

Abstract
Objective
Succinic Semialdehyde Dehydrogenase (SSADH) deficiency is a disorder of elevated gamma‐amino butyric acid (GABA) and gamma hydroxybutyric acid (GHB) and a complex neuropsychiatric profile. Adult reports suggest worsening epilepsy and high SUDEP risk.
Methods
Subjects with confirmed SSADH deficiency were recruited into a longitudinal study. Plasma thyroid hormone and total GABA/GHB were quantified by standard clinical chemistry methodologies and mass spectrometry, respectively.
Results
A total of 133 subjects with SSADH deficiency are enrolled in the registry; 49 participated in the longitudinal study. The age range of the population is 8 weeks to 63 years (median 7.75 year; 44% male). There is a significant difference in proportions among the age groups in subjects affected with hypotonia, compulsive behavior, sleep disturbances, and seizures. Epilepsy is present in 50% of the total population, and more prevalent in subjects 12 years and older (P = 0.001). The median age of onset for absence seizures was 2 years, and 12 years for generalized tonic‐clonic seizures (P < 0.01). The SUDEP rate in adults was 12% (4/33). There was a significant age‐dependent negative correlation between GABA and T3 levels.
Interpretation
There is an age‐dependent association with worsening of epilepsy, behavioral disturbances including obsessive‐compulsive behavior, and sleep disturbances with age in SSADH deficiency. There is a high risk of SUDEP. We have observed more absence seizures in younger patients, compared to tonic‐clonic in the older cohort, which correlates with age‐related changes in GABA and GHB concentration and thyroid function, as well as the natural history of seizures in the murine model.
Introduction
Succinic Semialdehyde Dehydrogenase (SSADH) deficiency is a rare autosomal recessive disease that interferes with the catabolism of the major inhibitory neurotransmitter gamma‐amino butyric acid (GABA) and furthermore leads to accumulation of various potential toxic metabolites, most prominently gamma hydroxybutyric acid (GHB). Increased endogenous levels of both GABA and GHB have been associated with the protean neurological manifestations, which typically include early life developmental impairment with cognitive deficiency and severe limitation in expressive language. Other neurological signs and symptoms are ataxia, hypotonia, hyporeflexia, behavioral dysregulation, and obsessive‐compulsive disorder.1, 2, 3, 4 Epilepsy is common, and a high SUDEP rate has been reported.5
Recent longitudinal studies have demonstrated changes in GABA and GHB levels with age. Highest levels are seen in the first years of life, with attainment of a nadir in GHB levels as measured in red blood cells by 10 years of age. Furthermore, GABA levels in patients reached control levels at 30–40 years of age.6 Hair analyses quantified for GHB levels from 10 patients with SSADH deficiency showed significantly elevated concentrations at ages 3–7 years with levels within the control range by 12–13 years.7
The mutant mouse model has shown a transition from absence to generalized convulsive seizures, culminating in lethal status epilepticus by three weeks of age.8, 9, 10, 11 Both animal and translational human studies in this disorder have demonstrated downregulation of both GABAA and GABAB receptors, attributed to chronic overexposure and possibly explaining the heightened epileptogenesis in this hyper‐GABAergic disorder.9, 10, 12, 13 Yet the evolution in the seizure manifestations over time in the mouse model remains unexplained. Furthermore, development of the GABA system, involving both GABAergic terminals and the excitatory to inhibitory maturation of GABA signaling, is dependent on the status of thyroid hormone.14 Both animal models and clinical studies of euthyroid and hypothyroid subjects have shown that GABA inhibits pituitary release of thyroid stimulating hormone (TSH). In mice models, GABA inhibits TSH‐stimulated thyroid hormone release.15 Based upon observed clinical manifestations and the established interactions between GABAergic and thyroid functions, we also investigated plasma T3, T4, and total GABA and GHB levels.
Methods
Subjects in our SSADH registry were enrolled into a longitudinal study approved by the Boston Children's Hospital Institutional Review Board (IRB), which obtains baseline and periodic six month follow‐up data. Data are collected and stored via REDCap (Research Electronic Data Capture) surveys. All subjects have confirmed SSADH deficiency based on gene sequencing and/or enzymatic quantification, in addition to clinical and biochemical phenotyping. Data obtained include age of onset and diagnosis, clinical manifestations including detailed seizure characteristics, diagnostic studies, and neuropsychiatric medication history. Developmental assessments for the longitudinal study are obtained utilizing the MACS (manual ability) instrument,16 GMFCS (gross motor),17 ABAS (adaptive behavior), and a quality of life inventory.
Data were analyzed in SPSS to compare the clinical manifestations, including seizure incidence, seizure types, and behavioral manifestations, between the pediatric (<age 12) and adolescent/adult (age 12+ years) cohorts for all subjects in the registry. Chi‐square analysis/Fisher exact test, depending on the cell count, was utilized to test for statistical significance. The longitudinal cohort was used to determine seizure frequency. Active epilepsy was defined as >2 seizures/year.
Quantification of plasma total GABA and GHB was performed as described previously6 and thyroid hormone measurements were done via standard clinical chemistry methodology.
Results
A total of 133 subjects with confirmed SSADH deficiency are enrolled in the registry; 49 are participating in the longitudinal study. The age range of the registry population is 8 weeks to 63 years (median 7.75 years; 44% male). The proportions of patients with common clinical features in the pediatric versus adolescent/adult groups are reported in the Table. Compulsive behaviors, sleep disturbances (based on difficulties with initiation or maintenance of sleep, or excessive daytime somnolence), hypotonia, and seizures show a statistically significant difference in proportion with age when comparing the pediatric to the adolescent/adult group (Table 1 and Fig. 1). Adolescent and adult subjects have a higher proportion of compulsive behavior, sleep disturbances, and seizures. Conversely, a smaller proportion of the older subjects have hypotonia compared to the childhood cohort (Table 1).
Table 1.
SSADH deficiency clinical features (N = 133)
| Pediatric (<12 years) N = 83 (%) | Adolescent/Adult (>12 years) N = 50 (%) | Odds ratio (95% CI) | P value | |
|---|---|---|---|---|
| Intellectual disability | 47 (56) | 29 (58) | 1.058 (0.520–2.151) | 0.877 |
| Fine motor delay | 55 (66) | 38 (76) | 1.612 (0.730–3.561) | 0.238 |
| Gross motor delay | 60 (72) | 38 (76) | 1.214 (0.541–2.722) | 0.638 |
| Speech delay | 63 (76) | 40 (80) | 1.270 (0.539–2.990) | 0.585 |
| Seizures | 31 (37) | 34 (68) | 3.565 (1.697–7.487) | 0.001a |
| Sleep disturbance | 28 (34) | 31 (62) | 3.205 (1.544–6.651) | 0.002a |
| Hypotonia | 65 (78) | 30 (60) | 0.415 (0.192–0.897) | 0.025a |
| Repetitive behavior/OCD | 17 (20) | 29 (58) | 5.361 (2.472–11.629) | <0.001a |
| Ataxia | 46 (55) | 23 (46) | 0.685 (0.339–1.386) | 0.293 |
| ADHD | 43 (52) | 28 (56) | 1.184 (0.585–2.396) | 0.639 |
P < 0.05
Figure 1.
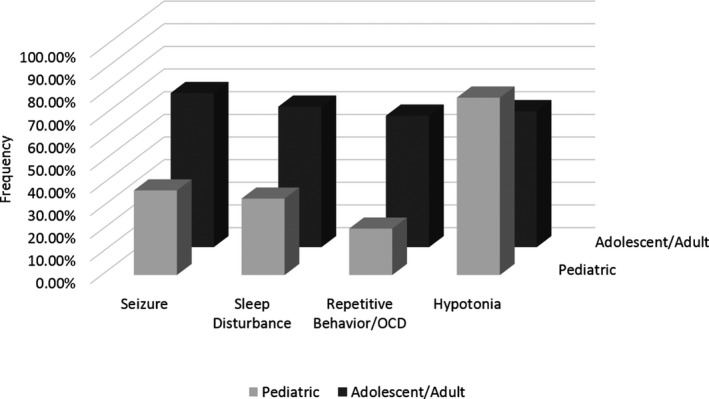
Proportion of comorbid clinical symptoms with age. Compulsive behaviors, sleep disturbances, hypotonia, and seizures show a statistically significant difference in proportion with age when comparing age the pediatric cohort to the adolescent/adult cohort.
Epilepsy is present in 50% of the total population, and more prevalent in the age 12 and older cohort (OR 3.565 [95% CI, 1.697–7.487]; P = 0.001). The most common seizure type was generalized tonic‐clonic (35/65), followed by myoclonic (21/65) and absence (20/65). Of the patients in the registry, the median age of onset was two years for absence seizures and 12 years generalized tonic‐clonic seizures (Fig. 2). Of subjects with a history of absence seizures, 94% had onset before age 12. In contrast, 55% of patients with generalized tonic clonic seizures had onset of this seizure type at age 12 or older (P < 0.01). Of the patients in the registry, 91 reported having had an EEG, of which 35% were abnormal (32/91), with generalized epileptiform discharges (12/32) and diffuse slowing (10/32) being the most common abnormalities.
Figure 2.
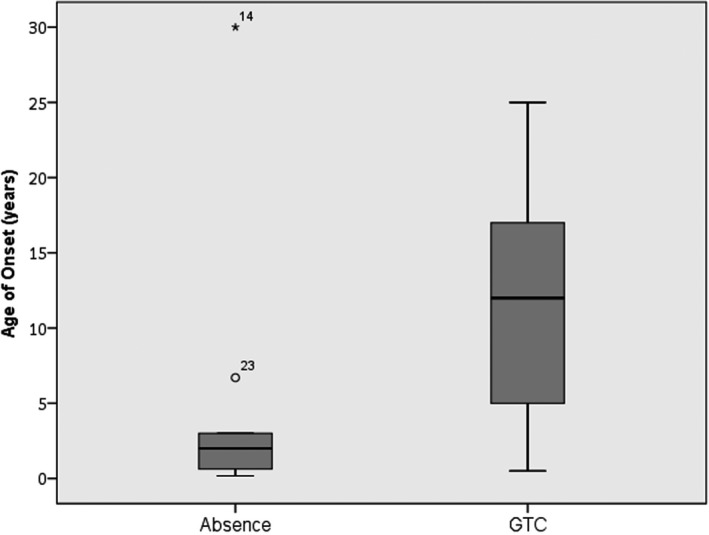
Box plot of age of onset versus seizure type in SSADH deficiency. Box plot of age of onset for type of seizures. The median age of onset is indicated by the solid black line inside the box (absence = 2 years old, GTC = 12 years old). The box signifies the 1st quartile (Q1) and the 3rd quartile (Q3). The vertical lines (whiskers) indicate the minimum and maximum values (range). Data falling well outside the Q1 and Q3 range are plotted as outliers of the data; the circle and asterisk indicate outliers. The circle represents an age of onset of absence seizures at 6.7 years, which is outside the Q3 value but still within age group <12 years. The asterisk represents an age of onset of absence seizures at 30 years old, which was reported during an EEG.
In the longitudinal group, 29 subjects are age 12+ and 20 are <12 years. Active epilepsy is more prevalent in the cohort of subjects 12 years and older (OR, 4.923 [95% CI, 1.318–18.385]; P = 0.018). To date we are aware of 4 SUDEP‐related deaths of SSADH‐deficient patients. Two were adult subjects in the registry, ages 63 and 33 identified as possible and probable SUDEP, respectively, and two were affected siblings confirmed postmortem, ages 19 and mid‐20s. With 31 adult subjects in the registry, this derives a potential SUDEP rate in the adult population of 12% (4/33).
Based upon observed clinical manifestations, and the established interactions between GABAergic and thyroid functions,14 we investigated plasma T3, T4, and total GABA in an archival set of SSADH‐deficient samples (age range, 2–30 years; 60% males). We observed a linear relationship between total GABA and both thyroid biomarkers, in addition to an age‐dependent negative correlation (Fig. 3). This correlation was not observed with plasma GHB levels.
Figure 3.
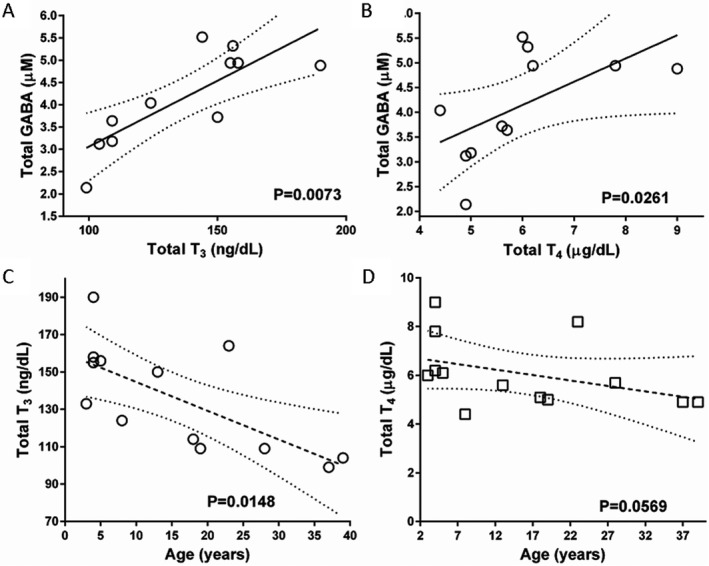
Correlations between thyroid hormones and total GABA in patient plasma (A and B) and thyroid hormones as a function of patient age (C and D). Dotted lines indicate 95th percentile. Confidence intervals (statistical analysis, Spearman correlation). Samples were derived from an archival collection of SSADH‐deficient plasma samples maintained at −80°C.
Discussion
SSADH deficiency is a lifelong neurometabolic disorder with onset in early childhood and evolving features over time. The ALDH5A1 null mouse model demonstrates an age‐specific onset of absence seizures followed in time by convulsive seizures and ultimately fatal status epilepticus by day P21.8, 18 The natural history of the condition is not well understood, including the basis for the evolution in the murine epilepsy. Downregulation of GABAergic activity has been demonstrated in murine and human studies, the latter utilizing flumazenil‐PET imaging and paired‐pulse transcranial magnetic stimulation for demonstration of GABAA and GABAB receptor activity, respectively. In addition, negative age correlations have been demonstrated with both GABA and GHB levels in patients, indicating high initial levels in the first decade that later reach a nadir and steady‐state.6, 7 The imbalance in GABA and GHB concentrations in this disorder appears to fluctuate with age.
Based on our registry of 133 patients, we noted that the proportion of subjects affected with hypotonia, compulsive behavior, sleep disturbances, and seizures changes over time. Subjects in the older cohort have an increased risk of compulsive behavior, sleep disturbances, and seizures compared to the cohort younger than age 12 years. Hypotonia was more likely to involve the younger group. While 50% are affected with epilepsy, the proportion with seizures is nearly 70% in those ages 12 and older, compared to 40% in the younger patients. The vast majority, over 90%, with absence seizures had onset under the age of 12. Of those with convulsive seizures, over half had onset over age 12. In addition, our longitudinal study has enabled assessment of seizure frequency. Those patients age 12 and over were more likely to have active epilepsy, that is 55% versus 20% (P < 0.02). Furthermore, there appears to be an extraordinary risk for SUDEP in this population.
The primary clinical features of SSADH deficiency (developmental impairment, seizures, altered sleep, and behavior disturbance), in addition to consideration of GABAergic mechanisms in both epilepsy and thyroid function,17, 19, 20, 21 suggested evaluation of thyroid function in SSADH deficiency (Fig. 4). Here, we demonstrate for the first time a linear relationship between GABA and T3/T4, as well as an age‐dependent negative correlation for T3 in SSADH‐deficient patients. These fluctuations are likely mediated via altered vagal nerve brainstem input.22 Previously, we have documented decreased GABAergic receptor density in both patients and the murine model, which will likely mitigate thyroid function and output. Importantly, co‐occurrence of age‐dependent negative correlations for both total GABA and thyroid hormones with age in patients6 could well underpin the evolving seizure phenotype in patients.22 It is recognized that observed changes in motor tone, seizure semiology, or psychiatric symptomatology may reflect the natural progression of neurological manifestations over the lifetime. Three adult patients (ages 23, 33, and 39 years) in our registry report taking thyroid hormone, but we have not systematically tested for clinical or laboratory hypothyroidism. This is planned for our upcoming NIH sponsored natural history study, as well as a multivariate analysis to compare the clinical manifestations over time with the levels of thyroid hormones, GABA, and related metabolites. Our longitudinal studies suggest a transition in symptomatology as well as a striking age‐dependent correlation with plasma GABA and thyroid function levels.
Figure 4.
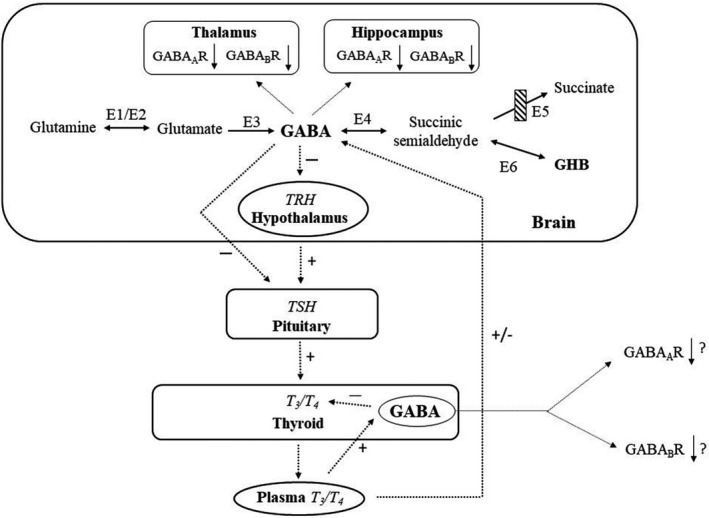
Schematic representation of potential interactions between the thyroid and GABA and the GABA catabolic pathway. Enzymes represented numerically: E1 and E2, glutaminase and glutamine synthetase; E3, glutamic acid decarboxylase; E4, GABA‐transaminase; E5, succinic semialdehyde dehydrogenase, site of the defect in patients with SSADH deficiency (crossed‐hatched box); and E6, aldo‐keto reductase 7a2. Additional abbreviations: GABA, γ‐aminobutyrate; GHB, γ‐hydroxybutyrate; TRH, thyrotropin releasing hormone; TSH, thyroid stimulating hormone; T3, liothyronine; T4, thyroxine. Plus (+) and minus (−) symbols refer to stimulation and inhibition, respectively. GABA is believed to downregulate thyroid activity at all levels of the hypothalamic‐pituitary‐thyroid endocrine axis, most likely effected via vagal innervation. Changes in plasma T3/T4 appear to differentially stimulate or depress GABAergic systems in brain and thyroid gland. Both GABA and GHB are elevated in brain of SSADH‐deficient patients and null mice. Plasma GABA concentrations did not correlate with circulating TSH (not shown). Also shown in the brain are the use‐dependent effects of GABA and GABAB subunit expression, as demonstrated in thalamus and hippocampus of SSADH‐deficient mice.9, 10 Downregulation of both receptor subtypes has been confirmed as well in SSADH‐deficient patients. (Figure adapted from Ref. 15). GHB, gamma hydroxybutyric acid; GABA, gamma‐amino butyric acid.
One may speculate that the ontogenic features of epilepsy and other clinical features in SSADH deficiency are related to developmental changes in the concentrations of GABA, GHB, their metabolites, and thyroid hormones over time. Our data support the evolution of epilepsy characteristics in patients with SSADH deficiency similar to the changes observed in the animal model.
Author Contributions
Melissa L. DiBacco (melissa.dibacco@childrens.harvard.edu) participated in conception and design of the study, acquisition and analysis of data, and drafting and revising the manuscript for content. Jean‐Baptiste Roullet (j.roullet@wsu.edu) participated in conception and design of the study, acquisition and analysis of data, and drafting and revising the manuscript for content. Kush Kapur (kush.kapur@childrens.harvard.edu) participated in analysis of data, and revising the manuscript for content. Madalyn N. Brown (madalyn.brown@wsu.edu) participated in revising the manuscript for content. Dana C. Walters (dcwalters@wsu.edu) participated in revising the manuscript for content. K. Michael Gibson (mike.gibson@wsu.edu) participated in conception and design of the study, acquisition and analysis of data, and drafting and revising the manuscript for content. Phillip L. Pearl (phillip.pearl@childrens.harvard.edu) participated in conception and design of the study, acquisition and analysis of data, and drafting and revising the manuscript for content.
Conflict of Interests
Melissa DiBacco reports no disclosures or conflicts of interest. Jean‐Baptiste Roullet reports no disclosures or conflicts of interest. Kush Kapur reports no disclosures or conflicts of interest. Madalyn N. Brown reports no disclosures or conflicts of interest. Dana C. Walters reports no disclosures or conflicts of interest. Michael Gibson reports no disclosures or conflicts of interest. Phillip Pearl reports no disclosures or conflicts of interest.
Acknowledgments
Erwin Jansen for measure of plasma GABA and GHB, and Mayo Medical Laboratories for measure of T3/T4.
Funding Information
Boston Intellectual and Development Disabilities Research Center 5U54HD090255
Funding Statement
This work was funded by Boston Intellectual and Development Disabilities Research Center grant 5U54HD090255.
References
- 1. Pearl PL, Novotny EJ, Acosta MT, et al. Succinic semialdehyde dehydrogenase deficiency in children and adults. Ann Neurol 2003;54:S73–S80. [DOI] [PubMed] [Google Scholar]
- 2. Pearl PL, Gibson KM, Acosta MT, et al. Clinical spectrum of succinic semialdehyde dehydrogenase deficiency. Neurology 2003;60:1413–1417. [DOI] [PubMed] [Google Scholar]
- 3. Pearl PL, Capp PK, Novotny EJ, Gibson KM. Inherited disorders of neurotransmitters in children and adults. Clin Biochem 2005;38:1051–1058. [DOI] [PubMed] [Google Scholar]
- 4. Gibson KM, Gupta M, Pearl PL, et al. Significant behavioral disturbances in succinic semialdehyde dehydrogenase (SSADH) deficiency (gamma‐hydroxybutyric aciduria). Biol Psychiatry 2003;54:763–768. [DOI] [PubMed] [Google Scholar]
- 5. Lapalme‐Remis S, Lewis EC, De Meulemeester C, et al. Natural history of succinic semialdehyde dehydrogenase deficiency. Neurology 2015;85:861–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jansen EE, Vogel KR, Salomons GS, et al. Correlation of blood biomarkers with age informs pathomechanisms in succinic semialdehyde dehydrogenase deficiency (SSADHD), a disorder of GABA metabolism. J Inherit Metab Dis 2016;39:795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Johansen SS, Wang X, Sejer Pedersen D, et al. Gamma‐Hydroxybutyrate (GHB) content in hair samples correlates negatively with age in succinic semialdehyde dehydrogenase deficiency. JIMD Rep 2017;36:93–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cortez MA, Wu Y, Gibson KM, Snead OC III. Absence seizures in succinic semialdehyde dehydrogenase deficient mice: a model of juvenile absence epilepsy. Pharmacol Biochem Behav 2004;79:547–553. [DOI] [PubMed] [Google Scholar]
- 9. Buzzi A, Wu Y, Frantseva MV, et al. Succinic semialdehyde dehydrogenase deficiency: GABAB receptor‐mediated function. Brain Res 2006;1090:15–22. [DOI] [PubMed] [Google Scholar]
- 10. Wu Y, Buzzi A, Frantseva M, et al. Status epilepticus in mice deficient for succinate semialdehyde dehydrogenase: GABAA receptor‐mediated mechanisms. Ann Neurol 2006;59:42–52. [DOI] [PubMed] [Google Scholar]
- 11. Vogel KR, Pearl PL, Theodore WH, et al. Thirty years beyond discovery–clinical trials in succinic semialdehyde dehydrogenase deficiency, a disorder of GABA metabolism. J Inherit Metab Dis 2013;36:401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pearl PL, Gibson KM, Quezado Z, et al. Decreased GABA‐A binding on FMZ‐PET in succinic semialdehyde dehydrogenase deficiency. Neurology 2009;73:423–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Reis J, Cohen LG, Pearl PL, et al. GABAB‐ergic motor cortex dysfunction in SSADH deficiency. Neurology 2012;79:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sawano E, Takahashi M, Negishi T, Tashiro T. Thyroid hormone‐dependent development of the GABAergic pre‐ and post‐synaptic components in the rat hippocampus. Int J Dev Neurosci 2013;31:751–761. [DOI] [PubMed] [Google Scholar]
- 15. Wiens SC, Trudea VL. Thyroid hormone and GABA interactions in neuroendocrine systems. Comp Biochem Physiol A Mol Integr Physiol 2006;144:334–344. [DOI] [PubMed] [Google Scholar]
- 16. Eliasson AC, Krumlinde‐Sundholm L, Rosblad B, et al. The Manual Ability Classification System (MACS) for children with cerebral palsy: scale development and evidence of validity and reliability. Dev Med Child Neurol 2006;48:549–554. [DOI] [PubMed] [Google Scholar]
- 17. Palisano R, Rosenbaum P, Walter S, et al. Development and reliability of a system to classify gross motor function in children with cerebral palsy. Dev Med Child Neurol 1997;39:214–223. [DOI] [PubMed] [Google Scholar]
- 18. Pearl PL, Gibson KM, Cortez MA, et al. Succinic semialdehyde dehydrogenase deficiency: lessons from mice and men. J Inherit Metab Dis 2009;32:342–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pereira JC Jr, Anderson ML. The role of thyroid hormone in sleep deprivation. Med Hypotheses 2014;82:350–355. [DOI] [PubMed] [Google Scholar]
- 20. Kincaid AE. Spontaneous circling behavior and dopamine neuron loss in a genetically hypothyroid mouse. Neuroscience 2001;105:891–898. [DOI] [PubMed] [Google Scholar]
- 21. Lee YJ, Nam SO, Kim KM, et al. Longitudinal change in thyroid hormone levels in children with epilepsy on a ketogenic diet: prevalence and risk factors. J Epilepsy Res 2017;7:99–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Marrosu F, Serra A, Maleci A, et al. Correlation between GABA(A) receptor density and vagus nerve stimulation in individuals with drug‐resistant partial epilepsy. Epilepsy Res 2003;55:59–70. [DOI] [PubMed] [Google Scholar]