

Abstract
NMDA receptors (NMDARs) are glutamate‐gated ion channels that are key mediators of excitatory neurotransmission and synaptic plasticity throughout the central nervous system. They form massive heterotetrameric complexes endowed with unique allosteric capacity provided by eight extracellular clamshell‐like domains arranged as two superimposed layers. Despite an increasing number of full‐length NMDAR structures, how these domains cooperate in an intact receptor to control its activity remains poorly understood. Here, combining single‐molecule and macroscopic electrophysiological recordings, cysteine biochemistry, and in silico analysis, we identify a rolling motion at a yet unexplored interface between the two constitute dimers in the agonist‐binding domain (ABD) layer as a key structural determinant in NMDAR activation and allosteric modulation. This rotation acts as a gating switch that tunes channel opening depending on the conformation of the membrane‐distal N‐terminal domain (NTD) layer. Remarkably, receptors locked in a rolled state display “super‐activity” and resistance to NTD‐mediated allosteric modulators. Our work unveils how NMDAR domains move in a concerted manner to transduce long‐range conformational changes between layers and command receptor channel activity.
Keywords: allostery, glutamate, ligand‐gated ion channel, NMDA, receptor
Subject Categories: Neuroscience, Structural Biology
Introduction
A leading concept in molecular biology is the notion of “allosteric interaction” or “communication over distance” by which topographically distinct sites in a macromolecular structure interact through a discrete and reversible conformational change referred to as the “allosteric transition” (Monod et al, 1965). Allostery is widespread in the protein world and fulfills essential function in cellular signaling and inter‐cellular communication (Changeux & Christopoulos, 2016; Foster & Conn, 2017). Such is the case of ligand‐gated ion channels (LGICs), which mediate fast neurotransmission in the brain (Lemoine et al, 2012; Smart & Paoletti, 2012). Ligand‐gated ion channels undergo a key allosteric transition that converts chemical energy (neurotransmitter binding) into mechanical work (opening of a transmembrane ion channel pore). LGICs also undergo intense regulation through allosteric modulatory sites, distinct from the agonist and pore sites, which allow tuning of receptor channel activity by small ligand molecules known as allosteric modulators (Lemoine et al, 2012; Changeux & Christopoulos, 2016). Because of their high druggability and molecular selectivity, allosteric modulatory sites are of prime importance in pharmacology and therapeutics (e.g., Mony et al, 2009; Taly et al, 2009; Foster & Conn, 2017). Oligomerization and long‐range conformational changes provide an adequate setting for allostery, as emphasized by the classical examples of pentameric receptor channels (Nemecz et al, 2016). NMDA receptors, members of the superfamily of ionotropic glutamate receptors (iGluRs) which mediate excitatory neurotransmission and synaptic plasticity, are no exception as they assemble and operate as tetramers and contain a rich variety of modulatory sites scattered throughout the receptor (Traynelis et al, 2010; Zhu & Paoletti, 2015). However, the vast majority of our current understanding of iGluR mechanisms has focused and been conceptualized around individual domains or dimers of domains (Traynelis et al, 2010; Kumar & Mayer, 2013; Dawe et al, 2015; Greger et al, 2017). To grasp properly how these important receptors work and are regulated, it is now essential to integrate more intact views of the receptor and reason within the context of the tetrameric complex. The boom in structural studies of iGluRs, including the recent decoding of full‐length X‐ray and cryo‐EM structures of NMDARs (Karakas & Furukawa, 2014; Lee et al, 2014; Tajima et al, 2016; Zhu et al, 2016; Lu et al, 2017), provides an exceptional structural framework to tackle this issue.
iGluRs form massive (> 400 kDa) molecular complexes that operate as homo‐ or hetero‐tetramers. Individual iGluR subunits display a modular architecture, consisting of four domains (Traynelis et al, 2010; Greger et al, 2017): two large bilobate (clamshell‐like) extracellular domains, the N‐terminal domain (NTD) and the agonist‐binding domain (ABD); a transmembrane domain (TMD) where the ion channel resides, and an intracellular carboxy‐terminal domain (CTD) involved in receptor trafficking and signaling. In a functional receptor, the four subunits adopt a dimer‐of‐dimers arrangement and give rise to a stratified protein with at the “top” the NTDs, at the bottom the TMD and CTD, and sandwiched in between the ABDs (Fig 1A). The region encompassing the ABD layer and the TMD forms the core gating machinery. Because two adjacent ABDs pair through their top lobes, clamshell closure induced upon glutamate (or glycine) binding translates into separation between the ABD bottom lobes, which in turn pulls upon the TMD, eventually leading to channel gate opening (Kumar & Mayer, 2013; Zhou & Wollmuth, 2017). Although all iGluRs share this general activation mechanism, full‐length structures of NMDARs reveal strikingly different arrangements of the extracellular region compared to most AMPA and kainate receptors. In the latter, the NTDs “float” above the ABDs making minimal interactions with the gating core region (Sobolevsky et al, 2009; Meyerson et al, 2016, 2014; Twomey et al, 2017; but see Herguedas et al, 2016 for a more compact conformation of heteromeric GluA2/A3 AMPA receptors). In contrast, in NMDARs, the NTDs literally sit on top of the ABDs resulting in extensive interdigitations between the NTD and ABD layers (Karakas & Furukawa, 2014; Lee et al, 2014; Tajima et al, 2016; Zhu et al, 2016; Lu et al, 2017). This tight NTD‐ABD coupling likely relates to the unique role of NMDAR NTDs in allosteric signaling. In NMDARs, NTDs allosterically control key gating properties (channel open probability, deactivation kinetics, Gielen et al, 2009; Yuan et al, 2009) and harbor several binding sites for small‐molecule allosteric modulators, providing powerful ways to regulate receptor function (Hansen et al, 2010; Zhu & Paoletti, 2015). Similar to the ABDs, NTDs operate as dimers and adopt various conformational states (Karakas et al, 2011). Allosteric modulators act by shifting the conformational equilibrium between “active” and “inactive” states of the NTD dimers, thus boosting or dampening receptor activity (Gielen et al, 2009; Mony et al, 2011; Zhu et al, 2013; Romero‐Hernandez et al, 2016; Tajima et al, 2016).
Figure 1. NMDAR activation requires conformational mobility at inter‐domain and inter‐layer interfaces.
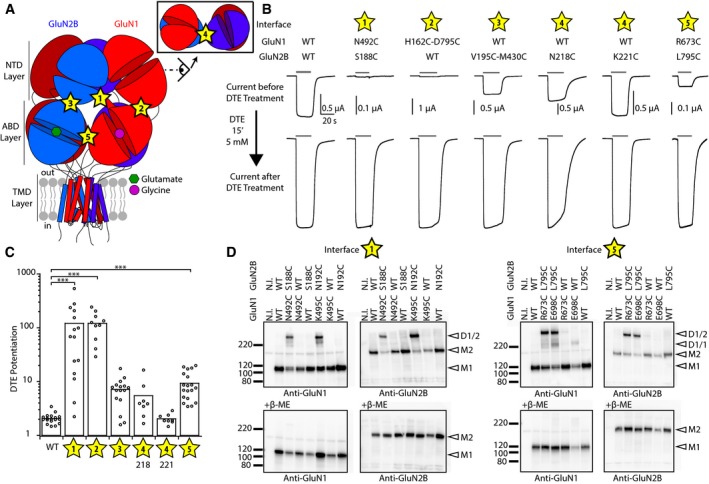
- Schematic representation of a full‐length GluN1/GluN2B NMDAR. The receptor displays a layered architecture. NTD, N‐terminal domain; ABD, agonist‐binding domain; TMD, transmembrane domain. The studied inter‐domain and inter‐layer interfaces are reported with numbered yellow stars. For clarity, interface 4 is shown from a side view (inset).
- Representative current traces from oocytes expressing wild‐type (WT) and double cysteine mutant NMDARs before and after DTE treatment. For each mutant, traces are normalized (in height, not in current amplitude) to the maximal response obtained after DTE treatment on wild‐type (WT) receptors.
- Summary of the DTE‐induced potentiation of WT and double cysteine mutants. Mean and n values are given in Appendix Table S1. ***P < 0.001, one‐way ANOVA on ranks (Kruskal–Wallis H test) followed by Bonferroni‐corrected Dunn's test.
- Immunoblots from Xenopus oocytes expressing either wt or mutant subunits. M1 indicates the GluN1 monomer (˜110 kDa); M2 the GluN2B monomer (˜180 kDa); D1/1 the GluN1 homodimer (˜220 kDa); and D1/2 the GluN1/GluN2B heterodimer (˜290 kDa). Lower panels: immunoblots in reducing conditions (+β‐mercaptoethanol). N.I., non‐injected oocytes.
Source data are available online for this figure.
While there is strong evidence that local conformational rearrangements within individual domains or dimers of domains are involved in NMDAR gating and modulation (Furukawa et al, 2005; Gielen et al, 2008; Kumar & Mayer, 2013; Regan et al, 2015), the importance, dynamics, and functional impact of contacts between the two constitutive dimers remains ill‐defined. Similarly, the long‐distance allosteric coupling between the membrane‐distal NTD layer and the downstream gating machinery, > 100 Å apart, is poorly understood. In this work, using a combination of macroscopic and single‐molecule electrophysiology, biochemical cross‐linking and computational approaches, we explore the dynamics of the complete NMDAR tetramer. We focused our analysis on the inter‐layer and inter‐dimer contacts revealed by the full‐length GluN1/GluN2B structures, aiming to understand the long‐range conformational interplay in the allosteric unit formed by the extracellular domains. We identify a rotation motion (or rolling) at the interface between the two ABD dimers as a critical quaternary reorganization during NMDAR gating. We show that trans‐layer communication converges through this rolling motion, which thus emerges as a key allosteric transition in the tetrameric complex. These results allow us to propose a first integrated view of NMDAR molecular operation, with important consequences on receptor physiology and drug action.
Results
NMDAR activation requires inter‐domain mobility
Recent crystal and cryo‐EM structures of full‐length NMDARs demonstrate broad interactions between the receptor's extracellular domains, with contacts both within and between subunits. Particularly striking is the extensive interface between the NTD and ABD layers (Karakas & Furukawa, 2014; Lee et al, 2014; Tajima et al, 2016; Zhu et al, 2016; Lu et al, 2017), providing molecular routes for the tight allosteric coupling between the NTDs and the downstream gating machinery (Gielen et al, 2009; Hansen et al, 2010). To assess the importance of inter‐domain contacts in NMDARs, we turned to a cross‐linking strategy based on engineered disulfide bridges and aiming at restraining the conformational mobility of the targeted interface. Guided by the full‐length structure of the inhibited GluN1/GluN2B receptor (in complex with glutamate, glycine and a GluN2B antagonist; TMD pore closed; Karakas & Furukawa, 2014; Lee et al, 2014), we introduced pairs of cysteines at positions close enough in space to disulfide bond (Cα‐Cα distance < 7 Å; Careaga & Falke, 1992). We targeted five sites, either in the NTD or ABD layer (sites 4 and 5, respectively; Fig 1A) or at the interface between the two (sites 1, 2 and 3; Fig 1A). We then expressed each GluN1/GluN2B double cysteine mutant receptor and, using electrophysiology, measured its activity and redox sensitivity (Fig 1B). Treatment with the reducing agent dithioerythritol (DTE) increased current amplitude of all mutant receptors. Levels of potentiation were particularly massive (> 100‐fold) at sites 1 (GluN1‐N492C/GluN2B‐S188C) and 2 (GluN1‐H162C‐D765C/GluN2B WT), contact zones between GluN1 ABD and the NTD layer. At both sites, a striking “awakening” phenotype was observed, which transformed almost completely silent receptors (currents barely measurable) prior DTE treatment to fully active receptors (currents similar in amplitude to that of WT receptors) following disulfide bond breakage (Fig 1B and C, and Appendix Table S1). Similar effects were observed on mutant receptors lacking the endogenous (and redox sensitive) GluN1 disulfide bridge (GluN1‐C744A‐C798A; Sullivan et al, 1994), thus demonstrating the critical role of the introduced disulfide bond (Appendix Fig S1A and Appendix Table S1). Marked potentiations (~10‐fold) were also observed at site 3, another contact region between the NTD and ABD layers, and at site 5, an ABD layer site where the two constitutive GluN1/GluN2 ABD dimers come in close proximity. In contrast, more modest effects were seen at site 4 (two different pairs of cysteines tested), a NTD layer site connecting the two constitutive GluN1 and GluN2 NTD dimers (Fig 1B and C, and Appendix Table S1).
Single‐mutant controls confirmed that both engineered cysteines are necessary to confer the enhanced redox sensitivity (Appendix Fig S1B and Appendix Table S1), while reversibility experiments using the oxidizing agent DTNB provided further support for the involvement of disulfide bonds (Appendix Fig S1C and Appendix Table S1). In the case of inter‐subunit interfaces (sites 1 and 5), we obtained direct evidence for the formation of disulfide links between GluN1 and GluN2B subunits by performing non‐reducing Western blots, and their respective controls using single cysteine mutants or reducing conditions (Fig 1D). The dramatic “unlocking” phenotypes observed at interfaces 1 and 2, along with the results obtained at positions 3 and 5, demonstrate that GluN1/GluN2B NMDARs can be trapped functionally in an inactive state, as represented by the currently available crystal structures. These results also reveal that conformational freedom at critical domain interfaces between the NTD and ABD layers, both within (sites 2 and 3) or between (site 1) subunits, is necessary for proper receptor activity. Screening of disulfide bridges in GluN1‐GluN2A receptors at homologous interfaces also revealed marked potentiation upon DTE release of the conformational constraint (Appendix Fig S1D), indicating shared mechanisms between GluN2A and GluN2B receptors.
Trapping a rolling motion at the ABD inter‐dimer interface results in super‐active receptors
We next sought to trap NMDARs in a high activity state. Although no structure of an active full‐length NMDAR is available, a complete cryo‐EM structure of the GluN1/GluN2B extracellular region (NTDs + ABDs) was recently described, likely capturing an active state as evidenced by the “compact” NTD dimer conformation (apposition of GluN1 NTD and GluN2B NTD lower lobes; Tajima et al, 2016). In this TMD‐lacking structure, the two ABD dimers undergo a conspicuous rotation relative to each other of ~13°. We coined this inter‐ABD dimer rotation “rolling”. We decided to investigate the functional effect of this motion by introducing cysteines to disulfide trap the inter‐ABD dimer interface and thus prevent rolling. In the inactive conformation, the two ABD dimers contact each other through a GluN1‐GluN2 inter‐subunit interface involving the short GluN1 α‐helix E and GluN2B α‐helix K. As shown in Fig 1, mutating GluN1‐R673 and GluN2B‐L795 to cysteines at this interface (site 5) locks the receptor in an inhibited state (Fig 1, interface 5). In the rolled conformation, these two residues move apart by several Angstroms, and a new interface is formed between GluN1 α‐helix G and GluN2B α‐helix K. A new duo of facing residues emerges, with GluN2B‐L795 switching partner from GluN1‐R673 to GluN1‐E698 (Cα‐Cα distance of 7.7 Å; Fig 2A). Inspired by this partner swap, we co‐expressed GluN2B‐L795C with GluN1‐E698C. Resulting receptors carried currents of particularly large amplitudes, while Western blots confirmed the spontaneous formation of a redox sensitive disulfide bond between the mutant GluN1 and GluN2B subunits (Fig 1D). Trace of GluN1‐E698C homodimers was also observed (see also Riou et al, 2012), prompting us to use the additional GluN1‐E698S control mutant in our functional tests. Using MK‐801 inhibition kinetics to assess the receptor channel activity (see Material and Methods), we discovered that trapped “rolled” GluN1‐E698C/GluN2B‐L795C receptors displayed a massive increase in channel open probability (channel Po), as evidenced by the 5.4‐fold ± 1.2 (n = 24) acceleration in MK‐801 inhibition on‐rate compared to wild‐type or single‐mutant control receptors (Fig 2B and C). The MK‐801 kinetics were in fact so fast that off‐rate MK‐801 washing kinetics, usually extremely slow, could also be easily quantified, revealing a pronounced 7.9 ± 2.1 (n = 23) fold speeding of recovery kinetics (Fig 2C). Similar effects were also observed at GluN1/GluN2A receptors (Appendix Fig S2A and B), although with lower amplitudes as expected from the higher basal maximal channel Po of GluN2A vs. GluN2B receptors (0.5 vs. 0.1; Erreger et al, 2005; Gielen et al, 2009). Whole‐cell patch‐clamp experiments performed on HEK cells fully confirmed the effects observed in Xenopus oocytes (Appendix Fig S2C–D). These results show that ABD dimer rolling motion profoundly impacts NMDAR activity. Receptors trapped in a rolled conformation are functionally switched to a super‐active state, in which agonist‐induced channel gate openings are greatly facilitated.
Figure 2. Rolling between the two constitutive ABD dimers boosts receptor activity.
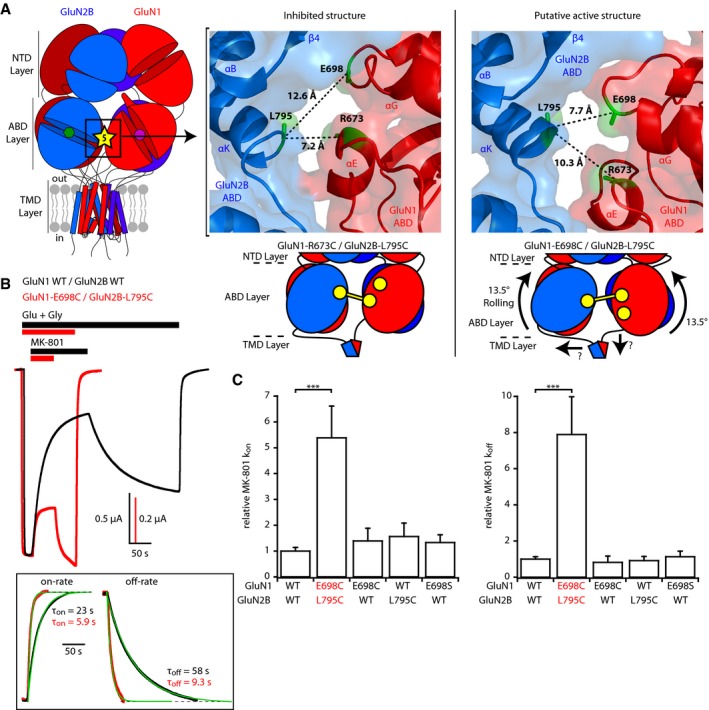
- Left, localization of site 5 at the interface between the two constitutive ABD dimers. Right top, crystal structures of the inhibited (PDB 5IOV; Zhu et al, 2016) and presumably active (5FXG; Tajima et al, 2016) states illustrating the differences in distances between GluN2B‐L795 and GluN1‐E698 or GluN1‐R673 at interface 5. Those three residues are colored green, while GluN1 and GluN2B subunits are colored red and blue, respectively. Right bottom, schematic representation of the GluN1‐R673C/GluN2B‐L795C disulfide cross‐link capturing an inhibited state of the receptor and of the GluN1‐E698C/GluN2B‐L795C disulfide cross‐link trapping the rolling motion.
- Assessment of receptor channel activity using MK‐801 inhibition kinetics. Representative current traces from oocytes expressing either wild‐type (WT) or GluN1‐E698C/GluN2B‐L795C mutant receptors in response to 10 nM MK‐801 during agonist application. Responses were scaled to the current amplitude obtained before MK‐801 application. Inset, mono‐exponential fits of MK‐801 wash‐in and wash‐out. Note the strikingly faster kinetics in mutant receptors, both at the onset and at the offset of MK‐801.
- Relative MK‐801 inhibition on‐ and off‐rate constants (k on and k off). All values were normalized to the value obtained for WT GluN1/GluN2B receptors. Mean and n values are given in Appendix Table S2. ***P < 0.001, one‐way ANOVA on ranks followed by Bonferroni‐corrected Dunn's test. Error bars, SD.
Super‐active receptors are resistant to NTD‐mediated allosteric modulation
Encouraged by the strong impact of “rolling” on the receptor gating, we subsequently investigated the influence of “rolling” on receptor pharmacology. For that purpose, we determined the sensitivity of receptors trapped in the rolled state (GluN1‐E698C/GluN2B‐L795C receptors; see above) to the agonist glutamate and glycine, and to various allosteric modulators. Exploiting the rich pharmacology conferred by the GluN2B NTD (Zhu & Paoletti, 2015; Fig 3A), we tested the sensitivity to ifenprodil, the prototypical GluN2B‐selective antagonist, and to spermine, zinc, and protons, all found endogenously in the brain and fine‐tuning NMDAR signaling (Traynelis et al, 2010; Paoletti, 2011; Zhu & Paoletti, 2015). We found that whatever the modulatory ligand, the “rolled” receptors displayed a striking phenotype of resistance to allosteric modulation. As assessed by measurements of IC50s from full dose–response curves, inhibition by zinc and ifenprodil was reduced by > 80‐fold and > 100‐fold, respectively (Fig 3B). The rightward shifts in sensitivity were as large as that obtained when removing the whole GluN2B NTD where both ligands bind (dashed curves, Fig 3B). We noticed, however, that shifts in zinc and ifenprodil sensitivity were also observed for the single‐mutant GluN1‐E698C/GluN2B receptor (but not for other single‐mutant controls; Appendix Fig S3A). Although homodimeric GluN1‐E698C cross‐linked subunits can form, they represent only a small fraction of total receptor number when co‐expressed with GluN2B‐L795C subunit (Fig 1D) and are thus unlikely to interfere with the observed phenotypes of the double cysteine mutant receptor. Proton inhibition of GluN1‐E698C/GluN2B‐L795C receptors was also markedly reduced (> 10‐fold; Fig 3B), while single‐mutant control receptors (including GluN1‐E698C/GluN2B) were unaffected (Appendix Fig S3A), further buttressing the exclusive role of the GluN1‐GluN2B cross‐link. The GluN1‐E698C/GluN2B‐L795C receptors are therefore largely insensitive to negative allosteric inhibition operated by the NTDs. Moreover, a spermine sensitivity test showed complete loss of spermine potentiation of “rolled” GluN2B receptors. Again, the situation was comparable to that observed on NTD‐deleted receptors (Fig 3C, and see Appendix Fig S3B for single‐mutant controls). Thus, rolled GluN2B receptors are also “immune” to NTD‐controlled allosteric potentiation. Recording from mutant GluN1/GluN2A receptors revealed that the resistance of “rolled” GluN2B receptors to allosteric modulation could be extended to GluN2A receptors (Appendix Fig S4A). In particular, the NTD‐mediated high‐affinity (nM) zinc sensitivity of GluN2A receptors was almost fully suppressed, with a rightward displacement of IC50 by 2 orders of magnitude. In contrast, “rolled” GluN2A receptors displayed unaltered inhibition by TCN‐201 (Appendix Fig S4B), a negative allosteric modulator that binds the local intra‐dimer ABD interface (Hansen et al, 2012; Yi et al, 2016). Hence, preventing the rolling–unrolling ABD motion has a major influence on the receptor's allosteric capacity, by functionally uncoupling the NTDs from the downstream gating machinery while more local gating transitions are preserved.
Figure 3. Super‐active receptors are insensitive to NTD‐mediated allosteric modulation.
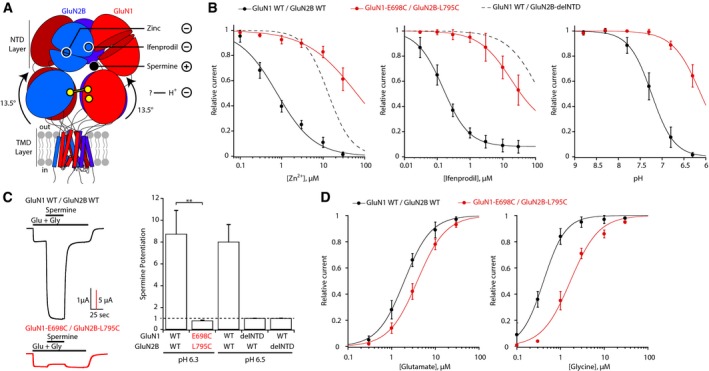
- Schematic representation of the super‐active GluN1‐E698C/GluN2B‐L795C receptor locked in the “rolled” conformation, showing the NTD‐binding sites for spermine, zinc, and ifenprodil, positive (+) and negative (−) allosteric modulators, respectively. Open circles indicate that the binding site is on the other side of the GluN2B NTD. The allosteric inhibitor H+ is also indicated, although its binding site remains ill‐defined.
- Zinc, ifenprodil, and pH dose–response curves of wild‐type (WT) GluN1/GluN2B receptors and mutant GluN1‐E698C/GluN2B‐L795C receptors. For comparison, zinc and ifenprodil dose–response curves of GluN1/GluN2B receptors lacking the whole GluN2B NTD (GluN1 WT/GluN2B‐delNTD) are also shown (dashed lines; data from Rachline et al, 2005). Values of IC50, maximal inhibition, Hill coefficient, and n are given in Appendix Table S3. Error bars, SD.
- Spermine (200 μM, pH 6.3) potentiation of WT GluN1/GluN2B and mutant GluN1‐E698C/GluN2B‐L795C receptors. Spermine (200 μM, pH 6.5) sensitivity is also shown for WT GluN1/GluN2B receptors and receptors lacking either the GluN1 (GluN1‐delNTD/GluN2B WT) or GluN2B (GluN1 WT/GluN2B‐delNTD) NTD (data from Mony et al, 2011). Mean values and n are given in Appendix Table S3. **P < 0.01, one‐way ANOVA on ranks followed by Bonferroni‐corrected Dunn's test. Error bars, SD.
- Glutamate and glycine dose–response curves of WT GluN1/GluN2B receptors and mutant GluN1‐E698C/GluN2B‐L795C receptors. Values of EC50, Hill coefficient, and n are given in Appendix Table S3. Error bars, SD.
We next investigated the sensitivity of rolled receptors to glutamate and glycine. While for GluN2B receptors, glutamate sensitivity was only slightly reduced (twofold increase in EC50), the effect on glycine sensitivity was stronger (fourfold increase in EC50; Fig 3D and see Appendix Figs S3C and S4C for control mutants and GluN2A receptors), potentially due to conformational constraints imposed by the disulfide bridge grafted onto the glycine‐binding GluN1 ABD (see Discussion). Measurement of glutamate deactivation kinetics also revealed a slightly faster off‐relaxation at rolled GluN2B receptors, in good agreement with the modest decrease in glutamate sensitivity observed under equilibrium conditions (Appendix Fig S3D and see Appendix Fig S4D for GluN2A receptors). Finally, rolled receptors underwent clear desensitization as their wild‐type counterparts, although steady‐state currents were larger on average (Appendix Figs S3E and S4E).
Rolling is coupled to NTD motions and favors pore opening
Our disulfide bridge results demonstrate that NMDARs are amenable to state trapping and that a simple conformational switch at the ABD tetrameric interface governs interconversion between active and inactive states of the receptor. They do not provide information on the conformational pathway between those states, however. Moreover, active NMDARs are still structurally ill‐defined since in the only “active” structure available, obtained using cryo‐EM (Tajima et al, 2016), the TMD region is missing preventing direct observation of receptor activation (i.e., pore opening). 3D modeling techniques have proven useful to study conformation transitions between various states of ion channels and receptors, including iGluRs (Dong & Zhou, 2011; Dutta et al, 2012, 2015; Dai & Zhou, 2013; Krieger et al, 2015; Pang & Zhou, 2017; Zheng et al, 2017). Accordingly, we first produced a full‐length model of the wild‐type GluN1‐GluN2B NMDAR (lacking the C‐terminus) combining the information of the inhibited state crystal structures (in complex with agonists and a GluN2B allosteric inhibitor; Karakas & Furukawa, 2014; Lee et al, 2014) and reconstructing the missing loops (see Material and Methods). We then modeled the transitions between the different structures of the receptor using iMODfit (Lopez‐Blanco & Chacon, 2013 and see Material and Methods), a program allowing flexible fitting of atomic structures into EM maps based on Normal Mode Analysis, and that has proven useful to study concerted motions of biomolecular structures (e.g., Gatsogiannis et al, 2016; Newcombe et al, 2018; Poepsel et al, 2018).
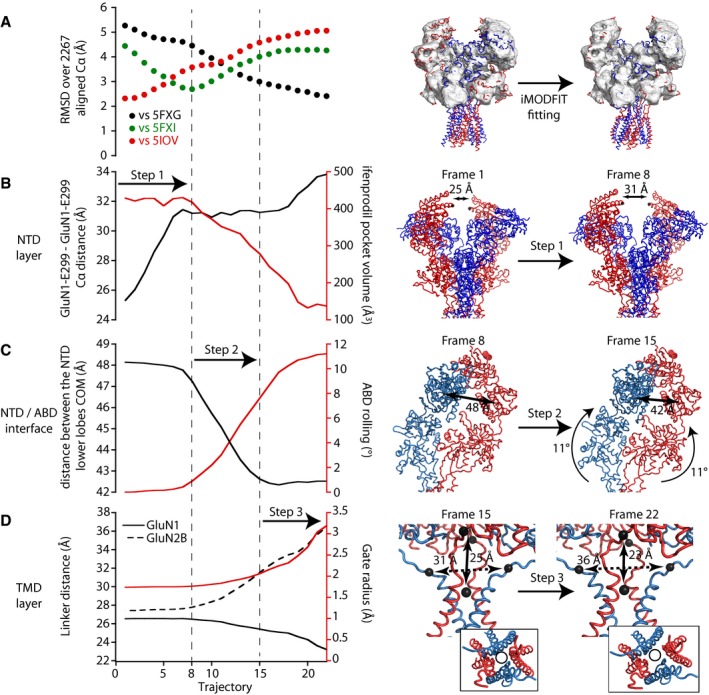
When fitting our full‐length model of the inhibited state into the TMD‐missing “active” state EM map, several features caught our attention (Fig 4, Movies EV1 and EV2). First, the RMSD between our model and the target structure (agonist‐bound “active” state, 5FXG; Tajima et al, 2016) dropped from 5.3 to 2.4 Å (2,267 aligned Cα), indicating a satisfactory fit (Fig 4A). Second, when comparing the fitting intermediates with the structure of the GluN2B NMDAR captured in a non‐active state (agonist bound, no antagonist; pdb 5FXI; Tajima et al, 2016), we noticed a minimum at 2.7 Å (Fig 4A), revealing that the trajectory passes by this experimentally determined structural state, even though this later is not used as input (Appendix Fig S5A). This structural match, observed under various fitting conditions (see Material and Methods and Appendix Fig S6), supports the realistic nature of our modeling. Third, the fitting trajectory could be consistently decomposed into three distinct and subsequent steps (Fig 4B–D). Step 1, resulting in a 5FXI‐like structure, involves mostly the membrane‐distal NTD layer with the two NTD dimers behaving as rigid bodies and moving apart from each other (Fig 4B). In the following step (Step 2), the two constitutive NTD dimers adopt a more compact conformation (with the GluN1 and GluN2 NTDs getting closer), while rolling of the two constitutive ABD dimers occurs. These structural rearrangements appear highly concerted (Fig 4C). Eventually, in the last part of the run (Step 3), major changes occur in the linking segments joining the pore TM3 helices to the ABDs with GluN1 linkers pushing down in a vertical movement and GluN2B linkers pulling toward the sides away from the channel fourfold axis of symmetry. Those motions are accompanied by a conspicuous dilation of the pore, as manifested by the splaying apart of the upper end of the TM3 helices (Fig 4D, right panel). Measurements of the pore radius revealed a wide enlargement of the ion conduction pathway around the gate region (Appendix Fig S5B), reminiscent of the iris‐type pore opening observed at AMPARs (Twomey et al, 2017). Although the fitting is realized on the extracellular domain (ECD) density only (sole density available), the elastic network model used for NMA maintains a realistic TMD and provides insight into the missing part of the structure (i.e., the transmembrane channel). Overall, our iMODfit analysis illuminates the fundamental role of ABD rolling in signal propagation in intact NMDARs. On one hand, it controls the energetics of the channel gate by acting on the TMD linkers as a gating switch; on the other, it transmits structural rearrangements of the “upper” NTD layer to the downstream gating machinery, thus permitting inter‐layer allosteric coupling.
Figure 4. Rolling mediates inter‐layer coupling and facilitates pore opening.
-
AiMODfit fitting of the GluN1/GluN2B inhibited state into the cryo‐EM map EMD‐3352 (target structure in a putatively active state; TMD not resolved). Left, evolution of the RMSD calculated against PDBs 5FXG (putative active state), 5FXI (non‐active state), and 5IOV (inhibited state) over 2,267 aligned Cα. The trajectory is divided into three steps (vertical dashed lines). Right, illustration of the fitting by superposition of the target EM density map (envelope representation) and the initial and final frames of the trajectory (line representation). GluN1 and GluN2B subunits are colored red and blue, respectively.
-
B–DLeft, evolution of selected collective variables during the iMODfit simulation. Each plot illustrates two collective variables, each with its own y‐axis (black or red). The upper panel (B) focuses on the NTD region (distance between the two GluN1 NTDs at the “apex” of the receptor; volume of the ifenprodil binding pocket at the interface between GluN1 and GluN2B NTDs), the middle panel (C) on the distance between NTD lower lobes and the ABD rolling motion, and the lower panel (D) on the ABD‐TMD connection and the M3 channel gate dilation. Right, illustration of the three steps of the fitting showing in step 1 (frame 1–8) the reduction of the inter NTD dimer distance; in step 2 (frame 8–15), the NTD lower lobes getting closer and the associated rolling of the ABD dimers; in step 3 (frame 15–22), the distance changes in the ABD‐M3 linker and the channel gate radius. The inset highlights a top view of the pore lining helixes M2 (P‐loop) and M3 with a same‐size circle emphasizing pore dilation during step 3. The GluN1 linker distance represents the distance between the center of masses (COM) of both GluN1‐R663 residues and the COM of the M3 SYTANLAAF sequence residues, and GluN2B linker distance represents the distance between both GluN2B‐E658 residues.
Structural rearrangements of an inter‐layer loop during rolling
NMDA receptors stand out from AMPA and kainate receptors by the extensive contacts between the NTD and ABD layers. In particular, in the inhibited state GluN1/GluN2B structure (Karakas & Furukawa, 2014; Lee et al, 2014), a GluN1‐specific loop (known as loop 2) which protrudes from GluN1 ABD upper lobe wedges in between GluN1 and GluN2B NTD lower lobes, making direct interaction with GluN2B NTD α4 helix. When covalently attaching this contact, receptors are fully silenced, in agreement with their immobilization in an inhibited state (Fig 1B, site 1 GluN1‐N492C/GluN2B‐S188C). Our modeling results reveal that loop 2 undergoes important structural rearrangements upon rolling, by sliding “downwards” GluN2B α4 helix. Accordingly, residues mutated to capture the inhibited state are moving away while GluN1‐K495 and GluN2B‐N192 are coming close (Fig 5A and B, and Movies EV1 and EV2). The functional significance of the sliding motion was tested experimentally via the mutation of those two residues into cysteines aiming to trap an active state at this inter‐layer interface. Assessment of channel activity using MK‐801 revealed a marked (twofold) acceleration of MK‐801 inhibition kinetics, indicative of an increased channel Po (Fig 5C). Western blots confirmed the formation of a disulfide bridge (Fig 1D), thus further buttressing the dynamic nature of NTD‐ABD interactions during gating of intact NMDARs.
Figure 5. Structural mobility of an inter‐layer GluN1 protruding loop during rolling.
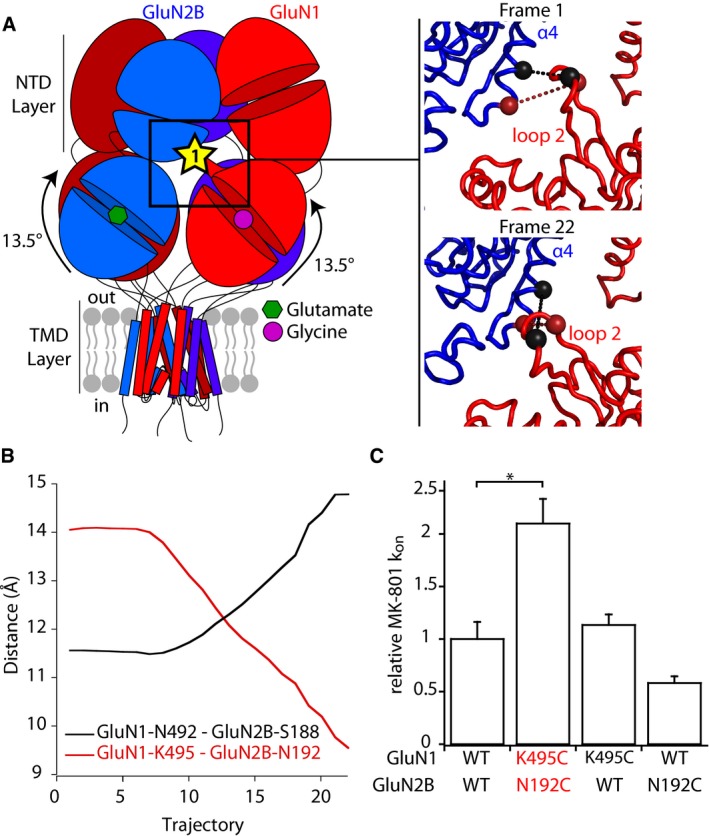
- Left, localization of interface 1 between GluN1 ABD and GluN2B NTD. This interface involves a GluN1‐specific loop that protrudes from GluN1 ABD upper lobe toward GluN2B NTD lower lobe. Right, initial and final frames of the fitting presented in Fig 4 at interface 1. The two pairs of residues targeted for cysteine mutations GluN1‐N492/GluN2B‐S188 and GluN1‐K495/GluN2B‐N192 are shown as spheres linked by a dotted line colored black and red, respectively.
- Evolution of Cα‐Cα distances of the two pairs of residues GluN1‐N492/GluN2B‐S188 and GluN1‐K495/GluN2B‐N192 during the fitting.
- MK‐801 inhibition kinetics. On‐rate constants (k on) of inhibition by 10 nM MK‐801 on wild‐type (WT) and mutant receptors. All values are normalized to that obtained for WT GluN1/GluN2B receptors. Values of Mean and n are given in Appendix Table S2. *P < 0.05, one‐way ANOVA on ranks followed by Bonferroni‐corrected Dunn's test. Error bars, SD.
Rolling controls channel flipping to open states
To gain further insights into the functional significance of the rolling mechanism and assess how it impacts NMDAR activation, we turned to single‐channel recordings. Recordings of individual receptor molecules provide a powerful tool to capture prominent features of NMDAR gating energetics and kinetics (Popescu & Auerbach, 2004; Erreger et al, 2005; Zhou & Wollmuth, 2017). We recorded single NMDAR channels either wild‐type receptors (GluN1/GluN2A or GluN1/GluN2B) or disulfide‐locked super‐active receptors (GluN1‐E698C/GluN2A‐L794C or GluN1‐E698C/GluN2B‐L795C). For GluN2A and GluN2B, the super‐active construct significantly enhanced receptor gating (Fig 6A and Appendix Fig S7A and B), in agreement with the results obtained at the macroscopic level. For GluN1/GluN2B receptors, equilibrium channel open probability (Po) increased nearly 2.5‐fold, matching very well the 2.81‐fold change in MK‐801 inhibition kinetics measured on whole oocytes in similar conditions (pH 8.0; Appendix Fig S7C and D). The increase in single‐channel Po reflected a significant increase in mean open time as well as a significant decrease in mean closed time (Fig 6B and Appendix Table S4). Super‐active GluN2B receptors spent most of the time in an active open channel state (mean equilibrium Po ± SEM of 0.74 ± 0.07 [n = 5]), with individual Po reaching values close to unity (0.94). In contrast, wild‐type GluN2B spent most of the time inactive (mean equilibrium Po of 0.29 ± 0.05 [n = 5]; highest individual Po value of 0.39). Given the pH insensitivity of super‐active receptors (but not of wild‐type receptors; see Fig 3B), this boost in Po is expected to be magnified even further at physiological pH (Fig 6B, dashed lines).
Figure 6. Influence of rolling on single‐receptor gating kinetics.
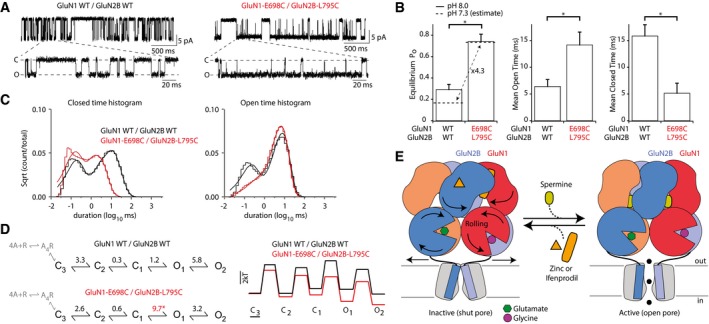
- Representative recordings of patches containing one single wild‐type (WT) or GluN1‐E698C/GluN2B‐L795C receptor. The bottom trace is an expanded view. C, closed channel; O, open channel.
- Single‐channel properties (equilibrium channel open probability, mean open time, mean closed time) of the WT and super‐active GluN1‐E698C/GluN2B‐L795C receptors. Mean and n values are given in Appendix Table S4. *P < 0.05, two‐tailed Student's t‐test, unpaired. Error bars, SEM.
- Closed (left) and open (right) time histograms for WT (black) or super‐active GluN1‐E698C/GluN2B‐L795C (red) receptors. Closed time histograms were best fit with five exponentials, whereas open time histograms were best fit with two exponentials (see Materials and Methods and Appendix Fig S8). Smooth lines are associated exponential fits.
- Kinetic schemes and equilibrium constants for WT (upper panel) or super‐active GluN1‐E698C/GluN2B‐L795C (lower panels) receptors. Records were analyzed at equilibrium. Ligand‐binding steps are shown in gray. Note that for simplicity, long‐lived desensitized steps are not shown. *P < 0.05 relative to WT GluN1/GluN2B (two‐tailed Student's t‐test, unpaired).
- Proposed mechanism for the conformational switch in full‐length tetrameric GluN1/GluN2B receptor: Inter‐dimer rolling in the ABD layer is coupled to entry of the NTD layer in its active state. The positive allosteric modulator spermine, which binds the interface between GluN1 and GluN2B NTD lower lobes, enhances receptor activity by stabilizing the NTD compact form and therefore rolling. Conversely, the negative allosteric modulators zinc and ifenprodil inhibit receptor activity by stabilizing an expanded form of the NTDs (lower lobes further apart) thus preventing rolling. The rolling motion facilitates pore opening by acting on the ABD‐TMD connecting linkers.
Following agonist binding, NMDARs undergo a series of kinetically resolvable transitions before pore opening. At least three non‐conducting and two conducting states are apparent from fully liganded receptors (Popescu & Auerbach, 2003; Erreger et al, 2005; Kazi et al, 2014). To further define how ABD rolling influences NMDAR function along the activation pathway, we fit our single‐channel dwell‐time histograms to a previously validated kinetic model of NMDAR activation (Kazi et al, 2014). Compared with wild‐type receptors, super‐active receptors showed marked reduction of short openings and complete disappearance of long closures (Fig 6C and Appendix Fig S8). This translated into clear changes in the energy profile on the pathway to receptor activation, resulting in enhanced fractional occupancies of open states (Fig 6D and Appendix Table S5). Remarkably, these effects were best accounted by an eightfold shift of a single equilibrium constant (K eq), that of the opening isomerization from C1 to O1 (C1–O1). These results identify the inter‐dimer ABD rolling motion as a key structural event closely associated with the flipping of closed NMDAR channels into a conductive and functionally active open state.
Discussion
The fundamental role of domain dimerization in iGluR activation, assembly, and regulation is firmly established (Traynelis et al, 2010; Herguedas et al, 2013; Kumar & Mayer, 2013; Dawe et al, 2015; Greger et al, 2017). Although dimer models of iGluRs have proved immensely useful to forge our understanding of how these receptors work, they provide simplified views and lack critical information about how the two pairs of dimers dynamically interact in the full‐length tetrameric assembly. Another key unanswered question concerns communication between layers within an intact receptor, an issue most relevant for NMDARs that display strong allosteric coupling between the membrane‐distal NTDs and the downstream ABD‐TMD gating core (Karakas et al, 2011; Mony et al, 2011; Zhu et al, 2013; Tajima et al, 2016). Thus, there are gaps in knowledge both in the receptor's lateral dimension, between the two constitutive dimers, and vertically, between layers. In this work, we help fill these gaps. Using a combination of biochemical, functional, and structural modeling analysis, we identify a rolling motion between the two constitutive ABD dimers as a key structural mechanism in NMDAR activation and NTD‐mediated allosteric modulation. Our single‐channel kinetics analysis shows that this reorientation of the two ABD dimers precipitates pre‐open closed channels to switch into the active open state. Moreover, we establish that inter‐dimer ABD rolling provides a conformational route by which structural changes within the NTD layer (Gielen et al, 2009; Karakas et al, 2011; Mony et al, 2011; Zhu et al, 2013; Krieger et al, 2015; Tajima et al, 2016) are transduced into rearrangement of the downstream gating machinery. Altogether, our results broaden our views of NMDAR operation from a dimeric to a more realistic tetrameric framework and provide integrated views of long‐distance domain coupling and dynamics in an intact NMDAR. Our work also highlights the potential of the iGluR inter‐dimer interfaces as novel sites for pharmacological manipulations.
As evidenced by the complete silencing of receptor activity, our disulfide trapping experiments reveal that conformational mobility at the NTD‐ABD layer interface is a prerequisite for receptor gating. This silencing phenotype differs strikingly from previous results showing that conformational freezing of individual domains within a given layer alters but not prevent receptor activation (Gielen et al, 2008; Mony et al, 2011; Paganelli et al, 2013; Zhu et al, 2013; Tajima et al, 2016). This essential inter‐layer mobility involves both inter‐ and intra‐subunit conformational rearrangements. Bridging the ABD to the NTD within the same GluN1 subunit (site 2, Fig 1) produced particularly large effects, highlighting the critical importance of structural rearrangements of the obligatory GluN1 subunit during receptor activation. This result corroborates previous findings that the GluN1 NTD, together with the GluN2 NTD, undergoes large‐scale conformational dynamics during receptor gating involving interlobe opening‐closure and twist–untwisting motions (Zhu et al, 2013, 2016; Krieger et al, 2015; Tajima et al, 2016). Although NMDARs devoid of NTDs are able to gate (Rachline et al, 2005; Qiu et al, 2009; Mony et al, 2011; Ogden & Traynelis, 2013), in intact receptors, NTDs are not static and activation of the receptor requires concerted conformational rearrangements between the NTDs and ABDs (see Movies EV1 and EV2). The tight packing of the NMDAR extracellular region (Karakas & Furukawa, 2014; Lee et al, 2014) compared to the more loosely organization in most AMPA and kainate receptors likely imposes strong structural constraints and inter‐dependence on NMDAR extracellular modules. Interestingly, the NMDAR NTD‐ABD interface is an important locus for allosteric modulation, harboring potential binding sites for small‐molecule drug compounds (Khatri et al, 2014), as well as hosting GluN1 subunit exon‐5 splice motif (Regan et al, 2018). Alterations in the stability of the NTD‐ABD interactions emerge as an effective mechanism to influence NMDAR activity. In AMPA receptors, similar mechanisms may also be at play at GluA2/A3 heteromers which have been proposed to adopt a more tightly packed NMDAR‐like conformation (Dutta et al, 2015; Herguedas et al, 2016).
Recent cryo‐EM data obtained on the GluN1/GluN2B receptor suggest that the two ABD dimers undergo a significant rotation movement when the receptor transit from the non‐active to the active conformation (Tajima et al, 2016). Based on cross‐linking mutagenesis, we now provide functional evidence that this rolling motion between the two pairs of ABD heterodimers is an essential step in the receptor gating mechanism. Although cross‐linking domain–domain interfaces may alter receptor structure and function in unforeseen ways, targeted loss‐ and gain‐of‐function phenotypes together with modeling results pinpoint rolling as the most parsimonious explanation for the observed effects. We propose that inter‐dimer ABD rolling acts as a gating switch that controls the energetics of channel opening but also as a pivotal allosteric transition, that structurally and functionally couples the “upper” NTD region with the receptor's gating core. Reorientation of the two GluN1/GluN2 ABD dimers in the tetrameric receptor provides a simple and powerful physical mechanism for translating structural changes of the NTD region to alterations of the ion channel gate (Fig 6E and Movies EV1 and EV2). In this scheme, close apposition of the NTD lower lobes, as observed in the “active” structure of the isolated GluN1/GluN2B NTD dimer (Tajima et al, 2016), favors ABD rolling which in turn increases channel activity. This is presumably the conformation stabilized by the positive allosteric modulator spermine, which would act as a molecular “glue” between GluN1 and GluN2B NTD lower lobes (Mony et al, 2011). Accordingly, super‐active mutant receptors, locked in a rolled state, display an exceptionally high channel Po, and are insensitive to spermine because there are already maximally activated (channel Po > 0.7). On the other hand, moving apart of the NTD lower lobes, as occurring when the NTD dimer transits to an “inactive” state (Tajima et al, 2016), promotes an unrolled state of the two ABD dimers, which in turn decreases channel activity. Our data indicate that negative allosteric modulators such as zinc, ifenprodil, or protons inhibit receptor activity by stabilizing this state. Super‐active mutants are thus unresponsive to NTD‐mediated allosteric inhibition because the two ABD pairs covalently linked cannot escape from their trapped rolled state.
Our 3D modeling analysis based on an original NMA fitting approach allows us to reconstruct the unresolved TMD of the cryo‐EM density (Tajima et al, 2016) and to explain how the ABD rolling motion may in turn influence the channel gate activity. It also provides the first insights onto NMDAR channel opening (Movies EV1 and EV2). Trajectory analysis indicates that ABD rolling directly translates into structural changes of the short linkers connecting the ABD to the ion channel. These linkers, which exert mechanical force to pull open the channel (Kazi et al, 2014), are differentially affected by ABD rolling whether the GluN1 or GluN2 subunit is considered. In GluN1, ABD rolling is coupled to a vertical movement that exerts compression forces on the downstream linkers. In contrast, in GluN2B, ABD rolling translates into a lateral separation of the linkers away from the ion channel central axis. This motion likely leads to an outward displacement of the upper end of the TM3 helices, eventually favoring pore dilation and opening. It is important to stress that ABD rolling by itself is insufficient to trigger pore opening. Indeed, super‐active mutant receptors locked in a rolled state are not constitutively active and still require agonists to gate. In agreement, “rolled” receptors are still sensitive to local perturbations of ABD clamshell conformations (as provided by the allosteric inhibitor TCN‐201) while NTD‐ABD coupling is essentially lost. Agonist‐induced closure of individual ABD clamshells and expansion of the ABD gating ring are the essential structural rearrangements necessary for channel gating (Furukawa et al, 2005; Twomey et al, 2017). ABD rolling provides a control mechanism that tunes the efficacy of chemical energy (agonist‐binding) conversion into mechanical forces (channel opening). Our single‐channel analysis reveals that ABD rolling massively and specifically affects a distinct intermediate state along the activation pathway: the late transition between a pre‐open closed state and an open state. Rolled receptors gate with high “efficacy” because they have a lower energy barrier to activation and their active open state is more stable. In studies examining how the NTD‐binding allosteric inhibitors zinc and ifenprodil affect the gating reaction of NMDARs, the same pre‐open to open transition is preferentially affected (Amico‐Ruvio et al, 2011, 2012). Single‐molecule FRET analysis also indicates that zinc inhibition of NMDARs occurs by uncoupling of the agonist‐induced changes at the ABD domains from the gating motions (Dolino et al, 2017). Our work provides the novel information that this uncoupling occurs through a quaternary rearrangement involving the unrolling of the two constitutive ABD dimers. We note that because of domain swapping (Sobolevsky et al, 2009) and tight packing between the NTD and ABD layers (Karakas & Furukawa, 2014; Lee et al, 2014), NMDAR NTDs are ideally positioned to affect how the two ABD pairs interact. Inter‐dimer ABD rotations also occur in AMPARs (Lau et al, 2013; Durr et al, 2014; Salazar et al, 2017), although the impact on receptor activation differs (Lau et al, 2013; Salazar et al, 2017). This is not unexpected given the much weaker NTD‐ABD interactions in AMPARs (at least in GluA2 homotetramers) and the gating specificities of each receptor subfamily.
Because of their high gating efficacy, we expected super‐active rolled NMDARs to display enhanced agonist sensitivity. This was not the case (see Fig 3). The ABD disulfide cross‐linking may impose structural constraints perturbing proper ABD clamshell closure, an effect that would oppose the enhanced gating efficacy. This effect may be particularly marked for the glycine‐binding GluN1 ABD where the disulfide bridge inserts on the lower lobe. Alternatively, as suggested by our macroscopic whole‐cell recordings, the conformational space explored by super‐active mutants may exclude some high agonist affinity desensitized states, rare states that may have been missed in our single‐channel recordings. NMDA receptors show a complex structural and energetic landscape (Dolino et al, 2017), translating into a particularly complex kinetic behavior with multiple intermediate closed steps (Popescu & Auerbach, 2004; Erreger et al, 2005; Schorge et al, 2005; Zhou & Wollmuth, 2017). There is obviously still much to be learned about the structure–function relationships underlying this elaborate behavior. It is thus unclear how the vertical motion of the GluN1 ABD‐TM3 linker impacts channel opening. What makes the NTD‐ABD linkers so critical in the receptor's allostery (Gielen et al, 2009; Yuan et al, 2009) is also unknown. Finally, how the inter‐dimer ABD rolling described here on GluN2B NMDARs compares to intra‐dimer ABD motions previously described on GluN2A NMDARs (Gielen et al, 2008; Borschel et al, 2011) remains to be clarified. The availability of additional full‐length structures of NMDARs, including in an active state, would certainly provide key insights into these questions.
Materials and Methods
Molecular biology
The pcDNA3‐based expression plasmids for rat GluN1‐1a (named GluN1 herein), rat GluN2A and mouse ε2 (named GluN2B herein) subunits, site‐directed mutagenesis strategy, and sequencing procedure have been described previously (Gielen et al, 2008). GluN1* represents the GluN1‐C744A‐C798A double mutant.
Two‐electrode voltage clamp (TEVC)
Recombinant NMDARs were expressed in Xenopus laevis oocytes after coinjection of 37 nl of a mixture of cDNAs or mRNAs (at 30–60 ng/μl; nuclear injection for cDNAs) coding for various GluN1‐1a and GluN2 subunits (ratio 1:1). mRNAs were obtained using mMESSAGE mMACHINE™ T7 Transcription Kit (Ambion™). Oocytes were prepared, injected, perfused, and voltage‐clamped as previously described (Gielen et al, 2008). Data were collected and analyzed using pClamp 10.5 (Molecular Devices) and fitted using Sigmaplot 11.0 (SSPS) or KaleidaGraph 4.0 (Synergy software). Unless otherwise mentioned, error bars represent the s.d. of the mean value.
The standard external solution contained (in mM) 100 NaCl, 2.5 KCl, 0.3 BaCl2, and 5 HEPES, pH adjusted to 7.3 with NaOH. In all “0” zinc solutions, 10 μM DTPA was added to chelate contaminating zinc (Paoletti et al, 1997). Recordings were performed at a holding potential of −60 mV and at room temperature. Unless otherwise noted, NMDAR‐mediated currents were induced by simultaneous application of saturating concentrations of l‐glutamate and glycine (100 μM each). Glutamate and glycine dose–response curves (DRC) were performed in the presence of 100 μM glycine and 100 μM glutamate, respectively. Agonists dose–response curves were fitted with the following Hill equation: I rel = 1/(1 + (EC50/[A])nH), where I rel is the mean relative current, [A] the agonist concentration, and n H the Hill coefficient. EC50 and n H were fitted as free parameters. For zinc DRC on GluN1/GluN2A receptors, 10 mM tricine was used to buffer zinc (Paoletti et al, 1997), and the following relationship was used to calculate the free zinc concentrations (Fayyazuddin et al, 2000; Gielen et al, 2008): [Zn]free = [Zn]added/200. No zinc buffer was used for zinc DRC on GluN1/GluN2B receptors. Ifenprodil and zinc DRC were fitted with the following Hill equation: I rel = 1−a/(1 + (IC50/[B]nH), where I rel is the mean relative current, [B] the ifenprodil or free zinc concentration, (1−a) the maximal inhibition and nH the Hill coefficient. IC50, a, and n H were fitted as free parameters except when fitting zinc sensitivity of GluN1/GluN2B receptors (a fixed to 1). For proton DRC, an external solution with increased HEPES concentration (40 mM) was used to insure proper pH buffering, and corrections for small shifts in the reference potential of bath electrodes and analysis of the data were performed as previously described (Gielen et al, 2008). Spermine sensitivity was assessed at pH 6.3 to maximize the spermine‐induced potentiation (Mony et al, 2011). For TCN‐201 experiments, glutamate was used at 100 μM while glycine concentrations were adjusted to ensure similar occupancy of the co‐agonist GluN1 ABD glycine site between WT and mutant GluN2A receptors (2 μM vs. 11 μM, respectively).
Redox treatment
To induce cleavage of disulfide bonds, oocytes were incubated for at least 15 min with DTE (dithioerythritol, 5 mM) in Barth solution (pH 8). To promote formation of disulfide bonds, oocytes were incubated for at least 10 min in DTNB (5,5’‐dithio‐bis‐[2‐nitrobenzoic acid], 0.5 mM) in Barth solution supplemented with DTPA (10 μM). All redox treatments on whole oocytes were performed “offline” (on non‐impaled oocytes). Reversibility was calculated as (I DTE − I DTNB)/(I DTE−I Initial) with I Initial the initial current level, I DTE the current level after DTE treatment, and I DTNB the current level after DTE and DTNB treatment.
Patch‐clamp recordings
Whole‐cell recordings
Recordings were performed using HEK293 cells (obtained from ATCC Inc.) transfected with GluN1, GluN2A, or GluN2B and a separate GFP construct (0.5 μg/ml each) using polyethylenimine (Polyscience). All experiments were performed 24–48 h post‐transfection at room temperature. Currents were sampled at 10 kHz and low‐pass‐filtered at 2 kHz using an Axopatch 200B amplifier and Clampfit 10.5 (Molecular Devices). Pipettes (thick‐wall, borosilicate, Hilgenberg) were pulled and fire‐polished achieving 3–5 MΩ resistances. At −60 mV, seal resistance ranged between 2 and 20 GΩ. The external solution contained (in mM): 140 NaCl, 10 HEPES, 2.8 KCl, 1 CaCl2, 20 sucrose, 0.01 DTPA, pH 7.3 (290–300 mOsm). The pipette solution contained (in mM): 110 D‐gluconate, 110 CsOH, 30 CsCl, 5 HEPES, 4 NaCl, 0.5 CaCl2, 2 MgCl2, 5 BAPTA, 2 Na+‐ATP, 0.3 Na+‐GTP, pH 7.35 (270–280 mOsm). Agonists (100 μM glycine and 100 μM glutamate) and MK‐801 (1 μM) were applied using a multi‐barrel fast perfusion exchanger (RSC‐200; BioLogic). Recordings were performed at a holding potential of −60 mV. Glutamate deactivation kinetics were fitted by single exponentials.
Single‐channel recordings
Recordings were performed on HEK293 cells (cell‐attached mode) transfected with GluN1 and GluN2A or GluN2B and a separate peGFP‐Cl construct (at a ratio N1:N2:eGFP of 4:1.5:1 for GluN2A or 4:2.5:1 for GluN2B) using X‐tremeGene HP (Roche). All experiments were performed 24–48 h post‐transfection at 21–24°C. Currents were recorded using an integrating patch‐clamp amplifier (Axopatch 200B, Molecular Devices), analog‐filtered at 10 kHz (four‐pole Bessel filter), and digitized at 50 kHz (ITC‐16 interfaced with PatchMaster, HEKA). Patch pipettes (thick‐wall, borosilicate, Sutter Instruments) were pulled and fire‐polished achieving resistances between 10 and 20 MΩ when measured in the bath. At −100 mV, seal resistance ranged between 2 and 20 GΩ. Cells were transferred to coverslips in a bath solution containing (in mM): 150 NaCl, 10 HEPES, 2.5 KCl, pH 8.0. Patch pipettes were filled with bath solution and 1 mM glutamate and 0.1 mM glycine. 0.05 mM EDTA was added to minimize gating effects of divalent ions. Inward currents were elicited by applying a pipette potential of +100 mV.
Single‐channel analysis
Analysis of single‐channel records was comparable to Kazi et al (2014). Briefly, after recordings were complete, data were exported from PatchMaster to QuB (https://qub.mandelics.com) for processing (removable of unstable regions, defining baseline) and analysis. Processed data were idealized using the segmental k‐means (SKM) algorithm. Kinetic analysis was performed using the maximum interval likelihood (MIL) algorithm in QuB with a dead time of 20 μs. We used a linear, fully liganded state model containing three closed states, two desensitized states, and two open states to fit the data (see Kazi et al, 2014 for details). For each individual record, state models with increasing closed (3–6) and open (2–4) states were constructed and fitted to the recordings until log‐likelihood (LL) values improved by less than 10 LL units/added state or if the next added state showed 0% occupancy. All constructs and records were best fit by 5 closed states and 2 open states. Time constants and the relative areas of each component, the transition rate constants (forward, k f and reverse, k r), as well as mean closed time (MCT) and mean open time (MOT), were averaged for each receptor. All analysis was done on patches that contained a single channel, which was clearly definable for all constructs given their high open probability. Kinetic analysis was done only on patches that had a minimum of 10,000 events and had low noise.
Thermodynamic effects
We quantified the Gibbs free energy of each transition using the following:
where k f and k r refer to the forward and reverse rates of that transition, R is the empirical gas constant (1.987 × 10−3 kcal/mol), and T is the recording temperature (295 Kelvin).
MK‐801 inhibition
MK‐801 is an open channel blocker (that is, blocker action requires prior channel gate opening) with slow reversibility, and consequently, the rate at which MK‐801 inhibits NMDAR responses depends on the level of channel activity, that is, channel open probability (Po; Huettner & Bean, 1988; Jahr, 1992; Rosenmund et al, 1993). Based on this principle, MK‐801 is classically used to estimate the level of NMDAR channel activity in macroscopic whole‐cell recordings (see, for instance, Gielen et al, 2009; Talukder et al, 2010; Hansen et al, 2013; Zhu et al, 2013). Note, however, that MK‐801 inhibition kinetics can index relative, not absolute, Po. Final solutions containing MK‐801 (10 nM at pH 7.3 and 30 nM at pH 8 for oocytes experiments, 1 μM for HEK cells) were prepared by dilution of the stock solutions into the agonist‐containing solution. MK‐801 inhibition time constants (τon) were obtained by fitting currents with a single‐exponential component within a time window corresponding to 10–90% of the maximal inhibition. Recovery from inhibition was also measured for GluN1‐E698C/GluN2B‐L795C and GluN1‐E698C/GluN2A‐L794C because its rate was high enough for substantial recovery in a reasonable delay. On‐rate (k on) and off‐rate (k off) constants were then calculated with a first‐order reaction scheme: k on = 1/([MK‐801]*τon) and k off = 1/τoff. Each constant was then normalized to the mean constant of the corresponding wild‐type receptors measured in the same conditions on the same day.
Immunoblotting
For each condition, sixteen oocytes were selected based on their high level of NMDAR expression as assessed by TEVC recordings. Selected oocytes were instantly frozen in liquid nitrogen for later use. Oocytes expressing “silent” mutant receptors were selected based on redox treatment of other oocytes of the same batch revealing high expression of “silent” receptors. Each mix of 16 oocytes was then homogenized on ice by back and forth pipetting with 160 μl of lysis buffer (20 mM Tris pH 8.0, 50 mM NaCl, 1% DDM, 1/20 of a complete protease inhibitor cocktail tablet, Roche Complete, Mini) until a homogenous suspension was obtained. The samples were then centrifuged (15,000 g for 8 min at 4°C), re‐homogenized, and centrifuged again. Supernatants enriched in membrane proteins were collected and separated in two equal volumes for subsequent Western blotting experiments in non‐reducing and reducing (9% vol/vol β‐mercaptoethanol added in the loading buffer) conditions. Samples were separated on 3–8% SDS–PAGE gradient gels (4 oocytes per lane), semi‐dry transferred to nitrocellulose membrane, and immunoblotted with anti‐GluN1 antibody (1:750, mouse monoclonal MAB1586 clone R1JHL, Millipore) or anti‐GluN2B antibody (1:500, mouse monoclonal 75‐101 clone N59/36, NeuroMab). Protein bands were visualized using secondary goat peroxidase‐linked anti‐mouse antibodies (1:10,000, Jackson ImmunoResearch catalog number #115‐035‐003), with the SuperSignal™ West Pico Chemiluminescent Substrate (Thermo Scientific™).
Chemicals
Salts (including ZnCl2), DTPA, tricine, DTE, DTNB, and spermine were purchased from Sigma‐Aldrich. A stock solution of DTNB was prepared at 50 mM in 500 mM HEPES pH 8.0. DTPA was prepared as 10 mM stock aliquots at pH 7.3. Ifenprodil (generous gift from Synthélabo, France) was prepared as 10 mM stock aliquots in 1% HCl. MK‐801 (Ascent Scientific) was prepared as 50 μM stock aliquots. TCN‐201 (Tocris) was prepared as 10 mM stock aliquots in DMSO.
Molecular modeling and fitting
A 3D model of full‐length GluN1/GluN2B NMDAR representing an inhibited state based on the best resolved parts of the pdbs 4TLL, 4TLM (Lee et al, 2014), and 4PE5 (Karakas & Furukawa, 2014) was initially produced using Modeller (Eswar et al, 2006), with rat (GluN1) and mouse (GluN2B) protein sequences and reconstruction of missing loops and sidechains. To obtain complete models of different states of the receptor, the model was then fitted using iMODfit (Lopez‐Blanco & Chacon, 2013) to the following target cryo‐EM maps: EMD‐8098 (Zhu et al, 2016) for the agonist‐bound inhibited state (in complex with glutamate, glycine, and the GluN2B antagonist Ro‐256981), EMD‐8106 (Zhu et al, 2016) for the “resting‐like” DCKA/D‐APV‐bound state, EMD‐3352 (Tajima et al, 2016) for the putative active state (in complex with glutamate and glycine; TMD region not resolved), and EMD‐3354 (Tajima et al, 2016) for the agonist‐bound non‐active state (in complex with glutamate and glycine but ion channel pore is closed).
To gain insight into the transitions between those four states, each one of the four templates obtained was then fitted again using the cryo‐EM maps of the three other states as target. The transition from the inhibited state to EMD‐3352 is presented extensively. Replication of the calculations of this transition with identical iMODfit parameters yielded highly similar trajectories (Appendix Fig S6A), demonstrating the robustness over the randomization in the choices of modes made at each step. Control runs modifying the EM density map threshold (<cutoff> parameter), the range of modes (‐n option), or fixing secondary elements dihedral (‐S option) also showed that results were reproducible and robust (Appendix Fig S6B–D), and that the best fitting was obtained with default program options. In all runs targeting EMD‐3352, we used the volume eraser option of UCSF Chimera (Pettersen et al, 2004) to erase the density of the poorly resolved TMD region. This procedure allowed to avoid detergent density influence in fitting, and therefore, only the density of the extracellular part of the receptor as target could be used. We performed control fittings by erasing the TMD on cryo‐EM maps of other states to ensure that pore opening was not a mere artifact of TMD density erasing.
Trajectory analysis was performed using the R package Bio3D (Grant et al, 2006). 43 collective variables were measured among which (i) the ABD‐TMD linker “tension”, measured for GluN1 as the distance between the center of masses of both GluN1‐R663 residues and the center of masses of the four SYTANLAAF sequences, and for GluN2B as the GluN2B‐E658–GluN2B‐E658 distance; (ii) the ABD “rolling”, measured as the dihedral angle between a vector defined by the center of masses of an ABD domain lower and upper lobes and the equivalent vector of the opposite domain. The volume of the ifenprodil binding pocket (Karakas et al, 2011) was calculated as the volume of the non‐clashing part of a 7 Å sphere centered at equal distance of GluN1‐G112 and GluN2B‐Q110. Because of the twofold symmetry of the receptor extracellular region, distance between the center of masses of the lower lobes of the NTD dimers, volume of the ifenprodil pocket, extent of ABD rolling, and distances between the GluN1 ABD loop and the GluN2 NTD can be measured two times independently on the tetrameric receptor. A mean value of both measures was taken after verifying that they were not significantly different.
Structure illustrations
The figures were prepared with PyMOL (http://pymol.org) using either our whole‐receptor (‐CTD) model (based on PDB 4PE5, 4TLL, and 4TLM; Karakas & Furukawa, 2014; Lee et al, 2014), iMODfit‐based simulations (targeting cryo‐EM map EMD‐3352; Tajima et al, 2016), or cryo‐EM‐based PDBs 5FXG (Tajima et al, 2016) and 5IOV (Zhu et al, 2016).
Statistical analysis
All statistical tests and n numbers are specified in the main text or figure legends. Results are presented as mean ± SD (standard deviation of the mean) unless otherwise noted. To assess statistical significance, unpaired Student's t‐test or one‐way ANOVA was used as appropriate.
Author Contributions
J‐BE, DS, and TG designed and performed all experiments except the single‐channel recordings which were performed by KC. J‐BE, DS, and AT performed the iMODfit analysis. MD provided technical help in molecular biology. KC and LPW analyzed the single‐channel recordings. DS, AT and PP supervised the work and participated in data analysis. J‐BE and PP wrote the manuscript, with help from all authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Movie EV1
Movie EV2
Review Process File
Source Data for Figure 1
Acknowledgements
This work was supported by the French government (“Investissements d'Avenir” ANR‐10‐LABX‐54 MEMO LIFE, ANR‐11‐IDEX‐0001‐02 PSL Research University, ANR‐11‐LABX‐0011‐01), the European Research Council (ERC Advanced Grant #693021 to PP), and grants from the National Institutes of Health (NS 088479 to LPW and F30‐MH115618 to KC). We thank Jose Ramon Lopez Blanco for his assistance with iMODFIT and Jérôme Hénin for his assistance in the measure of dihedral angles.
The EMBO Journal (2019) 38: e99894
Contributor Information
Antoine Taly, Email: antoine.taly@ibpc.fr.
Pierre Paoletti, Email: pierre.paoletti@ens.fr.
References
- Amico‐Ruvio SA, Murthy SE, Smith TP, Popescu GK (2011) Zinc effects on NMDA receptor gating kinetics. Biophys J 100: 1910–1918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amico‐Ruvio SA, Paganelli MA, Myers JM, Popescu GK (2012) Ifenprodil effects on GluN2B‐containing glutamate receptors. Mol Pharmacol 82: 1074–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borschel WF, Murthy SE, Kasperek EM, Popescu GK (2011) NMDA receptor activation requires remodelling of intersubunit contacts within ligand‐binding heterodimers. Nat Commun 2: 498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Careaga CL, Falke JJ (1992) Thermal motions of surface alpha‐helices in the D‐galactose chemosensory receptor. Detection by disulfide trapping. J Mol Biol 226: 1219–1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Changeux JP, Christopoulos A (2016) Allosteric modulation as a unifying mechanism for receptor function and regulation. Cell 166: 1084–1102 [DOI] [PubMed] [Google Scholar]
- Dai J, Zhou HX (2013) An NMDA receptor gating mechanism developed from MD simulations reveals molecular details underlying subunit‐specific contributions. Biophys J 104: 2170–2181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawe GB, Aurousseau MR, Daniels BA, Bowie D (2015) Retour aux sources: defining the structural basis of glutamate receptor activation. J Physiol 593: 97–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolino DM, Chatterjee S, MacLean DM, Flatebo C, Bishop LDC, Shaikh SA, Landes CF, Jayaraman V (2017) The structure‐energy landscape of NMDA receptor gating. Nat Chem Biol 13: 1232–1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Zhou HX (2011) Atomistic mechanism for the activation and desensitization of an AMPA‐subtype glutamate receptor. Nat Commun 2: 354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durr KL, Chen L, Stein RA, De Zorzi R, Folea IM, Walz T, McHaourab HS, Gouaux E (2014) Structure and dynamics of AMPA receptor GluA2 in resting, pre‐open, and desensitized states. Cell 158: 778–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta A, Shrivastava IH, Sukumaran M, Greger IH, Bahar I (2012) Comparative dynamics of NMDA‐ and AMPA‐glutamate receptor N‐terminal domains. Structure 20: 1838–1849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta A, Krieger J, Lee JY, Garcia‐Nafria J, Greger IH, Bahar I (2015) Cooperative dynamics of intact AMPA and NMDA glutamate receptors: similarities and subfamily‐specific differences. Structure 23: 1692–1704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erreger K, Dravid SM, Banke TG, Wyllie DJ, Traynelis SF (2005) Subunit‐specific gating controls rat NR1/NR2A and NR1/NR2B NMDA channel kinetics and synaptic signalling profiles. J Physiol 563: 345–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N, Webb B, Marti‐Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A (2006) Comparative protein structure modeling using Modeller. Curr Protoc Bioinformatics Chapter 5: Unit‐5.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fayyazuddin A, Villarroel A, Le Goff A, Lerma J, Neyton J (2000) Four residues of the extracellular N‐terminal domain of the NR2A subunit control high‐affinity Zn2+ binding to NMDA receptors. Neuron 25: 683–694 [DOI] [PubMed] [Google Scholar]
- Foster DJ, Conn PJ (2017) Allosteric modulation of GPCRs: new insights and potential utility for treatment of schizophrenia and other CNS disorders. Neuron 94: 431–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa H, Singh SK, Mancusso R, Gouaux E (2005) Subunit arrangement and function in NMDA receptors. Nature 438: 185–192 [DOI] [PubMed] [Google Scholar]
- Gatsogiannis C, Merino F, Prumbaum D, Roderer D, Leidreiter F, Meusch D, Raunser S (2016) Membrane insertion of a Tc toxin in near‐atomic detail. Nat Struct Mol Biol 23: 884–890 [DOI] [PubMed] [Google Scholar]
- Gielen M, Le Goff A, Stroebel D, Johnson JW, Neyton J, Paoletti P (2008) Structural rearrangements of NR1/NR2A NMDA receptors during allosteric inhibition. Neuron 57: 80–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gielen M, Siegler Retchless B, Mony L, Johnson JW, Paoletti P (2009) Mechanism of differential control of NMDA receptor activity by NR2 subunits. Nature 459: 703–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant BJ, Rodrigues AP, ElSawy KM, McCammon JA, Caves LS (2006) Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics 22: 2695–2696 [DOI] [PubMed] [Google Scholar]
- Greger IH, Watson JF, Cull‐Candy SG (2017) Structural and functional architecture of AMPA‐type glutamate receptors and their auxiliary proteins. Neuron 94: 713–730 [DOI] [PubMed] [Google Scholar]
- Hansen KB, Furukawa H, Traynelis SF (2010) Control of assembly and function of glutamate receptors by the amino‐terminal domain. Mol Pharmacol 78: 535–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KN, Ogden KK, Traynelis SF (2012) Subunit‐selective allosteric inhibition of glycine binding to NMDA receptors. J Neurosci 32: 6197–6208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KB, Tajima N, Risgaard R, Perszyk RE, Jorgensen L, Vance KM, Ogden KK, Clausen RP, Furukawa H, Traynelis SF (2013) Structural determinants of agonist efficacy at the glutamate binding site of N‐methyl‐D‐aspartate receptors. Mol Pharmacol 84: 114–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herguedas B, Garcia‐Nafria J, Cais O, Fernandez‐Leiro R, Krieger J, Ho H, Greger IH (2016) Structure and organization of heteromeric AMPA‐type glutamate receptors. Science 352: aad3873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herguedas B, Krieger J, Greger IH (2013) Receptor heteromeric assembly‐how it works and why it matters: the case of ionotropic glutamate receptors. Prog Mol Biol Transl Sci 117: 361–386 [DOI] [PubMed] [Google Scholar]
- Huettner JE, Bean BP (1988) Block of N‐methyl‐D‐aspartate‐activated current by the anticonvulsant MK‐801: selective binding to open channels. Proc Natl Acad Sci USA 85: 1307–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahr CE (1992) High probability opening of NMDA receptor channels by L‐glutamate. Science 255: 470–472 [DOI] [PubMed] [Google Scholar]
- Karakas E, Simorowski N, Furukawa H (2011) Subunit arrangement and phenylethanolamine binding in GluN1/GluN2B NMDA receptors. Nature 475: 249–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakas E, Furukawa H (2014) Crystal structure of a heterotetrameric NMDA receptor ion channel. Science 344: 992–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazi R, Dai J, Sweeney C, Zhou HX, Wollmuth LP (2014) Mechanical coupling maintains the fidelity of NMDA receptor‐mediated currents. Nat Neurosci 17: 914–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatri A, Burger PB, Swanger SA, Hansen KB, Zimmerman S, Karakas E, Liotta DC, Furukawa H, Snyder JP, Traynelis SF (2014) Structural determinants and mechanism of action of a GluN2C‐selective NMDA receptor positive allosteric modulator. Mol Pharmacol 86: 548–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger J, Bahar I, Greger IH (2015) Structure, dynamics, and allosteric potential of ionotropic glutamate receptor N‐terminal domains. Biophys J 109: 1136–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar J, Mayer ML (2013) Functional insights from glutamate receptor ion channel structures. Annu Rev Physiol 75: 313–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau AY, Salazar H, Blachowicz L, Ghisi V, Plested AJ, Roux B (2013) A conformational intermediate in glutamate receptor activation. Neuron 79: 492–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CH, Lu W, Michel JC, Goehring A, Du J, Song X, Gouaux E (2014) NMDA receptor structures reveal subunit arrangement and pore architecture. Nature 511: 191–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemoine D, Jiang R, Taly A, Chataigneau T, Specht A, Grutter T (2012) Ligand‐gated ion channels: new insights into neurological disorders and ligand recognition. Chem Rev 112: 6285–6318 [DOI] [PubMed] [Google Scholar]
- Lopez‐Blanco JR, Chacon P (2013) iMODFIT: efficient and robust flexible fitting based on vibrational analysis in internal coordinates. J Struct Biol 184: 261–270 [DOI] [PubMed] [Google Scholar]
- Lu W, Du J, Goehring A, Gouaux E (2017) Cryo‐EM structures of the triheteromeric NMDA receptor and its allosteric modulation. Science 355: eaal3729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyerson JR, Kumar J, Chittori S, Rao P, Pierson J, Bartesaghi A, Mayer ML, Subramaniam S (2014) Structural mechanism of glutamate receptor activation and desensitization. Nature 514: 328–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyerson JR, Chittori S, Merk A, Rao P, Han TH, Serpe M, Mayer ML, Subramaniam S (2016) Structural basis of kainate subtype glutamate receptor desensitization. Nature 537: 567–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monod J, Wyman J, Changeux JP (1965) On the nature of allosteric transitions: a plausible model. J Mol Biol 12: 88–118 [DOI] [PubMed] [Google Scholar]
- Mony L, Kew JN, Gunthorpe MJ, Paoletti P (2009) Allosteric modulators of NR2B‐containing NMDA receptors: molecular mechanisms and therapeutic potential. Br J Pharmacol 157: 1301–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mony L, Zhu S, Carvalho S, Paoletti P (2011) Molecular basis of positive allosteric modulation of GluN2B NMDA receptors by polyamines. EMBO J 30: 3134–3146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemecz A, Prevost MS, Menny A, Corringer PJ (2016) Emerging molecular mechanisms of signal transduction in pentameric ligand‐gated ion channels. Neuron 90: 452–470 [DOI] [PubMed] [Google Scholar]
- Newcombe J, Chatzidaki A, Sheppard TD, Topf M, Millar NS (2018) Diversity of nicotinic acetylcholine receptor positive allosteric modulators revealed by mutagenesis and a revised structural model. Mol Pharmacol 93: 128–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogden KK, Traynelis SF (2013) Contribution of the M1 transmembrane helix and pre‐M1 region to positive allosteric modulation and gating of N‐methyl‐D‐aspartate receptors. Mol Pharmacol 83: 1045–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paganelli MA, Kussius CL, Popescu GK (2013) Role of cross‐cleft contacts in NMDA receptor gating. PLoS One 8: e80953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang X, Zhou HX (2017) Structural modeling for the open state of an NMDA receptor. J Struct Biol 200: 369–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P, Ascher P, Neyton J (1997) High‐affinity zinc inhibition of NMDA NR1‐NR2A receptors. J Neurosci 17: 5711–5725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P (2011) Molecular basis of NMDA receptor functional diversity. Eur J Neuorsci 33: 1351–1365 [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25: 1605–1612 [DOI] [PubMed] [Google Scholar]
- Poepsel S, Kasinath V, Nogales E (2018) Cryo‐EM structures of PRC2 simultaneously engaged with two functionally distinct nucleosomes. Nat Struct Mol Biol 25: 154–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popescu G, Auerbach A (2003) Modal gating of NMDA receptors and the shape of their synaptic response. Nat Neurosci 6: 476–483 [DOI] [PubMed] [Google Scholar]
- Popescu G, Auerbach A (2004) The NMDA receptor gating machine: lessons from single channels. Neuroscientist 10: 192–198 [DOI] [PubMed] [Google Scholar]
- Qiu S, Zhang XM, Cao JY, Yang W, Yan YG, Shan L, Zheng J, Luo JH (2009) An endoplasmic reticulum retention signal located in the extracellular amino‐terminal domain of the NR2A subunit of N‐Methyl‐D‐aspartate receptors. J Biol Chem 284: 20285–20298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachline J, Perin‐Dureau F, Le Goff A, Neyton J, Paoletti P (2005) The micromolar zinc‐binding domain on the NMDA receptor subunit NR2B. J Neurosci 25: 308–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regan MC, Romero‐Hernandez A, Furukawa H (2015) A structural biology perspective on NMDA receptor pharmacology and function. Curr Opin Struct Biol 33: 68–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regan MC, Grant T, McDaniel MJ, Karakas E, Zhang J, Traynelis SF, Grigorieff N, Furukawa H (2018) Structural mechanism of functional modulation by gene splicing in NMDA receptors. Neuron 98: 521–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riou M, Stroebel D, Edwardson JM, Paoletti P (2012) An alternating GluN1‐2‐1‐2 subunit arrangement in mature NMDA receptors. PLoS One 7: e35134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero‐Hernandez A, Simorowski N, Karakas E, Furukawa H (2016) Molecular basis for subtype specificity and high‐affinity zinc inhibition in the GluN1‐GluN2A NMDA receptor amino‐terminal domain. Neuron 92: 1324–1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Clements JD, Westbrook GL (1993) Nonuniform probability of glutamate release at a hippocampal synapse. Science 262: 754–757 [DOI] [PubMed] [Google Scholar]
- Salazar H, Eibl C, Chebli M, Plested A (2017) Mechanism of partial agonism in AMPA‐type glutamate receptors. Nat Commun 8: 14327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorge S, Elenes S, Colquhoun D (2005) Maximum likelihood fitting of single channel NMDA activity with a mechanism composed of independent dimers of subunits. J Physiol 569: 395–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart TG, Paoletti P (2012) Synaptic neurotransmitter‐gated receptors. Cold Spring Harb Perspect Biol 4: a009662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobolevsky AI, Rosconi MP, Gouaux E (2009) X‐ray structure, symmetry and mechanism of an AMPA‐subtype glutamate receptor. Nature 462: 745–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan JM, Traynelis SF, Chen HS, Escobar W, Heinemann SF, Lipton SA (1994) Identification of two cysteine residues that are required for redox modulation of the NMDA subtype of glutamate receptor. Neuron 13: 929–936 [DOI] [PubMed] [Google Scholar]
- Tajima N, Karakas E, Grant T, Simorowski N, Diaz‐Avalos R, Grigorieff N, Furukawa H (2016) Activation of NMDA receptors and the mechanism of inhibition by ifenprodil. Nature 534: 63–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talukder I, Borker P, Wollmuth LP (2010) Specific sites within the ligand‐binding domain and ion channel linkers modulate NMDA receptor gating. J Neurosci 30: 11792–11804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taly A, Corringer PJ, Guedin D, Lestage P, Changeux JP (2009) Nicotinic receptors: allosteric transitions and therapeutic targets in the nervous system. Nat Rev Drug Discovery 8: 733–750 [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R (2010) Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 62: 405–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twomey EC, Yelshanskaya MV, Grassucci RA, Frank J, Sobolevsky AI (2017) Channel opening and gating mechanism in AMPA‐subtype glutamate receptors. Nature 549: 60–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi F, Mou T‐C, Dorsett KN, Volkmann RA, Menniti FS, Sprang SR, Hansen KB (2016) Structural basis for negative allosteric modulations of GluN2A‐containing NMDA receptors. Neuron 91: 1316–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Hansen KB, Vance KM, Ogden KK, Traynelis SF (2009) Control of NMDA receptor function by the NR2 subunit amino‐terminal domain. J Neurosci 29: 12045–12058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Wen H, Iacobucci GJ, Popescu GK (2017) Probing the structural dynamics of the NMDA receptor activation by coarse‐grained modeling. Biophys J 112: 2589–2601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HX, Wollmuth LP (2017) Advancing NMDA receptor physiology by integrating multiple approaches. Trends Neurosci 40: 129–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Paoletti P (2015) Allosteric modulators of NMDA receptors: multiple sites and mechanisms. Curr Opin Pharmacol 20: 14–23 [DOI] [PubMed] [Google Scholar]
- Zhu S, Stein RA, Yoshioka C, Lee CH, Goehring A, McHaourab HS, Gouaux E (2016) Mechanism of NMDA receptor inhibition and activation. Cell 165: 704–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Stroebel D, Yao CA, Taly A, Paoletti P (2013) Allosteric signaling and dynamics of the clamshell‐like NMDA receptor GluN1 N‐terminal domain. Nat Struct Mol Biol 20: 477–485 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Movie EV1
Movie EV2
Review Process File
Source Data for Figure 1