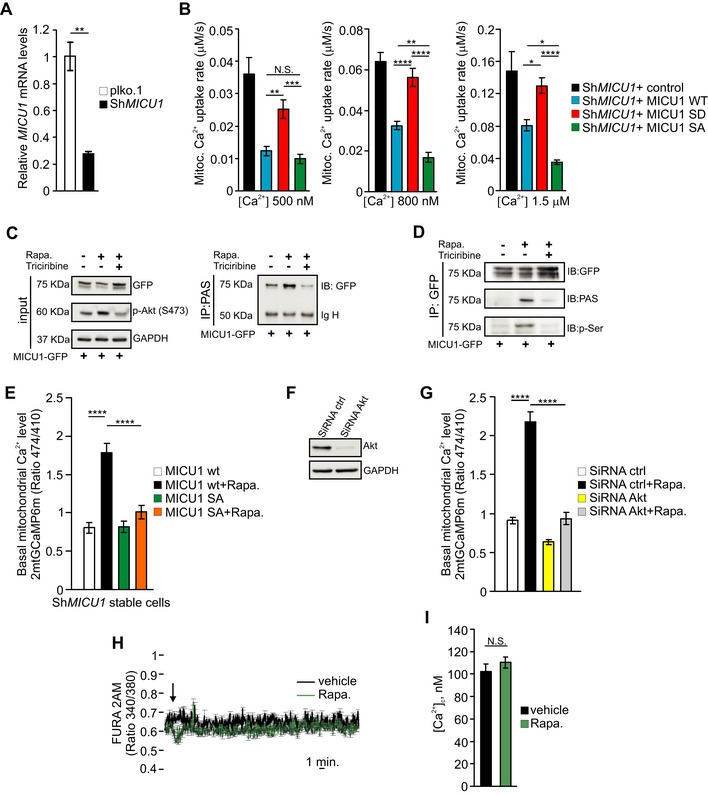
- MICU1 mRNA levels in plko.1 and ShRNA MICU1 HeLa stable clone cells (n = 3 independent experiments).
- Analysis of aequorin‐based [Ca2+]m measurements in permeabilized ShRNA MICU1‐HeLa cells transfected with the indicated constructs and challenged with 500, 800 nM or 1.5 μM buffered [Ca2+] (n = 3 independent experiments).
- HEK293 cells were transfected with GFP‐tagged wild‐type (WT) MICU1 and then treated with 1 μM rapamycin (Rapa.) alone for 4 h or in combination with the Akt inhibitor triciribine (10 μM). The cell lysates were immunoprecipitated with PAS (phospho‐Akt substrate) antibody and then analyzed by Western blotting. IgH: immunoglobulin heavy chain.
- HEK293 cells were transfected with either GFP‐tagged wild‐type (WT) MICU1 or MICU1 S124A‐GFP mutant and treated as in (C). MICU1‐GFP immuno complexes were precipitated with a GFP antibody and analyzed with PAS (phospho‐Akt substrate) and phosphoserine (p‐Ser) antibodies by Western blotting.
- Resting mitochondrial calcium levels, evaluated through ratiometric imaging of the mitochondrial‐targeted GCaMP6m, in ShRNA MICU1 HeLa stable clone cells transfected with the indicated constructs, treated with vehicle or 1 μM rapamycin for 4 h (n = 3 independent experiments; 27–30 cells).
- Western blot analysis of Akt levels in HeLa cells silenced with scramble (ctrl) or Akt siRNAs.
- Resting mitochondrial calcium levels, evaluated through ratiometric imaging of the mitochondrial‐targeted GCaMP6m, in HeLa cells silenced with the indicated constructs, treated with vehicle or 1 μM rapamycin for 4 h (n = 3 independent experiments; 40–63 cells).
- FURA‐2 AM ratiometric measurements in HeLa cells upon stimulation with vehicle (DMSO) or 1 μM rapamycin (n = 3 independent experiments; 46–75 cells).
- Calibrated FURA‐2 AM cytosolic [Ca2+] in HeLa cells treated with vehicle or 1 μM rapamycin for 4 h (n = 3 independent experiments; 50 cells).
To further demonstrate this concept, we analyzed the basal mitochondrial Ca2+ levels in intact HeLa cells. Overexpression of the MICU1 WT or MICU1 SA mutant caused no significant changes in the basal mitochondrial Ca2+ levels compared with those found in untransfected control cells. However, the basal mitochondrial Ca2+ content in the MICU1 SD‐overexpressing cells was significantly increased compared to that in the MICU1 WT‐ and MICU1 SA‐expressing cells (Appendix Fig S1A). These data are independent from changes in the mitochondrial membrane potential (Appendix Fig S1B) and were confirmed using both TBHQ and CPA inhibitors (Appendix Fig S1C–F).
Because the protein levels of MICU1 and MCU and their stoichiometry are crucial aspects in regulating [Ca2+]m (Paillard et al, 2017), we reconstituted the MICU1 WT and mutants in a MICU1‐null background, generated using a CRISPR/CAS9‐mediated approach (Fig 1I). Using the mito‐GCaMP6m indicator, we observed higher Ca2+ levels only in MICU1‐null and MICU1 SD‐expressing cells (Fig 1J). Similar findings were obtained with an aequorin‐based approach upon stimulation with either TBHQ or CPA (Fig 1K–N).
Therefore, the results achieved in three different cell lines and using three different techniques to measure [Ca2+]m, namely (i) in untreated intact cells, (ii) upon treatment of intact cells with SERCA antagonists, and (iii) upon Ca2+ addition to permeabilized cells, indicate that phosphorylation of MICU1 at position 124 limits its negative effect on MCU and thus induces Ca2+ accumulation inside the mitochondrial matrix. Based on these results, we became interested in identifying the kinase responsible for MICU1 phosphorylation.
MICU1 phosphorylation is mediated by mitochondrial Akt
The MICU1 sequence surrounding Ser124 matches the Akt consensus phosphorylation motif, R‐X‐R‐X‐X‐S/T (Manning & Cantley, 2007), which is highly conserved among different species (Fig 2A). We observed that upon treatment with rapamycin, which is a known activator of Akt, a significant number of cells displayed an obvious mitochondrial localization of activated (Ser473 phosphorylated) Akt (Fig 2B and C and Appendix Fig S2A). Subcellular fractionation followed by immunoblot analyses suggested that the treatment of HEK293T cells with rapamycin resulted in an increased Akt activation/phosphorylation to mitochondria (Appendix Fig S2B). This effect was observed not only in the crude mitochondrial fractions (Appendix Fig S2B), but also highly purified mitochondria without any detectable plasma membrane contamination (Fig 2D). Consistent with these results, pure mitochondria extracted from mouse livers after in vivo exposure to rapamycin also contained higher levels of Akt with phosphorylated Ser473 (Fig 2E). Having established the existence of a rapamycin‐induced pool of active Akt in mitochondria, we sought to determine its submitochondrial localization. Proteinase K (PK) digestion of purified mitochondria that were subjected to selective outer membrane permeabilization by osmotic swelling (i.e., via the removal of sucrose) or complete lysis with Triton X‐100 revealed that MICU1 behaved similarly to the inner mitochondrial membrane (IMM)–intermembrane space (IMS) protein TIM23 (both of which became susceptible to proteolysis after outer membrane permeabilization), in contrast to the matrix proteins HSP60 and MCU, which only became digested when the detergent was added (Fig 2F). This finding indicates that MICU1 is located at the outer surface of the IMM, as previously suggested (Csordas et al, 2013; Tsai et al, 2016). Importantly, in response to rapamycin, active Akt located predominantly at the IMS and, to a lesser extent, in the matrix compartment (Fig 2F). Alkaline carbonate extraction of isolated HEK293T cell mitochondria revealed that active Akt is loosely attached to the IMM, sharing this characteristic with cytochrome c (Fig 2G). Taken together, these results demonstrate that active Akt localizes in the mitochondria in a membrane‐unbound state and accumulates in the same submitochondrial compartment as MICU1.
Figure 2. Mitochondrial Akt phosphorylates MICU1 at the Ser124 position.
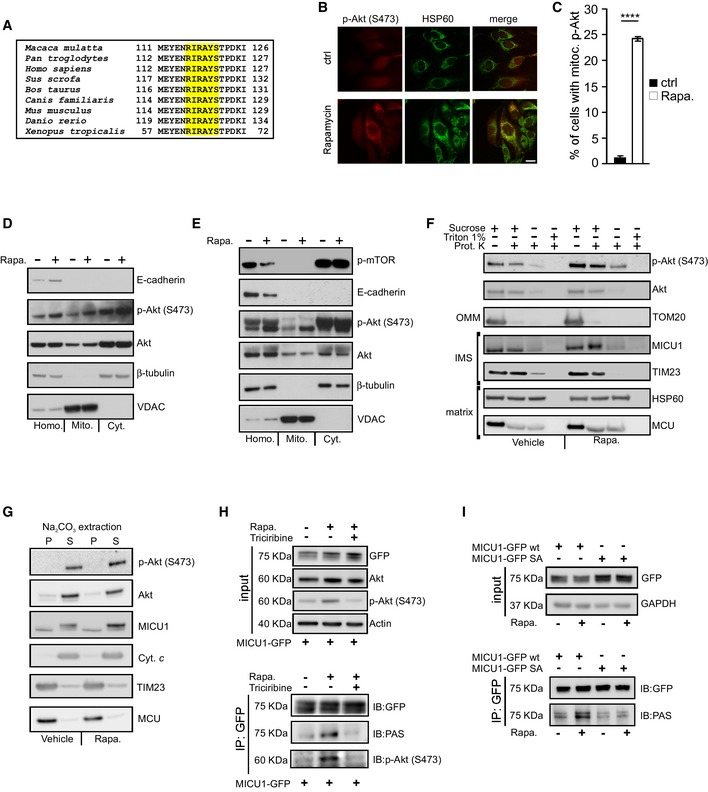
- Sequence alignment of the MICU1 protein from nine vertebrate species. The Akt consensus phosphorylation motif, R‐X‐R‐X‐X‐S/T, is marked in yellow.
- HeLa cells treated with vehicle or 1 μM rapamycin for 4 h were stained for phosphorylated (S473) Akt (p‐Akt) or HSP60 (mitochondrial marker). Merged images are indicated (merge). Scale bar 10 μm.
- Analysis of the number of cells, expressed as a percentage, showing obvious mitochondrial staining of activated (S473 phosphorylated) Akt (p‐Akt) (n = 3 independent experiments; 350‐355 cells). Means ± SEM. ****P < 0.0001 (Student's t‐test).
- HEK293T cells treated with vehicle or 1 μM rapamycin (Rapa.) for 4 h were fractionated into cytosol (Cyt.) or mitochondrial (Mito.) extracts and analyzed by Western blotting. E‐cadherin: plasma membrane marker; β‐tubulin: cytosolic marker; VDAC: mitochondrial marker; Homo: cell homogenate.
- Mouse livers treated with vehicle or 1 μM rapamycin (Rapa.) for 16 h by an intraperitoneal injection were fractionated into cytosol (Cyt.) or mitochondrial (Mito.) extracts and analyzed by Western blotting. Phosphorylated (S2468) mTOR (p‐mTOR) was used to assess the rapamycin activity. E‐cadherin: plasma membrane marker; β‐tubulin: cytosolic marker; VDAC: mitochondrial marker; Homo: cell homogenate.
- Mitochondria isolated from HEK293T cells were subjected to the indicated treatments and analyzed by Western blotting against Akt, phosphorylated (S473) Akt (p‐Akt), and mitochondrial proteins with known localizations. Osmotic swelling through the removal of sucrose from the buffer was used to induce OMM rupture. OMM: outer mitochondrial membrane; IMS: intermembrane space.
- Western blot analysis of the supernatant (S) and insoluble pellet (P) fractions of vehicle‐ or rapamycin (Rapa., 1 μM for 4 h)‐treated HEK293T cell mitochondria following carbonate extraction at pH 11.5. Akt, phosphorylated (S473) Akt (p‐Akt), MICU1, the established integral membrane proteins TIM23 and MCU, and the soluble protein cytochrome c (Cyt. c) was analyzed.
- HEK293T cells were transfected with the GFP‐tagged wild‐type (WT) MICU1 and then treated with 1 μM rapamycin (Rapa.) alone for 4 h or in combination with the Akt inhibitor triciribine (10 μM). GFP‐MICU1 immunocomplexes were precipitated with a GFP antibody and analyzed with PAS (phospho‐Akt substrate) and phosphorylated (S473) Akt (p‐Akt) antibodies by Western blotting.
- HEK293T cells were transfected with either the GFP‐tagged wild‐type (WT) MICU1 or MICU1 S124A‐GFP mutant. MICU1‐GFP immunocomplexes were precipitated with a GFP antibody and analyzed with PAS (phospho‐Akt substrate) antibody by Western blotting.
Source data are available online for this figure.
We then immunoprecipitated WT MICU1‐GFP and analyzed its phosphorylation status following rapamycin treatment with an antibody that recognizes the Akt consensus phosphorylation protein sequence, R‐X‐R‐X‐X‐S/T (anti‐PAS, phospho‐Akt substrate antibody). The MICU1‐GFP immune complexes reacted with the PAS antibody preferentially in the rapamycin‐treated cells, and triciribine, which is an allosteric inhibitor of Akt activation (Yang et al, 2004; Littlepage et al, 2012), counteracted this reaction (Fig 2H). Accordingly, the immune complexes precipitated with the anti‐PAS antibody upon rapamycin stimulation of Akt (Fig EV1C). Moreover, rapamycin increased the reactivity of the MICU1‐GFP immune complexes with an antibody that detects proteins with phosphorylated serine residues (anti‐p‐Ser) (Fig EV1D), suggesting that the Akt‐mediated phosphorylation of MICU1 occurs at a specific serine residue. To verify that Ser124 is the Akt target site on MICU1, we immunoprecipitated the GFP‐tagged MICU1 WT and MICU1‐S124A mutant and then analyzed the MICU1 phosphorylation status using the PAS antibody. As shown in Fig 2I, rapamycin induced phosphorylation of the WT MICU1 but failed to stimulate the phosphorylation of the defective mutant S124A. Notably, quantitation of the basal [Ca2+]m content using the mito‐GCaMP6m probe revealed that rapamycin increased [Ca2+]m in the WT MICU1‐transfected cells, but the corresponding increase in the MICU1 SA mutant‐transfected cells was decreased (Fig EV1E). The knockdown of Akt in HeLa cells (Fig EV1F) significantly attenuated the rapamycin‐induced [Ca2+]m elevation (Fig EV1G), further confirming that Akt is critical for this pathway. Moreover, rapamycin did not induce Ca2+ release from the stores (Fig EV1H) and consequent cytosolic Ca2+ elevation (Fig EV1I), suggesting that the rapamycin‐mediated [Ca2+]m rise is related to Akt mitochondrial activity.
Overall, these findings indicate that rapamycin promotes the translocation of activated Akt to mitochondria, thereby inducing Akt‐mediated N‐terminal MICU1 phosphorylation and a consequent increase in [Ca2+]m.
Mitochondrion‐targeted Akt increases the basal [Ca2+]m content in an MICU1‐dependent manner
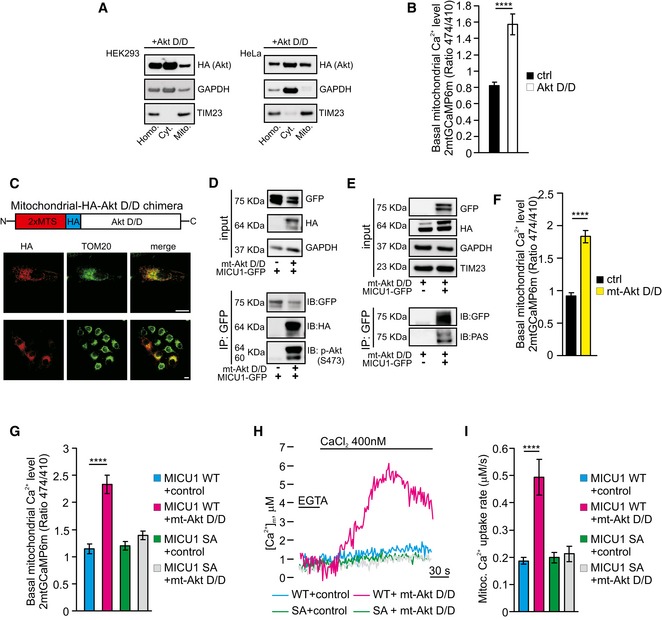
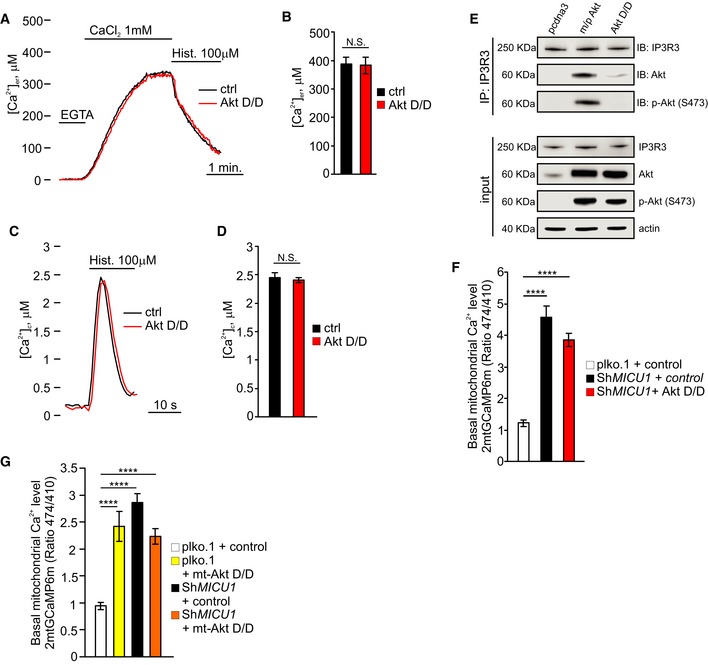
To further investigate the role of Akt in the regulation of mitochondrial Ca2+ dynamics, we used a constitutively active Akt form (Akt D/D), which has previously been reported to localize inside the mitochondrial compartment (Barksdale & Bijur, 2009). Transfection‐mediated Akt D/D overexpression caused a significant pool of the Akt construct to be located in mitochondria (Fig 3A), with a concomitant increase in [Ca2+]m under resting conditions (Fig 3B). To investigate whether the Akt D/D mutant might specifically regulate mitochondrial Ca2+ homeostasis, we measured the Ca2+ concentrations in different intracellular compartments, including the ER and cytosol, using the aequorin technique. Control cells and Akt D/D‐expressing cells revealed similar ER Ca2+ kinetics (Fig EV2A and B) and cytosolic Ca2+ levels (Fig EV2C and D), which is in line with the notion that the alteration in [Ca2+]m induced by Akt D/D expression is indeed due to local effects. Importantly, a membrane‐targeted active form of Akt kinase (m/p‐Akt), which has been described to inhibit ER Ca2+ release through IP3R binding and phosphorylation (Szado et al, 2008; Marchi et al, 2012), interacted with type 3 IP3R, whereas Akt D/D failed to do so (Fig EV2E). These data are consistent with prior reports indicating that the Akt D/D construct, which is active without membrane targeting (Dufner et al, 1999), does not act at the level of the ER and is unable to phosphorylate IP3R (Khan et al, 2006). Moreover, the expression of Akt D/D in the MICU1‐silenced cells did not further increase the basal [Ca2+]m levels (Fig EV2F), suggesting that its effects are largely dependent on MICU1.
Figure 3. Mitochondrial Akt augments the basal Ca2+ levels in a MICU1‐dependent manner.
-
AMitochondrial preparations of both HEK293T (left) and HeLa (right) cells transiently transfected with an HA‐tagged constitutively active Akt T308D/S473D (Akt D/D) construct were analyzed by Western blotting. GAPDH: cytosolic marker; TIM23: mitochondrial marker.
-
BResting mitochondrial calcium levels in the control (ctrl) and Akt D/D‐expressing HeLa cells evaluated by ratiometric imaging of the mitochondrial‐targeted GCaMP6m (n = 4 independent experiments; 44–49 cells).
-
CSchematic model of the HA‐tagged constitutively active Akt form targeted to the mitochondrial intermembrane space (mt‐Akt D/D). Both the expression and the mitochondrial localization of the chimera were assessed by immunofluorescence. TOM20 was used as a mitochondrial marker. Merged images are indicated (merge). Scale bar 10 μm.
-
DHEK293T cells were transfected with the indicated constructs. The GFP‐tagged MICU1 was immunoprecipitated from the whole‐cell lysate, and the precipitated proteins were immunoblotted with the HA and phosphorylated (S473) Akt (p‐Akt) antibodies.
-
EHEK293T cells were transfected with the indicated constructs. MICU1‐GFP immunocomplexes were precipitated with a GFP antibody and analyzed with PAS (phospho‐Akt substrate) antibody by Western blotting.
-
FResting mitochondrial calcium levels in control (ctrl) and mitochondrial‐targeted Akt D/D (mt‐Akt D/D)‐expressing HeLa cells, evaluated through ratiometric imaging of the mitochondrial‐targeted GCaMP6m (n = 4 independent experiments; 43–47 cells).
-
GResting mitochondrial calcium levels in ShRNA MICU1 HeLa stable cells transfected with the indicated constructs and evaluated through ratiometric imaging of the mitochondrial‐targeted GCaMP6m (n = 3 independent experiments; 38–50 cells).
-
H, IRepresentative kinetics (H) and analysis (I) of aequorin‐based [Ca2+]m measurements in permeabilized ShRNA MICU1 HeLa stable cells transfected with the indicated constructs and challenged with 400 nM buffered [Ca2+] (n = 3 independent experiments).
Figure EV2. The constitutively active Akt D/D construct does not alter ER calcium homeostasis.
-
A, BRepresentative kinetics (A) and analysis (B) of aequorin‐based endoplasmic reticulum ([Ca2+]ER) measurements in intact HeLa cells transfected with the indicated constructs (n = 3 independent experiments).
-
C, DRepresentative kinetics (C) and analysis (D) of aequorin‐based cytosolic ([Ca2+]c) measurements in intact HeLa cells transfected with the indicated constructs (n = 3 independent experiments).
-
EHEK293T cells were transfected with the indicated constructs. IP3 receptor type 3 (IP3R3) was immunoprecipitated from whole‐cell lysate and the precipitated proteins were immunoblotted with Akt and phosphorylated (S473) Akt (p‐Akt) antibodies.
-
FResting mitochondrial calcium levels, evaluated through ratiometric imaging of the mitochondrial‐targeted GCaMP6m, plko.1 vector and ShRNA MICU1 HeLa stable cells transfected or not a constitutively active form of Akt (Akt D/D) (n = 3 independent experiments).
-
GResting mitochondrial calcium levels, evaluated through ratiometric imaging of the mitochondrial‐targeted GCaMP6m, plko.1 vector, and ShRNA MICU1 HeLa stable cells transfected or not mitochondrial‐targeted Akt D/D (mt‐Akt D/D) (n = 4 independent experiments; 51–68 cells).
To further explore the Ca2+‐related activities of Akt inside mitochondria, we generated a HA‐tagged active Akt chimera targeted to the mitochondrial IMS (mt‐Akt D/D) (Fig 3C). The construct correctly localized to this mitochondrial compartment (Fig 3C) and promoted MICU1 phosphorylation (Fig 3D and E). Measurements of basal [Ca2+]m revealed that mt‐Akt D/D overexpression evoked higher Ca2+ levels in unstimulated cells (Fig 3F). Consistently, this increase in [Ca2+]m by mt‐Akt D/D did not occur in MICU1‐depleted cells (Fig EV2G). Therefore, mt‐Akt D/D is sufficient to elevate basal [Ca2+]m even in the absence of rapamycin through an effect that requires MICU1.
To confirm that serine 124 in the MICU1 sequence is the target residue of the mitochondrial Akt kinase activity, we analyzed the [Ca2+]m levels upon expression of the mt‐Akt D/D chimera together with different MICU1 mutants. mt‐Akt D/D was capable of elevating the resting [Ca2+]m levels in the presence of the WT MICU1 form but not with the nonphosphorylatable MICU1 SA mutant (Fig 3G). Moreover, in MICU1 SD‐expressing cells, mt‐Akt D/D had no additional effects on [Ca2+]m (Appendix Fig S3A). Second, in a permeabilized cell‐based experiment, the reconstitution of MICU1‐silenced cells with the WT MICU1, but not with the MICU1 SA mutant, yielded a higher Ca2+‐stimulated mitochondrial increase in [Ca2+]m in the presence of mt‐Akt D/D (Fig 3H and I). The role of mitochondrial Akt in the regulation of basal Ca2+ levels is strictly dependent on its activation, since a mitochondrial‐targeted Akt WT construct (mt‐Akt WT) (Appendix Fig S3B) induced a marginal increase in [Ca2+]m (Appendix Fig S3C).
Taken together, these data support a model in which active Akt that is located at the mitochondrial IMS promotes higher [Ca2+]m through the inhibitory phosphorylation of MICU1.
Phosphorylation affects MICU1 processing and destabilizes the MICU1‐MICU2 heterodimer
We focused on the molecular alterations induced by Akt‐mediated MICU1 phosphorylation. When we reconstituted the essential uniporter complex by co‐transfecting MCU and EMRE with different MICU1 forms, we observed a notable reduction in the mature MICU1 levels only when the MICU1 SD mutant was present, which appeared to accumulate in an unprocessed form (Fig 4A and B). It has been proposed that MICU1 undergoes multiple cleavage steps to become completely mature (Petrungaro et al, 2015). We wanted to investigate whether phosphorylation could affect the correct processing of MICU1. We hypothesized that MICU1 could fall into the first class of IMS proteins, which are first cleaved by a matrix protease, integrated in the IMM and then subjected to a second cleavage event that ensures the release of the mature protein into the IMS (Herrmann & Hell, 2005). Thus, defects in the second stage of processing result in an accumulation of the intermediate form, which is locked into the IMM. Mature WT MICU1 and the SA mutant were recovered in the supernatant fraction following alkaline extraction, whereas SD accumulated in the intermediate form and was more tightly associated with membranes (Fig 4C). Consistently, larger forms of MICU1 SD were mainly recovered in the pellet fractions and were detected only in the supernatant fraction at pH 12.5 (Fig 4D). To test the function of Akt in this process, we performed immunoprecipitation experiments on sodium carbonate extracts from MICU1 WT‐expressing cells, treated or not with rapamycin. We observed a strong PAS reactivity only in the rapamycin‐treated pellet fraction (Fig EV3A), indicating a key role of Akt in the regulation of MICU1 processing and membrane association. Notably, the expression of active mitochondrial Akt chimera inhibited MICU1 maturation, which was only slightly affected by the mt‐Akt WT form (Fig EV3B). Moreover, mt‐Akt D/D induces accumulation of larger forms of MICU1 in the pellet fractions upon alkaline extraction of mitochondrial membranes, whereas MICU1 WT alone is easily released by sodium carbonate (Fig EV3C).
Figure 4. Akt‐mediated phosphorylation of MICU1 affects protein maturation and stability.
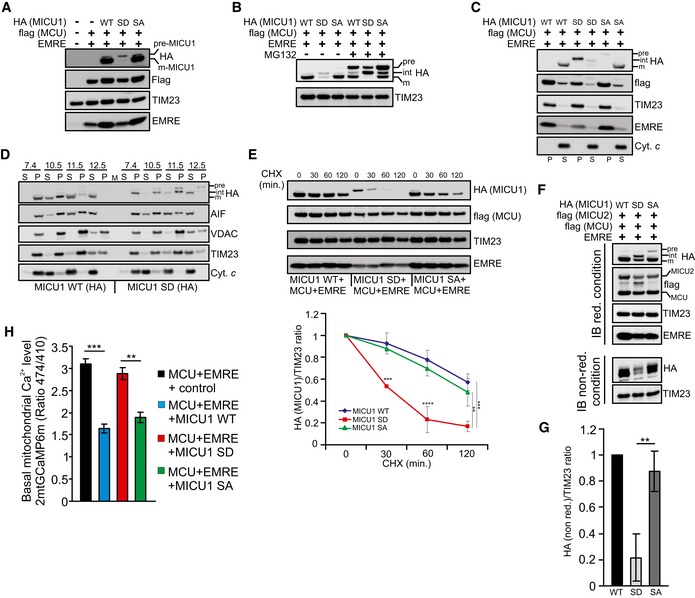
- Western blot analysis of ShRNA MICU1 HeLa stable cells transfected with the indicated constructs. pre‐MICU1: precursor form of MICU1; m‐MICU1: mature form of MICU1.
- Western blot analysis of ShRNA MICU1 HeLa stable cells transfected with the indicated constructs. Where indicated, the cells were treated with 5 μM MG132 for 4 h. pre: precursor; int: intermediate; m: mature form.
- Western blot analysis of the supernatant (S) and insoluble pellet (P) fractions of ShRNA MICU1 HeLa cell mitochondria transfected with the indicated constructs, following carbonate extraction at pH 10. The established integral membrane protein TIM23 and the soluble protein cytochrome c (Cyt. c) were analyzed as markers. The cells were treated with 5 μM MG132 for 4 h before fractionation.
- Western blot analysis of the supernatant (S) and insoluble pellet (P) fractions of ShRNA MICU1 HeLa cell mitochondria transfected with MICU1 WT‐HA or MICU1 SD‐HA, together with MCU‐flag and EMRE constructs, following carbonate extraction at pH 7.4, pH 10.5, pH 11.5, or pH 12.5. The cells were treated with 5 μM MG132 for 4 h before fractionation.
- ShRNA MICU1 HeLa stable cells were transfected with the indicated constructs. Cells were incubated with 50 μg/ml cycloheximide (CHX) for the indicated times, collected, and subjected to Western blotting analysis as indicated. In the graph, the amount of HA (MICU1), both WT and mutants, is represented relative to the amount at time 0 (n = 3 independent experiments).
- ShRNA MICU1 HeLa stable cells were transfected with the indicated constructs. Cells were harvested, and total protein was extracted and subjected to Western blotting analysis with the indicated antibodies. Immunoblotting was performed under reducing (with the presence of DTT) or non‐reducing (non‐red) conditions (without DTT). Cells were treated with 5 μM MG132 for 4 h before lysis.
- Quantification of HA levels in non‐reducing condition showed in (E) (n = 3 independent experiments).
- Resting mitochondrial calcium levels, evaluated through ratiometric imaging of the mitochondrial‐targeted GCaMP6m, in ShRNA MICU1 HeLa stable clone cells transfected with the indicated constructs (n = 3 independent experiments; 46–54 cells).
Figure EV3. MICU1 phosphorylation affects MICU2 levels.
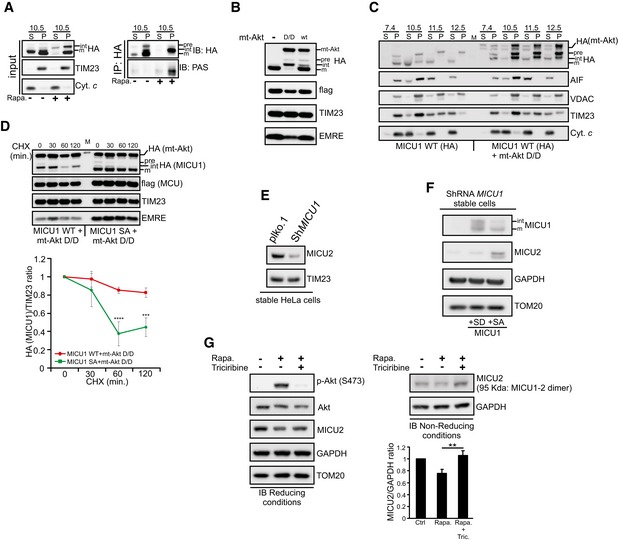
- HEK293T cells were transfected with the HA‐tagged wild‐type (WT) MICU1, treated with 1 μM rapamycin (Rapa.) for 4 h and then subjected to carbonate extraction at pH 10.5. MICU1‐HA immunocomplexes were precipitated in both supernatant (S) and insoluble pellet (P) fractions using a HA antibody and analyzed with PAS (phospho‐Akt substrate) antibody by Western blotting. pre: precursor; int: intermediate; m: mature form.
- Western blot analysis of ShRNA MICU1 HeLa stable cells transfected with flag‐tagged MCU, EMRE and HA‐tagged MICU1 WT, together with and mitochondrial‐targeted Akt D/D (mt‐Akt D/D) or WT (mt‐Akt WT). pre: precursor; int: intermediate; m: mature form.
- Western blot analysis of the supernatant (S) and insoluble pellet (P) fractions of ShRNA MICU1 HeLa cell mitochondria transfected with flag‐tagged MCU, EMRE and HA‐tagged MICU1 WT, in the presence or absence of and mitochondrial‐targeted Akt D/D (mt‐Akt D/D), following carbonate extraction at pH 7.4, pH 10.5, pH 11.5 or pH 12.5. The cells were treated with 5 μM MG132 for 4 h before fractionation. pre: precursor; int: intermediate; m: mature form.
- ShRNA MICU1 HeLa stable cells were transfected with flag‐tagged MCU, EMRE, and HA‐tagged MICU1 WT or SA mutant, together with and mitochondrial‐targeted Akt D/D (mt‐Akt D/D). Cells were incubated with cycloheximide 50 μg/ml (CHX) for the indicated times, collected, and analyzed by Western blotting as indicated. In the graph, the amount of HA (MICU1) is represented relative to the amount at time 0 (n = 3 independent experiments). pre: precursor; int: intermediate; m: mature form. M: marker.
- Western blot analysis of MICU2 levels in plko.1 vector and ShRNA MICU1 HeLa stable cells.
- Western blot analysis of MICU2 levels in ShRNA MICU1 HeLa stable cells where either MICU1 S124D (SD) or MICU1 S124A (SA) has been stably expressed.
- Analysis of MICU2 levels in HeLa cells treated with 1 μM rapamycin (Rapa.) alone for 4 h or in combination with the Akt inhibitor triciribine (10 μM). Cells were harvested and total protein was extracted and subjected to Western blotting analysis with the indicated antibodies. Immunoblot was performed in reducing conditions (with the presence of DTT) or in non‐reducing (non‐red) conditions (without DTT). Quantification of non‐red MICU2 has been reported (n = 3 independent experiments).
It has been proposed that partial proteolysis would be required to convert the unstable precursor intermediates into stable mature proteins (Vogtle et al, 2009, 2011). Therefore, we investigated whether the phosphorylation step in the MICU1 protein might impact MICU1 stability. When protein synthesis was inhibited with cycloheximide (CHX) to measure the half‐life of MICU1, the MICU1 SD mutant degraded more rapidly than the WT protein and the SA mutant (Fig 4E). Moreover, co‐expression of the MICU1 WT with the mitochondrial Akt construct reduced its half‐life (Appendix Fig S4A), whereas MICU1 SA mutant maintains higher stability also in the presence of mito‐Akt D/D (Fig EV3D).
It has previously been reported that MICU2 requires MICU1 for its stability but not vice versa (Plovanich et al, 2013). Therefore, we sought to determine whether the relative instability of phosphorylated MICU1 might entail a concomitant reduction in MICU2 stability. Consistent with the recent literature, we observed very low amounts of MICU2 in the MICU1‐depleted cells (Fig EV3E). Importantly, the stable expression of the MICU1 SA mutant, but not that of the MICU1 SD mutant, in ShMICU1 cells restored the MICU2 levels (Fig EV3F). These findings were not affected by alterations in MICU1‐2 binding because WT MICU1 and the two phosphomimetic and phosphorylation‐resistant mutants similarly co‐immunoprecipitated with flag‐tagged MICU2 (Appendix Fig S4B). Immunoblot analysis in non‐reducing conditions revealed that MICU1 SD expression strongly reduced the formation of the disulfide‐linked MICU1‐2 dimer (Fig 4F and G). These results were also observed in rapamycin‐treated cells, and this effect was abolished by concomitant addition of the Akt inhibitor triciribine (Fig EV3G).
Finally, we analyzed the Ca2+ levels upon reconstitution of MICU1 plus the minimal uniporter components. The expression of MCU and EMRE in a MICU1‐KD background increased the basal mitochondrial [Ca2+] content, which was significantly lowered by the MICU1 WT and SA mutant, but not by the SD construct (Fig 4H).
Altogether, we concluded that the Akt‐mediated phosphorylation of MICU1 affects its maturation process, resulting in instability of MICU1 and the MICU1‐2 dimer and defective mitochondrial Ca2+ regulation.
Inhibition of MICU1 maturation recapitulates all the effects observed upon MICU1 SD expression
To substantiate the crucial role of MICU1 processing in mitochondrial Ca2+ regulation, we planned to inhibit MICU1 maturation by preventing the second cleavage event, thereby inducing the accumulation of the intermediate form (Fig EV4A). We investigated the processing peptidases mitochondrial inner membrane protease subunit 1 (IMMP1L) and IMMP2L, which have been indicated to promote protein maturation and activation by removing hydrophobic sorting signals from proteins that are sorted to the IMS, usually after these proteins have been first cleaved by matrix peptidases (Quiros et al, 2015). IMMP1L‐ and, to a lesser extent, IMMP2L‐silencing (Appendix Fig S5A) inhibited MICU1 processing (Fig 5A and B), increasing the accumulation of the intermediate form and its association with membranes (Fig 5C and D). As previously observed for SD mutant expression, affecting MICU1 maturation promoted its degradation (Fig 5E and Appendix Fig S5B) and decreased the stability of the MICU1‐2 dimer (Appendix Fig S5C). Finally, we measured the basal mitochondrial [Ca2+] upon IMMP1L depletion. Both aequorin‐based (Fig 5F and G) and GCaMP‐based measurements (Fig 5H) showed higher Ca2+ levels in IMMP1L‐silenced cells.
Figure EV4. Glucose deprivation promotes MICU1 degradation.
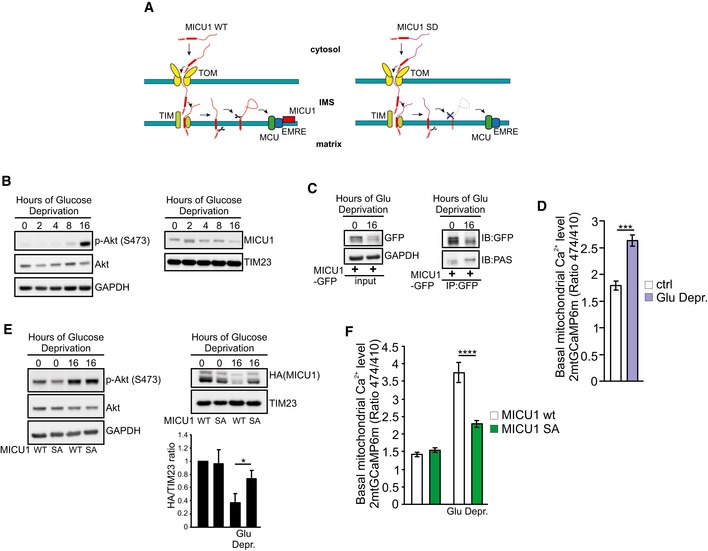
- Schematic model of MICU1 processing and maturation. Phosphorylated MICU1 (MICU1 SD mutant, in purple) cannot be subjected to the second cleavage event, which occurs in the IMS, leading to its rapid degradation. IMS: intermembrane space.
- Western blot analysis of phosphorylated (S473) Akt (p‐Akt) and MICU1 levels in HeLa cells glucose‐deprived for the indicated times.
- HeLa cells were transfected with the GFP‐tagged WT MICU1 and then glucose‐deprived for 16 h. MICU1‐GFP immunocomplexes were precipitated with a GFP antibody and analyzed with PAS (phospho‐Akt substrate) antibody by Western blotting.
- Resting mitochondrial calcium levels in control (ctrl) and glucose‐deprived HeLa cells, evaluated through ratiometric imaging of the mitochondrial‐targeted GCaMP6m (n = 3 independent experiments; 49–58 cells).
- Western blot analysis of HeLa cells transfected with the indicated MICU1 constructs, before and after 16 h of glucose deprivation. Quantification of HA (MICU1) has been reported (n = 3 independent experiments).
- Resting mitochondrial calcium levels in HeLa cells transfected with the indicated constructs, evaluated through ratiometric imaging of the mitochondrial‐targeted GCaMP6m before and after 16 h of glucose deprivation (n = 3 independent experiments; 45–72 cells).
Figure 5. IMMP1L‐silencing alters MICU1 processing and basal [Ca2+]m levels.
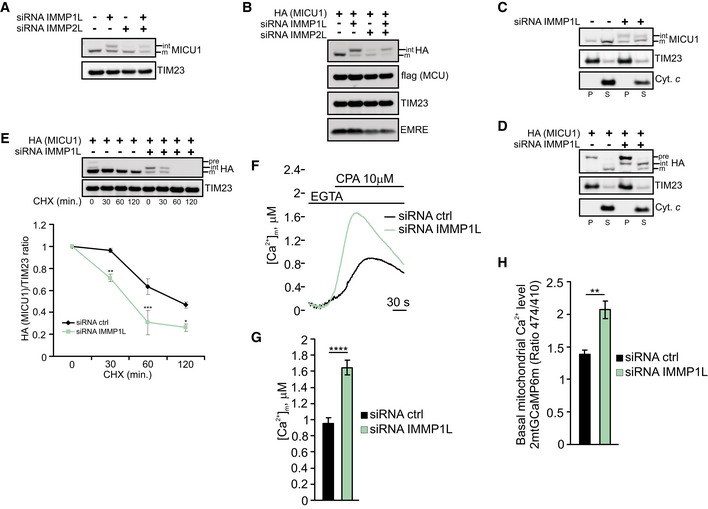
-
AWestern blot analysis of endogenous MICU1 levels in HeLa cells upon silencing of IMMP1L and/or IMMP2L. int: intermediate; m: mature form.
-
BWestern blot analysis of HeLa cells transfected with the indicated constructs and silenced with IMMP1L and/or IMMP2L. int: intermediate; m: mature form. Cells were treated with 5 μM MG132 for 4 h before lysis.
-
CWestern blot analysis of the supernatant (S) and insoluble pellet (P) fractions of control or IMMP1L‐silenced HeLa cell mitochondria following carbonate extraction at pH 10. The established integral membrane protein TIM23 and the soluble protein cytochrome c (Cyt. c) were analyzed as markers.
-
DWestern blot analysis of the supernatant (S) and insoluble pellet (P) fractions of control or IMMP1L‐silenced HeLa cell mitochondria transfected with HA‐tagged MICU1 WT following carbonate extraction at pH 10. The established integral membrane protein TIM23 and the soluble protein cytochrome c (Cyt. c) were analyzed as markers. pre: precursor; int: intermediate; m: mature form. Cells were treated with 5 μM MG132 for 4 h before lysis.
-
EControl or IMMP1L‐silenced HeLa cells were transfected with HA‐tagged MICU1 WT. Cells were incubated with 50 μg/ml cycloheximide (CHX) for the indicated times, collected and subjected to Western blotting analysis as indicated. In the graph, the amount of HA (MICU1) is represented relative to the amount at time 0 (n = 3 independent experiments).
-
F, GRepresentative kinetics (F) and analysis (G) of aequorin‐based [Ca2+]m measurements in control or IMMP1L‐silenced HeLa cells challenged with 10 μM cyclopiazonic acid (CPA) in the presence of 100 μM EGTA (n = 3 independent experiments).
-
HResting mitochondrial calcium levels, evaluated through ratiometric imaging of the mitochondrial‐targeted GCaMP6m, in control or IMMP1L‐silenced HeLa cells (n = 3 independent experiments; 37–55 cells).
Overall, these data reveal that the accumulation of unprocessed MICU1 by reducing IMMP1L levels mimics the effects obtained with MICU1 SD expression, thus suggesting that the impairment of MICU1 processing affects mitochondrial Ca2+ homeostasis.
Akt‐MICU1 phosphorylation axis in ROS production and tumor development
We aimed to identify putative patho‐physiological scenarios in which the MICU1 regulation by Akt could play an essential role. Glucose deprivation, a common form of metabolic stress, can induce Akt activation (Gao et al, 2014), and it has been recently showed that glucose withdrawal could regulate a series of molecular pathways by evoking Ca2+ elevations (Lee et al, 2018). Thus, we investigated whether the Akt‐MICU1 signaling is involved in the cellular response to glucose starvation. Prolonged glucose deprivation (16 h) induced strong Akt activation, which is associated with reduced MICU1 levels (Fig EV4B). Accordingly, MICU1 appeared highly phosphorylated by Akt upon glucose starvation (Fig EV4C) and the basal mitochondrial [Ca2+] significantly increased, compared to cells cultured in normal conditions (Fig EV4D). Finally, the MICU1 nonphosphorylatable SA mutant showed higher stability under glucose deprivation compared to the WT form (Fig EV4E), and MICU1 SA mutant expression in MICU1‐depleted cells restored the normal mitochondrial Ca2+ levels (Fig EV4F), sustaining the importance of Akt‐MICU1 interplay in the glucose deprivation‐mediated Ca2+ elevations.
Akt is overactivated in a wide range of human malignancies. Thus, we sought to determine whether mitochondrial Akt‐MICU1 signaling might contribute to cancer cell proliferation and tumor growth in vivo. We compared the phosphorylation of MICU1 in three paired sets of tumor cell lines with and without Akt activation (Fig 6A–F and Appendix Fig S6A). The A549 lung cancer cell line displayed higher phospho‐Akt levels than the mucoepidermoid carcinoma cell line H292, correlating with reduced expression of phosphatase and the tensin homolog (PTEN) (Fig 6A). An analysis of the MICU1 phosphorylation status using the PAS antibody showed more phosphorylated MICU1 in A549 cells (Fig 6B). Importantly, lower levels of both MICU1 and MICU2 proteins have been detected in Akt‐positive cells (Fig 6A and Appendix Fig S6A). The PTEN‐null melanoma cells (WM793b) (Paraiso et al, 2011) showed greater MICU1 phosphorylation levels (Fig 6C and D) and reduced amounts of MICU1 and 2 (Fig 6C and Appendix Fig S6A) than WT PTEN cells (451Lu). Similarly, the astrocytoma cell line U87‐MG, which displayed high Akt activity due to the inactivating mutations in PTEN, showed reduced expression of the MCU regulators compared with the T98G glioblastoma cells (Fig 6E and Appendix Fig S6A), which is linked to the high bulk of phospho‐MICU1 (Fig 6F). These findings correlate with higher basal mitochondrial [Ca2+], which was observed in both Akt‐positive lung (Appendix Fig S6B and C), melanoma (Appendix Fig S6D and E), and glioblastoma (Appendix Fig S6F and G) cancer cells. Importantly, reduction of Akt activity in Akt‐positive cells (U87MG), or increasing it in Akt‐negative (T98G cells), affected the levels of MICU1‐2 (Appendix Fig S6H), as well as cell proliferation and colony formation (Appendix Fig S6I and J).
Figure 6. The mitochondrial Akt‐MICU1 axis is critical for tumor growth.
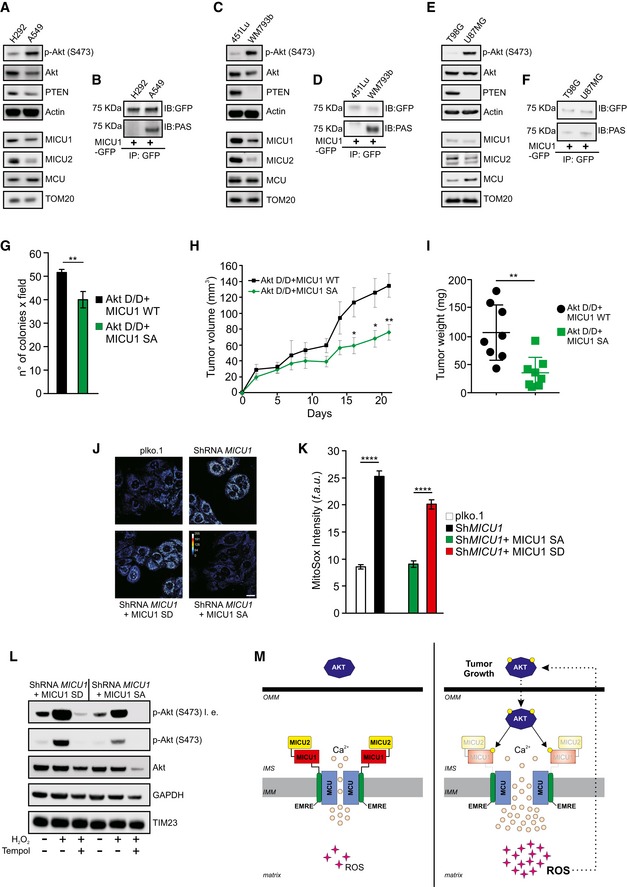
-
AWestern blot analysis of MICU1 and MICU2 levels in lung cancer cells with low or high Akt activity.
-
BLung cancer cells were transfected with the GFP‐tagged WT MICU1. MICU1‐GFP immunocomplexes were precipitated with a GFP antibody and analyzed with a PAS (phospho‐Akt substrate) antibody by Western blotting.
-
CWestern blot analysis of MICU1 and MICU2 levels in melanoma cells with low or high Akt activity.
-
DMelanoma cells were transfected with the GFP‐tagged WT MICU1. MICU1‐GFP immunocomplexes were precipitated with a GFP antibody and analyzed with PAS (phospho‐Akt substrate) antibody by Western blotting.
-
EWestern blot analysis of MICU1 and MICU2 levels in glioblastoma cells with low or high Akt activity.
-
FGlioblastoma cells were transfected with the GFP‐tagged WT MICU1. MICU1‐GFP immunocomplexes were precipitated with GFP antibody and analyzed with PAS (phospho‐Akt substrate) antibody by Western blotting.
-
G–INumber of soft agar colonies (n = 3 independent experiments) (G), xenograft tumor volumes (H), and tumor weights at day 21 (n = 8 mice for each group) (I) formed by Rat2 cells stably expressing the indicated constructs.
-
J, KRepresentative images (J) and analysis (K) of MitoSOX‐based ROS measurements in ShRNA MICU1 HeLa stable cells stably expressing either MICU1 S124D (SD) or MICU1 S124A (SA) (n = 3 independent experiments). Scale bar 10 μm. f.a.u.: fluorescence arbitrary unit.
-
LShRNA MICU1 HeLa stable cells stably expressing either MICU1 S124D (SD) or MICU1 S124A (SA) were treated with 500 μM H2O2 alone or in combination with the ROS scavenger Tempol and analyzed by Western blotting as indicated. l. e.: long exposure.
-
MSpeculative model of the mitochondrial Akt‐MICU1 axis in the regulation of tumor growth.
However, by comparing the MCF‐10CA1h breast carcinoma cell line with an activating H1047R mutation in PIK3CA and MDA‐MB231 cells lacking constitutive Akt activation (Fig EV5A) (Kadota et al, 2010), we detected no Akt‐mediated phosphorylation of MICU1 (Fig EV5B). We reasoned that the lack of an effect of Akt on MICU1 that was observed in the MCF‐10CA1h cells might be attributed to PTEN activity. A subcellular fractionation analysis revealed that MCF‐10CA1h contained a discrete pool of PTEN located in mitochondria and very low levels of mitochondrial activated Akt (Fig EV5C). To further explore this process, we used pten gene‐knockout (KO) MEFs (Fig EV5D). The PTEN KO cells expressed lower amounts of both the MICU1 and MICU2 proteins (Fig EV5E) and exhibited enhanced phosphorylation of MICU1 (Fig EV5F) compared with WT MEFs. The treatment of PTEN KO MEFs with the Akt inhibitor triciribine enhanced the stability of the MICU1‐2 heterodimers (Fig EV5G). Collectively, these data indicate that in different tumor types, the activation of Akt is associated with the phosphorylation of MICU1 and a reduced MICU1‐2 gatekeeping activity, supporting the notion that the N‐terminal phosphorylation of MICU1 might be a key feature in human tumors bearing activated Akt. This aspect is, at least partially, dependent on the PTEN status, which might differentially affect mitochondrial Akt‐MICU1 signaling based on its subcellular localization.
Figure EV5. Akt‐mediated MICU1 phosphorylation is regulated by PTEN.
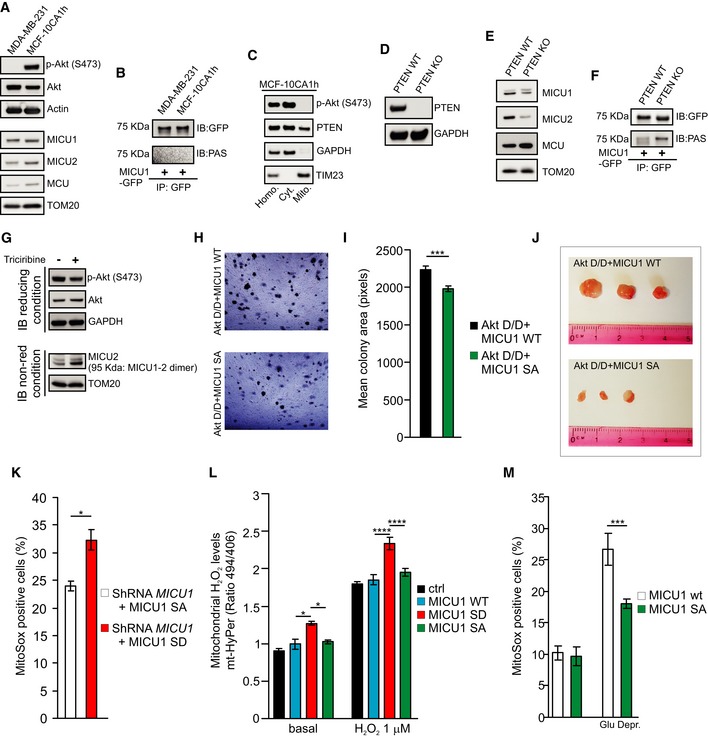
-
AWestern blot analysis of MICU1 and MICU2 levels in breast cancer cell lines with low or high activities of Akt.
-
BBreast cancer cells were transfected with the GFP‐tagged WT MICU1. MICU1‐GFP immunocomplexes were precipitated with a GFP antibody and analyzed with PAS (phospho‐Akt substrate) antibody by Western blotting.
-
CMCF‐10CA1h (Akt positive) breast cancer cells were fractionated into cytosol (Cyt.) or “crude” mitochondrial (Mito.) extracts and analyzed by Western blotting. GAPDH: cytosolic marker; TIM23: mitochondrial marker. Homo.: cell homogenate.
-
DWestern blot analysis of PTEN levels in WT and PTEN KO MEFs.
-
EWestern blot analysis of MICU1 and MICU2 levels in WT and PTEN KO MEFs.
-
FWT and PTEN KO MEFs were transfected with the GFP‐tagged WT MICU1. MICU1‐GFP immunocomplexes were precipitated with a GFP antibody and analyzed with PAS (phospho‐Akt substrate) antibody by Western blotting.
-
GWestern blot analysis of WT and PTEN KO MEFs treated with the Akt inhibitor Triciribine (10 μM). Cells were harvested, and total protein was extracted and subjected to Western blotting analysis with the indicated antibodies. Immunoblot was performed in reducing conditions (with the presence of DTT) or in non‐reducing (non‐red) conditions (without DTT).
-
H, IRepresentative images (H) and analysis (I) of the soft agar colonies size, expressed in pixels, formed by Rat2 cells stably transfected with the indicated constructs (n = 3 independent experiments).
-
JRepresentative images of tumors formed in NOD SCID® mice at 21 days from subcutaneous injection after necropsy and tumor dissection.
-
KAnalysis of MitoSox‐based ROS measurements in ShRNA MICU1 HeLa stable cells stably transfected with the indicated constructs, using the TALI®TM image‐based cytometer (n = 3 independent experiments).
-
MAnalysis of mitochondrial H2O2 levels in HeLa cells transfected with the indicated constructs using the genetically encoded ratiometric H2O2 sensor mt‐HyPer, before and after stimulation with low doses (1 μM) of H2O2 (n = 4 independent experiments; 72‐104 cells).
-
NAnalysis of MitoSox‐based ROS measurements in ShRNA MICU1 HeLa stable cells transfected with the indicated constructs before and after 16 h of glucose deprivation using the TALI®TM image‐based cytometer (n = 3 independent experiments).
To examine the role of Akt‐mediated MICU1 phosphorylation in Akt‐driven tumor growth, we generated stable cells that overexpressed the constitutively active Akt D/D form together with either WT MICU1 or the nonphosphorylatable MICU1 S124A mutant. In an anchorage‐independence growth assay, both the number and the size of the colonies formed by the Akt D/D‐expressing cells were significantly reduced by the co‐expression of MICU1 SA (Figs 6G and EV5H and I). Importantly, both Akt D/D‐expressing cells formed palpable tumors in immunodeficient mice, but the tumor growth rate and the tumor mass weight at necropsy were reduced for the clones expressing the MICU1 SA mutant (Figs 6H and I, and EV5J). Therefore, the expression of a mutant of MICU1 that cannot be phosphorylated by Akt lowers the tumorigenic potential of transformed cells and decreases the tumor growth rate and size.
Reportedly, Akt is able to elevate the reactive oxygen species (ROS) levels in multiple cellular contexts, and this Akt‐mediated ROS generation is essential for Akt‐driven tumor progression (Govindarajan et al, 2007; Nogueira et al, 2008; Okoh et al, 2013). Moreover, the basal mitochondrial Ca2+ overload observed upon MICU1 deficiency causes higher ROS production (Mallilankaraman et al, 2012). Thus, we investigated the possibility that MICU1 phosphorylation by Akt kinase would result in elevated mitochondrial ROS production. As expected, the ShMICU1 cells exhibited a higher basal level of mitochondrial ROS (Fig 6J and K). The stable introduction of the MICU1 SA mutant into MICU1‐silenced cells abrogated the ROS elevation, whereas the expression of the phosphomimic MICU1 SD form failed to restore the normal ROS levels (Figs 6J and K, and EV5K). Measurements of mitochondrial hydrogen peroxide production using the ratiometric genetically encoded mitochondria–targeted HyPer probe (mito−HyPer) revealed that the MICU1 SD mutant enhanced ROS generation both in the resting condition and after the stimulation of cells with a low dose (1 μM) of H2O2 (Fig EV5L). Finally, glucose deprivation stimulates ROS production (Marambio et al, 2010), which is limited by expression of MICU1 SA mutant (Fig EV5M).
Exogenous H2O2 stimulates Akt phosphorylation, suggesting that Akt activation is redox‐sensitive (Wang et al, 2000). Thus, we investigated the possibility that the increased production of ROS, which is mediated by phospho‐MICU1, might affect Akt activation. Indeed, MICU1 SD‐expressing cells exhibited higher levels of phosphorylated Akt than cells expressing the MICU1 SA mutant (Fig 6L). Consistently, oxidative stress strongly enhanced the Akt activation in the MICU1 SD cells, whereas application of the antioxidant compound Tempol abrogated Akt activation (Fig 6L). Altogether, we conclude that the mitochondrial Akt‐mediated phosphorylation of MICU1 promotes tumor progression through deregulation of the MCU complex, thereby increasing the basal mitochondrial Ca2+ levels and ROS production, which, in turn, generates a positive feedback loop that sustains Akt activation (Fig 6M).
Discussion
In this study, we showed that MICU1, which is one of the pivotal regulators of the MCU channel, could be subjected to an Akt‐mediated phosphorylation event that severely affects its regulatory functions on MCU. Its crucial role as a molecular gatekeeper of the uniporter complex identifies MICU1 as an attractive target modulator of mitochondrial Ca2+ homeostasis. Accordingly, the protein arginine methyl transferase 1 (PRMT1) has been described to mediate the methylation of MICU1, which, in turn, desensitizes this protein to Ca2+ and reduces the mitochondrial Ca2+ uptake (Madreiter‐Sokolowski et al, 2016). Moreover, the essential oxidoreductase Mia40/CHCHD4 can bind MICU1 and facilitate its dimerization with MICU2 (Petrungaro et al, 2015). Our present data underscore the importance of the N‐terminal phosphorylation of the MICU1 protein in the regulation of the basal mitochondrial Ca2+ content. The localization of this modification is quite distant from the two EF‐hand domains in the MICU1 primary sequence; thus, a mechanistic explanation of the altered functions of phospho‐MICU1 could not be found in the intrinsic modification of its Ca2+ affinity. Our findings reveal that Akt can affect MICU1 maturation through direct phosphorylation, leading to intrinsic instability of the uniporter‐associated dimer and altered mitochondrial Ca2+ accumulation. These data are supported by experiments in IMMP1L‐silenced cells, which displayed higher levels of unprocessed MICU1 and increased [Ca2+]m contents (Fig 5). Although IMMP1‐2L exists in a heterodimeric complex (Gakh et al, 2002), our results revealed that IMMP2L appears to play a marginal role in MICU1 processing (Fig 5A and B), suggesting that each subunit recognizes different substrates.
It is important to note that MICU1 phosphorylation appears to have an impact on abundance of both MCU and EMRE (Figs 4 and EV3), suggesting that the anomalous MICU1 processing and localization might affect the stability of other binding partners. In line with this evidence, loss of MICU1 correlates with low levels of MCU (Plovanich et al, 2013) and EMRE (Liu et al, 2016) in different cellular systems. Moreover, forced expression of MCU extended the half‐life of MICU1 (Petrungaro et al, 2015), and a MICU1 mutant that cannot interact with MICU2 (MICU1 C463A) showed a shorter half‐life, as well as a reduced expression of MCU, compared to MICU1 WT (Petrungaro et al, 2015). Thus, structural modifications in a single uniporter's component or the stoichiometry of MCU, EMRE, MICU1, and MICU2 influence the stability of the whole complex.
An aberrant Ca2+ regulation is currently thought to be a general hallmark of cancer cells, and multiple lines of evidence have noted the crucial role of Ca2+ homeostasis deregulation in tumor cell proliferation, apoptosis resistance, tumor development, and metastasis (Marchi & Pinton, 2016). Reduced mitochondrial Ca2+ uptake protects from apoptosis mediated by sustained ER Ca2+ release, an aspect could be especially relevant in conferring chemoresistance, or play an essential role in a specific cancer type or tumor stage. However, the correlation between high [Ca2+]m and ROS production is emerging as a prominent signaling route for the regulation of cancer progression (Tosatto et al, 2016; Ren et al, 2017).
MICU1 has been associated with different types of cancer, but its role remains controversial. A correlation between MICU1 overexpression and a better prognosis has been reported in the context of mammary carcinomas, suggesting that MICU1 might function as a tumor‐suppressor gene (Hall et al, 2014). Moreover, MICU1 appears to be a favorable prognostic marker in renal cancer (https://www.proteinatlas.org/ENSG00000107745-MICU1/pathology). In contrast, high MICU1 levels confer chemoresistance to ovarian cancer cells (Chakraborty et al, 2017), and MICU1 downregulation has been associated with a higher sensitivity to apoptotic stress and impaired endothelial cell migration (Mallilankaraman et al, 2012). However, an accelerated proliferation of MICU1‐knockdown hepatocytes has been recently observed following treatment with NIM811 (Antony et al, 2016), which is an inhibitor of the mitochondrial permeability transition pore (mPTP), the channel responsible for the initiation of mitochondrial cell death programs (Bonora et al, 2015). Through inhibition of its primary target, cyclophilin D (CypD), which is a component of the mPTP, NIM811 desensitizes the mPTP to Ca2+, thereby preventing cell killing (Waldmeier et al, 2002). Indeed, Akt activity has been widely associated with mPTP inhibition, and it was recently shown that mitochondrial Akt directly phosphorylates CypD, thereby limiting apoptosis (Ghosh et al, 2015). Thus, when the mPTP opening is inhibited by mitochondrial Akt or pharmacological approaches, higher mitochondrial Ca2+ levels might confer proliferative advantages to cancer cells without triggering apoptotic responses. Of note, our scenario agrees with recent data showing that cancer cells require basal mitochondrial Ca2+ uptake for survival (Cardenas et al, 2016).
The mitochondrial compartment has emerged as a novel critical subcellular district in which Akt exerts its kinase activity. Akt signaling regulates cellular energy metabolism and apoptosis via mechanisms that converge on mitochondria (Stiles, 2009), via its association with HK2 and VDAC (Gottlob et al, 2001), or via the phosphorylation of key proteins, such as BAD (Datta et al, 1997). In addition, Akt acts inside mitochondria, where it can phosphorylate compartment‐specific substrates, including the previously cited CypD (Ghosh et al, 2015) and PDK1 (Chae et al, 2016). The intra‐mitochondrial Akt activity contributes to apoptosis resistance and tumor growth and might provide a novel adaptive mechanism for tumor resistance to PI3K‐based therapy (Ghosh et al, 2015). Our findings provide additional clues for understanding the still elusive intra‐organellar activities of Akt, showing that phosphorylation of the MCU complex regulator MICU1 could sustain cancer progression by augmenting the basal mitochondrial Ca2+ content and ROS production (Fig 6M). Elevated levels of ROS have been detected in almost all cancers, including Akt‐positive tumors (Liou & Storz, 2010). We observed reduced tumor growth with the expression of the nonphosphorylatable mutant MICU1 SA (Fig 6H), correlating with reduced mitochondrial ROS generation (Fig 6J and K), which suggests that ROS might play a pivotal role in the regulation of cell malignancy by mitochondrial Ca2+ accumulation. However, Ca2+‐related ROS generation might offer a specific therapeutic opportunity. Therefore, our data could integrate previous observations showing that cancer cells with hyperactive Akt displayed high levels of ROS and that treatment with rapamycin, which further increases Akt activation, in combination with an oxidative stress agent could be a valuable strategy for selectively eradicating Akt‐positive tumor cells (Nogueira et al, 2008, 2018). Intriguingly, expression of a constitutively active Akt form in cells displaying very low Akt activity increased the susceptibility to glucose withdrawal‐induced death (Elstrom et al, 2004).
We found that the bulk of PTEN located at the mitochondria might, at least partially, counteract the action of mitochondrial Akt and hence prevent MICU1 phosphorylation. This finding is consistent with the ability of mito‐PTEN to inhibit Akt activation (Shen et al, 2015).
In conclusion, the results presented herein expand the functions of the mitochondrial pool of Akt, which are not limited to the regulation of apoptosis and mitochondrial bioenergetics but also involve the control of mitochondrial Ca2+ homeostasis. Moreover, this study provides the first demonstration of the crucial role played by post‐translational modifications of the MCU complex subunit MICU1 in the regulation of Akt kinase‐driven tumorigenesis. Our data note the strategic importance of the association between aberrant mitochondrial Ca2+ levels and tumor development and highlight as alteration in the organization and activity of the MCU complex could represent critical aspects that contribute to the aggressiveness of certain cancers.
Materials and Methods
Cell cultures and transfections
HeLa, HEK293T, T98G, U87‐MG (ATCC), and Rat2 (Sigma‐Aldrich; passage > 70) cells, as well as PTEN WT and KO MEFs, were maintained in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% fetal bovine serum (FBS; Thermo Fisher Scientific), 2 mM l‐glutamine, 100 U/ml penicillin, and 100 mg/ml streptomycin (all from Euroclone). MDA‐MB‐231 and H292 cells (ATCC) were maintained in RPMI supplemented with 10% FBS, l‐glutamine, and penicillin/streptomycin. MCF‐10CA1h cells were maintained in 1:1 DMEM:F12 supplemented with penicillin/streptomycin, 5% horse serum (Invitrogen), hEGF at 20 ng/ml (Lonza), hydrocortisone at 0.5 ng/ml (Sigma), Cholera toxin at 100 ng/ml (Sigma), insulin at 10 ng/ml (Gibco), and 10% FBS. A549 cells were maintained in F12 medium supplemented with 10% FBS, l‐glutamine, penicillin/streptomycin. WM793B and 451Lu cells (Coriell) were maintained in 4:1 keratinocyte‐SFM:Leibovitz L‐15 (Invitrogen) supplemented with penicillin/streptomycin, insulin at 5 ng/ml, and 5% FBS.
HeLa plko and ShMICU1 stable clones were maintained in DMEM supplemented with 10% FBS, l‐glutamine, penicillin/streptomycin, and 2 μg/ml of Puromycin.
For glucose starvation, cells were washed twice with PBS and then incubated in DMEM without glucose and pyruvate, supplemented with 10% dialyzed FBS (Thermo Fisher Scientific).
For HeLa and HEK293T cells, transient transfections were performed with a standard calcium‐phosphate procedure. For the other cell lines, transient transfections were performed using JetPEI (Polyplus transfection™) or Lipofectamine 2000 (Thermo Fisher Scientific) as transfecting reagents, according to the manufacturer's instructions.
For aequorin‐, GCaMP6m‐ and HyPer‐based experiments, the cells were transiently transfected with a 3:1 cDNA ratio, where 1 is the luminescent/fluorescent indicator. The ratio of 3:1 in favor of the protein of interest versus the indicator guarantees that all the cells expressing the specific probe (and thus the only recorded by the instruments) are also expressing the protein of interest.
For TMRM‐based experiments, the cells were also transfected with a 3:1 ratio, where 3 is the amount of the construct of interest and 1 is mitochondrial‐targeted GFP (mt‐GFP). Only cells expressing mt‐GFP have been analyzed for mitochondrial membrane potential.
pcDNA3.1 (referred as “control”) was used as a control for transfection unless otherwise indicated.
For silencing experiments, transfections were performed using Lipofectamine RNAiMax (Thermo Fisher Scientific) as transfecting reagents, according to the manufacturer's instructions. Akt and negative (scramble) siRNAs (final concentration 40 nM) were purchased from Cell Signaling Technology (CST). IMMP1L, IMMP2L, and negative (scramble) siRNAs (final concentration 40 nM) were purchased from Sigma‐Aldrich.
Lentiviral transduction and generation of stable cell lines
Lentiviruses were produced by transfecting shRNA‐targeting plasmids together with helper plasmids pCMV–VSVG and pCMV–dR8.74 into HEK293T cells. Cell supernatants were collected 48 h after transfection and were either used to infect cells or stored at −80°C. To obtain stable cell lines, cells were infected at low confluence (20%) for 24 h with lentiviral supernatants diluted 1:1 with normal culture media in the presence of 5 ng/ml of Polybrene (Sigma‐Aldrich). Cells were subjected to Puromycin selection 24 h after infection.
Lentiviral ShRNAs were obtained from Sigma (MISSION ShRNA). For MICU1 silencing, 5 shRNAs were identified and tested, TRCN0000053368 (Sh‐MICU1 #1), TRCN0000053369 (Sh‐MICU1 #2), TRCN0000053370 (Sh‐MICU1 #3), TRCN0000053371 (Sh‐MICU1 #4), and TRCN0000053372 (Sh‐MICU1 #5). Among them, Sh‐MICU1 #4 showed high knockdown efficiency in HeLa cells with the lowest homology with mouse sequence (NM_144822) and it was thus selected for the generation of HeLa ShMICU1 stable cell line.
HeLa cells stably overexpressing Sh‐MICU1 #4 were transfected with both MICU1 mutant versions and selected with 500 μg/ml G418 for at least 4 weeks before assaying. The resulting ShMICU1 HeLa cells stably overexpressing MICU1 S124D or MICU1 S124A were maintained in complete DMEM plus 2 μg/ml of Puromycin and 500 μg/ml G418.
Gene knockout by CRISPR/Cas9
Gene knockout by CRISPR/Cas9 was performed using the published protocol (Ran et al, 2013). To prevent off‐targets, the RNA guides (gRNA) were designed using Broad Institute's CRISPR Design tool (http://crispr.mit.edu/) uploading the sequence of the early 5′ exon of the gene of interest. Two different gRNAs targeting the 5′ of the gene were designed and used to rule out off‐target effects.
The 20‐nucleotide RNA guide sequence was transfected into HeLa cells with the S. pyogenes Cas9 (IDT) using Lipofectamine RNAiMAX Transfection Reagent (ThermoFisher). After two days of incubation, single cells were isolated by serial dilutions and expanded for 5 weeks. After one round of expansion, one clone with the lowest expression of protein was selected by Western blot and was transfected with a second round of gRNA and Cas9 protein to achieve the complete KO. Gene KO was assessed by Western blot.
Constructs and MICU1 mutants
Murine pcDNA3.1 MICU1‐HA, MICU1‐GFP, MICU2‐flag, MCU‐flag, and untagged EMRE plasmids were gently gift by Rosario Rizzuto Lab.
Creation of MICU1 mutants
The S124D and S124A mutations were introduced in the MICU1‐HA and MICU1‐GFP constructs by site‐directed mutagenesis using the QuickChange® II Site‐Directed Mutagenesis Kit (QuickChange® II XL Site‐Directed Mutagenesis Kit; Agilent Technologies, Santa Clara, CA, USA), essentially as previously described (Branchini et al, 2013). The following forward oligonucleotides were used: 5′‐GAATCCGAGCCTACGACACACCAGACAA‐3′ (S124D) and 5′‐GAATCCGAGCCTACGCGACACCAGACAA‐3′ (S124A); the modified nucleotide triplets are underlined. Reverse oligonucleotides were complementary to the forward ones. All plasmids have been validated by sequencing.
Creation of pCMV5‐2xMTS‐HA‐Akt D/D chimera
MTS (Mitochondrial Target Sequence from mouse MICU2) was amplified by PCR from MICU2 vector using the mMICU2mtsBglII‐F (5′‐AAAAGATCTATGGCGGCGGCTGCGGGAAG‐3′) and mMICU2mtsSalI‐R (5′‐AAAGTCGACGAGCCCGCGCCGCAGCCTTC‐3′) oligonucleotides under standard conditions (restriction sites underlined). MTS was further digested with BglII and SalI restriction enzymes and then cloned into pCMV5‐HA‐Akt D/D, previously cut with the same enzymes, to give the pCMV5‐MTS‐HA‐Akt D/D plasmid. In order to maintain the appropriate amino acid frame downstream of MTS in the resulting chimaeric protein, SalI site and the following triplet were removed from the pCMV5‐MTS‐HA‐Akt D/D construct by using site‐directed mutagenesis and the forward oligonucleotide 5′‐CTGCGGCGCGGGCTCATGGCTTACCCATAC‐3′. Reverse oligonucleotide was complementary to the forward one. MTS was then amplified again from MICU2 vector using oligonucleotides mMICU2mtsBglII‐F and mMICU2mtsBglII‐R (5′‐AAAAGATCTGAGCCCGCGCCGCAGCCTTC‐3′) under standard conditions. The resulting fragment was further digested with BglII restriction enzyme and cloned into pCMV5‐MTS‐HA‐Akt D/D, previously cut with the same enzyme, to give the final pCMV5‐2xMTS‐HA‐Akt D/D chimaeric construct. The same cloning procedures were applied to pCMV5‐HA‐Akt WT to obtain the pCMV5‐2xMTS‐HA‐Akt WT construct.
All expression vectors have been validated by sequencing.
Immunoblot
For immunoblotting, cells were washed with ice‐cold phosphate‐buffered saline (PBS), scraped, and lysed, for 30 min on ice, in a buffer containing 50 mM Tris–HCl pH 7.4, 150 mM NaCl, 1% Triton X‐100, 0.2% SDS, protease, and phosphates inhibitor cocktail. After centrifugation at 16,000 g for 10 min, proteins were quantified by the Lowry method and 10 μg of proteins, denatured for 5 min at 100°C in LDS sample buffer (Thermo Fisher Scientific) and Sample Reducing Agent (Thermo Fisher Scientific), was loaded on a Novex NuPage Bis‐Tris 4–12% precast gel (Thermo Fisher Scientific). For non‐denaturing conditions, the same amount of proteins were dissolved in LDS sample buffer (Thermo Fisher Scientific) and heated for 5 min at 70°C before loading. After electrophoretic separation, proteins were transferred onto PVDF membranes. To saturate unspecific binding sites, the membranes were incubated with TBS–Tween‐20 (0.05%) supplemented with 5% non‐fat powdered milk for 1 h at RT, and then incubated overnight with primary antibodies. The revelation was assessed by specific horseradish peroxidase‐labeled secondary antibodies (Thermo Fisher Scientific), followed by detection by chemiluminescence (Thermo Fisher Scientific), using ImageQuant LAS 4000 (GE Healthcare). Western blots shown in figures are representative of at least 3 different independent experiments.
Immunoprecipitation
For immunoprecipitation experiments, cells were seeded in 10‐cm Petri dishes, transfected as indicated and lysed in an appropriate volume of lysis buffer [20 mM Tris–HCl pH 8, 137 mM NaCl, 10% Glycerol, 1% NP‐40, 2 mM EDTA, protease inhibitor mixture, and Phos‐STOP Phosphatase Inhibitor Cocktail (Roche Applied Science)]. The same amount of proteins from the whole‐cell lysate for each condition was incubated overnight with the specific primary antibody. The immunocomplexes were captured with the appropriate protein A or protein G Sepharose (GE Healthcare). Beads were pelleted and washed three times. The bait was eluted in Laemmli sample buffer and denatured for 5 min at 100°C. The whole‐cell lysate (input) and the immunoprecipitated (IP) samples were separated by SDS–PAGE, transferred to PVDF membranes, incubated with the appropriate antibodies, and developed according to standard procedures (see above).
Antibodies and reagents
Anti‐HA (sc‐805; sc‐7392. WB 1:5,000; IF 1:200; IP 1:100), anti‐HSP60 (sc‐13115. WB 1:1,000; IF 1:200), anti‐TOM20 (sc‐17764. WB 1:5,000; IF 1:200), anti‐EMRE (sc‐86337. WB 1:500), were purchased from Santa Cruz Biotechnology; anti‐E‐cadherin (#14472. WB 1:1,000), anti‐p‐Akt (Ser473) (#9271. WB 1:1,000; IF 1:100), anti‐Akt (#9272. WB 1:5,000), anti‐p‐mTOR (Ser2448) (#5536. WB 1:1,000), anti‐PAS (Phospho‐Akt Substrates) (#9614. WB 1:1,000; IP 1:100), anti‐GAPDH (#2118. WB 1:5,000), anti‐PTEN (#9559. WB 1:1,000) from Cell Signaling; anti‐β‐tubulin (T5201. WB 1:5,000), anti‐β‐actin (A1978. WB 1:10,000), anti‐flag (F7425. WB 1:1,000), anti‐LC3B (L7543. WB 1:1,000), anti‐MICU1 (HPA037480. WB 1:1,000), anti‐MCU (HPA016480. WB 1:1,000), from Sigma‐Aldrich; anti‐Cytochrome c (556433. WB 1:1,000), anti‐TIM23 (611222. WB 1:1,000), anti‐p‐Ser (612546. WB 1:1,000), anti‐IP3R3 (610312. WB 1:1,000; IP 1:100) from BD Transduction Laboratories; anti‐MICU2 (ab1011465. WB 1:2,000), anti‐AIF (ab1998. WB 1:1,000), anti‐GFP (ab6556; WB 1:5,000; IP 1:100) and anti‐VDAC1 (ab15895. WB 1:1,000) from Abcam; anti‐GFP (No. 11814460001. WB 1:5,000; IP 1:100) from Roche. Secondary, HRP‐conjugated antibodies (#31430; #31460. WB, 1:10,000) were purchased from Thermo Fisher Scientific. Secondary, AlexaFluor‐conjugated antibodies (IF, 1:500) were purchased from Thermo Fisher Scientific.
2,5‐di‐tert‐butylhydroquinone (TBHQ), Triciribine hydrate, Histamine, Cycloheximide, Cyclopiazonic Acid (CPA), MG132, and MitoTempol were purchased from Sigma‐Aldrich. Rapamycin was obtained from Calbiochem and MitoSox from Thermo Fisher Scientific.
Mitochondrial membrane potential (∆Ψm) measurements
Mitochondrial ∆Ψ was measured by loading cells with 20 nM tetramethyl rhodamine methyl ester (TMRM, Invitrogen) for 30 min at 37°C. Successively, cells were imaged with Nikon Swept Field Confocal equipped with CFI Plan Apo VC60XH objective (n.a. 1.4) and an Andor DU885 EM‐CCD camera, controlled by the NIS‐Elements 3.2. Basal levels were normalized on fluorescence in the presence of FCCP (carbonyl cyanide p‐trifluoromethoxyphenylhydrazone, 10 μM), a strong uncoupler of oxidative phosphorylation.
Aequorin measurements
For the experiments with the chimeric aequorins targeted to the cytosol (cytAEQ) and mitochondria (mtAEQ WT and mtAEQmut), cells were seeded onto 13‐mm glass coverslips and transfected as indicated. Before the measurement, cells were incubated with 5 μM coelenterazine for 1.5 h in Krebs–Ringer modified buffer (KRB; 125 mM NaCl, 5 mM KCl, 1 mM Na3PO4, 1 mM MgSO4, 5.5 mM glucose, and 20 mM 4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid [HEPES], pH 7.4, at 37°C) supplemented with 1 mM CaCl2, and then transferred to the perfusion chamber. To reconstitute the chimeric aequorins targeted to the ER (erAEQmut) with high efficiency, the ER luminal [Ca2+] first had to be reduced, by incubation for 45 min at 4°C in KRB supplemented with 5 μM coelenterazine, 5 μM Ca2+ ionophore ionomycin (Sigma‐Aldrich), and 600 μM EGTA. Then, cells were extensively washed with KRB supplemented with 2% bovine serum albumin and 2 mM EGTA, and then transferred to the perfusion chamber. All aequorin measurements were carried out in KRB supplemented with either 1 mM CaCl2 (cytAEQ and mtAEQmut) or 100 μM EGTA (erAEQmut), and the agonist was added to the same medium as indicated in figure legends. The experiments were terminated by lysing cells with Triton X‐100 in a hypotonic Ca2+‐rich solution (10 mM CaCl2 in H2O), thus discharging the remaining aequorin pool. The light signal was collected and calibrated into [Ca2+] values by an algorithm based on the Ca2+ response curve of aequorin at physiological conditions of pH, [Mg2+], and ionic strength, as previously described (Bonora et al, 2013).
The experiments with permeabilized cells were performed with a buffer mimicking the cytosolic ionic composition (IB): 130 mM KCl, 10 mM NaCl, 2 mM K2HPO4, 5 mM succinic acid, 5 mM malic acid, 1 mM MgCl2, 20 mM HEPES, and 1 mM pyruvate (pH 7) at 37°C. IB was supplemented with either 100 μM EGTA (IB/EGTA) or a 2 mM EGTA‐buffered [Ca2+] of the indicated concentration (IB/Ca2+). Calculated [Ca2+]free was predicted with CHELATOR software (Schoenmakers et al, 1992) and confirmed fluorimetrically with the Fura2 free acid form. Cells were permeabilized by a 1‐min perfusion with 100 μM digitonin (added to IB/EGTA) during luminescence measurements. Mitochondrial Ca2+ uptake rate was calculated as the first derivative by using the OriginLab® software. The higher value reached during Ca2+ addition represents the maximal Ca2+ uptake rate.
Mitochondrial‐targeted GCaMP6m measurements
The measurements were performed as previously described (Patron et al, 2014). Briefly, cells were grown on 24‐mm coverslips and transfected with 2mtGCaMP6m‐encoding plasmids and the indicated constructs. Imaging was performed on Cell^R multiple wavelength high‐resolution fluorescence microscopy system. Cells were alternatively illuminated at 474 and 410 nm and fluorescence was collected through a 515/30‐nm band‐pass filter. Exposure time was set to 200 ms at 474 nm and to 400 ms at 410 nm, in order to account for the low quantum yield at the latter wavelength. At least ten fields were collected per coverslip, and each field was acquired for 30 s (1 frame/s). Analysis was performed with the Fiji open source software. Both images were background corrected frame by frame by subtracting mean pixel values of a cell‐free region of interest. Data are presented as the mean of the averaged ratio of all time points.
Fura‐2 AM measurements
Cytosolic free Ca2+ concentration ([Ca2+]c) was evaluated using fluorescent Ca2+ indicator Fura‐2 AM. Briefly, cells were incubated in medium supplemented with 2.5 μM Fura‐2 AM for 30 min, washed with KRB to remove extracellular probe, supplied with preheated KRB (supplemented with 1 mM CaCl2), and placed in a thermostated incubation chamber at 37°C on the stage of an inverted fluorescence microscope (Cell^R system). [Ca2+]c was calculated as previously described (Marchi et al, 2008).
Immunofluorescence
Cells, grown onto 13‐mm coverslips and transfected as described, were washed with phosphate‐buffered saline (PBS) and fixed in 4% formaldehyde for 10 min at room temperature. After washing three times with PBS, cells were permeabilized with 0.1% Triton X‐100 in PBS (PBS‐T) for 10 min at room temperature and then blocked with PBS‐T containing 5% BSA at room temperature for 1 h. Cells were then incubated with primary antibodies overnight at 4°C, washed 3 times with PBS‐T, and incubated with the appropriate isotype matched, AlexaFluor‐conjugated secondary antibodies (Thermo Fisher Scientific). Coverslips were mounted with ProLong Gold Antifade reagent (Thermo Fisher Scientific) at room temperature, and images were acquired with confocal microscope (Zeiss LSM510) using a 63 × 1.4 NA Plan‐Apochromat oil‐immersion objective. Acquired images were then analyzed by using open source software Fiji.
ROS measurements
Detection of mitochondrial superoxide was performed by using MitoSOX™ Red, according to manufacturer's instructions (Molecular ProbesTM, Invitrogen). Briefly, cells grown to 50–80% confluence on glass coverslips were incubated with 5 μM MitoSOX in Hank's Buffered Salt Solution containing calcium and magnesium (HBSS), for 15 min at 37°C in a CO2 incubator and then washed three times with HBSS. Microscopy imaging was performed on the confocal microscope Zeiss LSM510, using a 63 × 1.4 NA Plan‐Apochromat oil‐immersion objective, 514 nm excitation laser, and a 580 ± 30 nm fluorescence detection range. Alternatively, MitoSOX™ Red fluorescence intensity were also measured using Tali® Image‐based Cytometer (Molecular ProbesTM, Invitrogen).
For mitochondrial hydrogen peroxide level measurements, cells were cultured on 24‐mm glass coverslips and transfected with the ratiometric fluorescent probe with mitochondrial localization pHyPer‐dMito (mt‐HyPer) (Marchi et al, 2015). After 24 h of expression, cells were maintained in Krebs–Ringer buffer (KRB) supplemented with 1 mM CaCl2, and placed in an open Leyden chamber on a 37°C thermostated stage. The experiments were performed on Cell^R Olympus multiple wavelength high‐resolution epi‐fluorescence microscope. 494/406‐nm excitation filters and a 500‐nm long‐pass beam splitter were used, and an image pair was obtained in every 200 ms with a 40× objective. The fluorescence data collected were expressed as emission ratios.
Real‐time PCR
Total RNA was extracted with TRIzol® Reagent (Invitrogen), RNAs were purified by using RNeasy Mini Kit (Qiagen), and DNase digestion was performed with RNase‐Free DNase Set (Qiagen). The RNA quality and concentration were measured using the NanoDropTM ND‐1000 (Thermo Fisher Scientific). For the first‐strand cDNA synthesis, 1,000 ng of total RNA of each sample was reverse‐transcribed with M‐MLV Reverse Transcriptase (Invitrogen), following the manufacturer's protocol. Human primers were selected for each target gene with Primer 3 software. Real‐time PCRs were carried out using the designed primers at a concentration of 300 nM and FastStart SYBR Green Master (Roche Diagnostics) on a Rotor‐Gene 3000 (Corbett Research). Thermal cycling conditions were as follows: 10‐min denaturation at 95°C, followed by 40 cycles of denaturation for 10 s at 95°C; annealing for 20 s at 60°C; and elongation for 30 s at 72°C. Values were normalized to the expression of the glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH), whose abundance did not change under our experimental conditions.
Mitochondria isolation and topology analysis
Both crude and pure mitochondria fractions from cells and mouse livers were prepared as previously described (Wieckowski et al, 2009).
For the localization study, 100 μg of crude mitochondria isolated from Hek293T was resuspended in a mitochondrial buffer containing 0.5 mM EGTA‐Tris and 10 mM Tris‐MOPS (pH 7.4), with (intact mitochondria) or without (to induce OMM rupture) 250 mM sucrose, or with 1% Triton X‐100 for inducing IMM rupture. All conditions were treated with 100 μg/ml Proteinase K (PK) for 30′ at 4°C, and then, 5 mM PMSF was added to stop the digestion. Mitochondrial proteins were precipitated following the TCA/Acetone method, and 5 μg of each fractions was used for Western blot analysis.
For the alkaline carbonate extraction study, 200 μg of crude mitochondria was pelleted and resuspended in 400 μl of 100 mM Na2CO3 buffer adjusted to different pHs (ranging from 7.4 to 12.5). After incubation on ice for 30 min, samples were centrifuged at 100,000 g for 30 min at 4°C. Mitochondrial proteins were precipitated following the TCA/Acetone method.
Soft agar colony formation assay
Assays were performed in triplicate in 6‐well plates. Base agar consisted of 0.5% agar (Sigma‐Aldrich), 1× DMEM, 500 μg/ml G418, and 10% FBS. Top agar consisted of 0.3% agar, 1× DMEM, 500 μg/ml G418, and 10% FBS with 2.5 × 104 Rat2 cells per well. Cells were incubated in 5% CO2 at 37°C for 4 weeks and then stained with 0.005% crystal violet. To quantify the colonies, five randomly selected images of each replicate were captured using a Leica DM IL LED microscope with a 4× magnification. Colonies were counted automatically using a custom made macro in the Fiji software. Briefly, dark objects were thresholded using the Yen algorithm and counted through the analyze particles tool.
Tumor Xenograft studies
To measure tumor formation by Rat2 fibroblasts stably expressing a constitutively active Akt form together with MICU1 WT or MICU1 S124A mutant, 6‐week‐old NOD SCID® mice (Charles River) were shaved on the right flank one day prior to injection and injected subcutaneously with 1 × 107 tumor cells in 200 μl of PBS. 8–9 mice per cell line were injected. Tumor growth was monitored by daily measurements of tumor length (L) and width (W), and tumor volume was estimated using the formula: tumor volume = ½(L × W2). After 21 days, mice were sacrificed and the tumors were dissected and weighed.
Statistical analysis
Quantitative data are presented as means ± SEM if not indicated otherwise. Statistical analyses were performed using an unpaired two‐tailed t‐test (two groups) or one‐way ANOVA with Tukey's correction (for groups of three or more). For grouped analyses, two‐way ANOVA with correction for multiple comparisons using the Holm–Sidak method was performed. Normal distribution of data was assessed by applying a D'Agostino & Pearson omnibus normality test. F‐test was used to compare variances between groups. A P‐value < 0.05 was considered significant.
Author contributions
SMa and PP designed the research. AB and MF generated Akt chimeras and MICU1 mutants, with the guidance of MPi. SMi and MPe performed in vivo experiments and clonogenic assays. SMa, MC, VAMV, and GM performed all other experiments. SMa, CG, MPi, LG, GK, and PP interpreted the data. SMa, GK, and PP wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
The authors thank Dr. Silvia Pignani for assistance in CRISPR/Cas9 clones generation. We are grateful for Dr. Ilio Vitale for critically reviewing the manuscript. PP is grateful to Camilla degli Scrovegni for continuous support. PP is supported by Telethon (GGP15219/B); the Italian Association for Cancer Research (AIRC: IG‐18624); and by local funds from the University of Ferrara. SMa is supported by “Fondazione Umberto Veronesi” and the Italian Ministry of Health (GR‐2016‐02364602). CG is supported by local funds from the University of Ferrara, the Italian Association for Cancer Research (AIRC: IG‐19803), the Italian Ministry of Health (GR‐2013‐02356747), and by a Fondazione Cariplo grant. GK is supported by the Ligue Contre le Cancer Comité de Charente‐Maritime (Équipe Labelisée); Agence National de la Recherche (ANR)—Projets Blancs; ANR Under the Frame of E‐Rare‐2, the ERA‐Net for Research on Rare Diseases; Association pour la Recherche sur le Cancer (ARC); Cancéropôle Ile‐de‐France; Institut National du Cancer (INCa); Institut Universitaire de France; Fondation pour la Recherche Médicale (FRM); the European Commission (ArtForce); the European Research Council (ERC); the LeDucq Foundation; the LabEx Immuno‐Oncology; the RHU Torino Lumière, the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); the SIRIC Cancer Research and Personalized Medicine (CARPEM); and the Paris Alliance of Cancer Research Institutes (PACRI).
The EMBO Journal (2019) 38: e99435
Contributor Information
Saverio Marchi, Email: saverio.marchi@unife.it.
Paolo Pinton, Email: paolo.pinton@unife.it.
References
- Antony AN, Paillard M, Moffat C, Juskeviciute E, Correnti J, Bolon B, Rubin E, Csordas G, Seifert EL, Hoek JB, Hajnoczky G (2016) MICU1 regulation of mitochondrial Ca(2+) uptake dictates survival and tissue regeneration. Nat Commun 7: 10955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barksdale KA, Bijur GN (2009) The basal flux of Akt in the mitochondria is mediated by heat shock protein 90. J Neurochem 108: 1289–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher‐Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK (2011) Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476: 341–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonora M, Giorgi C, Bononi A, Marchi S, Patergnani S, Rimessi A, Rizzuto R, Pinton P (2013) Subcellular calcium measurements in mammalian cells using jellyfish photoprotein aequorin‐based probes. Nat Protoc 8: 2105–2118 [DOI] [PubMed] [Google Scholar]
- Bonora M, Wieckowsk MR, Chinopoulos C, Kepp O, Kroemer G, Galluzzi L, Pinton P (2015) Molecular mechanisms of cell death: central implication of ATP synthase in mitochondrial permeability transition. Oncogene 34: 1608 [DOI] [PubMed] [Google Scholar]
- Boroughs LK, DeBerardinis RJ (2015) Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol 17: 351–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branchini A, Campioni M, Mazzucconi MG, Biondo F, Mari R, Bicocchi MP, Bernardi F, Pinotti M (2013) Replacement of the Y450 (c234) phenyl ring in the carboxyl‐terminal region of coagulation factor IX causes pleiotropic effects on secretion and enzyme activity. FEBS Lett 587: 3249–3253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardenas C, Muller M, McNeal A, Lovy A, Jana F, Bustos G, Urra F, Smith N, Molgo J, Diehl JA, Ridky TW, Foskett JK (2016) Selective vulnerability of cancer cells by inhibition of Ca(2+) transfer from endoplasmic reticulum to mitochondria. Cell Rep 14: 2313–2324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae YC, Vaira V, Caino MC, Tang HY, Seo JH, Kossenkov AV, Ottobrini L, Martelli C, Lucignani G, Bertolini I, Locatelli M, Bryant KG, Ghosh JC, Lisanti S, Ku B, Bosari S, Languino LR, Speicher DW, Altieri DC (2016) Mitochondrial Akt regulation of hypoxic tumor reprogramming. Cancer Cell 30: 257–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty PK, Mustafi SB, Xiong X, Dwivedi SKD, Nesin V, Saha S, Zhang M, Dhanasekaran D, Jayaraman M, Mannel R, Moore K, McMeekin S, Yang D, Zuna R, Ding K, Tsiokas L, Bhattacharya R, Mukherjee P (2017) MICU1 drives glycolysis and chemoresistance in ovarian cancer. Nat Commun 8: 14634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordas G, Golenar T, Seifert EL, Kamer KJ, Sancak Y, Perocchi F, Moffat C, Weaver D, de la Fuente Perez S, Bogorad R, Koteliansky V, Adijanto J, Mootha VK, Hajnoczky G (2013) MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca(2)(+) uniporter. Cell Metab 17: 976–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danese A, Patergnani S, Bonora M, Wieckowski MR, Previati M, Giorgi C, Pinton P (2017) Calcium regulates cell death in cancer: roles of the mitochondria and mitochondria‐associated membranes (MAMs). Biochim Biophys Acta 1858: 615–627 [DOI] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME (1997) Akt phosphorylation of BAD couples survival signals to the cell‐intrinsic death machinery. Cell 91: 231–241 [DOI] [PubMed] [Google Scholar]
- De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R (2011) A forty‐kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476: 336–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufner A, Andjelkovic M, Burgering BM, Hemmings BA, Thomas G (1999) Protein kinase B localization and activation differentially affect S6 kinase 1 activity and eukaryotic translation initiation factor 4E‐binding protein 1 phosphorylation. Mol Cell Biol 19: 4525–4534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM, Thompson CB (2004) Akt stimulates aerobic glycolysis in cancer cells. Cancer Res 64: 3892–3899 [DOI] [PubMed] [Google Scholar]
- Frezza C (2014) The role of mitochondria in the oncogenic signal transduction. Int J Biochem Cell Biol 48: 11–17 [DOI] [PubMed] [Google Scholar]
- Gakh O, Cavadini P, Isaya G (2002) Mitochondrial processing peptidases. Biochim Biophys Acta 1592: 63–77 [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Kepp O, Kroemer G (2012) Mitochondria: master regulators of danger signalling. Nat Rev Mol Cell Biol 13: 780–788 [DOI] [PubMed] [Google Scholar]
- Gao M, Liang J, Lu Y, Guo H, German P, Bai S, Jonasch E, Yang X, Mills GB, Ding Z (2014) Site‐specific activation of AKT protects cells from death induced by glucose deprivation. Oncogene 33: 745–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaude E, Frezza C (2014) Defects in mitochondrial metabolism and cancer. Cancer Metab 2: 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh JC, Siegelin MD, Vaira V, Faversani A, Tavecchio M, Chae YC, Lisanti S, Rampini P, Giroda M, Caino MC, Seo JH, Kossenkov AV, Michalek RD, Schultz DC, Bosari S, Languino LR, Altieri DC (2015) Adaptive mitochondrial reprogramming and resistance to PI3K therapy. J Natl Cancer Inst 107: dju502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgi C, Marchi S, Pinton P (2018) The machineries, regulation and cellular functions of mitochondrial calcium. Nat Rev Mol Cell Biol 19: 713–730 [DOI] [PubMed] [Google Scholar]
- Gottlieb E, Tomlinson IP (2005) Mitochondrial tumour suppressors: a genetic and biochemical update. Nat Rev Cancer 5: 857–866 [DOI] [PubMed] [Google Scholar]
- Gottlob K, Majewski N, Kennedy S, Kandel E, Robey RB, Hay N (2001) Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev 15: 1406–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindarajan B, Sligh JE, Vincent BJ, Li M, Canter JA, Nickoloff BJ, Rodenburg RJ, Smeitink JA, Oberley L, Zhang Y, Slingerland J, Arnold RS, Lambeth JD, Cohen C, Hilenski L, Griendling K, Martinez‐Diez M, Cuezva JM, Arbiser JL (2007) Overexpression of Akt converts radial growth melanoma to vertical growth melanoma. J Clin Invest 117: 719–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall DD, Wu Y, Domann FE, Spitz DR, Anderson ME (2014) Mitochondrial calcium uniporter activity is dispensable for MDA‐MB‐231 breast carcinoma cell survival. PLoS ONE 9: e96866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144: 646–674 [DOI] [PubMed] [Google Scholar]
- Herrmann JM, Hell K (2005) Chopped, trapped or tacked–protein translocation into the IMS of mitochondria. Trends Biochem Sci 30: 205–211 [DOI] [PubMed] [Google Scholar]
- Hill JM, De Stefani D, Jones AW, Ruiz A, Rizzuto R, Szabadkai G (2014) Measuring baseline Ca(2+) levels in subcellular compartments using genetically engineered fluorescent indicators. Methods Enzymol 543: 47–72 [DOI] [PubMed] [Google Scholar]
- Kadota M, Yang HH, Gomez B, Sato M, Clifford RJ, Meerzaman D, Dunn BK, Wakefield LM, Lee MP (2010) Delineating genetic alterations for tumor progression in the MCF10A series of breast cancer cell lines. PLoS ONE 5: e9201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MT, Wagner L II, Yule DI, Bhanumathy C, Joseph SK (2006) Akt kinase phosphorylation of inositol 1,4,5‐trisphosphate receptors. J Biol Chem 281: 3731–3737 [DOI] [PubMed] [Google Scholar]
- Lee HY, Itahana Y, Schuechner S, Fukuda M, Je HS, Ogris E, Virshup DM, Itahana K (2018) Ca(2+)‐dependent demethylation of phosphatase PP2Ac promotes glucose deprivation‐induced cell death independently of inhibiting glycolysis. Sci Signal 11: eaam7893 [DOI] [PubMed] [Google Scholar]
- Liou GY, Storz P (2010) Reactive oxygen species in cancer. Free Radic Res 44: 479–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littlepage LE, Adler AS, Kouros‐Mehr H, Huang G, Chou J, Krig SR, Griffith OL, Korkola JE, Qu K, Lawson DA, Xue Q, Sternlicht MD, Dijkgraaf GJ, Yaswen P, Rugo HS, Sweeney CA, Collins CC, Gray JW, Chang HY, Werb Z (2012) The transcription factor ZNF217 is a prognostic biomarker and therapeutic target during breast cancer progression. Cancer Discov 2: 638–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JC, Liu J, Holmstrom KM, Menazza S, Parks RJ, Fergusson MM, Yu ZX, Springer DA, Halsey C, Liu C, Murphy E, Finkel T (2016) MICU1 serves as a molecular gatekeeper to prevent in vivo mitochondrial calcium overload. Cell Rep 16: 1561–1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan CV, Szabadkai G, Sharpe JA, Parry DA, Torelli S, Childs AM, Kriek M, Phadke R, Johnson CA, Roberts NY, Bonthron DT, Pysden KA, Whyte T, Munteanu I, Foley AR, Wheway G, Szymanska K, Natarajan S, Abdelhamed ZA, Morgan JE et al (2014) Loss‐of‐function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat Genet 46: 188–193 [DOI] [PubMed] [Google Scholar]
- Madreiter‐Sokolowski CT, Klec C, Parichatikanond W, Stryeck S, Gottschalk B, Pulido S, Rost R, Eroglu E, Hofmann NA, Bondarenko AI, Madl T, Waldeck‐Weiermair M, Malli R, Graier WF (2016) PRMT1‐mediated methylation of MICU1 determines the UCP2/3 dependency of mitochondrial Ca(2+) uptake in immortalized cells. Nat Commun 7: 12897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallilankaraman K, Doonan P, Cardenas C, Chandramoorthy HC, Muller M, Miller R, Hoffman NE, Gandhirajan RK, Molgo J, Birnbaum MJ, Rothberg BS, Mak DO, Foskett JK, Madesh M (2012) MICU1 is an essential gatekeeper for MCU‐mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell 151: 630–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD, Cantley LC (2007) AKT/PKB signaling: navigating downstream. Cell 129: 1261–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marambio P, Toro B, Sanhueza C, Troncoso R, Parra V, Verdejo H, Garcia L, Quiroga C, Munafo D, Diaz‐Elizondo J, Bravo R, Gonzalez MJ, Diaz‐Araya G, Pedrozo Z, Chiong M, Colombo MI, Lavandero S (2010) Glucose deprivation causes oxidative stress and stimulates aggresome formation and autophagy in cultured cardiac myocytes. Biochim Biophys Acta 1802: 509–518 [DOI] [PubMed] [Google Scholar]
- Marchi S, Rimessi A, Giorgi C, Baldini C, Ferroni L, Rizzuto R, Pinton P (2008) Akt kinase reducing endoplasmic reticulum Ca2+ release protects cells from Ca2+ ‐dependent apoptotic stimuli. Biochem Biophys Res Commun 375: 501–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi S, Marinello M, Bononi A, Bonora M, Giorgi C, Rimessi A, Pinton P (2012) Selective modulation of subtype III IP(3)R by Akt regulates ER Ca(2)(+) release and apoptosis. Cell Death Dis 3: e304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi S, Lupini L, Patergnani S, Rimessi A, Missiroli S, Bonora M, Bononi A, Corra F, Giorgi C, De Marchi E, Poletti F, Gafa R, Lanza G, Negrini M, Rizzuto R, Pinton P (2013) Downregulation of the mitochondrial calcium uniporter by cancer‐related miR‐25. Curr Biol 23: 58–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi S, Corricelli M, Trapani E, Bravi L, Pittaro A, Delle Monache S, Ferroni L, Patergnani S, Missiroli S, Goitre L, Trabalzini L, Rimessi A, Giorgi C, Zavan B, Cassoni P, Dejana E, Retta SF, Pinton P (2015) Defective autophagy is a key feature of cerebral cavernous malformations. EMBO Mol Med 7: 1403–1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi S, Pinton P (2016) Alterations of calcium homeostasis in cancer cells. Curr Opin Pharmacol 29: 1–6 [DOI] [PubMed] [Google Scholar]
- Nogueira V, Park Y, Chen CC, Xu PZ, Chen ML, Tonic I, Unterman T, Hay N (2008) Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell 14: 458–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogueira V, Patra KC, Hay N (2018) Selective eradication of cancer displaying hyperactive Akt by exploiting the metabolic consequences of Akt activation. Elife 7: e32213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okoh VO, Felty Q, Parkash J, Poppiti R, Roy D (2013) Reactive oxygen species via redox signaling to PI3K/AKT pathway contribute to the malignant growth of 4‐hydroxy estradiol‐transformed mammary epithelial cells. PLoS ONE 8: e54206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paillard M, Csordas G, Szanda G, Golenar T, Debattisti V, Bartok A, Wang N, Moffat C, Seifert EL, Spat A, Hajnoczky G (2017) Tissue‐specific mitochondrial decoding of cytoplasmic Ca(2+) signals is controlled by the stoichiometry of MICU1/2 and MCU. Cell Rep 18: 2291–2300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paraiso KH, Xiang Y, Rebecca VW, Abel EV, Chen YA, Munko AC, Wood E, Fedorenko IV, Sondak VK, Anderson AR, Ribas A, Palma MD, Nathanson KL, Koomen JM, Messina JL, Smalley KS (2011) PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res 71: 2750–2760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patron M, Checchetto V, Raffaello A, Teardo E, Vecellio Reane D, Mantoan M, Granatiero V, Szabo I, De Stefani D, Rizzuto R (2014) MICU1 and MICU2 finely tune the mitochondrial Ca2 + uniporter by exerting opposite effects on MCU activity. Mol Cell 53: 726–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, Mootha VK (2010) MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature 467: 291–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrungaro C, Zimmermann KM, Kuttner V, Fischer M, Dengjel J, Bogeski I, Riemer J (2015) The Ca(2+)‐dependent release of the Mia40‐induced MICU1‐MICU2 Dimer from MCU regulates mitochondrial Ca(2+) uptake. Cell Metab 22: 721–733 [DOI] [PubMed] [Google Scholar]
- Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, Li AA, Girgis HS, Kuchimanchi S, De Groot J, Speciner L, Taneja N, Oshea J, Koteliansky V, Mootha VK (2013) MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS ONE 8: e55785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prevarskaya N, Skryma R, Shuba Y (2011) Calcium in tumour metastasis: new roles for known actors. Nat Rev Cancer 11: 609–618 [DOI] [PubMed] [Google Scholar]
- Quiros PM, Langer T, Lopez‐Otin C (2015) New roles for mitochondrial proteases in health, ageing and disease. Nat Rev Mol Cell Biol 16: 345–359 [DOI] [PubMed] [Google Scholar]
- Raffaello A, De Stefani D, Sabbadin D, Teardo E, Merli G, Picard A, Checchetto V, Moro S, Szabo I, Rizzuto R (2013) The mitochondrial calcium uniporter is a multimer that can include a dominant‐negative pore‐forming subunit. EMBO J 32: 2362–2376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR‐Cas9 system. Nat Protoc 8: 2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren T, Zhang H, Wang J, Zhu J, Jin M, Wu Y, Guo X, Ji L, Huang Q, Yang H, Xing J (2017) MCU‐dependent mitochondrial Ca(2+) inhibits NAD(+)/SIRT3/SOD2 pathway to promote ROS production and metastasis of HCC cells. Oncogene 36: 5897–5909 [DOI] [PubMed] [Google Scholar]
- Sancak Y, Markhard AL, Kitami T, Kovacs‐Bogdan E, Kamer KJ, Udeshi ND, Carr SA, Chaudhuri D, Clapham DE, Li AA, Calvo SE, Goldberger O, Mootha VK (2013) EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 342: 1379–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenmakers TJ, Visser GJ, Flik G, Theuvenet AP (1992) CHELATOR: an improved method for computing metal ion concentrations in physiological solutions. Biotechniques 12: 870–874, 876‐9 [PubMed] [Google Scholar]
- Shen SM, Guo M, Xiong Z, Yu Y, Zhao XY, Zhang FF, Chen GQ (2015) AIF inhibits tumor metastasis by protecting PTEN from oxidation. EMBO Rep 16: 1563–1580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiles BL (2009) PI‐3‐K and AKT: onto the mitochondria. Adv Drug Deliv Rev 61: 1276–1282 [DOI] [PubMed] [Google Scholar]
- Szado T, Vanderheyden V, Parys JB, De Smedt H, Rietdorf K, Kotelevets L, Chastre E, Khan F, Landegren U, Soderberg O, Bootman MD, Roderick HL (2008) Phosphorylation of inositol 1,4,5‐trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proc Natl Acad Sci USA 105: 2427–2432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosatto A, Sommaggio R, Kummerow C, Bentham RB, Blacker TS, Berecz T, Duchen MR, Rosato A, Bogeski I, Szabadkai G, Rizzuto R, Mammucari C (2016) The mitochondrial calcium uniporter regulates breast cancer progression via HIF‐1alpha. EMBO Mol Med 8: 569–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MF, Phillips CB, Ranaghan M, Tsai CW, Wu Y, Willliams C, Miller C (2016) Dual functions of a small regulatory subunit in the mitochondrial calcium uniporter complex. Elife 5: e15545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogtle FN, Wortelkamp S, Zahedi RP, Becker D, Leidhold C, Gevaert K, Kellermann J, Voos W, Sickmann A, Pfanner N, Meisinger C (2009) Global analysis of the mitochondrial N‐proteome identifies a processing peptidase critical for protein stability. Cell 139: 428–439 [DOI] [PubMed] [Google Scholar]
- Vogtle FN, Prinz C, Kellermann J, Lottspeich F, Pfanner N, Meisinger C (2011) Mitochondrial protein turnover: role of the precursor intermediate peptidase Oct1 in protein stabilization. Mol Biol Cell 22: 2135–2143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldeck‐Weiermair M, Malli R, Parichatikanond W, Gottschalk B, Madreiter‐Sokolowski CT, Klec C, Rost R, Graier WF (2015) Rearrangement of MICU1 multimers for activation of MCU is solely controlled by cytosolic Ca(2+). Sci Rep 5: 15602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldmeier PC, Feldtrauer JJ, Qian T, Lemasters JJ (2002) Inhibition of the mitochondrial permeability transition by the nonimmunosuppressive cyclosporin derivative NIM811. Mol Pharmacol 62: 22–29 [DOI] [PubMed] [Google Scholar]
- Wang X, McCullough KD, Franke TF, Holbrook NJ (2000) Epidermal growth factor receptor‐dependent Akt activation by oxidative stress enhances cell survival. J Biol Chem 275: 14624–14631 [DOI] [PubMed] [Google Scholar]
- Wieckowski MR, Giorgi C, Lebiedzinska M, Duszynski J, Pinton P (2009) Isolation of mitochondria‐associated membranes and mitochondria from animal tissues and cells. Nat Protoc 4: 1582–1590 [DOI] [PubMed] [Google Scholar]
- Yang L, Dan HC, Sun M, Liu Q, Sun XM, Feldman RI, Hamilton AD, Polokoff M, Nicosia SV, Herlyn M, Sebti SM, Cheng JQ (2004) Akt/protein kinase B signaling inhibitor‐2, a selective small molecule inhibitor of Akt signaling with antitumor activity in cancer cells overexpressing Akt. Cancer Res 64: 4394–4399 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6