

Abstract
Objective
The Guillain–Barré syndrome (GBS) is an acute, immune‐mediated disease of peripheral nerves. Plasmablasts and plasma cells play a central role in GBS by producing neurotoxic antibodies. The standard treatment for GBS is high‐dose intravenous immunoglobulins (IVIg), however the working mechanism is unknown and the response to treatment is highly variable. We aimed to determine whether IVIg changes the frequency of B‐cell subsets in patients with GBS.
Methods
Peripheral blood mononuclear cells were isolated from 67 patients with GBS before and/or 1, 2, 4, and 12 weeks after treatment with high‐dose IVIg. B‐cell subset frequencies were determined by flow cytometry and related to serum immunoglobulin levels. Immunoglobulin transcripts before and after IVIg treatment were examined by next‐generation sequencing. Antiglycolipid antibodies were determined by ELISA.
Results
Patients treated with IVIg demonstrated a strong increase in plasmablasts, which peaked 1 week after treatment. Flow cytometry identified a relative increase in IgG2 plasmablasts posttreatment. Within IGG sequences, dominant clones were identified which were also IGG2 and had different immunoglobulin sequences compared to pretreatment samples. High plasmablast frequencies after treatment correlated with an increase in serum IgG and IgM, suggesting endogenous production. Patients with a high number of plasmablasts started to improve earlier (P = 0.015) and were treated with a higher dose of IVIg.
Interpretation
High‐dose IVIg treatment alters the distribution of B‐cell subsets in the peripheral blood of GBS patients, suggesting de novo (oligo‐)clonal B‐cell activation. Very high numbers of plasmablasts after IVIg therapy may be a potential biomarker for fast clinical recovery.
Introduction
Intravenous immunoglobulin (IVIg) is the first‐choice treatment for the Guillain‐Barré syndrome (GBS), which is a B‐cell and autoantibody‐mediated disease of the peripheral nerve. GBS is typically a monophasic postinfectious disease, characterized by progressive weakness of the limb and respiratory muscles. Other common symptoms include sensory deficits and autonomic dysfunction. The majority of GBS patients have serum antibodies directed against gangliosides and other glycolipids enriched in the peripheral nerve.1 These antibodies are able to activate the complement cascade leading to nerve damage.2 Activation of B cells and maturation into plasmablasts and plasma cells producing neurotoxic antibodies are therefore key pathogenic events in GBS.
IVIg consists of pooled human IgG purified from thousands of healthy individuals.3 Despite the demonstrated efficacy of high‐dose IVIg in GBS, about 25% of patients still deteriorate during treatment, become paralytic, or require artificial ventilation.4 It is not known why some GBS patients improve following IVIg treatment, whereas others deteriorate further. Although differences in pharmacokinetics may underlie the heterogeneous clinical response to therapy,5 it is also possible that host‐dependent differences in pharmacodynamic effects of IVIg play a role.
Several mechanisms for the therapeutic efficacy of IVIg have been described, including neutralization of autoantibodies, inhibition of complement and cytokines as well as interaction with (neonatal) Fc‐receptors.6 In addition, there are indications that IVIg treatment may result in selection or a “reset” of the immunoglobulin repertoire.7, 8 There is evidence that IVIg may directly affect B cell function by inducing B‐cell proliferation and immunoglobulin synthesis in vitro.9, 10 Correspondingly, in vivo, IVIg was found to induce an increase in the number of plasmablasts in the peripheral blood of patients with GBS, myasthenia gravis, and in chronic inflammatory demyelinating polyneuropathy (CIDP).11 Such a response was not observed in GBS patients treated with plasmapheresis. Importantly, the IVIg‐induced plasmacytosis was associated with improved clinical outcome in GBS.11 Further understanding of whether and how these plasmablasts contribute to clinical recovery may provide critical clues for further optimizing treatment for GBS and other antibody‐mediated diseases.
Here we determined the relation between IVIg and B‐cell differentiation into plasmablasts, serum immunoglobulin (Ig) levels, clinical outcome, and antiglycolipid antibody levels in a large cohort of GBS patients treated with IVIg. Furthermore, we used next‐generation sequencing to analyze subclass and gene segment usage as well as amino acid composition of the complementary determining region (CDR)‐3 of immunoglobulin sequences.
Our data indicate that IVIg treatment induced a de novo B‐cell response as evidenced by increased numbers of plasmablasts, changes in BCR specificity and subclass usage, and higher immunoglobulin levels. This response may have clinical impact as patients with a very high number of plasmablasts showed an earlier start of recovery even though antiganglioside GM1 antibody titers were higher at onset.
Methods
Ethics statement
Ethical approval to study GBS patients was granted by the Erasmus MC Institutional Review Board (MEC‐2009‐368). The use of biomaterials from healthy controls was approved (MEC‐2014‐305, MEC‐2016‐202 and MEC‐2016‐173). Written informed consent was obtained from all participants.
Patients and controls
All patients with GBS participated in a trial, in which patients with a poor prognosis, that is a score of ≥6 on the modified Erasmus GBS Outcome Score, assessed 1 week after the start of IVIg treatment (2 g/kg for 5 days) were randomized for a second course of IVIg or placebo.12 Trial medication was started within 24 h after randomization. Clinical data and blood samples were collected from all patients, regardless of randomization. Inclusion criteria for patients were: fulfilling the diagnostic criteria for GBS13 and first course of IVIg treatment started within 2 weeks after onset of weakness. Patients were excluded from participation if they were younger than the age of 12, pregnant, breastfeeding, known with IgA deficiency, or were unable to be monitored for 6 months. Also, patients with preexisting symptoms of a polyneuropathy or patients diagnosed with a malignancy, AIDS or with a known allergic reaction to blood products were excluded. Blood was collected pretreatment, if possible, and 1, 2, 4, and 12 weeks after IVIg treatment. Physicians assessing clinical outcome did not have access to data on B‐cell subsets and remained blinded to medication status at any time during the study.
Healthy controls participated in the Rotterdam Biobank for Inflammatory Neuropathies (ROBIN) and were recruited among persons who were accompanying patients at the Erasmus MC neurology outpatient clinic. Exclusion criteria were: existence of immune‐mediated diseases, recent infection, use of anti‐inflammatory drugs, and malignancy (not in remission).
Quantification of peripheral blood B‐cell subsets using flow cytometry
Peripheral blood mononuclear cells (PBMC) were isolated from 67 GBS patients and 14 healthy controls (HC) using CPT tubes and directly stained with CD19‐PE/Cy7, CD27‐APC, CD38‐PerCP, IgD‐PE, and IgM‐FITC to determine the relative distribution of naïve, memory, and natural effector B cells, as well as plasmablasts within the total B‐cell population. Cells were measured on a BD Canto flow cytometer.
Simultaneously, a 100‐μL whole‐blood sample from EDTA blood (GBS patients: n = 45, HC: n = 14) was stained to quantify the total number of B cells per mL peripheral blood using TruCount tubes (BD Biosciences). Cells were stained for 20 min on ice using CD16‐FITC, CD56‐PE, CD19‐PE/Cy7, CD45‐BV510, and CD3‐AF700. Next, erythrocytes were lysed using 1 mL 0.155 mol/L NH4Cl, 10 mmol/L KHCO3, and 0.1 mmol/L Na2 EDTA.2H2O for 15 min on ice and the remaining cells were measured on a BD‐LSR II flow cytometer.
The total number of B cells was calculated using the ratio between CD45+ CD19+ cells and the number of beads measured in the same tube. Next, the absolute numbers of B‐cell subsets were calculated using the relative frequencies within the total CD19+ B‐cell population as described above.
Antiganglioside antibody ELISA
Serum obtained from 67 patients was isolated and stored at −80°C and later thawed to measure IgG and IgM antiganglioside antibodies; GM1, GM2, GD1a, GD1b, and GQ1b. As previously described,14 with minor modifications, half‐area plates were coated with 150 pmol of the glycolipid per well and incubated with 1:100 diluted serum. Sera were tested in duplicate, the ∆OD490 nm calculated by subtracting the OD490 nm of uncoated wells from that of coated wells, and titrated if positive (according to previously defined cutoff values), starting at a 1:100 dilution with twofold dilutions. The antibody titer was defined as the lowest dilution with a ∆OD490 nm above the cutoff value.
Quantification of immunoglobulin levels
Serum total IgA, IgG, and IgM levels were determined by routine immunoturbidimetry using Tina‐quant assays and a Cobas c 501 analyzer (Roche Diagnostics). Samples from patients obtained after randomization for a second course of IVIg or placebo were not tested.
Analysis of Ig‐heavy and Ig‐light chain expression in B‐cell subsets
Cryoprotected PBMC from 20 GBS patients and 20 healthy controls were thawed and plated in V‐bottom plates at 106 cells/well (IgH staining) or 5 × 105 cells/well (IgL staining). Cells were subsequently washed in PBS and incubated with Live/Dead Aqua reagent (Molecular Probes, ThermoFisher Scientific) at room temperature for 30 min and washed. The antibody mix consisted of: IgD‐bio, CD27‐BV421, CD38‐APC/Cy7, and either CD19‐BV786 (IgH staining) or CD19‐PE/Cy7 (IgL staining). Cells were incubated with the antibody mix for 30 min on ice. Next, cells were stained with streptavidin‐PerCP. To perform intracellular Ig staining, cells were washed and fixed with 2% paraformaldehyde for 15 min on ice and subsequently washed and permeabilized using 0.5% saponin for 15 min on ice. Intracellular staining was then performed for 60 min on ice with the antibody mix diluted in 0.5% saponin. For the IgH staining, the mix consisted of IgG2‐AF488, IgG1‐PE, IgA‐PE/Vio770, IgG3‐AF647, and IgM‐AF700. For the IgL staining, lambda‐FITC and kappa‐PE were used. Lastly, cells were washed with 0.5% saponin and subsequently with PBS/2% FCS and measured on a BD‐LSR II flow cytometer.
B‐cell repertoire sequencing using next‐generation sequencing
RNA was extracted from frozen PBMC from three GBS patients before, and 1 week after IVIg treatment and from 5 age‐ and gender‐ matched healthy controls (GenElute Mammalian total RNA miniprep, Sigma Aldrich). After reverse transcription using Superscript II reverse transcriptase (Invitrogen) and random hexamer primers, IGH transcripts were amplified using VH1‐6 framework region (FR1; BIOMED‐2) forward and IGHA, IGHM, and IGHG reverse primers combined with unique multiplex identifier sequences per constant chain.15 After amplification, DNA was purified using gel electrophoresis and Ampure DNA purification. sscDNA was subsequently amplified using EmPCR and sequenced using the 454 GS with the Lib‐A V2 kit as previously described.15
B‐cell repertoire data analysis
Samples were demultiplexed based on their multiplex identified sequence and 10nt were trimmed from both ends to remove the primer sequences using IGGalaxy.16 FASTA files were subsequently uploaded into IMGT and further analyzed using the IGGalaxy tool ARGalaxy.17 Using ARGalaxy and excel, data on IgH subclass, CDR3 length, and CDR3 composition were obtained, filtering for productive sequences only. Data were further analyzed in two different ways to correct for biased amplification during the processing of the mRNA, being; (1) analysis based on all productive acquired sequences and (2) based on productive unique sequences where a unique sequence is identified as combination of a specific CDR3 nucleotide sequence, constant region, and V gene.
Statistical analysis
Data on B‐cell subsets were expressed as absolute numbers or percentages and analyzed using Graphpad Prism software (version 5). Time points were compared using Kruskal–Wallis tests followed by Dunn's multiple comparisons tests unless indicated otherwise. All correlation analyses were performed according to Spearman. Differences in clinical outcome were evaluated using Kaplan–Meier survival curves and log‐rank tests with SPSS software (version 24). Cutoff points were analyzed using Cutoff Finder.18 A P‐value of <0.05 was considered statistically significant.
Results
Peripheral blood plasmablasts are increased in GBS patients treated with IVIg
In order to investigate B‐cell subsets and their dynamics during treatment with IVIg, we performed flow cytometry on whole blood and on freshly isolated PBMC from GBS patients and healthy controls (see Fig. 1A for the gating strategy of B‐cell subsets).
Figure 1.
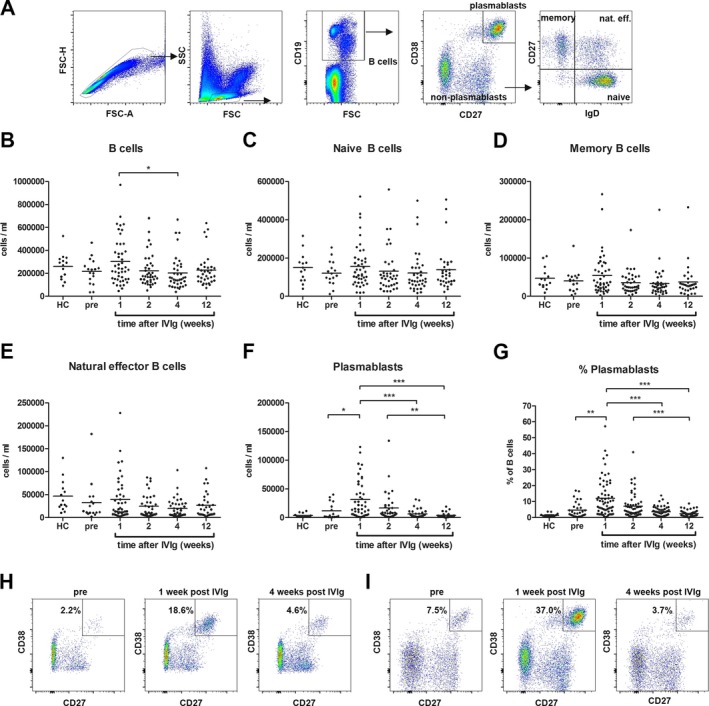
The number and frequency of plasmablasts are increased in the peripheral blood of GBS patients treated with IVIg. B‐cell subsets were gated as indicated (A). Total B cells (CD19+ cells; (B), naïve B cells (CD19+ IgD+ CD27−; (C), memory B cells (CD19+ IgD− CD27+; (D), natural effector B cells (CD19+ IgD+ CD27+; (E) and plasmablasts (CD19+/low CD27+ CD38+; (F) were quantified using flow cytometry. The percent plasmablasts of total B cells is shown in G. An increase in the percent of plasmablasts was also found in an IVIg‐treated patient with a spinal cord infarction (H). A representative IVIg‐treated GBS patients is shown in I. *P < 0.05; **P < 0.01; ***P < 0.001.
The number of peripheral blood B cells was not significantly different between healthy controls and GBS patients before treatment (Fig. 1B). One week after treatment, the number of B cells in GBS patients increased from 217 ± 112 cells/μL to 306 ± 203 cells/μL, reaching significance only when comparing the 1‐week to the 4‐week time point (P < 0.05). No differences were found in naïve, memory, and natural effector B cells between (pretreatment) patients and controls and treatment did not affect the absolute numbers of these B‐cell subsets (Fig. 1C–E). The number of plasmablasts, defined as CD19+/low CD38+ CD27+ cells, was elevated before treatment in approximately one‐third of the GBS patients compared to controls (>15,000 cells/mL, which is the mean plus three times the standard deviation of the controls). One week after treatment with IVIg, a significant increase was found in the number of plasmablasts (from 11.7 ± 14.2 cells/μL pretreatment to 31.6 ± 33.2 cells/μL posttreatment; P < 0.05; Fig. 1F). There was a significant correlation between the number of plasmablasts pretreatment and after 1 week of treatment (r s = 0.69; P = 0.004; n = 15). The number of plasmablasts decreased after 2 weeks, though not significantly, and further decreased 4 and 12 weeks after the start of IVIg treatment (P < 0.001). The percent of plasmablasts ranged from 0.29% to 57.2% of total B cells (mean 12.0%), 1 week after start of treatment (Fig. 1G). This was significantly higher compared to samples collected pretreatment (P < 0.01), and 4 and 12 weeks after treatment (P < 0.001). Before treatment, the percent of plasmablasts was elevated in one‐third of the GBS patients (range 0.24–16.9%) compared to controls (range 0.35–3.76%), but overall there was no significant difference.
The kinetics of the plasmablast appearance in the peripheral blood suggests that the plasmablasts are induced by IVIg. This is further supported by an increased number and percent of plasmablasts 1 week after IVIg treatment in a patient with acute‐onset CIDP (77,529 cells/mL; 28.3% of B cells) compared to 2 weeks after treatment (7734 cells/mL; 4.8% of B cells). In addition, in a patient with a GBS‐mimicking disease who was treated with IVIg but later diagnosed with a spinal cord infarction, plasmablasts increased from 2427 cells/mL (2.2% of B cells) before IVIg to 19,755 cells/mL (18.6% of B cells) 1 week after IVIg and then decreased again to 4098 (4.6% of B cells) 4 weeks after treatment (Fig. 1H), similar to GBS (Fig. 1I).
In summary, a transient increase in both absolute and relative numbers of plasmablasts is observed in patients with GBS after treatment with IVIg.
Plasmablasts after IVIg treatment are enriched for IgG2
The plasmablasts were further phenotyped by intracellular staining for IgM, IgA, IgG1, IgG2, IgG3, and kappa and lambda light chains (n = 20). All classes and subclasses were produced by plasmablasts derived from patients 1 week after treatment (Fig. 2A). No differences were observed in the frequency of IgM+, IgA+,and IgG+ plasmablasts between pretreatment and 1 week posttreatment time points (determined as percent of total plasmablasts; Fig. 2B–D). This implies that IgM+, IgA+, and IgG+ plasmablasts are all increased following IVIg treatment. However, within the IgG+ plasmablasts, we found a significant increase in the percent of IgG2+ cells and a significant decrease in the percent of IgG3+ cells 1 week posttreatment compared to pretreatment samples (Fig. 2E–G). No differences were found in the IgG subclass distribution of memory B cells (data not shown). The kappa/lambda ratio of plasmablasts analyzed from patients 1 week after IVIg treatment was less skewed but not significantly different compared to pretreatment plasmablasts (data not shown).
Figure 2.
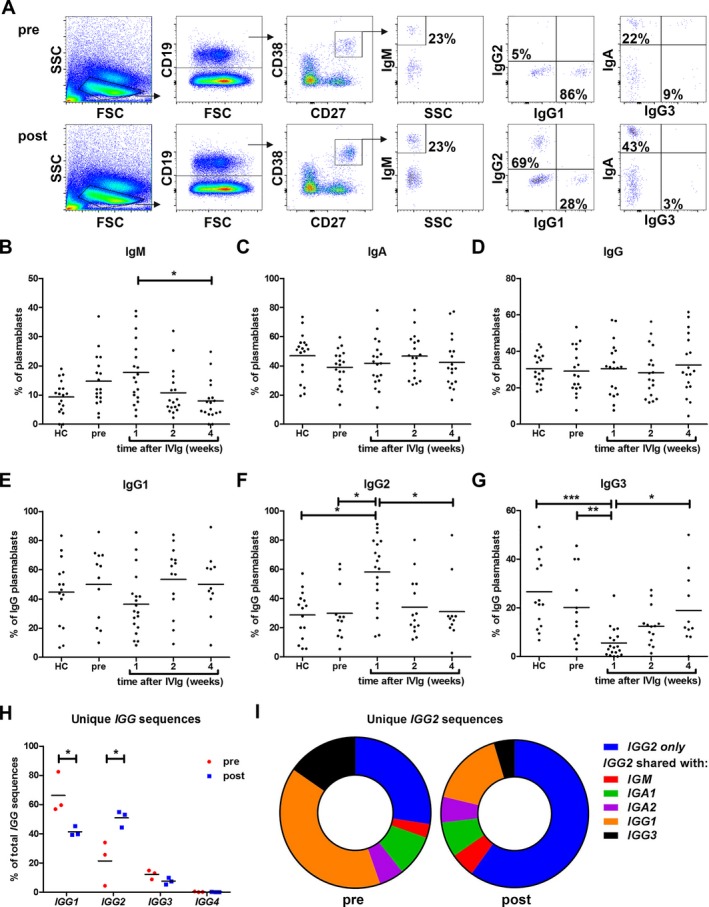
IVIg‐induced plasmablasts are enriched for IgG2. Intracellular staining of immunoglobulin classes and subclasses was performed on cryo‐preserved PBMC from GBS patients (n = 20) and healthy controls (n = 20). Plasmablasts (CD19+/low CD27+ CD38+) were gated and expression of all classes and subclasses was observed (A; top panels pretreatment and lower panels posttreatment). No difference was found in IgM (B), IgA (C), and IgG (D) positive plasmablasts before and 1 week after IVIg treatment. Quantification of IgG1 (E), IgG2 (F), and IgG3 (G) subclasses demonstrated a significant shift in IgG subclass usage 1 week after IVIg treatment. Samples with few plasmablasts were excluded. IGG transcripts analyzed by 454 sequencing also indicated a significant shift in unique IGG subclass sequences in GBS patients (n = 3) 1 week posttreatment (H; two‐way ANOVA followed by Bonferroni correction). The fraction of sequences found in IGG2 only, and not shared with other (sub)classes was also increased after treatment (I; data represent means of three patients). *P < 0.05; **P < 0.01; ***P < 0.001.
We next determined the sequences of immunoglobulin RNA transcripts from three patients before and after IVIg treatment using 454 sequencing. IgG subclass analysis showed that the percent of unique IgG2 transcripts was significantly increased in patients 1 week after IVIg treatment (Fig. 2H). Furthermore, after treatment with IVIg there was less overlap of unique IGG2 sequences with other classes and subclasses than before treatment (Fig. 2I), which further suggests that IVIg is skewing toward an IgG2 response.
Dominant B‐cell clones are present in GBS before and after treatment with IVIg
In order to identify possible B‐cell clones present in patients with GBS before and after treatment, we determined the percent of IgG sequences with the same CDR3 nucleotide length and identical V and D gene segment usage in three patients and five controls. Two dominant clones (>5% of all sequences) were found in one GBS patient before treatment (45% and 5% of all sequences; Fig. 3A) and in one out of five controls (Fig. 3B). Interestingly, patient 3 had elevated frequencies of VH4‐34 IgG sequences before treatment (15.3% of total IgG sequences), which may be related to the preceding infection with Mycoplasma pneumoniae, which is known to expand VH4‐34 expressing B cells.19 After treatment, dominant clones (6 in total) were observed in all GBS patients, the largest clone of each patient contributing to 7.3%, 12.6%, and 6.0% of the total sequences. These clones were predominantly of the IgG2 subclass. Analysis of CDR3 amino acid sequences revealed highly homogeneous sequences both before and after treatment (Fig. 3C). Analyzing the relationship between clones after and before treatment, we found two dominant clones after treatment that were already present before treatment. Of these, one was present as dominant clone (also IGG2), contributing to 5% of the sequences pretreatment (Fig. 3D). For the other clone (both IGG1 and IGG2 after treatment), only one sequence was found before treatment (IGG1). For both clones exactly the same CDR3 amino acid sequence was observed before and after treatment. Four other dominant clones after treatment were not present before treatment, suggesting that IVIg predominantly induces a de novo B‐cell response. For IgM sequences, persisting dominant clones were observed in two patients (Fig. S1).
Figure 3.
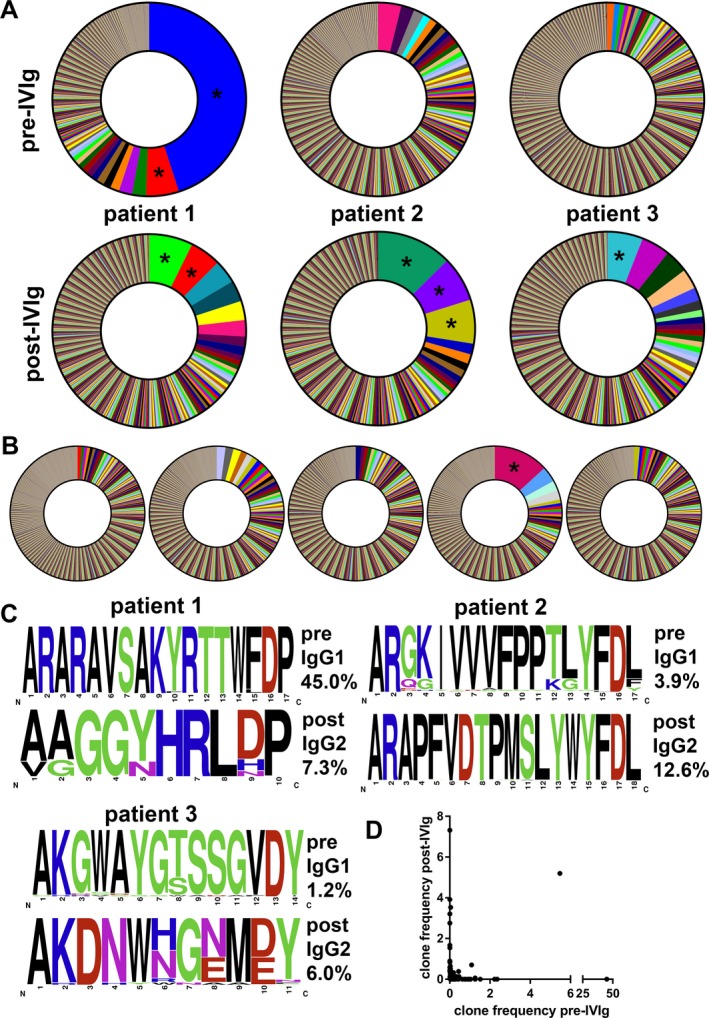
Dominant B‐cell clones are present in GBS patients after treatment with IVIg. RNA was isolated from PBMC before and 1 week after treatment with IVIg and immunoglobulin transcripts were sequenced. Potential clones were identified as the percent of (all) IGG sequences having the same V and D region usage and the same CDR3 length. Within each patient, unique clones are represented by a different color (only clones of >1%). Before treatment, a very dominant clone was identified in patient 1 (A). After treatment, the number of dominant clones (>5%, indicated with asterisks) increased in all patients and dominant clones were more common than in healthy controls (n = 5; B). Analysis of the CDR3 amino acid composition of the most prevalent clone of every patient before and after treatment identified common motifs (data shown as frequency; C). Note that after treatment the most prevalent clones were of the IgG2 subclass. Comparison of the IgG clone frequency from patient 1 before and after treatment revealed only little overlap between clones (D).
These results indicate that dominant B‐cell clones are present in patients with GBS and that IVIg treatment results in changes in immunoglobulin sequences suggesting (oligo‐)clonal activation of B cells.
Plasmablasts after IVIg treatment contribute to immunoglobulin synthesis
Since the increase in plasmablasts may be caused by apoptotic displaced plasmablasts due to FcγRIIb crosslinking,20 we investigated the functional capacity of plasmablasts to synthesize immunoglobulin in vivo. Immunoglobulin levels were determined in sera from 60 GBS patients and correlated to the number and frequency of plasmablasts, 1 week after treatment.
The serum IgM level and the percent of plasmablasts, both measured 1 week after IVIg treatment, were not significantly correlated (r s = 0.24; P = 0.07; Fig. 4A). However, a strong positive correlation was found between the percent of plasmablasts and the delta IgM levels (increase in IgM level posttreatment compared to pretreatment) 1 week after treatment (r s = 0.78; P < 0.0001; Fig. 4B) and 2 weeks after treatment (r s = 0.74; P < 0.0001; Fig. 4C). For IgG, a weak but significant correlation was found with the percent of plasmablasts 1 week after treatment (r s = 0.28; P = 0.03; Fig. 4D). The delta IgG 1 and 2 weeks after treatment correlated significantly with the percent of plasmablasts (r s = 0.54; P = 0.002 and r s = 0.44; P = 0.03, respectively; Fig. 4E and F). Regression analysis demonstrated that the delta IgG at 1 week, but not 2 weeks, was also significantly influenced (P = 0.014; β = 0.51) by the percent of plasmablasts 1 week after treatment when corrected for the IVIg dose. The delta IgM at 1 week, but not at 2 weeks also showed significant correlation with the absolute number of plasmablasts. For IgA, significant correlations were observed between the percent of plasmablasts and the delta IgA levels at 1 (r s = 0.41; P = 0.03) and 2 weeks (r s = 0.44; P = 0.03) after treatment (data not shown).
Figure 4.
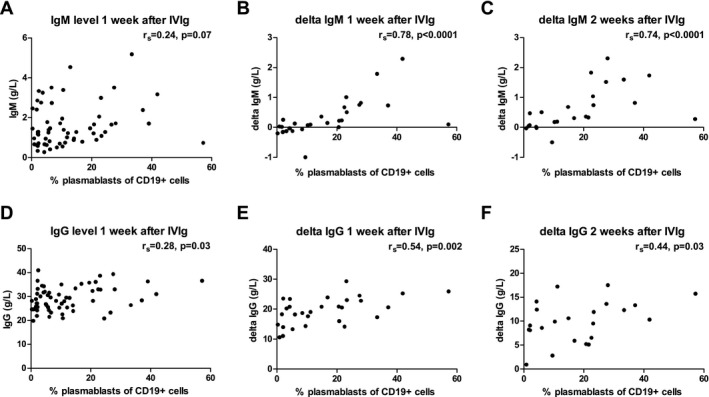
The percent of plasmablasts after 1 week of IVIg treatment correlates with the rise in serum IgM and IgG levels. Serum IgM (A–C) and IgG (D–F) levels were determined before and after IVIg treatment and related to the percent of plasmablasts measured after 1 week of IVIg treatment. Absolute IgM/G levels are shown in A and D, the delta IgM/G levels at 1 week in B and E, and the delta IgM/G level at 2 weeks are indicated in C and F. Delta IgM/G levels were calculated compared to pretreatment levels, which were not available for all patients. Patients randomized for a second course of IVIg or placebo were excluded from analysis in C and F.
Since IVIg does not contain IgM, the increase in IgM must be caused by increased endogenous production. Therefore, these data suggest that the observed plasmablasts are viable in vivo and contribute to immunoglobulin synthesis.
Patients with high numbers of plasmablasts after IVIg treatment start to improve earlier
Since the number of plasmablasts in the peripheral blood of GBS patients 1 week after the start of IVIg therapy was highly variable and not elevated in every patient, we determined the relation between the plasmablasts and clinical outcome.
No significant correlation was observed between the number of plasmablasts 1 week after IVIg and the GBS disability score at 4 weeks (P = 0.18, n = 43; Fig. 5A). Next, using the absolute counts of plasmablasts in GBS patients before the start of treatment as a cutoff point, we discriminated patients with a high (>50,000 cells/mL) and a low number (<50,000 cells/mL) of plasmablasts after IVIg therapy. Demographic and clinical characteristics were not significantly different between the two groups (Table 1), with the exception of the IVIg dose which was significantly higher in the group with high numbers of plasmablasts (198 ± 49 vs. 155 ± 30 g IVIg; P < 0.05 Mann–Whitney U test). All patients who were randomized for a second course of IVIg or placebo (n = 8) had a plasmablast count of <50,000/mL (mean 21880 ± 13716 cells/mL). This was not significantly different from nonrandomized patients in the group of <50,000 plasmablasts/mL (P = 0.11).
Figure 5.
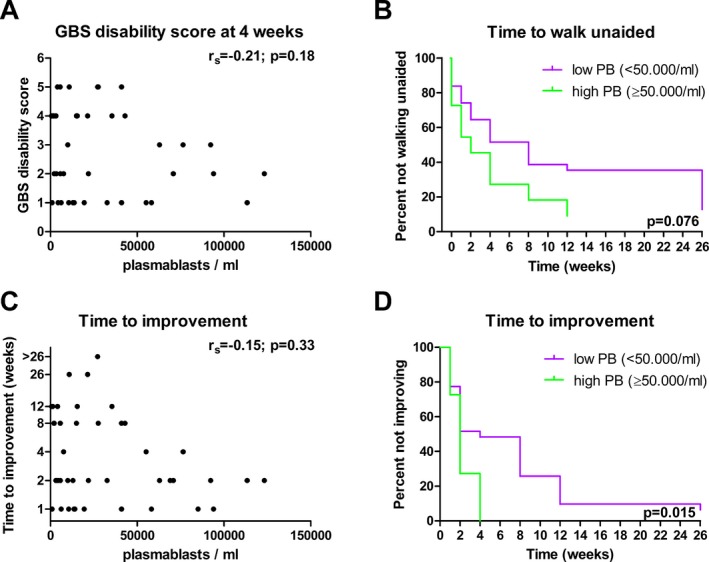
Plasmablasts after IVIg treatment in relation to clinical outcome in GBS. The absolute number of plasmablasts in relation to the GBS disability score determined 4 weeks after IVIg therapy (A) or the time to improve one grade on the GBS disability scale (C; y‐axis in log2 scale; n = 43). The time to reach independent walking (B) that is a GBS disability score of two, and the time to improvement of one grade on the GBS disability scale (D) was compared in patients with high (≥50,000 cells/mL) and low absolute numbers (<50,000 cells/mL) using logrank testing (n = 42).
Table 1.
Demographics, laboratory, and clinical characteristics of patients with high and low numbers of plasmablasts 1 week after IVIg treatment
| Plasmablasts after IVIg <50,000 (n = 33) | Plasmablasts after IVIg ≥50,000 (n = 11) | |
|---|---|---|
| Demographic characteristics | ||
| Female, n (%) | 10/33 (30) | 5/11 (45) |
| Age, median (range) | 60 (18–86) | 50 (18–80) |
| Antecedent events | ||
| Diarrhea, n (%) | 8/31 (26) | 5/11 (45) |
| URTI, n (%) | 11/31 (35) | 2/11 (18) |
| Diarrhea and URTI, n (%) | 3/31 (10) | 2/11 (18) |
| Other, n (%) | 2/31 (6) | 0/11 (0) |
| None, n (%) | 7/31 (23) | 2/11 (18) |
| Characteristics of GBS | ||
| Days of weakness before start of IVIg therapy, median (range) | 2 (0–12) | 2 (1–10) |
| GBS disability score at entry, median (range) | 4 (1–4) | 3 (1–4) |
| GBS disability score at nadir, median (range) | 4 (1–5) | 4 (2–4) |
| Mechanical ventilation, n (%) | 8/32 (25) | 1/11 (9) |
| Antiganglioside IgG antibodies, n (%) | 12/33 (36) | 5/11 (46) |
| Treatment | ||
| Total IVIg dose (g), mean (SD) | 155 (30) | 198 (49)a |
| Randomized for second course of IVIg or placebo, n (%) | 8/31 (26) | 0/11 (0) |
| Methylprednisolone, n (%) | 6/28 (21) | 3/10 (30) |
P < 0.05, Mann–Whitney U test.
Patients with a high absolute number of plasmablasts at 1 week after IVIg treatment, acquired the ability to walk unaided earlier than patients with a low absolute number of plasmablasts (mean 4.0 weeks vs. 11.4 weeks), although this difference was not statistically significant (P = 0.076, n = 42, logrank test, Fig. 5B). The time to improvement was defined as number of weeks until a one‐point improvement on the GBS disability scale was achieved. Overall, no significant correlation was found with the number of plasmablasts 1 week after treatment (P = 0.33, n = 43; Fig. 5C), however comparison of the two subgroups indicated that patients with a high number of plasmablasts started to improve earlier than patients with a low number of plasmablasts (P = 0.015, n = 42, logrank test, Fig. 5D). This significant difference was also observed when a range of cutoff values was applied (36,000–68,000 plasmablasts/mL), demonstrating the robustness of the cutoff value. After correction for age or GBS disability score at entry, the time to improvement was not significantly different between patients with high and low numbers of plasmablasts (P = 0.07 and P = 0.085, respectively; n = 41; Cox regression). The percent of plasmablasts 1 week after treatment was not related to clinical outcome, that is the time to acquire the ability to walk unaided, or the time to one‐point improvement on the GBS disability scale (data not shown).
In summary, patients with high plasmablast numbers after IVIg treatment started to recover earlier and regained the ability to walk earlier than patients with low plasmablast numbers, however this effect was lost after correction for other prognostic factors such as age and GBS disability score at entry.
Higher anti‐GM1 antibody titers in patients with a high number of plasmablasts
Finally, the relation between plasmablasts and anti‐ganglioside antibodies was determined by measuring the GM1 IgG titers pretreatment and at different time points after treatment. GM1 was chosen because it is the most frequent target for autoantibodies in GBS.
Sixteen patients with anti‐GM1 IgG antibodies were identified (24%). Patients with high numbers of plasmablasts posttreatment had higher titers of anti‐GM1 IgG antibodies pretreatment (P < 0.001; Fig. 6A). There was no significant difference in the GM1 IgG titers at later time points between patients with high or low frequencies of plasmablasts. However, it was observed that anti‐GM1 IgG titers remained high in patients with high numbers of plasmablasts who already started to recover. Subclass analysis of anti‐GM1 antibodies (Fig. 6B–E) showed that most patients had anti‐GM1 antibodies of the IgG1 and/or IgG3 subclass, as reported previously. IgG4 antibodies were also identified in a subset of patients. In the majority of the patients, no IgG2 antibodies reactive with GM1 developed following IVIg treatment. In only one patient, with a high number of plasmablasts posttreatment, IgG2 antibodies against GM1 were detected. In this patient anti‐GM1 IgG1 and IgG3 did not increase after IVIg treatment, suggesting that IVIg may promote an existing IgG2 anti‐GM1 antibody response.
Figure 6.
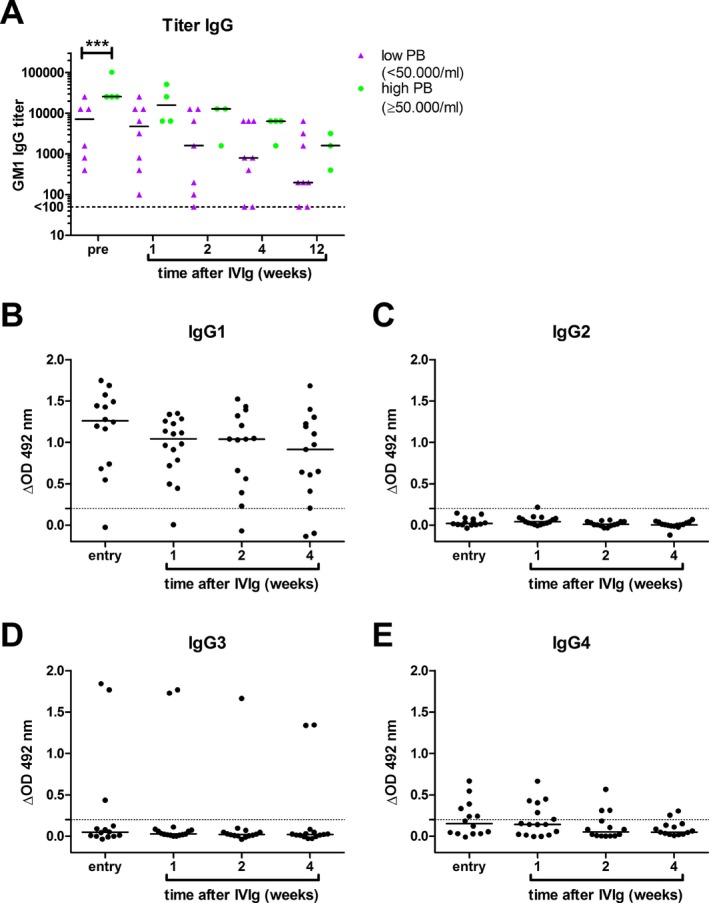
Patients with high plasmablast numbers after IVIg treatment have high GM1 IgG titers at onset. Pre‐ and posttreatment sera from GBS patients positive for GM1 IgG antibodies (n = 12) were titrated by ELISA. Data is shown stratified according to low or high plasmablast count assessed 1 week after IVIg treatment (A). A titer of <100 is considered negative. Two‐way ANOVA followed by Bonferroni correction (***P < 0.001). Subclass analysis of anti‐GM1 antibodies (B–E) demonstrated that the majority of patients do not produce anti‐GM1 antibodies of the IgG2 subclass; only one patient is positive for anti‐GM1 IgG2 1 week after treatment (C). The dotted line indicated the cutoff value (ΔOD = 0.2).
In summary, patients with a high number of plasmablasts have higher titers of anti‐GM1 antibodies at disease onset.
Discussion
GBS is a prototypic autoantibody‐mediated disease which is preferentially treated with IVIg. However, the effect of IVIg on B cells in GBS in relation to immunoglobulin levels, pathogenic antibodies, and clinical outcome is poorly understood. Here we demonstrate that peripheral blood plasmablasts are increased in GBS patients treated with IVIg. The plasmablasts are enriched for IgG2 subclass and actively contribute to immunoglobulin secretion in vivo. Molecular data indicate that IVIg induces a de novo B‐cell response, distinct from the pretreatment B‐cell response. High plasmablast numbers were associated with an earlier start of recovery even though anti‐GM1 antibody titers were higher at onset.
The increase in plasmablasts in GBS patients was transient, being the highest at 1 week after the start of IVIg treatment. Concurrent treatment with high‐dose methylprednisolone did not affect plasmablast counts. The kinetics are highly reminiscent of a vaccination response, in which newly formed plasmablasts peak in the peripheral blood on day 6–7 after vaccination.21, 22 This suggests that the increase in plasmablasts in GBS patients is a direct response to IVIg treatment. Correspondingly, the total dose of the IVIg infusions was significantly higher in patients who demonstrated a high number of plasmablasts 1 week after treatment. Moreover, an increase in plasmablasts was not only found in patients with GBS but also in a patient with acute‐onset CIDP, and in a patient with GBS‐mimicking disease who was later diagnosed with a spinal cord infarction both initially treated with IVIg. These findings are in line with the results of Mori et al.,11 showing that plasmablasts were also increased 1 week after IVIg treatment in other diseases and not in GBS patients who underwent plasmapheresis or received no treatment, indicating that the increase in plasmablasts after 1 week of treatment is induced by IVIg rather than GBS. Furthermore, our finding that IgG2+ plasmablasts are increased after IVIg treatment does not correspond with current concepts regarding GBS pathogenesis, since the pathogenic antibody response in GBS is not associated with the IgG2 subclass, but rather with IgG1 and IgG3. Lastly, the timing of the blood sampling seems critical because shortly after IVIg treatment (3 days), plasmablasts appear to be decreased in the peripheral blood of most CIDP and multifocal motor neuropathy patients treated with high‐dose IVIg,23 however no data were presented from samples obtained 1 week after treatment.
Our findings correspond with results from in vitro studies of de Grandmont et al. showing that IVIg enhances the differentiation of CD40‐activated B cells into plasmablasts and immunoglobulin synthesis and secretion.9 Importantly, in the latter study, only a small fraction (<15%) of B cells differentiated in response to IVIg. The antibodies were directed against diverse nonself‐ and self‐antigens, including F(ab)2 fragments but not Fc fragments, suggesting that the antibodies are induced through idiotype anti‐idiotype interactions. Additionally, B cells from patients with common variable immunodeficiency secrete increased levels of IgM in vitro in the presence of IVIg and anti‐IgM as B‐cell receptor crosslinker.10 In contrast, in the study by Heidt et al.,24 no effect of IVIg was observed on B‐cell proliferation or immunoglobulin production. Strong stimulation of B cells with CpG and cytokines in that study may possibly have rendered B cells insensitive to IVIg. Another study has reported that IVIg induces phosphorylation of extracellular signal‐regulated kinases (ERK)‐1 and 2, especially in IgG+ cells.25 Our data extend all these in vitro findings by demonstrating that IVIg can also promote immunoglobulin synthesis in vivo, as shown by the increase in IgM levels in patients with a high percent (>20%) of plasmablasts. Since IVIg does not contain IgM, the increase in IgM must be caused by increased endogenous production of immunoglobulins. This finding also indicates that the IVIg‐induced plasmablasts are viable in vivo and do not represent displaced apoptotic plasmablasts caused by FcγRIIb crosslinking.20 The increase in IgG levels compared to baseline (i.e., the delta IgG) at 1 and 2 weeks after IVIg treatment was also significantly correlated with the percent of plasmablasts, but the association was weaker than for IgM. This may be due to the high levels of infused IgG, potentially masking additional endogenous IgG production. IgG levels after IVIg therapy are highly diverse in patients with GBS5 and may be influenced by several complex mechanisms, including redistribution to tissues, target‐mediated clearance, and FcRn‐mediated recycling.26, 27 The exact role of endogenous IgG production by IVIg‐responsive plasmablasts to serum IgG levels can only be investigated using appropriate pharmacokinetic modeling correcting for known confounding factors which was beyond the scope of this study.
Recent in vivo studies in mice have demonstrated that human IVIg, but not monoclonal humanized antibodies used at the same dose, induce widespread T‐ and B‐cell activation.28 Interestingly, this response was only observed when animals were coinjected with adjuvant. Additionally, IVIg reduced the response to ovalbumin, which was also present in the adjuvant.28 Here we found that IVIg‐induced plasmablasts were the highest in patients with an ongoing humoral immune response before treatment (i.e., patients with a high number of plasmablasts). Likewise, patients with higher number of plasmablasts after treatment also had higher anti‐GM1 antibody titers pretreatment. This may suggest that an ongoing immune response favors a microenvironment with increased levels of survival factors for plasmablasts, such as myeloid cells that secrete B‐cell activating factor, a proliferation inducing ligand, or interferon gamma‐induced protein‐10.29
IgG subclass analysis using flow cytometry demonstrated that the IVIg‐induced plasmablasts more frequently produced IgG2 than plasmablasts before treatment. The sequence data also indicated that after treatment with IVIg the most dominant clones within IgG were of the IgG2 subclass. IgG2 antibodies are typically, but not exclusively, produced against carbohydrate antigens.30, 31 These antigens, often repetitive in nature (e.g., bacterial capsular polysaccharides), activate B cells through extensive crosslinking of the B‐cell receptor, and do not require cognate help from T cells.32 It is possible that high‐dose IVIg induces similar crosslinking of the BCR for example through idiotype anti‐idiotype interactions, as has been suggested from in vitro data.9 Alternatively, glycosylation or glycation of IVIg preparations,33, 34 may induce a preferential IgG2 response. Our data does not clarify whether (sequential) class switching has occurred or whether IgG2+ B cells are preferentially activated. However, IVIg did not induce IgG2 class switching of anti‐GM1 B cells, which were of the IgG1 and IgG3 subclass, as demonstrated previously.35 Our data support these findings as our antiganglioside ELISA showed no anti‐GM1 IgG2 responses in almost all patients. Moreover, mouse studies have indicated that memory B cells can be readily activated with particulate antigens in the absence of T cells to differentiate into plasmablasts.36
The results of the next‐generation sequencing demonstrated the presence of a dominant B‐cell clone before the start of treatment in one out of three GBS patients. This clone might represent a neuropathogenic clone since this patient was positive for anti‐GD1b IgG antibodies or may be related to the preceding infection. Furthermore, we noted dynamic changes in dominant clones before and after treatment with IVIg. The Ig sequences expressed by dominant clones after treatment were mostly absent or present at very‐low frequencies before treatment, suggesting that IVIg induces a de novo response, which may be a primary or a secondary response. The finding that dominant clones were present after treatment, appear to be increased, and have different sequences compared to the dominant clones before treatment, suggests that IVIg induces an (oligo‐)clonal expansion of IgG2+ B cells. It is still possible that this expansion occurs on top of a background of polyclonal activation, as flow cytometric analysis demonstrated that all Ig classes were produced by plasmablasts after IVIg therapy. However, the sequence data do not allow discrimination between polyclonal activation of B cells and normal genetic diversity which is present among resting cells. One other study investigated the effect of IVIg on the immunoglobulin repertoire. In patients under desensitization therapy prior to transplantation, no changes were observed in the frequency of unique sequences, mutation frequency, and the abundance of switched sequences.37 The pretransplant patients were treated with multiple, monthly courses of high‐dose IVIg, therefore the data suggest that IVIg does not change the immunoglobulin repertoire in the long term but leads only to a short wave of newly formed plasmablasts which disappear over time.
Do the clear biological effects of IVIg on B cells in GBS also translate into a beneficial effect on clinical recovery? Although we found that patients with a high number of plasmablasts after IVIg therapy started to improve significantly earlier and had a tendency to acquire the ability to walk earlier, no significant differences were found after correction for confounding factors like age and GBS disability score at entry, which are known prognostic factors for GBS. This is likely due to the relatively low number of patients with high plasmablast counts (n = 11), resulting in insufficient power for analysis with confounding factors. Further analysis is therefore required to determine to what extent IVIg‐induced plasmablasts can predict rapid clinical recovery when age, disease severity, other prognostic factors, and IVIg dose are taken into account.
Given that patients with high plasmablast numbers had higher antibody titers to GM1 at onset (all ≥ 25600), which are associated with poor recovery,38 it remains remarkable that these patients started to recover earlier than patients with low plasmablast numbers and lower anti‐GM1 IgG titers. This may be explained by differences in fine specificity and pathogenicity of the antibodies. In mice, it was recently demonstrated that if antiganglioside antibodies are not able to bind gangliosides in tissue, they remain elevated in the circulation because of a lack of target‐mediated clearance.26 Whether the anti‐GM1 antibodies in patients with a high titer and a fast recovery are pathogenic or nonpathogenic remains to be determined.
The improved clinical outcome of patients with high numbers of plasmablasts may also be caused by a higher dose of IVIg, since this was the only discriminative parameter between the two groups. The higher dose of IVIg may lead to improved neutralization of pathogenic antibodies, or induce more efficient immunomodulation through other mechanisms. That IVIg dosage affects clinical outcome in GBS is demonstrated by a trial in which high dose of IVIg (2.4 g/kg in 6 days) resulted in better clinical outcome of GBS than a lower dose (1.2 g/kg in 3 days).39
In conclusion, our study indicates that IVIg can promote humoral responses in patients with GBS, as reflected by an increase in peripheral blood plasmablasts, changes in BCR specificity and subclass distribution, and endogenous Ig production. A high number of plasmablasts after IVIg treatment were associated with an earlier start of recovery, despite higher titers of anti‐GM1 antibodies, and were related to a higher IVIg dose. Future studies should address the reactivity of the plasmablasts and further clarify the role of plasmablasts in the pharmacokinetics of IVIg in relation to clinical outcome.
Author Contributions
Conceived and designed the experiments: all; Acquired experimental and/or clinical data: MDB, WvR, WJRF, APTG, CW, RH; Analyzed the data: MDB, WvR, CW, HIJ, RH; Wrote the manuscript: MDB and RH; Critical revision of the manuscript: BCJ, WJRF, CW, PAvD, MvdB.
Conflict of Interest
B. Jacobs reports grants from Annexon, Prinses Beatrix Spierfonds, GBS‐CIDP Foundation International, CSL‐Behring, Grifols, Horizon 2020 and Baxter, outside the submitted work; membership of the Medical Advisory Board for the GBS‐CIDP Foundation International; membership of the Inflammatory Neuropathy Consortium. P. van Doorn reports grants from the Prinses Beatrix Spierfonds and Sanquin, during the conduct of the study; grants from Grifols and Shire, outside the submitted work; fees paid to the institution from Kedrion, CSL, Octapharma, Grifols, Kedrion, Baxter, Grifols, Hansa, outside the submitted work; Membership of the Medical Advisory board from the GBS‐CIDP Foundation International; Membership of the Inflammatory Neuropathy Consortium and being President‐Elect of the Peripheral Nervous Society. R. Huizinga reports grants from GBS‐CIDP Foundation International and Grifols outside the submitted work. Other authors have nothing to disclose.
Supporting information
Figure S1. Dominant IgM B‐cell clones are present in GBS patients before and after treatment with IVIg.
Acknowledgments
We are grateful for the enrolment of GBS patients by: R. Kleyweg and K. Kuitwaard (Albert Schweitzer Hospital, Dordrecht), C. Gijsbers and H. Kramers (Vlietland Hospital, Schiedam), J. Samijn (Maasstad Hospital, Rotterdam), F. Vermeij (Sint Fransicus Gasthuis, Rotterdam), J. Gilhuis (Reinier de Graaf Hospital, Delft), P. Wirtz (Haga Hospital, The Hague), M. van der Meulen (Antonius Hospital, Nieuwegein), J. Verschuuren (LUMC, Leiden), I. van Schaik and F. Eftimov (AMC, Amsterdam), L. Visser (St. Elisabeth Hospital, Tilburg), K. Jellema, S. Manschot and R. Groen (HMC, The Hague), B. van Engelen (UMC St. Radboud, Nijmegen), B. van Oosten (VUMC, Amsterdam), L. Ruts (Havenziekenhuis, Rotterdam) and W. Linssen (OLVG, Amsterdam). We thank B. van den Berg, A. Doets, J. Roodbol, J. Verboon and M. van Woerkom for assistance with collection of biomaterials and clinical data.
This work was supported by the Prinses Beatrix Spierfonds (grant number W.OR14‐16). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Funding Information
This work was supported by the Prinses Beatrix Spierfonds (grant number W.OR14‐16). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Funding Statement
This work was funded by Prinses Beatrix Spierfonds grant W.OR14‐16.
References
- 1. Willison HJ, Jacobs BC, van Doorn PA. Guillain‐Barré syndrome. Lancet 2016;388:717–727. [DOI] [PubMed] [Google Scholar]
- 2. McGonigal R, Rowan EG, Greenshields KN, et al. Anti‐GD1a antibodies activate complement and calpain to injure distal motor nodes of Ranvier in mice. Brain 2010;133:1944–1960. [DOI] [PubMed] [Google Scholar]
- 3. van der Meché FG, Schmitz PI. A randomized trial comparing intravenous immune globulin and plasma exchange in Guillain‐Barré syndrome. Dutch Guillain‐Barré Study Group. N Engl J Med 1992;326:1123–1129. [DOI] [PubMed] [Google Scholar]
- 4. van Doorn PA, Ruts L, Jacobs BC. Clinical features, pathogenesis, and treatment of Guillain‐Barré syndrome. Lancet Neurol 2008;7:939–950. [DOI] [PubMed] [Google Scholar]
- 5. Kuitwaard K, de Gelder J, Tio‐Gillen AP, et al. Pharmacokinetics of intravenous immunoglobulin and outcome in Guillain‐Barré syndrome. Ann Neurol 2009;66:597–603. [DOI] [PubMed] [Google Scholar]
- 6. Schwab I, Nimmerjahn F. Intravenous immunoglobulin therapy: how does IgG modulate the immune system? Nat Rev Immunol 2013;13:176–189. [DOI] [PubMed] [Google Scholar]
- 7. Kazatchkine MD, Dietrich G, Hurez V, et al. V region‐mediated selection of autoreactive repertoires by intravenous immunoglobulin (i.v.Ig). Immunol Rev 1994;139:79–107. [DOI] [PubMed] [Google Scholar]
- 8. Mitrevski M, Marrapodi R, Camponeschi A, et al. Intravenous Immunoglobulin and Immunomodulation of B‐Cell ‐ in vitro and in vivo Effects. Front Immunol 2015;6:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. de Grandmont MJ, Racine C, Roy A, et al. Intravenous immunoglobulins induce the in vitro differentiation of human B lymphocytes and the secretion of IgG. Blood 2003;101:3065–3073. [DOI] [PubMed] [Google Scholar]
- 10. Bayry J, Fournier EM, Maddur MS, et al. Intravenous immunoglobulin induces proliferation and immunoglobulin synthesis from B cells of patients with common variable immunodeficiency: a mechanism underlying the beneficial effect of IVIg in primary immunodeficiencies. J Autoimmun 2011;36:9–15. [DOI] [PubMed] [Google Scholar]
- 11. Mori I, Parizot C, Dorgham K, et al. Prominent plasmacytosis following intravenous immunoglobulin correlates with clinical improvement in Guillain‐Barré syndrome. PLoS ONE 2008;3:e2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Walgaard C, Jacobs BC, Lingsma HF, et al. Second IVIg Course in Guillain‐Barré Syndrome patients with poor prognosis (SID‐GBS trial): protocol for a double‐blind randomized, placebo‐controlled clinical trial. J Peripher Nerv Syst 2018; 10.1111/jns.12286. [DOI] [PubMed] [Google Scholar]
- 13. Asbury AK, Cornblath DR. Assessment of current diagnostic criteria for Guillain‐Barré syndrome. Ann Neurol 1990;27(Suppl):S21–S24. [DOI] [PubMed] [Google Scholar]
- 14. Kuijf ML, van Doorn PA, Tio‐Gillen AP, et al. Diagnostic value of anti‐GM1 ganglioside serology and validation of the INCAT‐ELISA. J Neurol Sci 2005;239:37–44. [DOI] [PubMed] [Google Scholar]
- 15. IJspeert H, van Schouwenburg PA, van Zessen D, et al. Evaluation of the antigen‐experienced B‐cell receptor repertoire in healthy children and adults. Front Immunol 2016;7:410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Moorhouse MJ, van Zessen D, IJspeert H, et al. ImmunoGlobulin galaxy (IGGalaxy) for simple determination and quantitation of immunoglobulin heavy chain rearrangements from NGS. BMC Immunol 2014;15:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. IJspeert H, vanSchouwenburg PA , vanZessen D , et al. Antigen receptor galaxy: a user‐friendly, web‐based tool for analysis and visualization of T and B cell receptor repertoire data. J Immunol 2017;198:4156–4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Budczies J, Klauschen F, Sinn BV, et al. Cutoff Finder: a comprehensive and straightforward Web application enabling rapid biomarker cutoff optimization. PLoS ONE 2012;7:e51862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chapman CJ, Spellerberg MB, Smith GA, et al. Autoanti‐red cell antibodies synthesized by patients with infectious mononucleosis utilize the VH4‐21 gene segment. J Immunol 1993;151:1051–1061. [PubMed] [Google Scholar]
- 20. Xiang Z, Cutler AJ, Brownlie RJ, et al. FcgammaRIIb controls bone marrow plasma cell persistence and apoptosis. Nat Immunol 2007;8:419–429. [DOI] [PubMed] [Google Scholar]
- 21. He XS, Sasaki S, Narvaez CF, et al. Plasmablast‐derived polyclonal antibody response after influenza vaccination. J Immunol Methods 2011;365:67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fink K. Origin and function of circulating plasmablasts during acute viral infections. Front Immunol 2012;3:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Korporal‐Kuhnke M, Haas J, Schwarz A, et al. Plasmacytosis is a common immune signature in patients with MMN and CIDP and responds to treatment with IVIg. J Neuroimmunol 2015;278:60–68. [DOI] [PubMed] [Google Scholar]
- 24. Heidt S, Roelen DL, Eijsink C, et al. Intravenous immunoglobulin preparations have no direct effect on B cell proliferation and immunoglobulin production. Clin Exp Immunol 2009;158:99–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dussault N, Ducas E, Racine C, et al. Immunomodulation of human B cells following treatment with intravenous immunoglobulins involves increased phosphorylation of extracellular signal‐regulated kinases 1 and 2. Int Immunol 2008;20:1369–1379. [DOI] [PubMed] [Google Scholar]
- 26. Cunningham ME, McGonigal R, Meehan GR, et al. Anti‐ganglioside antibodies are removed from circulation in mice by neuronal endocytosis. Brain 2016;139:1657–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fokkink W, Koch B, Ramakers C, et al. Pharmacokinetics and pharmacodynamics of intravenous immunoglobulin g maintenance therapy in chronic immune‐mediated neuropathies. Clin Pharmacol Ther 2017;102:709–716. [DOI] [PubMed] [Google Scholar]
- 28. Sordé L, Spindeldreher S, Palmer E, et al. Massive immune response against IVIg interferes with response against other antigens in mice: a new mode of action? PLoS ONE 2017;12:e0186046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xu W, Banchereau J. The antigen presenting cells instruct plasma cell differentiation. Front Immunol 2014;4:504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Barrett DJ, Ayoub EM. IgG2 subclass restriction of antibody to pneumococcal polysaccharides. Clin Exp Immunol 1986;63:127–134. [PMC free article] [PubMed] [Google Scholar]
- 31. von Gunten S, Smith DF, Cummings RD, et al. Intravenous immunoglobulin contains a broad repertoire of anticarbohydrate antibodies that is not restricted to the IgG2 subclass. J Allergy Clin Immunol 2009;123:1268–1276 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pollard AJ, Perrett KP, Beverley PC. Maintaining protection against invasive bacteria with protein‐polysaccharide conjugate vaccines. Nat Rev Immunol 2009;9:213–220. [DOI] [PubMed] [Google Scholar]
- 33. Fokkink WJ, Falck D, Santbergen TC, et al. Comparison of Fc N‐Glycosylation of pharmaceutical products of intravenous immunoglobulin G. PLoS ONE 2015;10:e0139828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Leblanc Y, Bihoreau N, Jube M, et al. Glycation of polyclonal IgGs: effect of sugar excipients during stability studies. Eur J Pharm Biopharm 2016;102:185–190. [DOI] [PubMed] [Google Scholar]
- 35. Ogino M, Orazio N, Latov N. IgG anti‐GM1 antibodies from patients with acute motor neuropathy are predominantly of the IgG1 and IgG3 subclasses. J Neuroimmunol 1995;58:77–80. [DOI] [PubMed] [Google Scholar]
- 36. Hebeis BJ, Klenovsek K, Rohwer P, et al. Activation of virus‐specific memory B cells in the absence of T cell help. J Exp Med 2004;199:593–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Beausang JF, Fan HC, Sit R, et al. B cell repertoires in HLA‐sensitized kidney transplant candidates undergoing desensitization therapy. J Transl Med 2017;15:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jacobs BC, van Doorn PA, Schmitz PI, et al. Campylobacter jejuni infections and anti‐GM1 antibodies in Guillain‐Barré syndrome. Ann Neurol 1996;40:181–187. [DOI] [PubMed] [Google Scholar]
- 39. Raphaël JC, Chevret S, Harboun M, et al. Intravenous immune globulins in patients with Guillain‐Barré syndrome and contraindications to plasma exchange: 3 days versus 6 days. J Neurol Neurosurg Psychiatry 2001;71:235–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Dominant IgM B‐cell clones are present in GBS patients before and after treatment with IVIg.