

Abstract
Objective
To validate whether serum Neurofilament Light‐chain (NfL) levels correlate with disease severity in CADASIL, and to determine whether serum NfL predicts disease progression and survival.
Methods
Fourty‐one (pre‐) manifest individuals with CADASIL causing NOTCH3 mutations and 22 healthy controls were recruited from CADASIL families. At baseline, MRI‐lesion load and clinical severity was determined and serum was stored. Disease progression was measured in 30/41 patients at 7‐year follow‐up, and survival of all individuals was determined at 17‐year follow‐up. Serum NfL levels were quantified using an ultra‐sensitive molecule array. Generalized estimated equation regression (GEE) was used to analyze association between serum NfL, MRI‐lesion load, disease severity, and disease progression. With GEE‐based Cox regression, survival was analyzed.
Results
At baseline, serum NfL levels correlated with MRI‐lesion load [lacune count (s = 0.64, P = 0.002), brain atrophy (r = −0.50, P = 0.001), and microbleed count (s = 0.48, P = 0.044)], cognition [CAMCOG (s = −0.45, P = 0.010), MMSE (r = −0.61, P = 0.003), GIT (r = −0.61, P < 0.001), TMT‐A (r = 0.70, P < 0.001)) and disability (mRS (r = 0.70, P = 0.002)]. Baseline serum NfL predicted 7‐year changes in disability (B = 0.34, P < 0.001) and cognition (CAMCOG B = −4.94, P = 0.032), as well as 17‐year survival. Higher NfL levels were associated with increased mortality (HR=1.8 per twofold increase in NfL levels, P = 0.006).
Interpretation
Serum NfL levels correlate with disease severity, disease progression and 17‐year survival in CADASIL patients. Serum NfL is a promising biomarker to monitor and predict disease course in CADASIL, as well as potentially assessing therapeutic response in future clinical trials.
Introduction
CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy) is the most prevalent hereditary form of cerebral small vessel disease (SVD), and is caused by mutations in the NOTCH3 gene.1 CADASIL patients suffer from recurrent ischemic strokes with a mean age at onset of 45–50 years, although age at first stroke is highly variable.1 Cognitive decline is a main but also variable feature, initially manifesting as executive dysfunction and progressing to global impairment in all cognitive domains and complete care dependency.2 Migraine with aura affects up to two‐thirds of patients.3 In CADASIL, mutant NOTCH3 proteins aggregate in the tunica media of small arteries,4, 5, 6 directly or indirectly compromising cerebrovascular reactivity.7, 8 On MRI, white matter hyperintensities can be observed in premanifest patients from the third decade, while lacunes, microbleeds and brain atrophy usually appear later in the disease course.1, 9, 10
Lacunes and brain parenchymal fraction (BPF) have been shown to be predictors of disease progression in CADASIL patients.11, 12, 13, 14, 15, 16, 17 However, a good fluid biomarker would have advantages above the more cumbersome neuroimaging markers and could ideally also be used as a marker for therapeutic response in future clinical trials. Various potential fluid biomarkers have been investigated for CADASIL,18, 19 but most do not seem to be feasible. Recently, serum Neurofilament light‐chain levels were shown to correlate with disease severity in CADASIL.20
Neurofilament light‐chain protein (NfL) levels in serum have made a recent surge in the field of biomarkers for brain disease.21, 22, 23, 24, 25, 26, 27 NfL is a component of the neurofilament complex, which has a scaffolding role in neuronal axons, and is released into the cerebrospinal fluid (CSF) and blood upon neuronal damage,21 in some studies remaining elevated in blood for several months.22, 23 NfL levels have been shown to correlate with disease severity and progression in Huntington's disease,24 Frontotemporal Dementia25, 26, and Amyotrophic Lateral Sclerosis.27 In Multiple Sclerosis, NfL levels in blood have even been shown to be responsive to treatment, showing its potential as a therapeutic response marker.28, 29 In sporadic SVD and CADASIL, serum NfL levels were shown to be associated cross‐sectionally with neuroimaging markers, processing speed and disability, but the associations between serum NfL and disease progression and survival were not investigated.20, 22
Here, we explored whether serum NfL, measured using the ultra‐sensitive single molecule array, may be a feasible fluid biomarker for monitoring CADASIL disease severity, predicting disease progression, and predicting long‐term survival. We used a well characterized cohort of CADASIL patients and controls, which has been uniquely followed‐up for 17 years.
Methods
Cohort
At baseline, in 2000, 41 patients and 22 controls, from a total of 15 CADASIL families of which 10 families had ≥2 participants, were enrolled in the study. Thirty‐two patients had a NOTCH3 mutation in exon 4 and nine patients had a mutation in exon 8, 11, 19 or 20. A full medical history, MRI, serum withdrawal, neuropsychological testing and genetic NOTCH3 testing was performed, as described earlier.30 The average age at baseline was 46 years in patients and 39 years in healthy relatives (Table 1). Seven years later this same cohort was invited to participate in a follow‐up study. Thirty patients consented to participate. Eleven participants were lost‐to‐follow‐up due to death (six patients) or severe disease (five patients). Five patients did not consent to MRI in the follow‐up study.15 Seventeen years after baseline, survival data from all participants of the baseline study was obtained from the Dutch population registry. These studies were approved by the medical ethics committee of the Leiden University Medical Center (P80/98) and each participant gave informed consent.
Table 1.
Cohort characteristics at baseline and follow‐up
| Cross‐sectional study at baseline | 7‐year follow‐up study in patients | |||
|---|---|---|---|---|
| Controls | Patients | Baseline | Follow‐up | |
| Mean (SD), median (range) or n (%) | Mean (SD), median (range) or n (%) | |||
| N | 22 | 41 | 30 | 30 |
| Age, y | 39.8 (12.5) | 45.8 (10.4) | 44.0 (9.8) | 51.1 (9.8) |
| Female | 12 (55%) | 22 (54%) | 17 (57%) | 17 (57%) |
| Prior stroke/TIA | 0 (0%) | 23 (56%) | 16 (53%) | – |
| Time since last stroke | – | 3.3 (0.1–17.7) | 3.5 (0.1–14.2) | – |
| Hypertension | 6 (27%) | 3 (7%) | 2 (7%) | – |
| Diabetes mellitus | 0 (0%) | 3 (7%) | 3 (10%) | – |
| Hyperlipidaemia | 4 (18%) | 15 (37%) | 10 (33%) | – |
| Current or past smoking | 14 (64%) | 26 (63%) | 19 (63%) | – |
| Serum NfL (pg/mL) | 1.66 (0.77–9.7) | 6.31 (1.22–107.7) | 5.21 (1.22–31.6) | – |
| MRI data available (n) a | 21 | 40 | 25 | 25 |
| Lacunes on MRI | ||||
| 0 lacunes | 21 (100%) | 8 (20%) | 6 (24%) | 5 (20%) |
| 1–10 lacunes | 15 (38%) | 14 (56%) | 12 (48%) | |
| >10 lacunes | 17 (42%) | 5 (20%) | 8 (32%) | |
| Brain atrophy (BPF) | 82.198 (2.977) | 82.648 (2.919) | 81.836 (3.065) | |
| WMHV | 0.003 (0–0.075) | 7.297 (0.009–19.209) | 4.303 (0.009–13.939) | 7.865 (0.005–19.806) |
| Microbleeds | ||||
| 0 microbleeds | 21 (100%) | 29 (73%) | 22 (88%) | 18 (72%) |
| 1–10 microbleeds | 9 (22%) | 2 (8%) | 5 (20%) | |
| >10 microbleeds | 2 (5.0%) | 1 (4%) | 2 (8%) | |
| Clinical score available (n)a | 22 | 41 | 30 | 30 |
| mRS | ||||
| score 0 | 22 (100%) | 13 (32%) | 12 (40%) | 9 (30%)b |
| score 1–2 | 17 (42%) | 15 (50%) | 10 (33%)b | |
| score ≥3 | 11 (27%) | 3 (10%) | 10 (33%)b | |
| CAMCOG | 94.2 (4.9) | 88.3 (15.0) | 93.5 (5.3) | 88.1 (13.7) |
| MMSE | 28.3 (1.5) | 26.2 (4.3) | 27.7 (1.6) | 26.2 (5.0) |
| GIT | 105.8 (11.7) | 99.8 (16.9)b | 103.8 (12.5) | 100.6 (18.6) |
| TMT‐A | 30 (21–90) | 37 (10–180)e | 36 (10–92)b | 40 (15–349)c |
| TMT‐B | 74.5 (38–216) | 84 (36–540)f | 82 (36–254)b | 75.5 (44–300)d |
At baseline, one patient and one control did not consent to radiological examination. At follow‐up, five patients did not consent to radiological examination.
Missing data for one patient.
Missing data for three patients.
Missing data for four patients.
Missing data for five patients.
Missing data for seven patients.
Neuroimaging and neuropsychological testing
MRI was performed on a 1.5 T MR system at baseline9 and at 7‐year follow‐up.15, 16 Lacune count, microbleed count, parenchymal volume, intracranial volume, and white matter hyperintensity volume were previously quantified.15, 16, 31 Parenchymal volume was normalized to intracranial volume to acquire the brain parenchymal fraction (BPF), as a measure for brain atrophy. White matter hyperintensity volume was normalized to parenchymal volume (WMHV). The neuropsychological test battery included the Cambridge Cognitive Examination (CAMCOG), Mini–Mental State Examination (MMSE), the Groningen Intelligence Test (GIT), and the Trail Making Test part A (TMT‐A) and part B (TMT‐B).32, 33, 34, 35 Disability was assessed using the modified Rankin scale (mRS).
Serum NfL measurement
At baseline, blood was withdrawn via standard vena puncture and centrifuged at 2750 g for 20 min at room temperature. The supernatant was aliquoted and stored at −80°C freezer until analysis. Serum NfL levels were quantified using an in‐house developed Simoa assay, as previously described.36 The assay was validated prior to use according to standardized international protocols.37 The lower level of quantification (LLOQ) was 0.77 pg/mL (10 times the standard deviation of 16 negative samples). Three of the 41 patients (age 22, 30, 39 years) and one of the 22 controls (age 24 years) had a serum NfL below the LLOQ and were excluded from the study. A sensitivity analysis, including these individuals and using half of the LLOQ as the measurement for these individuals, showed similar results.
Statistics
Nonnormally distributed data was transformed to obtain plausible normal distributions for further analyses: serum NfL was natural log (ln) transformed and to facilitate interpretation also log2 transformed for Cox‐regression; lacune count, WMHV and CAMCOG were square root transformed; TMT‐A was log10 transformed; and microbleed count was log10(1 + x) transformed. To determine the association between NfL levels and brain MRI lesion load and clinical outcomes at baseline, generalized estimated equation multivariable regression models (GEE), using an independent correlation structure and robust variance estimators, were used to enable correction for correlation within families, since 10 families had ≥2 participants in the study. Sex and age were included in all linear regression GEE analyses. To determine the association between age and NfL in both patients and controls, the GEE model also included mutation status, and the age‐mutation status interaction. To determine how NfL compared with MRI markers in predicting disease severity, forward and backward regular linear regression was performed with NfL, lacune count, brain atrophy, WMHV, and microbleed count as potential predictors. Potential predictors were added only if they significantly contributed to the model.
To determine whether NfL levels were associated with 7‐year changes in MRI lesion load and clinical deterioration, GEE models were used with either NfL and sex; or NfL, sex and baseline measurement of the tested outcome. For clarity, the correlations in the figures are presented using Pearson (r) or Spearman (s) correlation coefficients, together with p‐values obtained from GEE regression models. Hazard ratios for the relation of baseline NfL with survival at 17‐year follow‐up were calculated using a multivariate GEE‐based Cox regression analysis, including age and the covariate of interest. Area under the curve (AUC) and confidence intervals were calculated and compared, using DeLong tests in R package pROC. We did not formally prove equivalence in the prognostic value of NfL versus the neuroimaging markers, as formal equivalence testing would be under‐powered in the current data set. GEE‐based logistic regression analyses were performed with an independent correlation structure and were corrected for sex. All other statistical analyses were performed in IBM SPSS Statistics version 23.0.0.2. All statistical tests were two‐sided and the threshold for statistical significance was 0.05.
Results
NfL levels were higher in patients (median 6.31 pg/mL, range 1.22–107.7 pg/mL) than in controls (median 1.66 pg/mL, range 0.77 to 9.73 pg/mL) (P < 0.001). NfL levels positively correlated with age in both patients and controls. However, this age‐dependent increase was stronger in patients (7.6% increase per year, CI: 4.6–10.7%) than in controls (3.7% increase per year, CI: 2.2–5.3%)(P = 0.012). In contrast to controls, patients showed a steeper increase in NfL levels from age 40 onwards, which coincided with the presence of lacunes (Fig. 1).
Figure 1.
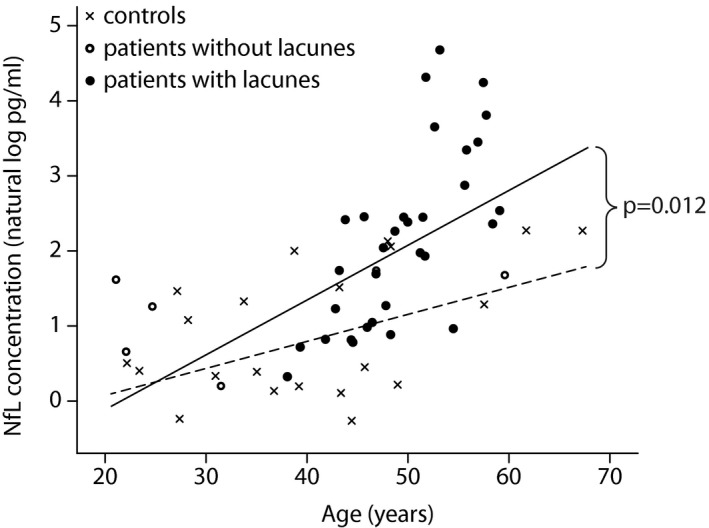
Serum NfL levels show an age‐dependent increase in CADASIL patients and controls. CADASIL patients with lacunes (black dots) show a stronger age‐dependent serum NfL increase than patients without lacunes (open circles) and controls (crosses).
Correlation of serum NfL with disease severity at baseline
After correcting for age and sex, NfL levels correlated strongly with MRI lacune count (s = 0.64, P = 0.002), brain parenchymal fraction (r = −0.50, P = 0.001), and microbleed count (s = 0.48, P = 0.044), but not with WMHV levels (r = 0.48, P = 0.894) (Fig. 2). Without correcting for age, NfL levels did correlate with WMHV (r = 0.48, P < 0.001). When including all MRI markers in the model, NfL levels independently correlated with lacune count (B = 0.20, P = 0.002), brain parenchymal fraction (B = −0.14, P < 0.001), and microbleed count (B = 0.56, P = 0.009). NfL levels did not correlate with time since last clinical stroke (data not shown).
Figure 2.
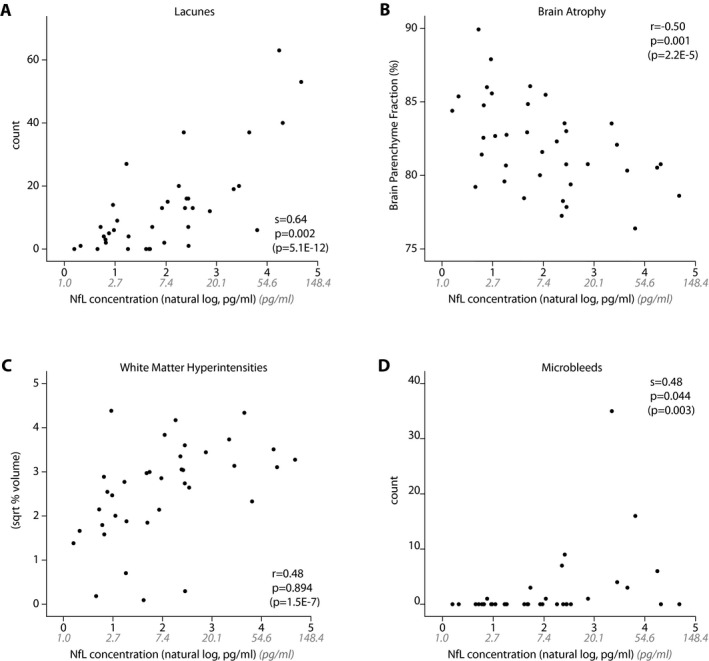
Cross‐sectional associations between serum NfL levels and MRI markers. After correction for age and sex, NfL levels significantly correlate with lacune count (A), brain atrophy (B), and microbleeds (D), but not with white matter hyperintensity volume (C). P‐values in parentheses represent P‐value before correction for age and sex. NfL levels were natural log transformed for analyses, nontransformed NfL levels are shown in gray. NfL, Neurofilament light‐chain.
Next, the relation of NfL with cognitive outcome measures was assessed. After correcting for sex and age, NfL levels correlated with disability scores of mRS (r = 0.70, P = 0.002), and cognitive function determined by CAMCOG (s = −0.45, P = 0.010), MMSE (r = −0.61, P = 0.003), GIT score (r = −0.61, P < 0.001), and TMT‐A (r = 0.70, P < 0.001), but not with TMT‐ B (s = 0.39, P = 0.115) (Fig. 3). Serum NfL correlated more strongly with cognitive function (CAMCOG, MMSE and GIT) than any of the MRI markers correlated with cognitive function. NfL was associated less strongly with disability (mRS) and executive function (TMT‐A and TMT‐B) than lacune count (Table 2).
Figure 3.
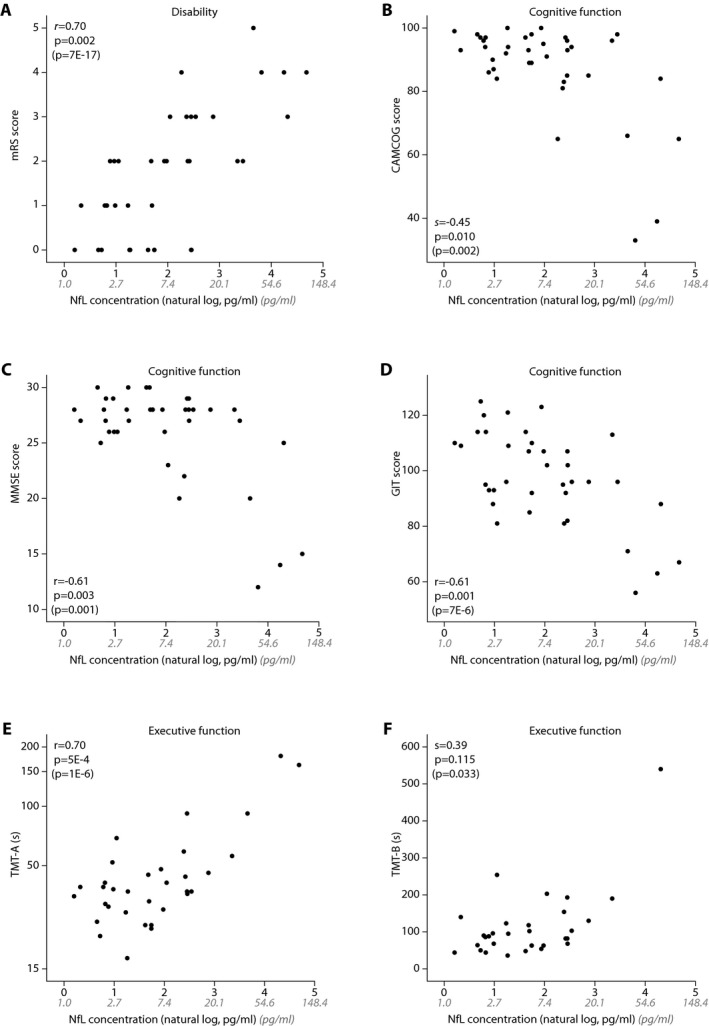
Cross‐sectional association between serum NfL levels and disability, cognitive function, and executive function. NfL levels correlate with disability score mRS (A), cognitive function scores CAMCOG (B), MMSE (C), GIT (D), and executive function score TMT‐A (E), but not with TMT‐B (F). P‐values in parentheses represent P‐value before correction for age and sex. Serum NfL levels were natural log transformed for analyses. Nontransformed NfL levels are shown in gray. NfL, Neurofilament light‐chain; mRS, modified Rankin Scale; CAMCOG, Cambridge Cognitive Examination; MMSE, Mini–mental state examination; GIT, Groningen Intelligence Test; TMT‐A, Trail Making Test part A; TMT‐B, Trail Making Test part B.
Table 2.
Independent cross‐sectional predictors for disability, cognition and executive function
| First predictor | Second predictor | ||||||
|---|---|---|---|---|---|---|---|
| Clinical scores | Covariate | Std. B | P | Covariate | Std. B | P | |
| Cognition | CAMCOG (sqrt) | NfL | −0.593 | <0.001* | |||
| Cognition | MMSE | NfL | −0.609 | <0.001* | |||
| Cognition | GIT | NfL | −0.611 | <0.001* | |||
| Executive function | TMT‐A (log) | Lacunes | 0.466 | 0.008* | NfL | 0.382 | 0.027* |
| Executive function | TMT‐B | Lacunes | 0.401 | 0.002* | NfL | 0.362 | 0.013* |
| Disability | mRS | Lacunes | 0.455 | 0.006* | NfL | 0.381 | 0.019* |
For clinical scores, the covariate with the best correlation is shown in the ‘first predictor’ column. A second predictor is only shown if it made a significant contribution. Results of forward regression are shown, but were validated with backward regression (not shown). NfL, serum concentration Neurofilament light‐chain; mRS, modified Rankin Scale; CAMCOG, Cambridge Cognitive Examination; MMSE, Mini–mental state examination; GIT, Groningen Intelligence Test; TMT‐A, Trail Making Test part A; TMT‐B, Trail Making Test part B. *P < 0.05.
Prediction of 7‐year disease progression
To determine whether serum NfL levels predict disease progression, we assessed whether baseline serum NfL levels correlated with changes over 7 years in MRI lesion load, disability and cognition. Baseline NfL levels correlated with increased disability (mRS; B = 0.34, P < 0.001), decline in cognitive function (CAMCOG; B = −4.94, P = 0.032), and decline in executive function (TMT‐B time; B = 24.55, P = 0.035)(Table 3A). After correction for baseline values of the clinical measures, the correlation between NfL and disability (mRS; B = 0.28, P = 0.044) and executive dysfunction (TMT‐B; B = 29.03, P = 0.025) remained significant, suggesting that NfL levels measured at any stage of the disease are associated with future disability.
Table 3.
Correlation between baseline serum NfL levels and 7‐year changes in neuroimaging markers and clinical scores in all patients (A) and in patients with disability at baseline (mRS>0) (B)
| Model 1, corrected for sex | Model 2, corrected for sex and baseline values of tested covariate | |||||
|---|---|---|---|---|---|---|
| NfL | Baseline | NfL | ||||
| B | P | B | P | B | P | |
| (A) All patients | ||||||
| MRI markers | ||||||
| ∆ Lacune count (sqrt) | 0.325 | 0.178 | 0.128 | 0.001* | −0.149 | 0.341 |
| ∆ BPF | −0.059 | 0.879 | −0.180 | 0.249 | −0.323 | 0.504 |
| ∆ WMHV (sqrt) | 0.074 | 0.593 | 0.487 | <0.001* | −0.165 | 0.291 |
| ∆ MB count (log) | 0.350 | 0.643 | 5.871 | 0.071 | −0.874 | 0.476 |
| Clinical scores | ||||||
| ∆ mRS score | 0.338 | <0.001* | 0.072 | 0.551 | 0.281 | 0.044* |
| ∆ CAMCOG score (sqrt) | −4.944 | 0.032* | 0.782 | 0.040* | −4.297 | 0.061 |
| ∆ MMSE score | −2.109 | 0.051 | −0.132 | 0.595 | −2.100 | 0.053 |
| ∆ GIT score | −1.714 | 0.462 | 0.157 | 0.538 | −1.073 | 0.727 |
| ∆ TMT‐A | 20.028 | 0.087 | 132.81 | 0.276 | 9.152 | 0.306 |
| ∆ TMT‐B | 24.551 | 0.035* | −0.175 | 0.421 | 29.025 | 0.025* |
| (B) Patients with disability at baseline (mRS>0) | ||||||
| MRI markers | ||||||
| ∆ Lacune count (sqrt) | 0.371 | 0.007* | 0.112 | 0.003* | −0.187 | 0.187 |
| ∆ BPF | −0.005 | 0.989 | 0.087 | 0.458 | 0.106 | 0.681 |
| ∆ WMHV (sqrt) | 0.102 | 0.576 | 0.774 | <0.001* | −0.273 | 0.115 |
| ∆ MB count (log) | 0.586 | 0.479 | 6.169 | 0.140 | −1.272 | 0.471 |
| Clinical scores | ||||||
| ∆ mRS score | 0.330 | <0.001* | −0.035 | 0.864 | 0.356 | 0.076 |
| ∆ CAMCOG score (sqrt) | −5.705 | 0.018* | 0.528 | 0.265 | −5.879 | 0.013* |
| ∆ MMSE score | −2.374 | 0.037* | −0.412 | 0.459 | −2.311 | 0.046* |
| ∆ GIT score | −2.280 | 0.397 | −0.038 | 0.847 | −2.347 | 0.422 |
| ∆ TMT‐A | 24.545 | 0.154 | 296.25 | 0.224 | 4.957 | 0.747 |
| ∆ TMT‐B | 33.488 | 0.030* | −0.548 | 0.054 | 53.286 | 0.001* |
Delta indicates the difference between baseline and 7‐year follow‐up for the respective MRI marker or clinical score; positive value indicate an increase in the score, while negative values indicate a decrease in the score. Data of all patients who participated at 7‐year follow‐up is shown (A), as well as the subset of patients with disability at baseline (B). In model 1, the prognostic value of baseline NfL for the differences in MRI markers and clinical scores are shown, after correcting for sex. In model 2, the prognostic value of baseline NfL and the baseline score of the tested variable are shown, after correcting for sex. NfL levels were natural log transformed. Behind MRI markers and clinical scores the type of transformation of the variable is indicated, if applicable. NfL, serum concentration Neurofilament light‐chain; mRS, modified Rankin Scale; CAMCOG, Cambridge Cognitive Examination; MMSE, Mini–mental state examination; GIT, Groningen Intelligence Test; TMT‐A, Trail Making Test A; TMT‐B, Trail Making Test B. *P < 0.05.
NfL was not associated with an increase in lesion load of any of the MRI measures at follow‐up. However, in patients with disability at baseline (mRS > 0), baseline NfL levels were associated with an increase in lacune count (B = 0.371, P < 0.001), as well as with increased disability (mRS; B = 0.33, P < 0.001), decline in cognitive function (CAMCOG; B = −5.71, P = 0.018; MMSE; B =−2.37, P = 0.037) and decline in executive function (TMT‐B time; B = 33.5, P = 0.030)(Table 3B). In patients who had no disability at baseline (mRS = 0), serum NfL did not associate with disease progression.
Prediction of 17‐year survival
After correction for age, serum NfL levels significantly predicted 17‐year survival (HR 2.3 per 1 unit natural log pg/mL NfL increase, CI:1.3–4.2, P = 0.006; corresponding to HR 1.8 per 2‐fold increase in absolute NfL levels, CI: 1.2–2.7, P = 0.006). In patients with the highest NfL levels at baseline (upper tertile; >11.2 pg/mL), the survival after 17 years was 25%, while survival was 50% and 75% in patients with NfL levels in the middle and lower tertile (<3.55 pg/mL) (log rank P = 0.012), respectively (Fig. 4). To determine whether NfL was better in predicting 17‐year survival than MRI markers or age, receiver operating characteristic (ROC) analyses were performed. The area under the curve (AUC) was similar for all parameters: 0.813 for serum NfL (CI: 0.680–0.947), 0.857 for lacune count (CI: 0.733–0.981), 0.850 for brain atrophy (CI: 0.735–0.965) and 0.843 for age (CI: 0.726–0.960). In both ROC analyses and logistic regression, the combination of NfL with any MRI marker or with age was not significantly better in predicating 17 year survival than NfL alone.
Figure 4.
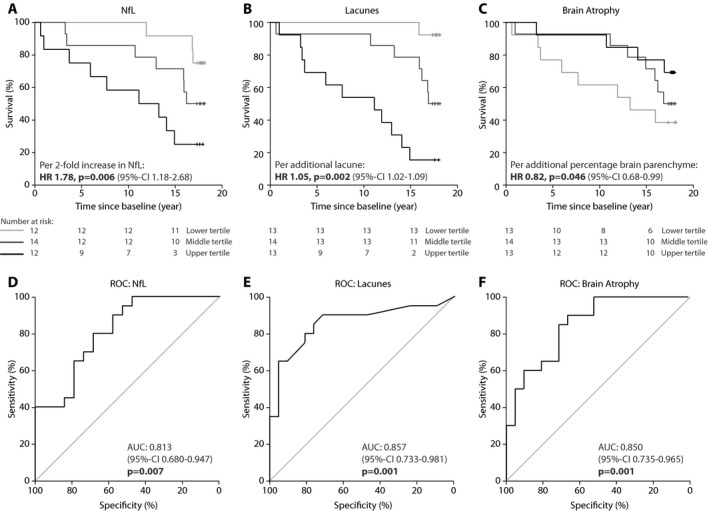
Association between serum NfL levels and 17‐year survival. Kaplan‐Meier plots show longitudinal survival divided into tertiles for serum NfL levels (upper tertile >11.2 pg/mL, middle tertile 3.53–11.22 pg/mL, lower tertile <3.53 pg/mL)(A), lacune count (upper tertile >13, middle tertile 4–13, lower tertile ≤3 )(B), and brain atrophy (upper tertile > 83.4%, middle tertile 80.7–83.4%, lower tertile <80.7%) (C). Receiver Operating Characteristic curves illustrate the prognostic ability for serum NfL levels (D), lacune count (E), and brain atrophy (F) at baseline to discriminate between live and deceased patients after 17‐year follow‐up. P‐values indicate testing with null‐hypothesis AUC 0.50 as indicated by the diagonal line. NfL = Neurofilament light‐chain; AUC = area under the curve; 95%‐CI = 95% confidence interval.
Discussion
Here, we validate a recent study showing that serum NfL levels correlate with disease severity in CADASIL. Moreover, we show that serum NfL levels at baseline also predict disease progression as well as 17‐year survival in a cohort of well‐characterized CADASIL patients.
We found that serum NfL levels correlated with all neuroimaging markers. After correction for age, serum NfL levels at baseline correlated independently with lacune count, brain atrophy, and to a lesser extent with microbleed count, but not with WMHV. These findings are in line with the recent study by Duering et al., showing serum NfL correlates with all neuroimaging markers in CADASIL.20
Although WMH are widely present in patients with SVD, such as CADASIL, and seem to correlate to some extent with NfL in serum or CSF,20, 22, 38 other neuroimaging markers, namely brain atrophy and lacunes, correlate more strongly with disease severity.11, 12, 13, 14, 15, 16, 17 In line with this, mean diffusivity from diffusion tensor imaging (DTI) was shown to be most strongly associated with serum NfL levels in CADASIL.20 Together with our finding that NfL independently correlates with lacunes and brain atrophy, this indicates that serum NfL reflects structural axonal damage, irrespective of the cause, and probably integrates the effect of lacunar infarcts and brain atrophy in a single measure, in a similar way as mean diffusivity does. This may implicate that serum NfL may be a suitable biomarker for manifest CADASIL, but not for the premanifest stages of CADASIL, i.e., when patients only have WMH. However, this needs to be further clarified in larger cohorts of premanifest patients.
In agreement with the study by Duering et al., serum NfL levels correlated with disease severity as reflected by disability scores, global cognitive function, and executive function. Global cognitive function correlated more strongly with serum NfL levels than with lacune count or BPF, which have been considered to be the best predictors of cognitive function and cognitive decline in CADASIL to date.11, 12, 13, 14, 15, 16, 17 Taken together, the correlation of serum NfL levels with MRI lesion load, cognition, and disability, suggests that serum NfL may serve as a feasible fluid biomarker to facilitate assessment of CADASIL disease severity in a single measure at a given time‐point.
Moreover, baseline serum NfL predicted 7‐year deterioration in disability (mRS), global cognitive function (CAMCOG), and executive function (TMT‐B), but did not correlate with 7‐year progression in MRI lesion load. Previous studies have shown that brain atrophy and lacunes predict disease progression in CADASIL,11, 12, 13, 14, 15, 16, 17 and both of these strongly correlate with serum NfL levels. Here, the lack of correlation between NfL and progressive MRI lesions at follow‐up is likely explained by the loss to follow up of more severely affected patients. Therefore, patients who were premanifest or mildly affected at baseline, were overrepresented at 7‐year follow‐up. Indeed, when only including in the analysis those patients who had disability at baseline, serum NfL did correlate with an increase in MRI lesion load (lacune count).
A general weakness of this study is that it was not originally designed for the purpose of a longitudinal study of serum biomarkers, and in this regard the cohort is relatively small.
In several studies, serum NfL has been shown to predict survival in neurodegenerative disease, but not yet in SVD.25, 27, 39 Here, we show that NfL also predicts survival in CADASIL, a pure model of SVD. However, as the follow‐up period was relatively long and our study lacks longitudinal assessment of serum NfL levels, we were not able to determine whether NfL levels can be used as a disease monitoring biomarker at shorter time intervals or as a potential marker for therapeutic response in future clinical trials. We do find that NfL correlates strongly with lacunes and BPF, which have both been shown to significantly change in a 3‐year time frame.14, 17 This suggests that serum NfL levels may also show significant changes at shorter intervals in manifest patients with progressive disease.
Like Duering et al., we used the ultra‐sensitive single‐molecule array (Simoa) for NfL measurements into the pg/mL range at high precision and sensitivity, which outperforms the conventional ELISA and chemiluminescence‐based methods.20, 36 Serum NfL levels were lower for both patients and controls in our study. A single‐measure serum biomarker such as NfL clearly has many advantages over more complex measures such as MRI markers or neuropsychological testing, which are currently used to assess CADASIL disease severity and predict disease progression.
In conclusion, we validate the recent finding that serum NfL levels correlate cross‐sectionally with relevant measures of disease severity in an independent CADASIL sample. Furthermore, we show that serum NfL is predictive of 7‐year deterioration in cognition and disability, and is associated with 17‐year survival. These findings suggest that serum NfL may be useful marker to monitor and predict disease course in this variable disorder, as well as potentially providing a feasible marker for therapeutic intervention in future clinical trials for CADASIL.
Authors Contribution
GG, JR, and SLO conceived and designed the study. ML and JG performed the MRI studies. IV performed the serum NfL quantification. GG, JR, SB, and SLO analyzed the data. GG, JR, and SLO drafted the manuscript. All authors reviewed the manuscript.
Conflict of Interest
We report no relevant conflict of interest. GG and JW reports grants from The Netherlands Brain Foundation during the conduct of the study. SLO reports grants from The Netherlands Brain Foundation, during the conduct of the study; grants from VIDI ZonMW, outside the submitted work. In addition, SLO has a patent Means and methods for modulating NOTCH3 protein expression and/or the coding region of NOTCH3 licensed to LUMC. IV reports a grant from Health~Holland via Alzheimer Nederland to support a research collaboration with Crossbeta Biosciences. Nonfinancial support in the form of research consumables was received from Crossbeta Biosciences for this same project, outside the submitted work. CT reports personal fees from the advisory board of Fujirebio and Roche, nonfinancial support from research consumables from ADxNeurosciences, others from performed contract research or received grants from the Janssen prevention center, Boehringer, EIP farma, Roche and Probiodrug, PeopleBio, Charles River, outside the submitted work. AAR reports grants from Netherlands Brain Foundation, during the conduct of the study; grants from EU, grants from ZonMw (Dutch Government), grants from Duchenne Parent Project, grants from Prinses Beatrix Spierfonds, outside the submitted work. In addition, AAR has a patent issued. JG and SB have nothing to disclose.
Acknowledgments
We acknowledge the support from the Netherlands Brain Foundation (HA2016‐02‐03). We thank the patients and their family members who participated in this study.
Funding Information
We acknowledge the support from the Netherlands Brain Foundation (HA2016‐02‐03).
Funding Statement
This work was funded by Netherlands Brain Foundation grant HA2016‐02‐03.
References
- 1. Chabriat H, Joutel A, Dichgans M, et al. Cadasil. Lancet Neurol 2009;8:643–653. [DOI] [PubMed] [Google Scholar]
- 2. Buffon F, Porcher R, Hernandez K, et al. Cognitive profile in CADASIL. J Neurol Neurosurg Psychiatry 2006;77:175–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tan RYY, Markus HS. CADASIL: migraine, encephalopathy, stroke and their inter‐relationships. PLoS ONE 2016;11:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Joutel A, Andreux F, Gaulis S, et al. The ectodomain of the Notch3 receptor accumulates within the cerebrovasculature of CADASIL patients. J Clin Invest 2000;105:597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Opherk C, Duering M, Peters N, et al. CADASIL mutations enhance spontaneous multimerization of NOTCH3. Hum Mol Genet 2009;18:2761–2767. [DOI] [PubMed] [Google Scholar]
- 6. Duering M, Karpinska A, Rosner S, et al. Co‐aggregate formation of CADASIL‐mutant NOTCH3: a single‐particle analysis. Hum Mol Genet 2011;20:3256–3265. [DOI] [PubMed] [Google Scholar]
- 7. Pfefferkorn T, von Stuckrad‐Barre S, Herzog J, et al. Reduced cerebrovascular CO(2) reactivity in CADASIL: a transcranial Doppler sonography study. Stroke 2001;32:17–21. [DOI] [PubMed] [Google Scholar]
- 8. Singhal S, Markus HS. Cerebrovascular reactivity and dynamic autoregulation in nondemented patients with CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy). J Neurol 2005;252:163–167. [DOI] [PubMed] [Google Scholar]
- 9. van den Boom R, Lesnik Oberstein SAJ, Ferrari MD, et al. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy: MR imaging findings at different ages–3rd‐6th decades. Radiology. 2003;229:683–690. [DOI] [PubMed] [Google Scholar]
- 10. Tikka S, Baumann M, Siitonen M, et al. CADASIL and CARASIL. Brain Pathol 2014;24:525–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liem MK, Van Der Grond J, Haan J, et al. Lacunar infarcts are the main correlate with cognitive dysfunction in CADASIL. Stroke 2007;38:923–928. [DOI] [PubMed] [Google Scholar]
- 12. Viswanathan A, Godin O, Jouvent E, et al. Impact of MRI markers in subcortical vascular dementia: a multi‐modal analysis in CADASIL. Neurobiol Aging 2010;31:1629–1636. [DOI] [PubMed] [Google Scholar]
- 13. Chabriat H, Hervé D, Duering M, et al. Predictors of clinical worsening in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy: prospective cohort study. Stroke 2016;47:4–11. [DOI] [PubMed] [Google Scholar]
- 14. Jouvent E, Duchesnay E, Hadj‐Selem F, et al. Prediction of 3‐year clinical course in CADASIL. Neurology 2016;87:1787–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liem MK, Lesnik Oberstein SAJ, Haan J, et al. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy: progression of MR abnormalities in prospective 7‐year follow‐up study. Radiology 2008;249:964–971. [DOI] [PubMed] [Google Scholar]
- 16. Liem MK, Haan J, Van Der Neut IL, et al. MRI correlates of cognitive decline in CADASIL. Neurology 2009;72:143–148. [DOI] [PubMed] [Google Scholar]
- 17. Ling Y, De Guio F, Jouvent E, et al. Clinical correlates of longitudinal MRI changes in CADASIL. J Cereb Blood Flow Metab 2018:0271678X1875787. 10.1177/0271678x18757875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Formichi P, Parnetti L, Radi E, et al. CSF biomarkers profile in CADASIL‐A model of pure vascular dementia: usefulness in differential diagnosis in the dementia disorder. Int J Alzheimers Dis 2010;2010:959257 10.4061/2010/959257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pescini F, Donnini I, Cesari F, et al. Circulating biomarkers in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy patients. J Stroke Cerebrovasc Dis 2017;26:823–833. [DOI] [PubMed] [Google Scholar]
- 20. Duering M, Konieczny MJ, Tiedt S, et al. Serum neurofilament light chain levels are related to small vessel disease burden. J Stroke 2018;20:228–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Teunissen CE, Dijkstra PC, Polman C. Biological markers in CSF and blood for axonal degeneration in multiple sclerosis. Lancet Neurol 2005;4:32–41. [DOI] [PubMed] [Google Scholar]
- 22. Gattringer T, Pinter D, Enzinger C, et al. Serum neurofilament light is sensitive to active cerebral small vessel disease. Neurology 2017;89:2108–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Thelin EP, Zeiler FA, Ercole A, et al. Serial sampling of serum protein biomarkers for monitoring human traumatic brain injury dynamics: a systematic review. Front Neurol 2017;8:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Byrne LM, Rodrigues FB, Blennow K, et al. Neurofilament light protein in blood as a potential biomarker of neurodegeneration in Huntington's disease: a retrospective cohort analysis. Lancet Neurol 2017;16:601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Meeter LH, Dopper EG, Jiskoot LC, et al. Neurofilament light chain: a biomarker for genetic frontotemporal dementia. Ann Clin Transl Neurol 2016;3:623–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rohrer JD, Woollacott IOC, Dick KM, et al. Serum neurofilament light chain protein is a measure of disease intensity in frontotemporal dementia. Neurology 2016;87:1329–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lu C‐H, Macdonald‐Wallis C, Gray E, et al. Neurofilament light chain: a prognostic biomarker in amyotrophic lateral sclerosis. Neurology 2015;84:2247–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Piehl F, Kockum I, Khademi M, et al. Plasma neurofilament light chain levels in patients with MS switching from injectable therapies to fingolimod. Mult Scler J. 2018;24:1046–1054. [DOI] [PubMed] [Google Scholar]
- 29. Disanto G, Barro C, Benkert P, et al. Serum Neurofilament light: a biomarker of neuronal damage in multiple sclerosis. Ann Neurol 2017;81:857–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lesnik Oberstein SA, van den Boom R, van Buchem MA, et al. Cerebral Microbleeds in CADASIL. Neurology 2001;57:1066–1070. [DOI] [PubMed] [Google Scholar]
- 31. vanden Boom R , Lesnik Oberstein SA, Spilt A, et al. Cerebral hemodynamics and white matter hyperintensities in CADASIL. J Cereb Blood Flow Metab 2003;23:599–604. [DOI] [PubMed] [Google Scholar]
- 32. Roth M, Tym E, Mountjoy CQ, et al. CAMDEX. A standardised instrument for the diagnosis of mental disorder in the elderly with special reference to the early detection of dementia. Br J Psychiatry 1986;149:698– 709. [DOI] [PubMed] [Google Scholar]
- 33. Folstein MF, Folstein SE, McHugh PR. “Mini‐mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189–198. [DOI] [PubMed] [Google Scholar]
- 34. Breteler MM, vanAmerongen NM , vanSwieten JC , et al. Cognitive correlates of ventricular enlargement and cerebral white matter lesions on magnetic resonance imaging. The Rotterdam Study. Stroke. 1994;25:1109–1115. [DOI] [PubMed] [Google Scholar]
- 35. Reitan RM. The relation of the trail making test to organic brain damage. J Consult Psychol 1955;19:393–394. [DOI] [PubMed] [Google Scholar]
- 36. Kuhle J, Barro C, Andreasson U, et al. Comparison of three analytical platforms for quantification of the neurofilament light chain in blood samples: ELISA, electrochemiluminescence immunoassay and Simoa. Clin Chem Lab Med 2016;54:1655–1661. [DOI] [PubMed] [Google Scholar]
- 37. Andreasson U, Perret‐Liaudet A, van Waalwijk van Doorn LJC, et al. A practical guide to immunoassay method validation. Front Neurol 2015;6(Aug):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jonsson M, Zetterberg H, Van Straaten E, et al. Cerebrospinal fluid biomarkers of white matter lesions ‐ Cross‐sectional results from the LADIS study. Eur J Neurol 2010;17:377–382. [DOI] [PubMed] [Google Scholar]
- 39. Gaiani A, Martinelli I, Bello L, et al. Diagnostic and prognostic biomarkers in amyotrophic lateral sclerosis: neurofilament light chain levels in definite subtypes of disease. JAMA Neurol 2017;74:525–532. [DOI] [PMC free article] [PubMed] [Google Scholar]