

Abstract
The E3 ubiquitin ligase Parkin is a key effector of the removal of damaged mitochondria by mitophagy. Parkin determines cell fate in response to mitochondrial damage, with its loss promoting early onset Parkinson's disease and potentially also cancer progression. Controlling a cell's apoptotic response is essential to co‐ordinate the removal of damaged mitochondria. We report that following mitochondrial damage‐induced mitophagy, Parkin directly ubiquitinates the apoptotic effector protein BAK at a conserved lysine in its hydrophobic groove, a region that is crucial for BAK activation by BH3‐only proteins and its homo‐dimerisation during apoptosis. Ubiquitination inhibited BAK activity by impairing its activation and the formation of lethal BAK oligomers. Parkin also suppresses BAX‐mediated apoptosis, but in the absence of BAX ubiquitination suggesting an indirect mechanism. In addition, we find that BAK‐dependent mitochondrial outer membrane permeabilisation during apoptosis promotes PINK1‐dependent Parkin activation. Hence, we propose that Parkin directly inhibits BAK to suppress errant apoptosis, thereby allowing the effective clearance of damaged mitochondria, but also promotes clearance of apoptotic mitochondria to limit their potential pro‐inflammatory effect.
Keywords: apoptosis, BAK, BAX, mitophagy, Parkin
Subject Categories: Autophagy & Cell Death; Post-translational Modifications, Proteolysis & Proteomics
Introduction
The RING‐between‐RING E3 ubiquitin ligase, Parkin and its upstream regulator PTEN‐induced Kinase 1 (PINK1) are involved in the removal of damaged mitochondria through a selective form of autophagy termed mitophagy (Nguyen et al, 2016; Harper et al, 2018; Pickles et al, 2018). The importance of Parkin‐mediated mitophagy is highlighted by the deletion or mutation of the gene encoding Parkin (PRKN/PARK2) as a driver of autosomal recessive juvenile Parkinson's disease (PD; Kitada et al, 1998) and also its emerging role in tumourigenesis (Poulogiannis et al, 2010; Veeriah et al, 2010; Bernardini et al, 2017). Hence, understanding how Parkin influences cell survival will provide important new insight in to how defects in Parkin promote neurodegenerative conditions and cancer (Kitada et al, 1998; Poulogiannis et al, 2010; Veeriah et al, 2010).
The proposed obligate activator of Parkin, PINK1, is constitutively expressed and is normally imported to the inner mitochondrial membrane where its N‐terminal region is cleaved by the presenilin‐associated rhomboid‐like protease (PARL; Jin et al, 2010; Deas et al, 2011) promoting its export and degradation by the 26S proteasome (Yamano & Youle, 2013). Following mitochondrial insult, including loss of the mitochondrial membrane potential or accumulation of misfolded mitochondrial proteins, PINK1 is stabilised on the mitochondrial outer membrane (Jin et al, 2010; Jin & Youle, 2013). Two key substrates of PINK1 are ubiquitin and Parkin itself, both of which are phosphorylated on Ser65 (Kondapalli et al, 2012; Shiba‐Fukushima et al, 2012; Kane et al, 2014; Kazlauskaite et al, 2014; Koyano et al, 2014; Ordureau et al, 2014; Wauer et al, 2015b). Binding of phospho‐ubiquitin is required for Parkin recruitment to mitochondria (Kane et al, 2014; Koyano et al, 2014; Shiba‐Fukushima et al, 2014). Indeed, a number of structural studies have elegantly demonstrated that phospho‐ubiquitin binding permits phosphorylation of Parkin (Kazlauskaite et al, 2015; Kumar et al, 2015; Sauve et al, 2015; Wauer et al, 2015a). During Parkin activation, a number of structural rearrangements occur, notably the release of the repressor element of Parkin (Wauer et al, 2015a; Pao et al, 2016; Kumar et al, 2017; Tang et al, 2017). Recent studies have also interrogated Parkin conformational changes following phosphorylation by PINK1, highlighting the importance of releasing the RING2 domain of Parkin (Gladkova et al, 2018; Sauve et al, 2018). Once conformationally active, Parkin then ubiquitinates a range of mitochondrial substrates that have been shown to modulate cellular metabolism, mitochondrial dynamics, cell cycling and apoptosis (Yoshii et al, 2011; Sarraf et al, 2013). Adaptor proteins that bind to ubiquitin on the mitochondrial surface in turn recruit and extend the autophagosome around the damaged mitochondrion (Wong & Holzbaur, 2014; Heo et al, 2015; Lazarou et al, 2015; Moore & Holzbaur, 2016; Richter et al, 2016). Whilst many of these proteomic studies have been performed in model systems over‐expressing Parkin (Sarraf et al, 2013; Cunningham et al, 2015), a recent study showed that endogenous Parkin ubiquitinates similar substrates in neurons, although some site and temporal differences were observed (Ordureau et al, 2018). Interestingly, recent studies have also shown that baseline mitophagy can proceed in a PINK1/Parkin‐independent fashion (Lee et al, 2018; McWilliams et al, 2018), perhaps implicating PINK1 and Parkin as emergency responders to mitochondrial damage.
Cellular stresses including DNA damage and cell matrix detachment induce apoptosis by upregulating or activating pro‐apoptotic BH3‐only proteins leading to activation of the downstream effectors of apoptosis: BCL‐2 Antagonist/Killer‐1 (BAK) and BCL‐2‐Associated X protein (BAX; Wei et al, 2001; Youle & Strasser, 2008). Activation promotes BAK (and BAX) homo‐dimerisation then multimerisation of homodimers that permeabilise the mitochondrial outer membrane (Dewson et al, 2008, 2012; Bleicken et al, 2010; Oh et al, 2010; Zhang et al, 2010; Czabotar et al, 2013; Aluvila et al, 2014; Subburaj et al, 2015). This releases apoptogenic factors including cytochrome c and commits a cell to death via initiation of a proteolytic caspase cascade (Wei et al, 2001). Activation of BAK and BAX by BH3‐only proteins, and their subsequent homo‐dimerisation, involves a hydrophobic surface groove comprising α‐helices 3–5 that is shared with pro‐survival BCL‐2 proteins (Petros et al, 2000; Dewson et al, 2008; Czabotar et al, 2013; Brouwer et al, 2014).
Parkin activity has been proposed to determine cellular fate. Limited mitophagic stress leads to effective mitochondrial clearance and a pro‐survival response, whilst excessive mitochondrial damage leads Parkin to induce apoptosis (Zhang et al, 2014). Among its substrates, Parkin ubiquitinates members of the B‐cell lymphoma‐2 (BCL‐2) family of proteins that regulate the intrinsic pathway of apoptosis (Sarraf et al, 2013). It has been argued that following mitochondrial damage, Parkin promotes apoptosis by targeting the pro‐survival BCL‐2 protein Myeloid Cell Leukemia1 (MCL1) for degradation (Carroll et al, 2014; Zhang et al, 2014). However, how Parkin limits apoptosis to permit clearance of damaged mitochondria is unclear. Parkin has been proposed to inhibit pro‐apoptotic BAX either by limiting recruitment of cytosolic BAX to mitochondria (Johnson et al, 2012; Charan et al, 2014) or by promoting degradation of dysregulated or mutated BAX (Cakir et al, 2017). However, this occurs at steady state without the mitochondrial damage thought necessary to promote PINK1‐dependent Parkin activity.
We have identified BAK as an important substrate of Parkin following mitophagic stress. Following mitochondrial depolarisation, Parkin ubiquitinated a conserved residue in the hydrophobic groove of BAK (Lys113), thereby impairing BAK activation, oligomerisation and apoptotic activity. Hence, Parkin is able to inhibit BAK and BAX by distinct mechanisms. We propose that BAK ubiquitination is a mechanism by which Parkin limits mitochondrial outer membrane permeabilisation, thus allowing the clearance of damaged mitochondria.
Results
Parkin inhibits BAX by preventing its mitochondrial localisation
Parkin‐mediated mitophagy is regarded as a pro‐survival response to mitochondrial damage, and activated Parkin has been previously reported to ubiquitinate and inhibit the pro‐apoptotic effector protein BAX (Fig 1A; Johnson et al, 2012). To test this, BAX −/− BAK −/− HeLa cells reconstituted with human BAX and ectopically expressing HA‐Parkin were treated with the mitochondrial electron transport chain uncouplers, antimycin A and oligomycin (AO), to induce Parkin activation and recruitment to mitochondria, followed by induction of apoptosis with the BH3 mimetics ABT‐737 (Oltersdorf et al, 2005) and S63485 (Kotschy et al, 2016). Cell death as assessed by Lactate Dehydrogenase (LDH) release was significantly reduced in Parkin‐expressing cells treated with AO suggesting that activated Parkin limits BAX‐mediated apoptosis (Fig 1B).
Figure 1. Parkin inhibits BAX‐mediated apoptosis by indirect ubiquitination.
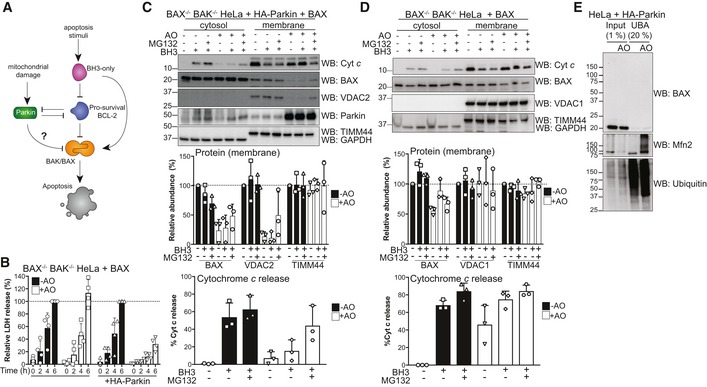
- Schematic of potential effects of activated Parkin on the BCL‐2 family of apoptosis regulators.
- BAX/BAK‐deficient HeLa cells reconstituted with BAX and where indicated HA‐Parkin were treated with antimycin A and oligomycin (AO) for 2 h and then 1 μM of each of the BH3 mimetics ABT‐737 and S63485 for up to 6 h. Cell death was assessed at the indicated times by LDH assay. Data are mean ± SD of four independent experiments.
- BAX/BAK‐deficient HeLa cells ectopically expressing BAX and HA‐Parkin were treated with antimycin A and oligomycin (AO, 3 h), MG132 (20 μM, 3.5 h) and BH3 mimetics (1 μM of each of ABT‐737 and S63485, 1 h). Cytosol and membrane fractions were immunoblotted as indicated. Graph shows densitometric analysis of non‐ubiquitinated membrane proteins (top, relative to untreated control) or cytochrome c release (bottom; cytosol/cytosol + membrane, relative to untreated controls) from three independent experiments. Error bars represent mean ± SD.
- BAX/BAK‐deficient HeLa cells ectopically expressing BAX were treated and analysed as in (C).
- UBA pull‐down of ubiquitinated proteins following antimycin A and oligomycin (AO) treatment of BAX/BAK‐deficient HeLa cells ectopically expressing HA‐Parkin and BAX for 3 h. Representative immunoblot from two independent experiments.
Source data are available online for this figure.
As BAX has been shown to be mislocalised from mitochondria as a result of Parkin's E3 ligase activity (Charan et al, 2014), we tested whether this contributed to the inhibition of apoptosis. BAX‐mediated permeabilisation of the mitochondrial outer membrane as measured by cytochrome c release into the cytosol was reduced following AO treatment of Parkin‐expressing HeLa cells (Fig 1C). Consistent with previous reports (Johnson et al, 2012; Charan et al, 2014), Parkin activation also reduced BAX levels at mitochondria (Fig 1C). Both the mitochondrial targeting of BAX and its ability to mediate cytochrome c release following activation of Parkin could be partially rescued with the addition of the proteasome inhibitor MG132 (Fig 1C). Consistent with BAX‐mediated cell death (Fig 1B), the effect of AO on BAX‐mediated cytochrome c release was dependent on Parkin (Fig 1D), although mitochondrial BAX was somewhat reduced upon AO treatment of Parkin‐null cells (Fig 1D).
To further elucidate whether BAX ubiquitination was responsible for its mislocalisation, we induced Parkin activity with AO in HeLa cells expressing Parkin and examined BAX ubiquitination (Fig 1E). Following induction of mitophagy, as expected, we observed Parkin‐dependent ubiquitination of the canonical substrate Mitofusin 2 (Mfn2) (Figs 1E and EV1A). However, we did not detect ubiquitinated forms of BAX in either whole cell lysates or in samples enriched for ubiquitinated proteins using recombinant GST‐Ubiquitin‐associated Domain (UBA) pull‐down (Fig 1E). These data suggested that the inhibition of BAX apoptotic function is likely indirect. As Parkin is auto‐inhibited until recruited to mitochondria, we reasoned that this might be through targeting a mitochondrial protein. BAX interacts with voltage‐dependent anion channel (VDAC) 2 at mitochondria and VDAC2 promotes BAX apoptotic activity (Yamagata et al, 2009; Ma et al, 2014). VDAC2 is one of the most prominent substrates of Parkin (Ordureau et al, 2018), suggesting that its ubiquitination could be responsible for the inability of BAX to mediate cytochrome c release. Consistent with this, the deubiquitination of VDAC2 observed following MG132 treatment correlated with the partial rescue of BAX localisation and cytochrome c release (Fig 1D). It is important to note that Parkin‐conjugated ubiquitin chains are commonly non‐degradative (Ordureau et al, 2018) and so BAX inhibition may not be due to VDAC2 degradation, but rather steric hindrance of the interaction between BAX and ubiquitinated VDAC2. Nevertheless, BAX‐mediated apoptotic activity is suppressed by Parkin in the absence of significant BAX ubiquitination.
Figure EV1. Ubiquitination of BAK by Parkin occurs in SH‐SY5Y and MEF cells on the conserved Lys113 in the hydrophobic surface groove (relates to Figs 1 and 2).
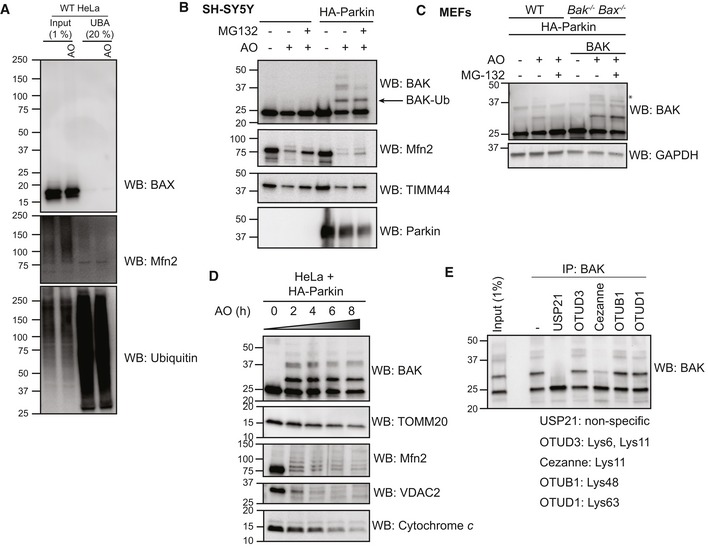
- UBA pull‐down of ubiquitinated proteins following antimycin A and oligomycin (AO) treatment of wild‐type (WT) HeLa cells for 3 h. Representative immunoblot from two independent experiments.
- Endogenous BAK is ubiquitinated in neuroblastoma cells upon mitochondrial damage. SH‐SY5Y cells ectopically expressing Parkin were treated with antimycin A and oligomycin (AO) for 2 h. Representative immunoblots of three independent experiments.
- Endogenous BAK is ubiquitinated in MEFs upon mitochondrial damage. Wild‐type MEFs (WT) or Bak −/− Bax −/− MEFs expressing hBAK and ectopically expressing HA‐Parkin were treated with antimycin A and oligomycin (AO) with or without MG132 for 2 h. Lysates were immunoblotted as described. Note that endogenous mouse BAK was ubiquitinated and that ubiquitinated forms of mouse and human BAK (*) were reduced with MG132. Representative immunoblots of three independent experiments.
- Timecourse of mitophagy induction. HeLa cells expressing HA‐Parkin were treated with antimycin A and oligomycin (AO) for the indicated times prior to immunoblotting of membrane fractions as described. Representative immunoblot from two independent experiments.
- BAK −/− BAX −/− HeLa cells expressing BAK and HA‐Parkin were treated with antimycin A and oligomycin (AO) for 2 h prior to immunoprecipitation of BAK. Immunoprecipitates were incubated with the indicated ubiquitin linkage‐specific DUB prior to analysis of BAK by immunoblotting. Data representative of three independent experiments.
Source data are available online for this figure.
Parkin ubiquitinates BAK during mitophagy
As BAK is constitutively localised to the outer mitochondrial membrane, whereas BAX is predominantly cytosolic (Fig 2A), we hypothesised that BAK could be a substrate for Parkin to limit apoptosis. Strikingly, upon activation of Parkin with AO we detected significant modification of BAK, consistent with its potential mono‐ and poly‐ubiquitination (Fig 2A). This modification of BAK was dependent on Parkin, as it was not detected in HeLa cells that lack endogenous Parkin (Fig 2B). At this early timepoint of mitochondrial damage, the ubiquitination of Parkin substrates Mfn2, VDAC1 and VDAC2 was detected (Fig 2B and C; Sarraf et al, 2013; Cunningham et al, 2015; McLelland et al, 2018). However, cytochrome c levels were unchanged, indicating that the ubiquitination of BAK preceded mitochondrial clearance (Fig 2B).
Figure 2. Parkin ubiquitinates BAK on a conserved lysine in the hydrophobic groove.
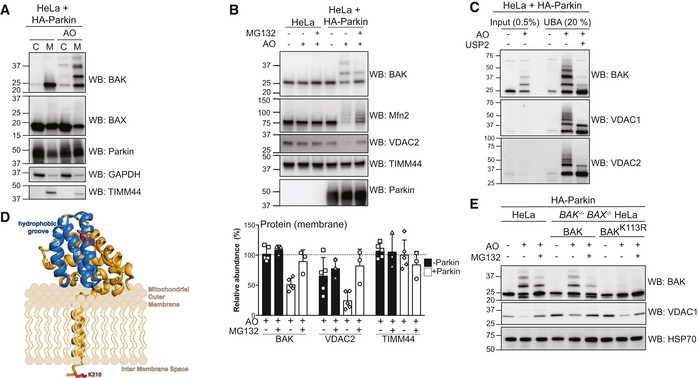
- Subcellular fractionation of cytosol and membrane fractions of HeLa cells expressing HA‐Parkin following 2 h of antimycin A and oligomycin (AO) treatment. Representative immunoblot of three independent experiments.
- Immunoblotting of whole cell lysates following treatment with antimycin A and oligomycin (AO) for 3 h with MG132 (20 μM). Graph shows densitometric analysis of non‐ubiquitinated proteins relative to untreated control from three independent experiments. Error bars represent mean ± SD.
- UBA pull‐down of ubiquitinated proteins following antimycin A and oligomycin (AO) treatment for 3 h and treatment with the non‐specific DUB USP2 for 30 min at 37°C, representative immunoblot from two independent experiments.
- Structure of human BAK (PDB 2IMS) with the hydrophobic binding groove comprising α‐helices 3–5 highlighted in blue. The ubiquitination site, K113, (red) resides in the binding groove. A modelled transmembrane anchor (α9) and the C‐terminal K210 localised to the inter‐membrane space are based on evidence that the BAK transmembrane anchor spans the mitochondrial outer membrane (Iyer et al, 2015). The orientation of the soluble portion (α‐helices 1–8) of BAK with respect to the mitochondrial outer membrane is hypothetical.
- Whole cell lysates of HA‐Parkin and BAK variant‐expressing HeLa cells following 3 h antimycin A and oligomycin (AO) treatment with 30 min pre‐treatment with MG132 (20 μM), representative immunoblots from three independent experiments.
Source data are available online for this figure.
To confirm that the modification on BAK was ubiquitin, we performed UBA pull‐down coupled with a deubiquitination assay. Following induction of mitophagy with AO, ubiquitinated forms of BAK could be enriched and the higher molecular weight forms were removed following treatment with the non‐specific deubiquitinase (DUB), ubiquitin‐specific protease‐2 (USP2; Fig 2C). To confirm that BAK ubiquitination was not a consequence of its overexpression, we also confirmed that endogenous BAK was a Parkin substrate in SH‐SY5Y neuroblastoma cells and mouse embryonic fibroblasts (MEFs; Fig EV1B and C).
Interestingly, in contrast to Mfn2 and VDAC1 that were rapidly poly‐ubiquitinated, BAK was predominantly mono‐ubiquitinated. In addition, poly‐ubiquitinated forms of BAK were persistent over time suggesting a non‐degradative ubiquitin chain linkage (Fig EV1D). Consistent with this, poly‐ubiquitinated BAK was significantly reduced with the K11‐specific DUB, Cezanne, whilst the K48‐specific OTUB1 had little effect (Fig EV1E). This suggests that BAK is modified predominantly with K11‐linked ubiquitin chains, in accord with the atypical chain linkages catalysed by Parkin during mitophagy (Cunningham et al, 2015). It is important to note, however, that the predominant form of modified BAK is the mono‐ubiquitinated form. As shown previously (Sarraf et al, 2013; Martinez et al, 2017), treatment with the proteasome inhibitor, MG132, enhanced detection of VDAC1 and ubiquitinated Mfn2 (Fig 2B). In contrast, ubiquitinated BAK was reduced upon MG132 treatment of HeLa and SH‐SY5Y (Figs 2B and EV1B) and to a lesser extent in MEFs (Fig EV1C). This indicates that ubiquitinated BAK was not targeted for proteasomal degradation and that BAK ubiquitination is potentially reversible, implicating a DUB that is stabilised in the presence of MG132. The reduction in poly‐ubiquitinated BAK following MG132 treatment was less evident in MEFs than in HeLa and SH‐SY5Y cells possibly due to differential expression of the potential BAK DUB (Fig EV1C).
Having established that BAK was ubiquitinated in a Parkin‐dependent fashion, we next determined which BAK residue(s) were modified following AO treatment. BAK contains only two lysine residues, one at position 113 within the α4 helix located at the hydrophobic surface groove (Fig 2D, PDB: 2IMT; Moldoveanu et al, 2006) and the second in the C‐terminus at position 210. We hypothesised that the highly conserved Lys113 residue (Fig EV2A) was the primary target of Parkin‐mediated ubiquitination as this residue is exposed to the cytosol, whereas Lys210 is localised to the mitochondrial inter‐membrane space (IMS) in this tail‐anchored protein (Iyer et al, 2015) and so would be likely inaccessible to Parkin. To test the role of Lys113, BAX/BAK‐deficient HeLa cells were retrovirally infected to express Parkin and either wild‐type BAK or BAK K113R (Fig EV2B). BAK K113R was expressed and localised to mitochondria like wild‐type BAK (Fig EV2C). Mutation of Lys113 prevented BAK ubiquitination induced by AO treatment, confirming that BAK has a single major site of ubiquitination at Lys113 (Fig 2E).
Figure EV2. Ubiquitinated BAK does not disassociate from mitochondria and persists post‐mitochondrial damage (relates to Fig 2).
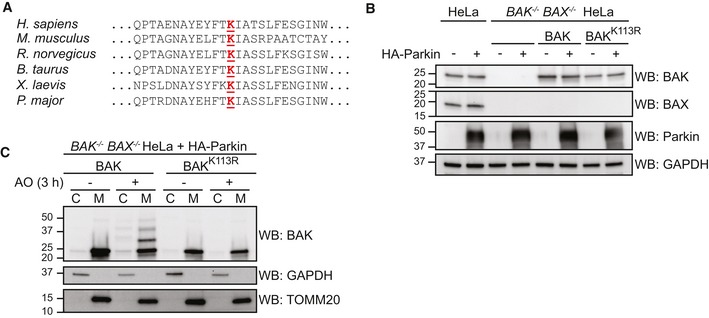
- Lys113 at the hydrophobic groove of BAK is evolutionarily conserved.
- Whole cell lysates of stable cell lines generated from BAK/BAX‐deficient HeLa re‐expressing BAK constructs and HA‐Parkin, immunoblotted as described.
- Ubiquitination of BAK does not significantly alter its subcellular localisation. BAK −/− BAX −/− HeLa cells expressing HA‐Parkin together with wild‐type BAK or BAK K113R were treated with antimycin A and oligomycin (AO) for 2 h prior to subcellular fractionation into cytosol (C) and mitochondria‐enriched heavy membrane pellet (M) fractions. Fractions were immunoblotted as described. Representative immunoblot of two independent experiments.
Source data are available online for this figure.
BAK is ubiquitinated by endogenous Parkin but not Parkinson's disease‐associated mutants
To test whether BAK ubiquitination by Parkin was restricted to AO treatment, we tested other mitophagy inducers. Treatment with the mitochondrial uncoupler carbonyl cyanide 3‐chlorophenylhydrazone (CCCP) or the mitochondrial targeted HSP90 inhibitor gamitrinib‐triphenylphosphonium (GTPP; Munch & Harper, 2016; Fiesel et al, 2017) likewise induced significant ubiquitination of BAK (Fig 3A).
Figure 3. BAK is ubiquitinated in response to various mitophagy stimuli and by endogenous Parkin, but is compromised in Parkinson's disease‐associated Parkin mutants.
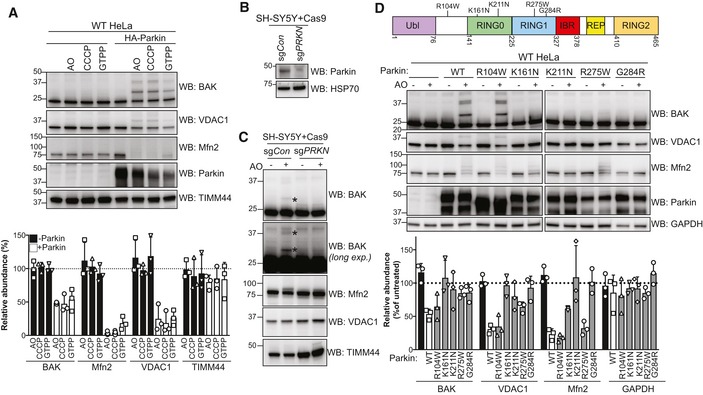
- HeLa cells with or without HA‐Parkin were treated with CCCP (10 μM), antimycin A and oligomycin (AO) for 2 h or GTPP (10 μM) for 6 h. Immunoblots are representative of three independent experiments. Graph shows densitometric analysis of non‐ubiquitinated proteins relative to untreated control from three independent experiments. Error bars represent mean ± SD.
- SH‐SY5Y neuroblastoma cells expressing Cas9 together with a sgRNA targeting PRKN (sgPRKN) or a non‐targeting control (sgCon) were assessed for Parkin expression by immunoblotting of cytosolic fractions.
- SH‐SY5Y cells generated in (B) were treated with antimycin A and oligomycin (AO) for 3 h prior to cell lysis and immunoblotting. Mono and di‐ubiquitinated BAK in Parkin‐expressing cells following AO are indicated (*). Data are representative of three independent experiments.
- Domain architecture of human Parkin with positions of selected PD‐associated mutations. Ubl, ubiquitin‐like; IBR, in‐between‐RING (Gladkova et al, 2018). HeLa cells expressing wild‐type Parkin or the indicated PD‐associated Parkin mutants were treated with antimycin A and oligomycin (AO) for 2 h. Immunoblots are representative of three independent experiments. Graph shows densitometric analysis of non‐ubiquitinated proteins in AO‐treated samples relative to untreated control from three independent experiments. Error bars represent mean ± SD. All Parkin constructs were N‐terminally FLAG‐tagged except R104W.
Source data are available online for this figure.
A recent study by Ordureau et al revealed that endogenous Parkin has a hierarchy of substrates based on the abundancy of their ubiquitinated forms in response to mitochondrial damage (Ordureau et al, 2018). To confirm that BAK was a substrate of endogenous Parkin, we analysed the SH‐SY5Y neuroblastoma cell line (Jiang et al, 2004; Vives‐Bauza et al, 2010). We first deleted endogenous Parkin in SH‐SY5Y cells using CRISPR/Cas9 gene editing and confirmed deletion in polyclonal populations by immunoblotting (Fig 3B). Following treatment with AO, we detected a modified form of BAK in SH‐SY5Y cells that was absent in cells lacking Parkin (Fig 3C). As with the canonical Parkin substrate Mfn2, ubiquitinated BAK in neuroblastoma cells was low compared with HeLa cells ectopically expressing Parkin (Fig 3C). In addition, consistent with its relative abundance in HeLa cells, the mono‐ubiquitinated form of BAK was predominant (Fig 3C).
Loss‐of‐function Parkin mutations cause approximately 50% of autosomal recessive Parkinson's disease (Arkinson & Walden, 2018). To test whether PD‐associated Parkin mutations compromise BAK ubiquitination, we stably expressed selected mutants in HeLa cells and assessed BAK ubiquitination following AO (Fig 3D). Those mutants with reduced ability to ubiquitinate canonical substrates VDAC1 and Mfn2 likewise did not efficiently ubiquitinate BAK (Fig 3D). Together, the data support BAK as a relevant substrate of Parkin whose ubiquitination would be impaired in PD patients with Parkin mutations.
Oligomerisation of BAK restricts its ubiquitination but promotes Parkin activity
Having observed that K113 was the primary site of BAK ubiquitination, we further tested whether occluding the hydrophobic groove by inducing BAK homo‐dimerisation was able to inhibit this ubiquitination event. To induce full activation and dimerisation of BAK, cells were treated with combined BH3 mimetics ABT‐737 and S63485, followed by induction of Parkin activity by AO. The majority of BAK dissociated from the VDAC2 complex and formed homodimers following BH3 mimetic treatment (Fig 4A; Ma et al, 2014). Notably, however, when BAK was activated and dimerised it was significantly less ubiquitinated by Parkin than inactive monomeric BAK (Fig 4A). In contrast, the targeting of canonical substrates Mfn2 and VDAC2 was unaffected by induction of apoptosis signalling (Fig 4A). Interestingly, enrichment of ubiquitinated proteins revealed limited ubiquitination of BAK and other mitochondrial proteins when mitochondrial damage was induced with BH3 mimetics even in the absence of AO treatment (Fig 4A and B). The ubiquitination of Mfn2 in response to BH3 mimetic‐induced mitochondrial permeabilisation was Parkin‐dependent (Fig 4B), suggesting that the loss of mitochondrial membrane potential via BAK/BAX apoptotic pores was sufficient to recruit and activate Parkin. We then tested whether this induced Parkin activity relied on PINK1. Although there was no detectable increase in PINK1 levels on mitochondria isolated from BH3 mimetic‐treated cells (Fig EV3A), deletion of PINK1 inhibited ubiquitination of Mfn2 by Parkin in response to BAK‐mediated mitochondrial damage (Figs 4C and D, and EV3B). These data suggest that mitochondrial damage during intrinsic apoptosis promotes a mitophagic response that is both PINK1‐dependent and Parkin‐dependent.
Figure 4. Oligomerisation of BAK restricts its ubiquitination and induces Parkin activity.
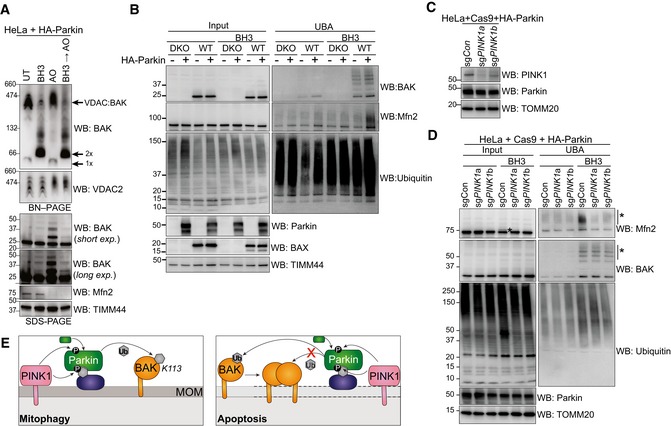
- BN–PAGE and SDS–PAGE of HeLa + HA‐Parkin cells treated with 1 μM of each of ABT‐737 and S63485 for 3 h (BH3), antimycin A and oligomycin (AO) for 2 h or a combination of the two. Experiment was performed in the presence of 10 μM QVD.oph and immunoblots are representative of three independent experiments.
- UBA enrichment of ubiquitinated proteins from HeLa + HA‐Parkin cells, BAX −/− BAK −/− (DKO) HeLa cells or wild‐type (WT) HeLa cells expressing HA‐Parkin in response to 1 μM of each of ABT‐737 and S63485 for 3 h (BH3) in the presence of QVD.oph (10 μM). Representative of three independent experiments.
- HeLa cells expressing HA‐Parkin and Cas9 were transduced with sgRNA targeting PINK1 or a non‐targeting sgRNA control, treated with AO (2 h) to stabilise PINK1 expression and immunoblotted for PINK1.
- Parkin activity induced by apoptotic mitochondrial damage is PINK1‐dependent. UBA enrichment of ubiquitinated proteins from HeLa cells generated in (C) in response to 1 μM of each of ABT‐737 and S63485 for 3 h (BH3) in the presence of QVD.oph (10 μM). (*) ubiquitinated protein. Representative of two independent experiments.
- Schematic showing Parkin ubiquitination of BAK monomer in non‐apoptotic cells, but not the BAK homo‐dimer in cells undergoing apoptosis. In response to mitophagy stimuli, PINK1 phosphorylates ubiquitin to recruit Parkin, which in turn is phosphorylated by PINK1 to become activated to ubiquitinate monomeric BAK on K113. Mitochondrial outer membrane (MOM) permeabilisation driven by BAK oligomers during apoptosis provokes Parkin activity in a PINK1‐dependent manner. Although Parkin can ubiquitinate mitochondrial outer membrane substrates (e.g. Mfn2) to promote mitochondrial clearance, it cannot ubiquitinate dimerised BAK on K113.
Source data are available online for this figure.
Figure EV3. BH3 mimetics induce mitochondrial damage in the absence of detectable PINK1 stabilisation (relates to Fig 4).
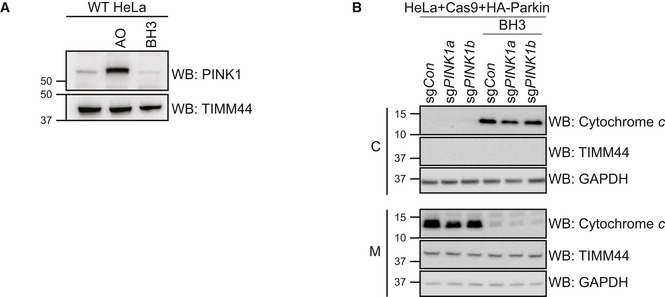
- HeLa cells were treated with antimycin A and oligomycin (AO) for 2 h or with 1 μM of each of ABT‐737 and S63485 for 3 h (BH3) in the presence of QVD.oph (10 μM). Representative of three independent experiments.
In contrast, to the ubiquitination of Mfn2, the limited ubiquitination of activated BAK was seemingly Parkin‐independent, thereby implicating another E3 ubiquitin ligase. In addition, as K113 is largely occluded in the dimeric form of BAK (Fig 4A), K210, which is normally inaccessible in the IMS, could be a ubiquitination site following the formation of BAK pores (Fig 4E). Whilst the modification of mitochondrial substrates is less prominent following BH3 mimetic treatment than with AO, these data suggest that Parkin can be recruited to sites of mitochondrial damage, regardless of the damaging agent.
Ubiquitination of BAK impairs its ability to permeabilise membranes
Having established that occluding the hydrophobic groove of BAK blocked its ubiquitination, we next tested whether ubiquitination of this site inhibited BAK apoptotic function. BAK and BAX fulfil at least partially redundant roles in apoptosis, yet whilst BAK is constitutively mitochondrial, BAX is predominantly cytosolic in healthy cells. Previously Parkin has been reported to promote cytosolic localisation of BAX to inhibit cell death (Charan et al, 2014). However, we found that ubiquitination did not significantly affect the localisation of BAK to mitochondria (Fig EV2C), excluding mislocalisation as a mechanism of Parkin‐mediated inhibition.
BAK activation induced by interaction with BH3‐only proteins such as BH3‐interacting domain death agonist (BID) promotes a conformational change allowing BAK to homo‐oligomerise and subsequently promote mitochondrial outer membrane permeabilisation. Both the initial activating interaction and self‐association involve a hydrophobic groove on the surface of BAK comprising α‐helices 3–5 (Dewson et al, 2008, Moldoveanu et al, 2013; Brouwer et al, 2014). Given that the site of BAK ubiquitination is in the α4 flanking this hydrophobic groove, we hypothesised that ubiquitinated BAK may have impaired apoptotic function due to inhibited activation or homo‐oligomerisation. To characterise the functional consequence of ubiquitination on the apoptotic function of BAK, a minimal model liposome system was used (Fig 5A). This liposome system mimics the mitochondrial outer membrane by incorporating the ratios of lipid components observed in the mitochondrial outer membrane (Kuwana et al, 2002). As full‐length BAK is unstable as a recombinant protein, BAK was engineered with a 6× histidine tail instead of its own hydrophobic transmembrane domain and also a truncated N‐terminus (∆N22, ∆C25, ∆Cys, 6‐His). This BAK variant can be targeted to liposomes through Ni‐NTA lipid headgroups and can permeabilise them upon activation with a BID BH3 peptide (Oh et al, 2010; Brouwer et al, 2017). This is achieved by the binding of the BID BH3 peptide to the hydrophobic groove of BAK and subsequent exposure of the BAK BH3 domain to promote formation of homodimers and subsequently oligomers that are able to permeabilise the liposome membrane. To model the ubiquitination, we expressed and purified ubiquitin with a cysteine instead of the terminal amino acid glycine (UbG76C). In addition, we designed a recombinant BAK construct with a single cysteine at position 113 on a Cys‐null template (C14S, C166S) that we have shown to be functional in cells (Dewson et al, 2008). This cysteine BAK mutant was recombinantly expressed allowing us to disulphide‐linked BAK with a single ubiquitin molecule specifically at this residue. Previous reports have used similar C‐terminal cysteine ubiquitin mutants in structural (Wiener et al, 2012) and protein–protein interaction studies (Borodovsky et al, 2002). The disulphide‐linked complex was purified and validated on non‐reducing SDS–PAGE (Figs 5B and EV4). We observed that BAK‐Ub had reduced liposome permeabilisation activity compared with unmodified BAK upon activation with BID BH3 peptide, suggesting that ubiquitination of BAK impairs its apoptotic function (Fig 5C). Examination of the BAK complexes formed on liposomes by blue native polyacrylamide gel electrophoresis (BN–PAGE) showed that dimerisation and higher‐order oligomerisation of BAK‐Ub was impaired (Fig 5D). To confirm that this inhibition was due to the conjugated ubiquitin, we reduced the linkage with the reducing agent Tris(2‐carboxyethyl) phosphine hydrochloride (TCEP). Although the kinetics of liposome permeabilisation and oligomerisation were somewhat slowed by TCEP, potentially due to impaired targeting of recombinant BAK‐6H to liposomes in a reducing environment, both the ability of BAK to permeabilise membranes and form oligomers was rescued when the conjugated ubiquitin was removed (Fig 5B–D).
Figure 5. Ubiquitin‐conjugated BAK at position 113 delays BAK permeabilisation of liposomes by impairing homo‐oligomerisation.
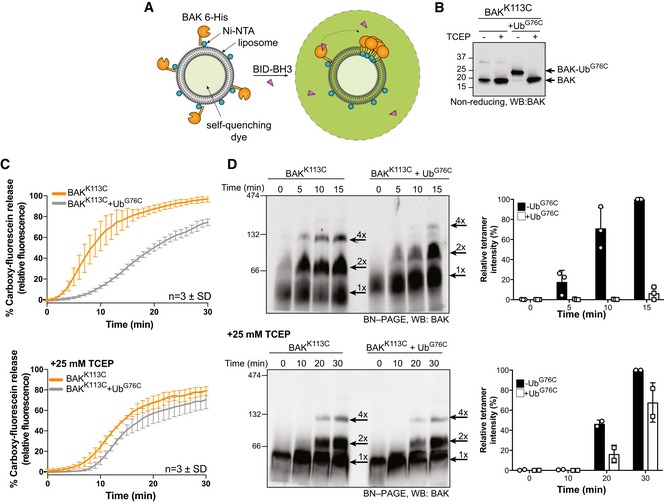
- Schematic of fluorescence‐based liposome assay to measure BAK activation on model membranes.
- Non‐reducing SDS–PAGE and immunoblot of recombinant BAK and BAK‐Ub following treatment with 25 mM TCEP.
- Timecourse monitoring fluorescence increase on liposomes following addition of 4 μM BID‐BH3 peptide to 50 nM BAK or BAK‐Ub in the presence or absence of 25 mM TCEP. Data are mean ± SD of three independent experiments.
- BN–PAGE of BAK and BAK‐Ub on liposomes following incubation with 4 μM BID‐BH3 peptide in the presence or absence of 25 mM TCEP, immunoblotted for BAK. Data representative of three independent experiments. Graphs shows densitometric analysis of the BAK tetramer (4×) from three or two independent experiments. Error bars represent mean ± SD for n = 3 or mean and range for n = 2.
Source data are available online for this figure.
Figure EV4. Purification of disulphide‐linked BAK‐Ub (relates to Fig 5).
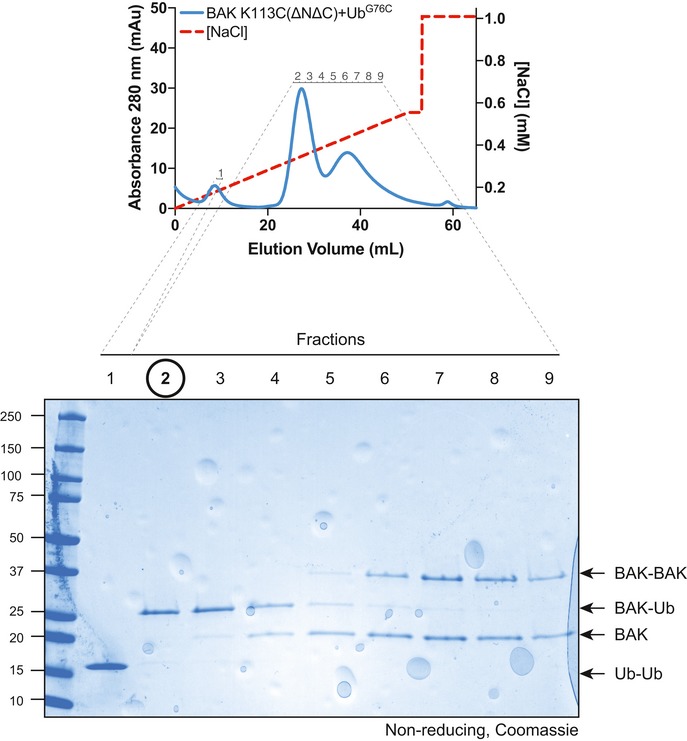
Recombinant human BAK K113C (∆N22/∆C25/6‐His) was combined with recombinant UbG76C and treated with oxidant to induce disulphide linkage. Disulphide‐linked protein was separated from non‐linked proteins by anion exchange chromatography. Fractions were run on a non‐reducing SDS–PAGE gel and Coomassie stained. Fraction #2 was used as the source of BAK‐Ub.Source data are available online for this figure.
Ubiquitination of BAK limits its ability to activate and permeabilise mitochondria
Given that artificial conjugation of ubiquitin onto BAK at this single site, analogous to mono‐ubiquitination, impaired the ability of BAK to permeabilise liposomes, we next sought to test whether ubiquitination of BAK had a role in limiting mitochondrial membrane permeabilisation. To mimic BAK ubiquitination on mitochondria, BAK K113C, or BAK ∆Cys as a control, were expressed in Bax −/− Bak −/− (DKO) MEFs (Fig 6A). We have previously shown that these variants retain apoptotic function when expressed in DKO MEFs (Dewson et al, 2008). Mitochondria‐enriched membrane fractions were isolated and incubated with recombinant UbG76C in the presence of oxidant copper phenanthroline (CuPhe) to induce disulphide bond formation and UbG76C conjugation onto mitochondrial proteins was confirmed (Fig EV5A). Importantly, conjugation of exogenous ubiquitin did not induce artefactual cytochrome c release to the supernatant fraction (Fig EV5A). We then tested whether artificial ubiquitination of BAK affected its ability to mediate mitochondrial outer membrane permeabilisation and cytochrome c release in response to recombinant cleaved BID (cBID; Fig 6B). Consistent with the liposome assays, conjugation of ubiquitin to BAK K113C abrogated its ability to mediate cytochrome c release in response to cBID compared to unmodified BAK (Fig 6B).
Figure 6. Ubiquitination of BAK on mitochondrial membranes and in cells inhibits its ability to mediate mitochondrial outer membrane permeabilisation and apoptosis.
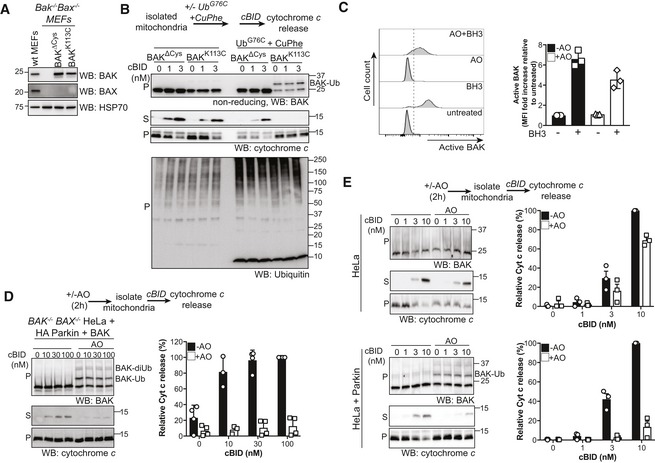
- Immunoblot of whole cell lysates of MEFs stably expressing ΔCys variants of BAK.
- Conjugation of ubiquitin to BAK K113C impairs BAK apoptotic function. Mitochondrially enriched membranes from MEFs expressing BAK or BAK K113C were cross‐linked (CuPhe) with UbG76C and treated with cBID for 30 min at 30°C. Supernatant (S) and pellet (P) fractions were then immunoblotted for BAK under non‐reducing conditions, or cytochrome c or ubiquitin under reducing conditions. Representative of three independent experiments. Note that the incorporation of recombinant UbG76C on higher molecular weight proteins is likely due to incomplete reduction on reducing SDS–PAGE.
- Parkin‐mediated ubiquitination limits BAK activation. HeLa + HA‐Parkin cells were treated with antimycin A and oligomycin (AO) for 2 h prior to induction of apoptosis with 1 μM of each of the BH3 mimetics ABT‐737 and S63485 (BH3 mim) for 1 h. Intracellular flow cytometry was performed using the conformation‐specific anti‐BAK antibody G317‐2. Representative histograms are shown and the fold increase in mean fluorescence intensity (MFI) is plotted from three independent experiments with error bars showing SD. Data are normalised to untreated control.
- Parkin‐mediated ubiquitination limits BAK apoptotic activity. BAK −/− BAX −/− HeLa cells expressing HA‐Parkin and BAK were treated with AO (2 h) prior to isolation of mitochondria and treatment with recombinant cBID and analysis of cytochrome c release. Immunoblots are representative of three independent experiments. Graph shows densitometric analysis of three independent experiments of cytochrome c in the supernatant fraction. Error bars represent mean ± SD.
- Parkin activity limits cytochrome c release. HeLa cells or HeLa cells expressing Parkin were treated and analysed as in (D). Immunoblots are representative of three independent experiments. Ubiquitinated BAK indicated (arrow). Graph shows densitometric analysis of three independent experiments showing cytochrome c release (supernatant/supernatant + membrane) relative to 100 nM cBID as 100% release. Error bars represent mean ± SD.
Source data are available online for this figure.
Figure EV5. cBID is targeted to damaged mitochondrial membranes (relates to Fig 6).
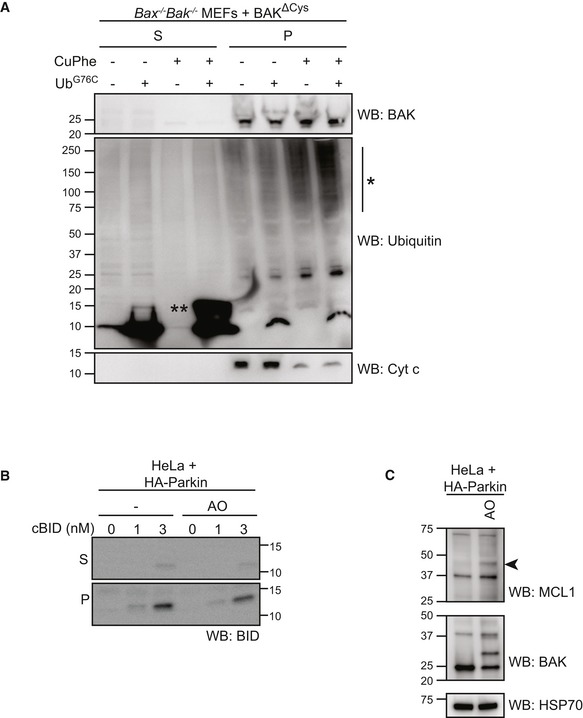
-
AMitochondria‐enriched membrane fraction from Bax −/− Bak −/− MEFs expressing BAK∆Cys were incubated with UbG76C (100 μM) in the presence or absence of oxidant copper phenanthroline (CuPhe, 1 mM) for 30 min prior to separation of supernatant and membrane fractions and immunoblotting as indicated. Note the incorporation of exogenous UbG76C into high molecular weight species (*) in the membrane fraction following treatment with oxidant. (**), UbG76C disulphide‐linked dimer.
-
BHeLa cells expressing HA‐Parkin were treated with antimycin and oligomycin (AO) for 2 h prior to fractionation into cytosol (C) and heavy membrane (M). Membrane fractions were then incubated with cBID as indicated. Incorporated cBID was then determined by centrifugation to separate the supernatant (S) from the membrane pellet (P) prior to immunoblotting.
-
CHeLa + HA‐Parkin cells were treated with antimycin and oligomycin (AO) for 2 h prior to harvesting. Samples were immunoblotted as described. Ubiquitinated MCL1 (arrowhead).
Source data are available online for this figure.
Parkin activity in response to mitochondrial damage has been argued to promote cell death by degrading the pro‐survival protein MCL1 (Carroll et al, 2014). However, our data revealed a potential pro‐survival influence of Parkin. So we next determined whether Parkin‐mediated ubiquitination of BAK induced by mitochondrial damage inhibited mitochondrial outer membrane permeabilisation, the point of no return in apoptosis. We first sought to test whether ubiquitination limited the ability for BAK to be activated. HeLa cells expressing Parkin were first treated with AO to induce Parkin‐mediated ubiquitination of BAK and subsequently treated cells with combined BH3 mimetics ABT‐737 and S63485. These cells were then measured for BAK activation using the conformation‐specific antibody (G317‐2) by intracellular flow cytometry (Alsop et al, 2015). The exposure of the normally buried N‐terminal epitope in BAK in response to BH3 mimetics was reduced following AO‐induced Parkin activity (Fig 6C). The data suggest that BAK activation (Fig 6C) and oligomerisation (Fig 5D) are impaired after ubiquitination and is consistent with the hydrophobic groove of BAK as an interface for BH3‐only proteins (Moldoveanu et al, 2013; Brouwer et al, 2014) and BAK oligomers. That BAK activation by flow cytometry was not completely blocked is consistent with the persistence of non‐ubiquitinated BAK under these conditions (Figs 2A–C and 6D).
To test whether this defect in activation inhibited BAK apoptotic function, we reconstituted BAX −/− BAK −/− HeLa cells with human BAK and Parkin and treated them with AO. Mitochondria isolated from these cells were then incubated with recombinant cBID, and cytochrome c release was assessed. AO pre‐treatment desensitised these mitochondria to cBID‐induced cytochrome c release (Fig 6D), indicating that ubiquitination of BAK was sufficient to limit mitochondrial outer membrane permeabilisation.
To test if this extended to endogenous BAK, we repeated these experiments with HeLa or HeLa cells expressing Parkin. As BAK is constitutively mitochondrial, whereas BAX is cytosolic in HeLa cells, mitochondrial outer membrane permeabilisation in these mitochondrial assays is likely mediated by BAK. As with mitochondria isolated from BAK‐reconstituted cells, mitochondria isolated from HeLa cells with endogenous BAK were more resistant to cytochrome c release when pre‐treated with AO (Fig 6E). This effect was Parkin‐dependent as AO treatment had only a modest effect on cytochrome c release from mitochondria isolated from wild‐type HeLa cells (Fig 6E). The protective effect was not due to impaired targeting of cBID to the mitochondrial outer membrane (Fig EV5B), indicating that BAK apoptotic activity was inhibited and that activated Parkin exerts a pro‐survival effect. Importantly, we observed little or no loss of MCL1 in this time frame (Fig EV5C). Together, our data highlight a key and specific role for BAK ubiquitination in limiting mitochondrial outer membrane permeabilisation in response to mitochondrial depolarisation.
Discussion
Parkin is a critical mediator of mitochondrial quality control. In order to efficiently clear damaged mitochondria, a properly co‐ordinated apoptosis response is essential. Hence, Parkin has been implicated in determining cell fate in the face of mitochondrial damage (Zhang et al, 2014). Deciphering how Parkin does this is paramount to reconciling how loss of Parkin promotes neuronal degeneration to cause early onset Parkinson's disease with the mounting and contrasting evidence that Parkin is a tumour suppressor (Bernardini et al, 2017). Our studies reveal that Parkin directly, and reversibly, modulates the intrinsic apoptosis machinery by suppressing the apoptotic activity of the mitochondrial apoptotic effector protein BAK. By limiting BAK and BAX apoptotic function, Parkin promotes cell survival and we propose that this facilitates the clearance of damaged mitochondria (Fig 7).
Figure 7. Model for Parkin‐mediated inhibition of intrinsic apoptosis.
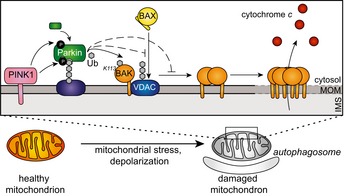
Model of the effect of Parkin‐mediated BAK ubiquitination on intrinsic apoptosis. Following mitochondrial damage, PINK1 is stabilised at the mitochondrial outer membrane (MOM) whereby it phosphorylates ubiquitin to recruit Parkin. Phosphorylation by PINK1 activates Parkin to ubiquitinate a variety of substrates including Mfn2 and VDAC. Parkin ubiquitinates BAK on a conserved lysine (K113) to inhibit BAK oligomerisation and its apoptotic function in releasing cytochrome c from the inter‐membrane space (IMS). Additionally, Parkin ubiquitinates VDAC2, that we propose limits BAX association with mitochondria. This limits apoptosis mediated by both BAK and BAX, thus enabling the effective engulfment of the mitochondrion by the autophagosome without errant activation of apoptosis.
Rather than promote BAK degradation, we found that ubiquitination directly inhibited BAK apoptotic activity by obscuring the hydrophobic surface groove. The hydrophobic groove of BAK is essential for its interaction with activating BH3‐only proteins and also for BAK homo‐dimerisation that is necessary for apoptotic pore formation and mitochondrial outer membrane permeabilisation (Dewson et al, 2008; Brouwer et al, 2014). Hence, ubiquitination on a conserved lysine on the periphery of the hydrophobic groove of BAK significantly impaired the ability of BAK to permeabilise liposomes and mediate cytochrome c release. The non‐degradative ubiquitination of BAK was supported by the predominantly K11‐linked ubiquitin chains, consistent with the profile of atypical linkages catalysed by Parkin during mitophagy (Cunningham et al, 2015) and also a recent study reporting that endogenous Parkin does not promote significant proteasomal turnover of mitochondrial substrates (Ordureau et al, 2018). Ubiquitination of BAK by the E3 ligase HERC1 was shown to promote its proteasomal degradation following irradiation of papillomavirus‐infected cells (Holloway et al, 2015). Whilst the same residue in BAK was ubiquitinated in these studies, it is likely that the type of chain linkage dictates whether BAK persists, as is the case following Parkin‐mediated ubiquitination, or is degraded, as with HERC1 (Holloway et al, 2015). The strength of the mitophagy stimulus may also determine whether BAK is degraded or not, with a strong mitophagy stimulus causing degradation of many outer mitochondrial membrane proteins (Chan et al, 2011). Our studies now reveal that ubiquitination of BAK may be an important regulatory event to limit cell death by specifically and reversibly impairing its apoptotic function.
Other BCL‐2 family proteins have been proposed as targets of Parkin either at steady state or following induction of mitophagy. The pro‐apoptotic protein BAX, which is predominantly localised in the cytosol of healthy cells, has been implicated as a Parkin substrate, thereby inhibiting its localisation to mitochondria (Johnson et al, 2012; Charan et al, 2014). Additionally, Parkin‐mediated ubiquitination has been proposed as a mechanism to degrade mutated mitochondrial BAX (Cakir et al, 2017). Both scenarios would result in a dampening of the apoptotic response, but, intriguingly, they would occur in the absence of induced mitochondrial damage when Parkin is thought to be largely inactive due to auto‐inhibition (Matsuda et al, 2010; Chaugule et al, 2011; Trempe et al, 2013; Wauer & Komander, 2013). Consistent with earlier findings, we found that constitutive and induced mitochondrial localisation of BAX was inhibited by Parkin. However, this was in the absence of detectable BAX ubiquitination either prior to or after induction of mitochondrial depolarisation, even under conditions of proteasomal inhibition. Although we cannot exclude degradation of BAX by alternative mechanisms, our data suggest that ubiquitination of VDAC2, an early and selective substrate of Parkin (Ordureau et al, 2018), may also contribute to the defect in BAX mitochondrial association. Furthermore, Parkin limited cytochrome c release in the absence of BAX, suggesting that the inactivation of BAK is also a key function of Parkin.
Our data uncover BAK as a novel substrate of Parkin. Whilst a number of ubiquitylomics studies have interrogated the proteome during mitophagy, BAK was not identified as a substrate of Parkin (Sarraf et al, 2013; Ordureau et al, 2018). This is likely due to the fact that when modified, K113 is present on a large 39 + 2 amino acid tryptic peptide (Y89 − R127 + GG) that makes identification using standard LC‐MS/MS workflows challenging. Our data show that ubiquitination of BAK inhibits its function in minimal model liposome systems and on mitochondrial membranes. Hence, the cyto‐protective role of Parkin likely involves inhibition of BAX, but also BAK.
Targeting Parkin with small molecules to induce its conformational change will likely lead to restrained apoptosis via inhibition of both BAK‐ and BAX‐mediated apoptosis. Alternatively, pharmacological inhibition of the Parkin antagonist, USP30, may also promote BAK ubiquitination and thus a higher apoptotic threshold. However, interestingly, USP30 depletion can sensitise cells to the BH3 mimetic, ABT‐737 (Liang et al, 2015), suggesting that imbalances in Parkin activity can have divergent effects on apoptotic response.
We also show that mitochondrial permeabilisation in response to BH3 mimetics can induce Parkin‐dependent ubiquitination of Mfn2. This may suggest a role for Parkin in silencing apoptotic mitochondria, which, if left unchecked, can promote inflammatory responses (Rongvaux et al, 2014; White et al, 2014; McArthur et al, 2018). Indeed, recent work has demonstrated that, under conditions of mitochondrial stress, ablation of mitophagy in Prkn −/− or Pink1 −/− mice promotes inflammation and drives the pathogenesis of PD (Sliter et al, 2018).
Contrasting with a pro‐survival role, Parkin has been reported to ubiquitinate and degrade the pro‐survival protein MCL1 and so promote cell death specifically in response to mitochondrial depolarisation (Carroll et al, 2014). Parkin‐mediated mitophagy is considered a pro‐survival mechanism in response to mitochondrial stress, and Parkin has been reported to prevent apoptosis in a variety of settings (Darios et al, 2003; Staropoli et al, 2003; Jiang et al, 2004; Wang et al, 2007; Berger et al, 2009). However, whether Parkin activity invokes a pro‐survival or a pro‐apoptotic response depends on the nature and extent of the mitochondrial damage (Zhang et al, 2014). Prolonged or excessive mitochondrial damage beyond the capacity of Parkin‐dependent repair leads to cell death driven by MCL1 degradation to remove the compromised cell. However, transient or limited mitochondrial damage, as is more likely to occur in vivo, promotes a pro‐survival response to enable Parkin‐dependent mitochondrial clearance. Our findings indicate that the initial and prevailing response to mitochondrial damage is Parkin‐dependent inhibition of BAK‐ and BAX‐mediated cytochrome c release to limit apoptosis. An inability to constrain BAK and BAX apoptotic activity may contribute to the apoptotic degeneration of Parkin‐deficient dopaminergic neurons in Parkinson's disease. Thus, impairing the pro‐apoptotic activity of these molecules, for example, by inhibiting the interaction between BAX and VDAC2 or inhibiting the activity of the BAK DUB, could be a novel strategy to prevent apoptosis.
Materials and Methods
Contact for reagent and resource sharing
Further information and requests for reagents may be directed to, and will be fulfilled by, the lead contact, Dr. Grant Dewson, Walter and Eliza Hall Institute of Medical Research, 1G Royal Parade, Parkville, Victoria 3052, Australia (dewson@wehi.edu.au).
Cell culture and induction of mitophagy
Mouse embryonic fibroblasts (MEFs) were isolated from embryos harvested at embryonic day 14.5 and transformed by transfecting with SV40‐large T antigen as described (Dewson et al, 2008). MEFs (generated by G. Dewson) were passaged in Dulbecco's modified Eagles medium (DMEM) supplemented with 8% foetal calf serum (FCS), 55 μM 2‐mercaptoethanol and 250 μM asparagine. HeLa (ATCC #CCL2), 293T (ATCC #CRL‐3216) and SH‐SY5Y (Jamie Fletcher, CCI, Sydney, Australia) neuroblastoma cells were passaged in DMEM supplemented with 8% FCS. Cells were cultured at 37°C and 10% CO2. Cells were routinely screened for mycoplasma using a MycoAlert Detection Kit as per manufacturer's instructions (Lonza, Switzerland). To induce mitophagy, cells were treated with 10 μM oligomycin and 4 μM antimycin A for times as indicated. Cell death was induced with 1 μM of ABT‐737 (Abbott, IL) 1 μM of S63485 (SYNthesis MedChem, Australia). Where indicated, cells were pre‐treated with the pan‐caspase inhibitor QVD.oph (MP Biomedicals, CA) at 10 μM or the proteasome inhibitor MG132 (Sigma‐Aldrich, MO) at 20 μM for 0.5 h each.
Retroviral infection
Parkin, BAX and BAK constructs were stably expressed in HeLa and MEF by retroviral infection. pMX‐IG, pMXs‐IH and pMXs‐IP retroviral constructs were first introduced into HEK293T cells by CaCl2 transfection. Filtered, virus‐containing supernatants were used to infect HeLa and MEF cells by spin inoculation (2,500 rcf centrifugation at 32°C for 45 min in the presence of 4 μg/ml polybrene; Sigma‐Aldrich, MO). Cells stably expressing constructs were selected for by culture in 2 μg/ml puromycin (Life Technologies, CA) and 300 ng/ml hygromycin (Sigma‐Aldrich, MO) or by FACS sorting for GFP‐positive cells.
CRISPR/Cas9 gene editing
Cells (HeLa, SH‐SY5Y) were lentivirally infected with constructs encoding Cas9 (mCherry) and single guide RNA (sgRNA, GFP) targeting early protein‐coding exons of human PRKN (GTGTCAGAATCGACCTCCAC), human PINK1 (sgRNAa, TGTGTCCGCCGGGAGCGTCC; sgRNAb, CCGGCCGGGCCTACGGCTTG) or a control non‐targeting sgRNA (ATTGGTGCCAATGCTCGGAT). Following selection of transduced cells by sorting double‐positive cells on a FACS AriaII flow cytometer (Becton Dickinson), cells were treated with 1 μg/ml doxycycline hyclate (Sigma) to induce sgRNA expression. Parkin or PINK1 deletion in polyclonal populations was confirmed by immunoblotting either before (Parkin) or after (PINK1) treatment with antimycin A and oligomycin. To generate BAK −/− BAX −/− HeLa cells, cells were transiently transfected with two sgRNAs targeting human BAK (GCATGAAGTCGACCACGAAG, GGCCATGCTGGTAGACGTGT) and two sgRNAs targeting human BAX (CTGCAGGATGATTGCCGCCG, TCTGACGGCAACTTCAACTG) cloned into PX458 plasmid (a kind gift from Feng Zhang (Ran et al, 2013) using XtremeGene; Sigma‐Aldrich, MO) according to the manufacturer's instructions. 48 h post‐transfection, cells were treated with 1 μM each of A‐1331852, ABT‐199 (Abbott, IL) and S63845 (SYNthesis MedChem, Aus). Surviving cells were screened for BAK and BAX deletion by immunoblotting.
Immunoblotting
Samples were generated by 1% Triton X‐100 lysis in ONYX buffer (20 mM Tris–HCl (pH7.4), 135 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 10% glycerol (v/v)) with cOmplete protease inhibitors (Calbiochem) on ice for 20 min. Samples were spun (17,000 rcf, 5 min, 4°C), and the concentration of protein in soluble supernatants was determined by BCA assay (Thermo Fisher, MA) as per manufacturer's instructions. Samples were normalised based on protein concentration and between 5 and 40 μg was loaded per well and run on SDS–PAGE. Gels were transferred to nitrocellulose or PVDF membranes. Non‐specific binding was blocked with 5% non‐fat milk in TBS‐T (20 mM Tris–HCl pH 7.6, 137 mM NaCl, 0.1% Tween‐20) for 1 h at room temperature. Membranes were washed with TBS‐T prior to incubation with primary antibodies (see Table EV1). Following membrane washing with TBS‐T, antibodies were incubated with appropriate secondary antibodies (see Table EV1) for 1 h at room temperature and developed with ECL solution (GE Lifesciences, MA) as per manufacturer's instructions.
Ubiquitin pull‐down and deubiquitinase treatment
Approximately 1 × 107 cells were treated to induce mitophagy as above. Cells were lysed in 1% Triton X‐100 (v/v) in ONYX buffer with cOmplete protease inhibitors and 10 mM N‐ethylmaleimide (NEM, Sigma‐Aldrich, MO). Lysates were incubated with glutathione sepharose beads (GE Healthcare, MA) pre‐equilibrated with recombinant 100 μg GST‐UBA for 4 h at 4°C. NEM was removed from beads with three washes of ONYX buffer with protease inhibitors. Beads were then equilibrated in DUB reaction buffer (50 mM Tris (pH 7.5), 50 mM NaCl and 5 mM DTT) and incubated with recombinant DUBs at previously established concentrations (Hospenthal et al, 2015). Samples were then incubated at 37°C for 30 min with agitation. Samples were eluted from the beads by boiling in SDS‐sample buffer and analysed by SDS–PAGE.
Subcellular fractionation and blue native PAGE
Cells were harvested and permeabilised at 1 × 107 cells/ml in modified egg lysis buffer (MELB, 20 mM HEPES pH 7.5, 100 mM sucrose, 2.5 mM MgCl2, 100 mM KCl) with protease inhibitors and 0.025% digitonin (w/v) for 10 min on ice. Membranes were enriched by centrifugation (17,000 rcf, 5 min, 4°C). For SDS–PAGE, 2× reducing SDS‐sample buffer was added to soluble supernatant fractions and pellet fractions were resuspended in an equal volume of 1× reducing SDS‐sample buffer and run on SDS–PAGE. For BN–PAGE, membrane fractions were resuspended in empty well buffer [EWB, 20 mM Bis‐Tris, pH 7.4, 50 mM NaCl, 10% glycerol (v/v)] with protease inhibitors, 10 μM DTT and 1% digitonin (w/v) for 30 min on ice. Soluble protein complexes were obtained by centrifugation (17,000 rcf, 5 min, 4°C) and supplemented with 10× BN‐loading dye (5% Coomassie Blue R‐250 (Bio‐Rad Laboratories, CA) in 500 mM 6‐aminohexanoic acid, 100 mM Bis‐Tris, pH 7.0) and run as previously described (Schagger & von Jagow, 1991).
Purification of recombinant protein
BAK K113C (∆N22 ∆C25 C166S C‐term‐His) was cloned into the pTYB1 vector. Plasmids were transformed into BL21 (DE3) E. coli and grown at 37°C until they reached an optical density (600 nm) of ~1.0 in Super Broth. 1 mM IPTG was then added and protein expressed for 16 h at 18°C. Cells were lysed in TSE buffer (20 mM Tris pH 8.0, 500 mM NaCl, 1 mM EDTA) and passed over a chitin binding column, washed with TSE buffer and then chemically cleaved with DTT (50 mM) on‐column for 48 h at 4°C. Proteins were further purified by gel filtration (Superdex75 10/300, GE Healthcare, MA) in TBS.
Ubiquitin G76C was cloned into the pGEX 6P3 vector, transformed into BL21 E. coli (DE3) and grown to an optical density (600 nm) of ~1.0 in Super Broth before protein expression was induced with 1 mM IPTG for 3 h at 37°C. Cells were lysed in TBS (20 mM Tris pH 8.0, 150 mM NaCl) supplemented with EDTA (2 mM), purified by GST affinity column purification, cleaved overnight with precision protease and then further purified by gel filtration (Superdex75 10/300) in TBS.
Disulphide linkage of cysteine mutants and cytochrome c release
Recombinant BAK K113C (ΔN22 ΔC25 C166S C‐term‐His) and UbiquitinG76C were incubated at 4°C in the presence of 1 mM copper(II)(1,10‐phenanthroline)3 (CuPhe) for 30 min. Heterodimeric products were purified by anion exchange on a HiTrap Sepharose Q column (GE, MA). For mitochondrial cross‐linking experiments, cells were harvested and permeabilised at 1 × 107 cells/ml in MELB with protease inhibitors and 0.025% digitonin (w/v) for 10 min on ice. Membranes were enriched by centrifugation (17,000 rcf, 5 min, 4°C) and resuspended with 100 μM UbG76C. CuPhe was added to a final concentration of 1 mM, and samples were incubated at room temperature for 30 min. The reaction was quenched by adding EDTA to a final concentration of 10 mM. Samples were centrifuged (17,000 rcf, 5 min, 4°C), and pellets were resuspended in MELB with protease inhibitors and varying doses of cBID. Samples were incubated at 30°C for 30 min. Samples were spun as above, and soluble supernatant fractions were supplemented with 2× reducing sample buffer. Pellets were resuspended in two volumes of 1× reducing sample buffer for cytochrome c blot and 1× non‐reducing sample buffer for BAK blot.
Fluorescence liposome assay, SDS–PAGE and BN–PAGE
Liposomes containing a combination of lipids that mimic the outer mitochondrial membrane (46% phosphatidylcholine (w/v), 25% phosphatidylethanolamine (w/v), 11% phosphatidylinositol (w/v), 10% phosphatidylserine (w/v) and 8% cardiolipin (w/v) encapsulating self‐quenching 5(6)‐carboxy‐fluorescein and supplemented with 5% nickel chelating lipid (1,2‐Dioleoyl‐sn‐Glycero‐3‐[N‐(5‐amino‐1‐carboxypentyl)iminodiacetic‐acid)‐succinyl] (w/v)) in chloroform and 0.01% butylated hydroxytoluene (w/v) were dried under N2 and then re‐suspended in 50 mM 5(6)‐carboxy‐fluorescein. Liposomes were then extruded through a 100 nm pore size membrane and passed over a PD10 column to remove excess dye. Liposomes (4 μg/ml) were incubated with either BAK (DN22 DC25 C‐term‐His) or cross‐linked BAK‐Ub at a concentration of 50 nM and 4 μM of BID‐BH3 peptide in SUV (small unilamellar vesicle/liposome) buffer (10 mM HEPES pH 7.5, 135 mM KCl) for 30 min at room temperature; 5(6)‐carboxy‐fluorescein is self‐quenching, and therefore, upon release from liposomes its fluorescence could be measured with an excitation wavelength of 485 nm and emission wavelength of 535 nm. BN–PAGE was performed on liposomes by solubilisation in 0.5% digitonin (w/v) on ice for 20 min followed by supplementing the samples with 10% glycerol (v/v) and BN‐loading dye and running on BN–PAGE as described above. SDS–PAGE was similarly performed by solubilising liposomes in SDS–PAGE sample buffer in the presence or absence of TCEP and running on SDS–PAGE.
BAK activation by flow cytometry
Cells were harvested following treatment with antimycin A and oligomycin (AO) for 2 h followed by 1 μM of each of the BH3 mimetics ABT‐737 and S63485 for 1 h. Cells were fixed and permeabilised using the eBioscience cell fixation and permeabilisation kit (Thermo Fischer, MA) according to manufacturer's instructions. Activated BAK was detected using anti‐BAK G317‐2 (1:100) and subsequently phycoerythrin‐conjugated anti‐mouse antibody (1:200), both in diluted permeabilisation buffer. Samples were analysed on a FACS‐Calibur (Becton Dickinson, NJ).
Cell death assessed by LDH assay
Of 1.5 × 106 HeLa cells were seeded in 10‐cm dishes overnight. Cells were pre‐treated with or without antimycin A and oligomycin (4 and 10 μM, respectively) for 2 h, prior to treatment with 1 μM of each of the BH3 mimetics ABT‐737 and S63845. Culture supernatants were collected at selected times and stored at −80°C overnight. Samples were thawed, and 50 μl of supernatant was assayed for LDH release using the CytoTox 96® Non‐Radioactive Cytotoxicity Assay (Promega, WI) according to manufacturer's instructions. Absorbance was quantified using a Chameleon plate reader (Noki Technologies, India) at a wavelength of 490 nm. Relative LDH release was calculated by subtracting the absorbance from an untreated control supernatant and normalising values to the 6 h BH3 mimetic only (set as 100%) for each cell line.
Cytochrome c release assay
Cells were permeabilised in 0.025% digitonin (w/v) in MELB with cOmplete protease inhibitors at a concentration of 1 × 107 cells/ml for 10 min on ice. Pelleted membrane fractions were obtained by centrifugation (17,000 rcf, 5 min, 4°C) and supplemented with cBID at concentrations indicated for 30 min at 30°C. Soluble supernatants were obtained by centrifugation (17,000 rcf, 5 min, 4°C) and supplemented with 2× reducing sample buffer. Insoluble pellets were resuspended in two volumes of 1× reducing sample buffer and samples were run on SDS–PAGE.
Densitometric analysis of immunoblots
Immunoblots were normalised to samples on the same membrane, at the same exposure using the ImageLab software package (Bio‐Rad, CA). Background signal was subtracted from equivalent bands, and samples were normalised to the untreated control lane. For mitophagy assays, the untreated samples were set at 100% and the signal of the appropriate bands normalised to this. For cytochrome c release assays, total release (%) was calculated by determining the intensity of cytochrome c in the cytosolic fraction over the sum intensity of cytochrome c in the membrane and cytosol fractions. Due to incomplete release of cytochrome c from the membrane fraction, the cytochrome c release assay shown in Fig 6D was determined from the intensity of cytochrome c in the supernatant fraction only, and normalising to the 100 nM dose of cBID in the absence of Parkin induction set at 100%. For native‐PAGE quantitation, the intensity of the BAK tetramer signal was determined and normalised to the end timepoint of the BAK K113C control sample in each respective assay.
Author contributions
J.P.B., J.M.B. and J.J.S. designed and performed experiments and interpreted data. I.K.L.T. and S.H. performed experiments. C.A.S. and A.B. provided reagents and assistance with deubiquitination experiments. C.D.R. provided reagents and assistance in generating BAK −/− BAX −/− cell lines. A.Z.W. provided assistance and reagents for liposome assays. P.E.C. designed and interpreted liposome experiments. M.L. designed experiments, provided advice, cell lines and reagents. G.D. supervised the study, designed experiments and interpreted data. J.P.B. and G.D. wrote the manuscript. All authors contributed to the production of the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We thank B. Reljic and M. Ryan for providing the VDAC2 antibody, D.C.S. Huang and D. Segal for cell lines and K. Scicluna for assistance with the manuscript. We thank Dario Altieri (Wistar Institute Cancer Center, Philadelphia) for the mitochondrial Hsp90 inhibitor GTPP. J.P.B. is supported by an Australian Government Research Training Program Scholarship and a Cancer Therapeutics CRC PhD Top Up Scholarship. M.L. is supported by the National Health and Medical Research Council (GNT1106471), and a fellowship from the Australian Research Council (FT160100063). P.E.C is supported by a fellowship from the National Health and Medical research Council (1079700). This work was supported by grants from the National Health and Medical Research Council (1078924), a fellowship from the Australian Research Council (FT100100791 to G.D.) and operational infrastructure grants through the Australian Government IRISS and the Victorian State Government OIS 9000220.
The EMBO Journal (2019) 38: e99916
References
- Alsop AE, Fennell SC, Bartolo RC, Tan IK, Dewson G, Kluck RM (2015) Dissociation of Bak alpha1 helix from the core and latch domains is required for apoptosis. Nat Commun 6: 6841 [DOI] [PubMed] [Google Scholar]
- Aluvila S, Mandal T, Hustedt E, Fajer P, Choe JY, Oh KJ (2014) Organization of the mitochondrial apoptotic BAK pore: oligomerization of the BAK homodimers. J Biol Chem 289: 2537–2551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkinson C, Walden H (2018) Parkin function in Parkinson's disease. Science 360: 267–268 [DOI] [PubMed] [Google Scholar]
- Berger AK, Cortese GP, Amodeo KD, Weihofen A, Letai A, LaVoie MJ (2009) Parkin selectively alters the intrinsic threshold for mitochondrial cytochrome c release. Hum Mol Genet 18: 4317–4328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardini JP, Lazarou M, Dewson G (2017) Parkin and mitophagy in cancer. Oncogene 36: 1315–1327 [DOI] [PubMed] [Google Scholar]
- Bleicken S, Classen M, Padmavathi PV, Ishikawa T, Zeth K, Steinhoff HJ, Bordignon E (2010) Molecular details of Bax activation, oligomerization, and membrane insertion. J Biol Chem 285: 6636–6647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borodovsky A, Ovaa H, Kolli N, Gan‐Erdene T, Wilkinson KD, Ploegh HL, Kessler BM (2002) Chemistry‐based functional proteomics reveals novel members of the deubiquitinating enzyme family. Chem Biol 9: 1149–1159 [DOI] [PubMed] [Google Scholar]
- Brouwer JM, Westphal D, Dewson G, Robin AY, Uren RT, Bartolo R, Thompson GV, Colman PM, Kluck RM, Czabotar PE (2014) Bak core and latch domains separate during activation, and freed core domains form symmetric homodimers. Mol Cell 55: 938–946 [DOI] [PubMed] [Google Scholar]
- Brouwer JM, Lan P, Cowan AD, Bernardini JP, Birkinshaw RW, van Delft MF, Sleebs BE, Robin AY, Wardak A, Tan IK, Reljic B, Lee EF, Fairlie WD, Call MJ, Smith BJ, Dewson G, Lessene G, Colman PM, Czabotar PE (2017) Conversion of Bim‐BH3 from activator to inhibitor of bak through structure‐based design. Mol Cell 68: 659–672.e9 [DOI] [PubMed] [Google Scholar]
- Cakir Z, Funk K, Lauterwasser J, Todt F, Zerbes RM, Oelgeklaus A, Tanaka A, van der Laan M, Edlich F (2017) Parkin promotes proteasomal degradation of misregulated BAX. J Cell Sci 130: 2903–2913 [DOI] [PubMed] [Google Scholar]
- Carroll RG, Hollville E, Martin SJ (2014) Parkin sensitizes toward apoptosis induced by mitochondrial depolarization through promoting degradation of Mcl‐1. Cell Rep 9: 1538–1553 [DOI] [PubMed] [Google Scholar]
- Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S, Chan DC (2011) Broad activation of the ubiquitin‐proteasome system by Parkin is critical for mitophagy. Hum Mol Genet 20: 1726–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charan RA, Johnson BN, Zaganelli S, Nardozzi JD, LaVoie MJ (2014) Inhibition of apoptotic Bax translocation to the mitochondria is a central function of parkin. Cell Death Dis 5: e1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaugule VK, Burchell L, Barber KR, Sidhu A, Leslie SJ, Shaw GS, Walden H (2011) Autoregulation of Parkin activity through its ubiquitin‐like domain. EMBO J 30: 2853–2867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham CN, Baughman JM, Phu L, Tea JS, Yu C, Coons M, Kirkpatrick DS, Bingol B, Corn JE (2015) USP30 and parkin homeostatically regulate atypical ubiquitin chains on mitochondria. Nat Cell Biol 17: 160–169 [DOI] [PubMed] [Google Scholar]
- Czabotar PE, Westphal D, Dewson G, Ma S, Hockings C, Fairlie WD, Lee EF, Yao S, Robin AY, Smith BJ, Huang DC, Kluck RM, Adams JM, Colman PM (2013) Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell 152: 519–531 [DOI] [PubMed] [Google Scholar]
- Darios F, Corti O, Lucking CB, Hampe C, Muriel MP, Abbas N, Gu WJ, Hirsch EC, Rooney T, Ruberg M, Brice A (2003) Parkin prevents mitochondrial swelling and cytochrome c release in mitochondria‐dependent cell death. Hum Mol Genet 12: 517–526 [DOI] [PubMed] [Google Scholar]
- Deas E, Plun‐Favreau H, Gandhi S, Desmond H, Kjaer S, Loh SH, Renton AE, Harvey RJ, Whitworth AJ, Martins LM, Abramov AY, Wood NW (2011) PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum Mol Genet 20: 867–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewson G, Kratina T, Sim HW, Puthalakath H, Adams JM, Colman PM, Kluck RM (2008) To trigger apoptosis, Bak exposes its BH3 domain and homodimerizes via BH3: groove interactions. Mol Cell 30: 369–380 [DOI] [PubMed] [Google Scholar]
- Dewson G, Ma S, Frederick P, Hockings C, Tan I, Kratina T, Kluck RM (2012) Bax dimerizes via a symmetric BH3: groove interface during apoptosis. Cell Death Differ 19: 661–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiesel FC, James ED, Hudec R, Springer W (2017) Mitochondrial targeted HSP90 inhibitor Gamitrinib‐TPP (G‐TPP) induces PINK1/Parkin‐dependent mitophagy. Oncotarget 8: 106233–106248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladkova C, Maslen SL, Skehel JM, Komander D (2018) Mechanism of parkin activation by PINK1. Nature 559: 410–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JW, Ordureau A, Heo JM (2018) Building and decoding ubiquitin chains for mitophagy. Nat Rev Mol Cell Biol 19: 93–108 [DOI] [PubMed] [Google Scholar]
- Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW (2015) The PINK1‐PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell 60: 7–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway A, Simmonds M, Azad A, Fox JL, Storey A (2015) Resistance to UV‐induced apoptosis by beta‐HPV5 E6 involves targeting of activated BAK for proteolysis by recruitment of the HERC1 ubiquitin ligase. Int J Cancer 136: 2831–2843 [DOI] [PubMed] [Google Scholar]
- Hospenthal MK, Mevissen TET, Komander D (2015) Deubiquitinase‐based analysis of ubiquitin chain architecture using Ubiquitin Chain Restriction (UbiCRest). Nat Protoc 10: 349–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer S, Bell F, Westphal D, Anwari K, Gulbis J, Smith BJ, Dewson G, Kluck RM (2015) Bak apoptotic pores involve a flexible C‐terminal region and juxtaposition of the C‐terminal transmembrane domains. Cell Death Differ 22: 1665–1675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Ren Y, Zhao J, Feng J (2004) Parkin protects human dopaminergic neuroblastoma cells against dopamine‐induced apoptosis. Hum Mol Genet 13: 1745–1754 [DOI] [PubMed] [Google Scholar]
- Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ (2010) Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol 191: 933–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SM, Youle RJ (2013) The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin‐mediated mitophagy of polarized mitochondria. Autophagy 9: 1750–1757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BN, Berger AK, Cortese GP, Lavoie MJ (2012) The ubiquitin E3 ligase parkin regulates the proapoptotic function of Bax. Proc Natl Acad Sci USA 109: 6283–6288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ (2014) PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol 205: 143–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K, Alessi DR, Knebel A, Trost M, Muqit MM (2014) Parkin is activated by PINK1‐dependent phosphorylation of ubiquitin at Ser65. Biochem J 460: 127–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazlauskaite A, Martinez‐Torres RJ, Wilkie S, Kumar A, Peltier J, Gonzalez A, Johnson C, Zhang J, Hope AG, Peggie M, Trost M, van Aalten DM, Alessi DR, Prescott AR, Knebel A, Walden H, Muqit MM (2015) Binding to serine 65‐phosphorylated ubiquitin primes Parkin for optimal PINK1‐dependent phosphorylation and activation. EMBO Rep 16: 939–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392: 605–608 [DOI] [PubMed] [Google Scholar]
- Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, Burchell L, Walden H, Macartney TJ, Deak M, Knebel A, Alessi DR, Muqit MM (2012) PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol 2: 120080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin‐Braizat G, Chanrion M, Kelly GL, Gong JN, Moujalled DM, Bruno A, Csekei M, Paczal A, Szabo ZB, Sipos S, Radics G, Proszenyak A, Balint B, Ondi L, Blasko G et al (2016) The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 538: 477–482 [DOI] [PubMed] [Google Scholar]
- Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, Endo T, Fon EA, Trempe JF, Saeki Y, Tanaka K, Matsuda N (2014) Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510: 162–166 [DOI] [PubMed] [Google Scholar]
- Kumar A, Aguirre JD, Condos TE, Martinez‐Torres RJ, Chaugule VK, Toth R, Sundaramoorthy R, Mercier P, Knebel A, Spratt DE, Barber KR, Shaw GS, Walden H (2015) Disruption of the autoinhibited state primes the E3 ligase parkin for activation and catalysis. EMBO J 34: 2506–2521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Chaugule VK, Condos TEC, Barber KR, Johnson C, Toth R, Sundaramoorthy R, Knebel A, Shaw GS, Walden H (2017) Parkin‐phosphoubiquitin complex reveals cryptic ubiquitin‐binding site required for RBR ligase activity. Nat Struct Mol Biol 24: 475–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD (2002) Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 111: 331–342 [DOI] [PubMed] [Google Scholar]
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524: 309–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EF, Czabotar PE, van Delft MF, Michalak EM, Boyle MJ, Willis SN, Puthalakath H, Bouillet P, Colman PM, Huang DC, Fairlie WD (2008) A novel BH3 ligand that selectively targets Mcl‐1 reveals that apoptosis can proceed without Mcl‐1 degradation. J Cell Biol 180: 341–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JJ, Sanchez‐Martinez A, Zarate AM, Beninca C, Mayor U, Clague MJ, Whitworth AJ (2018) Basal mitophagy is widespread in Drosophila but minimally affected by loss of Pink1 or parkin. J Cell Biol 217: 1613–1622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang JR, Martinez A, Lane JD, Mayor U, Clague MJ, Urbe S (2015) USP30 deubiquitylates mitochondrial Parkin substrates and restricts apoptotic cell death. EMBO Rep 16: 618–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma SB, Nguyen TN, Tan I, Ninnis R, Iyer S, Stroud DA, Menard M, Kluck RM, Ryan MT, Dewson G (2014) Bax targets mitochondria by distinct mechanisms before or during apoptotic cell death: a requirement for VDAC2 or Bak for efficient Bax apoptotic function. Cell Death Differ 21: 1925–1935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez A, Lectez B, Ramirez J, Popp O, Sutherland JD, Urbe S, Dittmar G, Clague MJ, Mayor U (2017) Quantitative proteomic analysis of Parkin substrates in Drosophila neurons. Mol Neurodegener 12: 29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, Kimura M, Komatsu M, Hattori N, Tanaka K (2010) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 189: 211–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArthur K, Whitehead LW, Heddleston JM, Li L, Padman BS, Oorschot V, Geoghegan ND, Chappaz S, Davidson S, San Chin H, Lane RM, Dramicanin M, Saunders TL, Sugiana C, Lessene R, Osellame LD, Chew TL, Dewson G, Lazarou M, Ramm G et al (2018) BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 359: eaao6047 [DOI] [PubMed] [Google Scholar]
- McLelland GL, Goiran T, Yi W, Dorval G, Chen CX, Lauinger ND, Krahn AI, Valimehr S, Rakovic A, Rouiller I, Durcan TM, Trempe JF, Fon EA (2018) Mfn2 ubiquitination by PINK1/parkin gates the p97‐dependent release of ER from mitochondria to drive mitophagy. eLife 7: e32866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWilliams TG, Prescott AR, Montava‐Garriga L, Ball G, Singh F, Barini E, Muqit MMK, Brooks SP, Ganley IG (2018) Basal mitophagy occurs independently of PINK1 in mouse tissues of high metabolic demand. Cell Metab 27: 439–449.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldoveanu T, Liu Q, Tocilj A, Watson M, Shore G, Gehring K (2006) The X‐ray structure of a BAK homodimer reveals an inhibitory zinc binding site. Mol Cell 24: 677–688 [DOI] [PubMed] [Google Scholar]
- Moldoveanu T, Grace CR, Llambi F, Nourse A, Fitzgerald P, Gehring K, Kriwacki RW, Green DR (2013) BID‐induced structural changes in BAK promote apoptosis. Nat Struct Mol Biol 20: 589–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore AS, Holzbaur EL (2016) Dynamic recruitment and activation of ALS‐associated TBK1 with its target optineurin are required for efficient mitophagy. Proc Natl Acad Sci USA 113: E3349–E3358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munch C, Harper JW (2016) Mitochondrial unfolded protein response controls matrix pre‐RNA processing and translation. Nature 534: 710–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TN, Padman BS, Lazarou M (2016) Deciphering the molecular signals of PINK1/parkin mitophagy. Trends Cell Biol 26: 733–744 [DOI] [PubMed] [Google Scholar]
- Oh KJ, Singh P, Lee K, Foss K, Lee S, Park M, Lee S, Aluvila S, Park M, Singh P, Kim RS, Symersky J, Walters DE (2010) Conformational changes in BAK, a pore‐forming proapoptotic Bcl‐2 family member, upon membrane insertion and direct evidence for the existence of BH3‐BH3 contact interface in BAK homo‐oligomers. J Biol Chem 285: 28924–28937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM et al (2005) An inhibitor of Bcl‐2 family proteins induces regression of solid tumours. Nature 435: 677–681 [DOI] [PubMed] [Google Scholar]
- Ordureau A, Sarraf SA, Duda DM, Heo JM, Jedrychowski MP, Sviderskiy VO, Olszewski JL, Koerber JT, Xie T, Beausoleil SA, Wells JA, Gygi SP, Schulman BA, Harper JW (2014) Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol Cell 56: 360–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordureau A, Paulo JA, Zhang W, Ahfeldt T, Zhang J, Cohn EF, Hou Z, Heo JM, Rubin LL, Sidhu SS, Gygi SP, Harper JW (2018) Dynamics of PARKIN‐dependent mitochondrial ubiquitylation in induced neurons and model systems revealed by digital snapshot proteomics. Mol Cell 70: 211–227.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao KC, Stanley M, Han C, Lai YC, Murphy P, Balk K, Wood NT, Corti O, Corvol JC, Muqit MM, Virdee S (2016) Probes of ubiquitin E3 ligases enable systematic dissection of parkin activation. Nat Chem Biol 12: 324–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petros AM, Nettesheim DG, Wang Y, Olejniczak ET, Meadows RP, Mack J, Swift K, Matayoshi ED, Zhang H, Thompson CB, Fesik SW (2000) Rationale for Bcl‐xL/Bad peptide complex formation from structure, mutagenesis, and biophysical studies. Protein Sci 9: 2528–2534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickles S, Vigie P, Youle RJ (2018) Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol 28: R170–R185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulogiannis G, McIntyre RE, Dimitriadi M, Apps JR, Wilson CH, Ichimura K, Luo F, Cantley LC, Wyllie AH, Adams DJ, Arends MJ (2010) PARK2 deletions occur frequently in sporadic colorectal cancer and accelerate adenoma development in Apc mutant mice. Proc Natl Acad Sci USA 107: 15145–15150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR‐Cas9 system. Nat Protoc 8: 2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, Zaffagnini G, Wild P, Martens S, Wagner SA, Youle RJ, Dikic I (2016) Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci USA 113: 4039–4044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rongvaux A, Jackson R, Harman CC, Li T, West AP, de Zoete MR, Wu Y, Yordy B, Lakhani SA, Kuan CY, Taniguchi T, Shadel GS, Chen ZJ, Iwasaki A, Flavell RA (2014) Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 159: 1563–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarraf SA, Raman M, Guarani‐Pereira V, Sowa ME, Huttlin EL, Gygi SP, Harper JW (2013) Landscape of the PARKIN‐dependent ubiquitylome in response to mitochondrial depolarization. Nature 496: 372–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauve V, Lilov A, Seirafi M, Vranas M, Rasool S, Kozlov G, Sprules T, Wang J, Trempe JF, Gehring K (2015) A Ubl/ubiquitin switch in the activation of Parkin. EMBO J 34: 2492–2505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauve V, Sung G, Soya N, Kozlov G, Blaimschein N, Miotto LS, Trempe JF, Lukacs GL, Gehring K (2018) Mechanism of parkin activation by phosphorylation. Nat Struct Mol Biol 25: 623–630 [DOI] [PubMed] [Google Scholar]
- Schagger H, von Jagow G (1991) Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal Biochem 199: 223–231 [DOI] [PubMed] [Google Scholar]
- Shiba‐Fukushima K, Imai Y, Yoshida S, Ishihama Y, Kanao T, Sato S, Hattori N (2012) PINK1‐mediated phosphorylation of the Parkin ubiquitin‐like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci Rep 2: 1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba‐Fukushima K, Arano T, Matsumoto G, Inoshita T, Yoshida S, Ishihama Y, Ryu KY, Nukina N, Hattori N, Imai Y (2014) Phosphorylation of mitochondrial polyubiquitin by PINK1 promotes Parkin mitochondrial tethering. PLoS Genet 10: e1004861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, Burman JL, Li Y, Zhang Z, Narendra DP, Cai H, Borsche M, Klein C, Youle RJ (2018) Parkin and PINK1 mitigate STING‐induced inflammation. Nature 561: 258–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staropoli JF, McDermott C, Martinat C, Schulman B, Demireva E, Abeliovich A (2003) Parkin is a component of an SCF‐like ubiquitin ligase complex and protects postmitotic neurons from kainate excitotoxicity. Neuron 37: 735–749 [DOI] [PubMed] [Google Scholar]
- Subburaj Y, Cosentino K, Axmann M, Pedrueza‐Villalmanzo E, Hermann E, Bleicken S, Spatz J, Garcia‐Saez AJ (2015) Bax monomers form dimer units in the membrane that further self‐assemble into multiple oligomeric species. Nat Commun 6: 8042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang MY, Vranas M, Krahn AI, Pundlik S, Trempe JF, Fon EA (2017) Structure‐guided mutagenesis reveals a hierarchical mechanism of Parkin activation. Nat Commun 8: 14697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trempe JF, Sauve V, Grenier K, Seirafi M, Tang MY, Menade M, Al‐Abdul‐Wahid S, Krett J, Wong K, Kozlov G, Nagar B, Fon EA, Gehring K (2013) Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science 340: 1451–1455 [DOI] [PubMed] [Google Scholar]
- Veeriah S, Taylor BS, Meng S, Fang F, Yilmaz E, Vivanco I, Janakiraman M, Schultz N, Hanrahan AJ, Pao W, Ladanyi M, Sander C, Heguy A, Holland EC, Paty PB, Mischel PS, Liau L, Cloughesy TF, Mellinghoff IK, Solit DB et al (2010) Somatic mutations of the Parkinson's disease‐associated gene PARK2 in glioblastoma and other human malignancies. Nat Genet 42: 77–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vives‐Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, Magrane J, Moore DJ, Dawson VL, Grailhe R, Dawson TM, Li C, Tieu K, Przedborski S (2010) PINK1‐dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci USA 107: 378–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Lu R, Ouyang X, Ho MW, Chia W, Yu F, Lim KL (2007) Drosophila overexpressing parkin R275W mutant exhibits dopaminergic neuron degeneration and mitochondrial abnormalities. J Neurosci 27: 8563–8570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wauer T, Komander D (2013) Structure of the human Parkin ligase domain in an autoinhibited state. EMBO J 32: 2099–2112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wauer T, Simicek M, Schubert A, Komander D (2015a) Mechanism of phospho‐ubiquitin‐induced PARKIN activation. Nature 524: 370–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wauer T, Swatek KN, Wagstaff JL, Gladkova C, Pruneda JN, Michel MA, Gersch M, Johnson CM, Freund SM, Komander D (2015b) Ubiquitin Ser65 phosphorylation affects ubiquitin structure, chain assembly and hydrolysis. EMBO J 34: 307–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292: 727–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MJ, McArthur K, Metcalf D, Lane RM, Cambier JC, Herold MJ, van Delft MF, Bedoui S, Lessene G, Ritchie ME, Huang DC, Kile BT (2014) Apoptotic caspases suppress mtDNA‐induced STING‐mediated type I IFN production. Cell 159: 1549–1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiener R, Zhang X, Wang T, Wolberger C (2012) The mechanism of OTUB1‐mediated inhibition of ubiquitination. Nature 483: 618–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YC, Holzbaur EL (2014) Optineurin is an autophagy receptor for damaged mitochondria in parkin‐mediated mitophagy that is disrupted by an ALS‐linked mutation. Proc Natl Acad Sci USA 111: E4439–E4448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata H, Shimizu S, Nishida Y, Watanabe Y, Craigen WJ, Tsujimoto Y (2009) Requirement of voltage‐dependent anion channel 2 for pro‐apoptotic activity of Bax. Oncogene 28: 3563–3572 [DOI] [PubMed] [Google Scholar]
- Yamano K, Youle RJ (2013) PINK1 is degraded through the N‐end rule pathway. Autophagy 9: 1758–1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshii SR, Kishi C, Ishihara N, Mizushima N (2011) Parkin mediates proteasome‐dependent protein degradation and rupture of the outer mitochondrial membrane. J Biol Chem 286: 19630–19640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, Strasser A (2008) The BCL‐2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 9: 47–59 [DOI] [PubMed] [Google Scholar]
- Zhang Z, Zhu W, Lapolla SM, Miao Y, Shao Y, Falcone M, Boreham D, McFarlane N, Ding J, Johnson AE, Zhang XC, Andrews DW, Lin J (2010) Bax forms an oligomer via separate, yet interdependent, surfaces. J Biol Chem 285: 17614–17627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Lee S, Peng Y, Bunker E, Giaime E, Shen J, Zhou Z, Liu X (2014) PINK1 triggers autocatalytic activation of Parkin to specify cell fate decisions. Curr Biol 24: 1854–1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6