

Abstract
The usefulness of non-steroidal anti-inflammatory drugs (NSAIDs) is hampered by their gastrointestinal side effects. Non-selective cyclooxygenases inhibitors interfere with both COX-1 and COX-2 isozymes. Since COX-1 mediates cytoprotection of gastric mucosa, its inhibition leads to the undesirable side effects. On the other hand, COX-2 is undetectable in normal tissues and selectively induced by inflammatory stimuli. Therefore, it is strongly believed that the therapeutic benefits derive from inhibition of COX-2 only. The presence of a strong connection between reported COX-2 inhibitors and cardiac toxicity encourages medicinal chemists to explore new scaffolds. In the present study, we introduced imidazopyrazolopyridines as new potent and selective COX-2 inhibitors that lack the standard pharmacophoric binding features to hERG. Starting from our lead compound 5a, structure-based drug-design was conducted and more potent analogues were obtained with high COX-2 selectivity and almost full edema protection, in carrageenan-induced edema assay, in case of compound 5e. Increased bulkiness around imidazopyrazolopyridines by adding a substituted phenyl ring(s) afforded less active compounds.
Keywords: aminopyrazolopyridine, anti-inflammatory, cyclooxygenase, hydrazonyl halides, selective inhibitors
1. Introduction
Non-steroidal anti-inflammatory drugs (NSAIDs) are commonly used for the treatment of inflammation, pain, and fever. From mechanistic point of view, NSAIDs exert their pharmacological action via inhibition of cyclooxygenase (COX) that catalyzes the conversion of arachidonic acid to the prostaglandins (PGs) [1]. PGs are hormone-like bio-substances that mediate different signaling pathways in many physiological and pathological processes. COX has been established to exist mainly in two distinct isoforms, COX-1 and COX-2. COX-1 is constitutively normally expressed in most tissues, and PGs controlled by COX-1 mediate cytoprotection of gastric mucosa and platelet aggregations in addition to some other physiological processes. On the other hand, COX-2 is undetectable in normal tissues and selectively induced locally by inflammatory stimuli; i.e., pro-inflammatory cytokines, leading to elevating PG levels at the site of inflammation [2,3]. Therefore, the therapeutic benefits derives from inhibition of the inducible isoform; i.e., COX-2, at the site of inflammation [1].
Overexpression of COX-2 is not limited by rheumatic inflammation but extends to mycobacterial pulmonary inflammation [4], vascular inflammation [5], cigarette-induced airway inflammation [6], and Helicobacter pylori induced gastritis [7]. It also plays a pivotal pathological role in intestinal inflammation and colorectal cancer [8]. Moreover, connection between COX-2 and different types of cancer is recently reported via different mechanisms [9,10,11,12,13,14].
Commonly used NSAIDs are non-selective in their action and inhibit both COX-1 and COX-2 explaining toxicity-mediated by inhibition of the non-regulated COX-1, in normal cells. However, NSAIDs are clinically effective in pain and inflammatory relief, their use is hampered by significant side-effects (mainly in GIT) due to inhibition of COX-1 [15]. In contrast to other NSAIDs, selective COX-2 inhibitors do not cause notable ulcers in the stomach or intestine, they are active as non-selective NSAIDs and inhibit PG synthesis in inflammatory cells [15,16,17]. So far, it was believed that “the more selective COX-2, the less side effects”.
Chemical structures of reported COX-2 inhibitors are highly diverse. In general, they are, unlike classical NSAIDs, lacking the free carboxylate and could be classified into: (1) Carbocycles and Heterocycles with Vicinal Aryl Moieties; (2) Diaryl or Aryl/Heteroaryl-Ether and -Thioether Derivatives; (3) cis-Stilbene Derivatives; (4) Diaryl and Aryl/Heteroaryl Ketones [18]. Regardless the relatively large number of approved COX-2 inhibitors, there is only one selective COX-2 inhibitor in the US market (Celecoxib) [19]. Recent market removal of some selective COX-2 inhibitors such as rofecoxib due to its cardiac toxicity [20] encourages medicinal chemists to explore other alternative scaffolds. The cardiotoxicity of the withdrawn COX-2 inhibitors was attributed mainly to their hERG affinity [21]. In our attempt to find a different selective COX-2 scaffold, we chose pyrazolopyridine that recently showed great inhibitory selectivity towards COX-2 [22,23,24]. One advantage of such new ring system is lacking the putative pharmacophoric criteria of hERG binders [25]. Virtual screening of our in-house library of fused pyrazolopyridines, pyrazolopyrimidines, imidazopyridines, and imidazopyrimidines, using Sybyl-X, furnished compound 5a as a potential hit structure. Compound 5a revealed an acceptable COX-2 inhibitory activity with 9.4 times selectivity towards the isozyme-2 (Table 1).
Table 1.
Cyclooxgenanse (COX-1 and COX-2) inhibitory activity.
| Compd. No. | Drug in μg/mL | Percentage Inhibition * | COX-2/COX-1 Selectivity | |
|---|---|---|---|---|
| COX-1 | COX-2 | |||
| 5a | 10 | 3.88 ± 0.0023 | 36.51 ± 0.35 | 9.4 |
| 5b | 10 | 3.84 ± 0.0034 | 59.11 ± 0.44 | 15.4 |
| 5c | 10 | 3.76 ± 0.0023 | 68.34 ± 0.53 | 18.2 |
| 5d | 10 | 3.80 ± 0.0032 | 67.72 ± 0.44 | 17.8 |
| 5e | 10 | 3.92 ± 0.0023 | 75.92 ± 0.36 | 19.4 |
| 5f | 10 | 3.96 ± 0.0034 | 25.31 ± 0.40 | 6.4 |
| 5g | 10 | 3.99 ± 0.0025 | 24.72 ± 0.59 | 6.2 |
| 5h | 10 | 3.62 ± 0.0036 | 20.85 ± 0.38 | 5.6 |
| 5i | 10 | 3.58 ± 0.0025 | 11.50 ± 0.47 | 3.2 |
| 5j | 10 | 3.54 ± 0.0034 | 22.15 ± 0.65 | 6.6 |
| 5k | 10 | 3.51 ± 0.0046 | 12.80 ± 0.74 | 3.6 |
| 5l | 10 | 3.69 ± 0.0027 | 9.58 ± 0.83 | 2.5 |
| Diclofenac | 10 | 56.78 | 7.88 | 0.13 |
| Valdecoxib | 10 | 12.15 | 76.15 | 6.26 |
| Ibuprofen | 10 | 78.89 | 5.23 | 0.06 |
Values were calculated from the mean values of data from three separate experiments and presented as mean ± S.E.M.; * The percentage Inhibition is the percent in inhibition of the either COX enzyme activities at the selected doses; All results are significant different from control values at p ≤ 0.005; All results are significant different from reference standard values at p ≤ 0.005.
Research Design: To explain the moderate COX-2 inhibitory activity and selectivity of the lead structure 5a, a modeling study was conducted using a co-crystal structure of a selective COX-2 inhibitor, celecoxib, within COX-2 active site (PDB ID:3ln1 [26]). Celecoxib was extracted and compound 5a was docked within COX-2 active pocket using GOLD (Figure 1a). Interestingly, our newly defined lead structure 5a showed great overlap with celecoxib binding conformation (Figure 1b); i.e., the imidazopyrazolopyridine was found to overlay the phenylpyrazole structure of celecoxib, while the phenyldiazene moiety of 5a extends parallel to benzenesulfonamide moiety of celecoxib as shown in Figure 1.
Figure 1.

The hypothetical binding mode of the lead compound 5a (colored yellow) within COX-2 active site (PDB ID:3ln1), co-crystalized with celecoxib (colored magenta). The important Arg 513 residue was highlighted as sticks in Figure 1a.
One key difference between the two cyclooxygenases is the size of their active pockets. COX-2 has a larger binding pocket, where three bulky amino acid residues: Ile523, His513, and Ile434 were replaced with less-sized residues (Val523, Arg513, and Val434) [27]. Therefore, all early discovered COX-2 inhibitors contain diaryl rings with cis-stilbene-like configuration to reduce binding affinity towards the less-volume binding site of COX-1 [18,28]. The higher degree of aromatic structure superposition between our lead compound and those of celecoxib might explain its COX-2 selectivity (Figure 1a). To improve the inhibitory potency and selectivity, medicinal chemists usually add a hydrogen-bond acceptor moiety to one aromatic ring as shown in Figure 2. This hydrogen-bond acceptor targets Arg513, which is replaced by a histidine residue in the case of COX-1 [27]. Analogously, we decided to explore the effect of tethering a hydrogen-bond acceptor to the phenyl ring of our newly discovered lead structure 5a. In addition, we also decided to increase the bulkiness degree around imidazopyrazolopyridine nucleus in order to improve compounds’ selectivity.
Figure 2.

Chemical structures of some selective COX-2 inhibitors (coxibs).
2. Results and Discussion
2.1. Chemistry
Generally, pyrazoplopyridines were reported to be prepared through two alternative routes, either from building up pyrazole moiety first, and then allowed it to react with activated nitriles [29]; the second route involved constructing polyfunctional pyridine nucleus, followed by reaction with various electrophilic reagents [30]. In the present work, we utilized aminopyrazolopyridine 1 as a key precursor for synthesis of our library of COX-2 inhibitors. Aminopyrazolopyridines 1 were chosen as starting building blocks because of their synthetic accessibility and affordable commercial availability of their starting materials.
When aminopyrazolopyridines 1 were allowed to react with a variety of hydrazonyl halides 2 in basic medium, a removal of hydrogen halide followed by cyclization through water loss led, finally, to the imidazopyrazolopyridines 5a–l (Scheme 1).
Scheme 1.
Synthesis of 2,7,9-trisubstituted-3-(aryldiazenyl)-3H-imidazo[1′,2′:1,5] pyrazolo[3,4-b]pyridine (5a–l).

| Compd. No. | R1 | R2 | X | Compd. No. | R1 | R2 | X |
|---|---|---|---|---|---|---|---|
| a | Me | Me | H | g | Ph | Me | OMe |
| b | Me | Me | Cl | h | Ph | Me | Cl |
| c | Me | Me | OMe | i | Ph | Me | Br |
| d | Me | Me | Br | j | Ph | Me | NO2 |
| e | Me | Me | NO2 | k | Ph | Ph | H |
| f | Ph | Me | H | l | Ph | 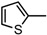 |
H |
Elucidation of all structures was assessed from their elemental analyses and spectral data. In general, the newly built imidazole ring was confirmed by disappearance of the NH2 signal characteristic to the starting material from 1H-NMR spectrum, as well as loss of the corresponding bands in the IR spectrum. In addition, no absorption bands in carbonyl group characteristic region were observed, which in turn confirmed the cyclization step. The products exist in two tautomeric forms (azo-hydrazo forms) with equilibrium shifted mainly towards the azo form as indicated by the 1H-NMR spectra. For example the 1H-NMR spectrum of 5a displayed a characteristic D2O-exchangeable singlet signal at 11.16 ppm attributable to imidazole NH proton (azo-structure). The 1H-NMR spectrum of compound 5g displayed two singlet signals at 2.26 and 3.81 for two methyl groups. In addition, the aromatic region showed aggregated peaks from 7.13–8.27 ppm integrating for 15 protons (one pyridine proton plus phenyl protons). The mass spectrum of this derivative gave the corresponding molecular ion peak at m/e = 458. Furthermore, when R1 = R2 = Ph and X = H (compound 5k), the 1H-NMR spectrum showed a nice singlet at δ = 6.95, which might be attributable to pyridine H at position-3. The aromatic region is integrating for 20 protons spread over a smaller chemical shift range from 7.01 to 7.38 ppm, because all aromatic protons are almost chemically equivalent (phenyl ones).
Alkylation of compound 1b with α-haloketones such as chloroacetone and phenacyl bromide in basic medium resulted in heterocyclization and afforded the tricycles compounds 7a,b. As compounds 7a,b were coupled to benzene diazonium chloride, they delivered the desired products 5f,k (Scheme 2); this could be considered as a chemical evidence for the structures of the products obtained from reaction of 1b with hydrazonyl halides. The mass spectra of compounds 7a,b gave the correct molecular ion peaks for the proposed structures, in addition, 1H-NMR spectrum of compound 7a showed a singlet at 2.37 ppm (CH3 protons) and a singlet signal at 11.71 ppm (NH).
Scheme 2.

Synthesis of imidazo[1′,2′:1,5]pyrazolo[3,4-b]pyridine (5f and 5k).
2.2. Biological Discussion
Cyclooxygenase Inhibition Activity: all synthesized compounds were subjected to colorimetric screening assay utilizing the peroxidase component of cyclooxygenases (COX-1 and COX-2). The peroxidase activity is assayed colorimetrically as detailed in the experimental section. The results were summarized in Table 1.
The lead compound 5a showed moderate COX-2 inhibitory activity and low affinity towards COX-1 (Table 1). Adding a hydrogen-bond acceptor para to the phenyl ring connected with the diaza moiety gave compounds 5b–e. The key idea behind inserting hydrogen-bond acceptors at this particular position is targeting Arg513, which is replaced by a histidine residue in the case of COX-1 [27], as detailed earlier. Again, targeting Arg513 was believed to ameliorate COX-2 selectivity. A Chlorine-containing derivative 5b showed enhanced COX-2 inhibitory activity; i.e., the percentile value of COX-2 inhibition jumped up from 36.5 in the case of 5a to 59.1 in the case of 5b, and selectivity towards COX-2 over COX-1 is also increased to 15 times (Table 1). Replacement chlorine atom with methoxy group afforded compound 5c that revealed better selectivity toward COX-2; i.e., 5c is 18 times more selective. Compound 5c was on par with the lead compound 5a in term of COX-1 inhibitory activity; however, its COX-2 suppression criteria was increased by factor of two (Table 1). The calculated binding mode represented in Figure 3 sheds some lights on the enhanced COX-2 affinity of the methoxy-containing derivative 5c. The methoxy group was calculated to from two potential hydrogen-bonds with the guanidine moiety of Arg513. Intriguingly, the nitro-containing analogue showed even better COX-2 inhibitory activity and selectivity as shown in Table 1. The hypothetical binding mode of compound 5e (Figure 3b) showed three possibilities of hydrogen-bonds formation between one oxygen atom of the nitro group and the guanido moiety of Arg513, where the oxygen atom was calculated to be 2.4 and 2.7 Å away from the two terminal nitrogen atoms of Arg513. The second oxygen atom of compound 5e nitro group was calculated to be in the vicinity of Arg513 and the carboxylate moiety of Glu524. Figure 3 details four different potential strong hydrogen-bonds between nitro group of compound 5e and the guanido group of Arg513 and the carboxylate moiety of Glu524.
Figure 3.

Hypothetical binding modes of compound 5c (a); and 5e (b) in COX-2 protein (PDB ID:3ln1). Dashed lines represent distances between heavy atoms.
As mentioned earlier, COX-2 has a larger binding pocket than its isozyme-1, where three bulky amino acid residues: Ile523, His513, and Ile434 were replaced with less-sized residues (Val523, Arg513, and Val434) [27]. Therefore, increasing the molecular volume of cyclooxygenase inhibitors is a well-established strategy to reduce the binding affinity towards the less-volume binding site of COX-1. So far, we managed to build new series of COX-2 inhibitors with remarkable COX-2 selectivity when compare with diclofenac (Table 1), but, on the other hand, the best obtained selectivity did not exceed 20 times (COX-2 selectivity of compound 5e was 19.4). This value is still less than that of celecoxib (selectivity = 30). In our attempts to improve the selectivity, we increased the bulkiness of the imidazopyrazolopyridine nucleus by replacing the methyl group(s) at positions 2, 7, and 9 with phenyl ones. Unfortunately, this approach gave less active and selective derivatives. Docking of 7,9-diphenyl analogues and 2,7,9-triphenyl analogues revealed unexpected binding modes. Figure 4 represents the hypothetical binding mode of compound 5i, as a representative example of this series. The diazaphenyl moiety of compound 5i was docked far away from the Arg513 residue. Unlike compound 5e, its 7,9-diphenyl analogue has no change to interact with Arg513. That might explain why compounds 5f–l have lower COX-2 inhibitory activity than their corresponding 7,9-dimethyl analogues.
Figure 4.

Hypothetical binding modes of compound 5i in COX-2 protein (PDB ID:3ln1).
Spectrophotometric Assay of Recombinant Human COX-2
Enzymatic activity of the purified COX-2 was measured using a chromogenic assay based on the oxidation of N,N,N9,N9-tetramethylp-phenylenediamine (TMPD) during the reduction of PGG2 to PGH2 [31]. The assay mixture (180 mL) contains 100 mM sodium phosphate, pH 6.5, 1 mM hematin, 1 mg/mL gelatin, 2 to 5 mg/mL of purified COX-2, and 4 mL of the test compound in dimethyl sulfoxide (DMSO). The assay was also performed in the presence of the detergent Genapol X-100 (CalBiochem, San Diego, CA, USA) at a final concentration of 2 mM. The mixture was preincubated at room temperature (22 °C) for 15 min before initiation of the enzymatic reaction by the addition of 20 mL of a solution of 1 mM arachidonic acid and 1 mM TMPD in assay buffer (without enzyme or hematin).
For assays in the presence of Genapol, the arachidonic acid and TMPD solution was prepared in 50% aqueous ethanol. The enzyme activity was measured by estimation of the initial velocity of TMPD oxidation over the first 36 s of the reaction as followed from the increase in absorbency at 610 nm. A low rate of nonenzymatic oxidation was observed in the absence of COX-2 and was subtracted before the calculation of the percentage of inhibition.
Anti-inflammatory activity: in the same vein the anti-inflammatory activity of all prepared compounds were assayed using Rats Paw Test, where groups of adult albino rats were orally administrated with tested compounds at two different dose levels (2.5–5 mg/kg) one hour before the carrageenan challenge. Four hours after drug administration, the average weight of edema was examined, and the percentage of edema inhibition was evaluated. The well-known potassium diclofenac was used as a positive control to which the tested compounds were compared (Table 2).
Table 2.
Anti-inflammatory effect.
| Compd. No. | Dose [mg/kg] | % Protection against Edema | % Inhibition of Plasma PGE2 |
|---|---|---|---|
| 5a | 2.5 | 31.76 ± 0.66 | 72.19 ± 0.45 |
| 5 | 40.05 ± 0.77 | 73.78 ± 0.43 | |
| 5b | 2.5 | 69.10 ± 0.85 | 82.65 ± 0.56 |
| 5 | 71.42 ± 0.96 | 84.25 ± 0.45 | |
| 5c | 2.5 | 78.65 ± 0.87 | 83.59 ± 0.68 |
| 5 | 88.96 ± 0.78 | 85.19 ± 0.79 | |
| 5d | 2.5 | 67.32 ± 0.89 | 83.13 ± 0.80 |
| 5 | 70.60 ± 0.90 | 84.72 ± 0.97 | |
| 5e | 2.5 | 92.88 ± 0.97 | 91.73 ± 0.53 |
| 5 | 97.15 ± 0.86 | 93.31 ± 0.46 | |
| 5f | 2.5 | 31.36 ± 0.55 | 61.28 ± 0.43 |
| 5 | 33.74 ± 0.64 | 62.84 ± 0.56 | |
| 5g | 2.5 | 41.82 ± 0.75 | 80.82 ± 0.44 |
| 5 | 44.21 ± 0.66 | 82.38 ± 0.21 | |
| 5h | 2.5 | 42.28 ± 0.57 | 75.49 ± 0.45 |
| 5 | 54.68 ± 0.75 | 77.13 ± 0.89 | |
| 5i | 2.5 | 40.45 ± 0.63 | 75.96 ± 0.96 |
| 5 | 42.80 ± 0.54 | 77.61 ± 0.89 | |
| 5j | 2.5 | 26.46 ± 0.45 | 66.45 ± 0.83 |
| 5 | 28.71 ± 0.56 | 68.11 ± 0.83 | |
| 5k | 2.5 | 19.99 ± 0.67 | 66.93 ± 0.83 |
| 5 | 22.34 ± 0.78 | 68.60 ± 0.98 | |
| 5l | 2.5 | 38.10 ± 0.69 | 75.01 ± 0.65 |
| 5 | 40.29 ± 0.77 | 76.65 ± 0.34 | |
| Diclofenac sodium | 2.5 | 69.59 ± 0.44 | 55.49 ± 0.80 |
| 5 | 72.19 ± 0.54 | 70.59 ± 0.80 | |
| Valdicoxib | 2.5 | 78.89 ± 0.33 | 60.09 ± 0.86 |
| 5 | 87.19 ± 0.23 | 81.11 ± 0.21 |
Values were calculated from the mean values of data from three separate experiments; All results are significant different from control values at p ≤ 0.005; All results are significant different from reference standard values at p ≤ 0.005.
The lead compound 5a showed moderate anti-inflammatory activity in rats paw assay (Table 2). A notable correlation between COX-2 inhibitory activity (Table 1) and anti-inflammatory properties (Table 2) was observed. In general, all compounds carry hydrogen-bond acceptors para to the phenyl group attached to the diaza moiety revealed higher anti-inflammatory effect than lead compound 5a. The most potent COX-2 inhibitor in this study (compound 5e) revealed almost full edema protection in carrageenan-induced edema assay. The corresponding 7,9-diphenyl and 2,7,9-triphenyl analogues were less active than the corresponding 7,9-dimethylated derivatives.
Purity ≥ 95% for all synthesized compounds and this was obtained by preparative thin layer chromatography.
3. Experimental Protocols
3.1. Chemistry
3.1.1. General
Melting points were measured on Electrothermal IA 9000 series digital melting point apparatus (Weiss-Gallenkamp, London, UK). The IR spectra were recorded in potassium bromide discs on a Pye Unicam SP 3300 and Shimadzu FT IR 8101 PC infrared spectrophotometer (Mattson Instruments Inc., Madison, WI, USA). 1H-NMR and 13C-NMR spectra were recorded in deuterated dimethyl sulfoxide (DMSO-d6) using a Varian Gemini 300 NMR spectrometer (300 MHz for 1H-NMR and 75 MHz for 13C-NMR, Varian, Palo Alto, CA, USA). Mass spectra were recorded on a Shimadzu GCMS-QP1000 EX mass spectrometer (Shimadzu, Tokyo, Japan) at 70 eV. Elemental analysis was carried out at the Microanalytical Centre of Cairo University, Giza, Egypt. All reactions were followed by TLC (Silica gel, Merck). 3-Amino-4,6-dimethyl-1H-pyrazolo[3,4-b]pyridine 1a [32], 3-amino-4,6-diphenyl-1H-pyrazolo[3,4-b]pyridine 1b [33] and hydrazonoyl halides 2 [34,35] were prepared as reported in the literature.
3.1.2. General Method for the Synthesis of 2,7,9-Trisubstituted-3-(aryldiazenyl)-3H-imidazo[1′,2′:1,5] pyrazolo[3,4-b]pyridine (5a–l)
A mixture of pyrazolopyridine 1a,b (1 mmol) and the appropriate hydrazonoyl halides 2 (1 mmol) in dioxane (20 mL) containing TEA (0.5 mL) was refluxed for 4–8 h (monitored by TLC), allowed to cool and the solid formed was filtered off, washed with EtOH, dried and recrystallized from DMF to give 5a–l.
2,7,9-Trimethyl-3-(phenyldiazenyl)-1H-imidazo[1′,2′:1,5]pyrazolo[3,4-b]pyridine (5a). Red solid, 73% yield; m.p. 233–234 °C; IR (KBr) υ 3426 (NH), 1597 (C=N) cm−1; 1H-NMR (300 MHz, DMSO-d6): δ 2.53 (s, 3H, CH3), 2.78 (s, 3H, CH3) , 2.84 (s, 3H, CH3), 7.18 (s, 1H, pyridine H), 7.44–8.19 (m, 5H, Ar-H), 11.16 (s, 1H, NH); 13C-NMR (DMSO-d6): δ 12.6 (CH3), 25.8 (CH3), 26.7 (CH3), 116.9, 128.6, 128.7, 128.8, 129.2, 129.8, 132.6, 132.8, 138.1, 149.6, 156.8, 162.3 (Ar-C) ppm; MS, m/z (%) 304 (M+, 81), 286 (97), 247 (92), 203 (97), 171 (88), 107 (100). Anal. calcd for C17H16N6 (304.35): C, 67.09; H, 5.30; N, 27.61; found: 66.90; H, 5.18; N, 27.46.
3-[(4-Chlorophenyl)diazenyl]-2,7,9-trimethyl-1H-imidazo[1′,2′:1,5]pyrazolo [3,4-b]pyridine (5b). Red solid, 71% yield; m.p. 251–253 °C; IR (KBr) υ 3423 (NH), 1591 (C=N) cm−1; 1H-NMR (300 MHz, DMSO-d6): δ 2.56 (s, 3H, CH3), 2.78 (s, 3H, CH3) , 2.88 (s, 3H, CH3), 7.26 (s, 1H, pyridine H), 7.70 (d, 2H, J = 8.4 Hz, Ar-H), 8.21 (d, 2H, J = 8.4 Hz, Ar-H), 11.26 (s, 1H, NH); 13C-NMR (DMSO-d6): δ 12.7 (CH3), 26.6 (CH3), 27.4 (CH3), 125.9, 127.2, 127.8, 128.2, 128.9, 129.5, 133.6, 137.1, 144.5, 149.5, 153.1, 161.9 (Ar-C) ppm; MS, m/z (%) 340 (M+ + 2, 11), 338 (M+, 31), 302 (55), 227 (54), 198 (87), 93 (100). Anal. calcd for C17H15ClN6 (338.79): C, 60.27; H, 4.46; N, 24.81; found: C, 60.07; H, 4.30; N, 24.70.
3-[(4-Methoxyphenyl)diazenyl]-2,7,9-trimethyl-1H-imidazo[1′,2′:1,5]pyrazolo[3,4-b]pyridine (5c). Red solid, 73% yield; m.p. 197–199 °C; IR (KBr) υ 3388 (NH), 1601 (C=N) cm−1; 1H-NMR (300 MHz, DMSO-d6): δ 2.58 (s, 3H, CH3), 2.72(s, 3H, CH3), 2.89 (s, 3H, CH3), 3.82 (s, 3H, OCH3), 7.20 (s, 1H, pyridine H), 7.61–8.21 (m, 4H, Ar-H), 11.18 (s, 1H, NH); MS, m/z (%) 334 (M+, 67), 318 (77), 303 (59), 214 (52), 199 (57), 76 (100). Anal. calcd for C18H18N6O (334.38): C, 64.66; H, 5.43; N, 25.13; found: C, 64.84; H, 5.57; N, 25.28.
3-[(4-Bromophenyl)diazenyl]-2,7,9-trimethyl-1H-imidazo[1′,2′:1,5]pyrazolo[3,4-b]pyridine (5d). Red solid, 70% yield; m.p. 212–214 °C; IR (KBr) υ 3388 (NH), 1599 (C=N) cm−1; 1H-NMR (300 MHz, DMSO-d6): δ 2.38 (s, 3H, CH3), 2.53 (s, 3H, CH3), 2.83 (s, 3H, CH3), 7.13 (s, 1H, pyridine H), 7.37 (d, 2H, J = 8.4 Hz, Ar-H), 8.01 (d, 2H, J = 8.4 Hz, Ar-H), 11.07 (s, 1H, NH); 13C-NMR (DMSO-d6): δ 16.6 (CH3), 26.6 (CH3), 27.4 (CH3), 126.9, 128.6, 128.7, 128.8, 129.8, 132.5, 132.8, 138.1, 149.6, 156.8, 162.3, 166.6 (Ar-C) ppm; MS, m/z (%) 384 (M+ + 2, 80), 382 (M+, 85), 345 (100), 301 (50), 227 (55), 213 (52), 76 (52). Anal. calcd for C17H15BrN6 (382.05): C, 53.28; H, 3.95; N, 21.93; found: C, 53.45; H, 3.70; N, 21.70.
2,7,9-Trimethyl-3-[(4-nitrophenyl)diazenyl]-1H-imidazo[1′,2′:1,5]pyrazolo[3,4-b]pyridine (5e). Red solid, 72% yield; m.p. 237–239 °C; IR (KBr) υ 3424 (NH), 1593 (C=N) cm−1; 1H-NMR (300 MHz, DMSO-d6): δ 2.54 (s, 3H, CH3), 2.73 (s, 3H, CH3), 2.88 (s, 3H, CH3), 7.24 (s, 1H, pyridine H), 8.41–8.48 (m, 4H, Ar-H), 11.73 (s, 1H, NH); 13C-NMR (DMSO-d6): δ 13.1 (CH3), 25.6 (CH3), 26.4 (CH3), 120.6, 123.7, 126.6, 127.4, 127.9, 128.3, 128.9, 129.4, 129.5, 131.6, 136.2, 154.9 (Ar-C) ppm; MS, m/z (%) 349 (M+, 63), 304 (71), 293 (100), 288 (92), 213 (46), 157(82), 108 (80). Anal. calcd for C17H15N7O2 (349.13): C, 58.45; H, 4.33; N, 28.07; found: C, 58.25; H, 4.21; N, 28.25.
2-Methyl-7,9-diphenyl-3-(phenyldiazenyl)-1H-imidazo[1′,2′:1,5]pyrazolo[3,4-b]pyridine (5f). Brown solid, 78% yield; m.p. 211–213 °C; IR (KBr) υ 3425 (NH), 1604 (C=N) cm−1; 1H-NMR (300 MHz, DMSO-d6): δ 2.35 (s, 3H, CH3), 7.33–8.26 (m, 16H, Ar-H and pyridine H), 11.50 (s, 1H, NH); 13C-NMR (DMSO-d6): δ 13.0 (CH3), 116.2, 117.5, 118.7, 122.0, 125.6, 126.70, 126.1, 126.8, 128.1, 128.2, 128.9, 129.6, 129.7, 130.7, 132.1, 135.1, 137.0, 140.1, 152.7, 158.3 (Ar-C) ppm; MS, m/z (%) 428 (M+, 100), 351 (65), 274 (45), 259 (74), 182 (69), 77 (92). Anal. calcd for C27H20N6 (428.17): C, 75.68; H, 4.70; N, 19.61; found: C, 75.55; H, 4.78; N, 19.56.
3-[(4-Methoxyphenyl)diazenyl]-2-methyl-7,9-diphenyl-1H-imidazo[1′,2′:1,5]pyrazolo[3,4-b]pyridine (5g). Brown solid, 78% yield; m.p. 206–208 °C; IR (KBr) υ 3412 (NH), 1607 (C=N) cm−1; 1H-NMR (300 MHz, DMSO-d6): δ 2.24 (s, 3H, CH3), 3.81 (s, 3H, OCH3), 7.13–8.27 (m, 15H, Ar-H and pyridine H), 10.95 (s, 1H, NH); MS, m/z (%) 458 (M+, 88), 427 (69), 412 (85), 350 (100), 273 (90) 175 (47), 77 (95). Anal. calcd for C28H22N6O (458.51): C, 73.35; H, 4.84; N, 18.33; found: C, 73.50; H, 4.94; N, 18.42.
3-((4-Chlorophenyl)diazenyl)-2-methyl-7,9-diphenyl-1H-imidazo[1′,2′:1,5]pyrazolo[3,4-b]pyridine (5h). Brown solid, 70% yield; m.p. 257–259 °C; IR (KBr) υ 3408 (NH), 1599 (C=N) cm−1; 1H-NMR (300 MHz, DMSO-d6): δ 2.32 (s, 3H, CH3), 7.19–8.05 (m, 15H, Ar-H and pyridine H), 11.67 (s, 1H, NH); 13C-NMR (DMSO-d6): δ 13.9 (CH3), 122.0, 122.4, 127.0, 127.3, 175.5, 128.4, 129.0, 130.7, 137.7, 138.7, 140.0, 142.3, 143.8, 144.5, 145.1, 146.6, 148.1, 161.3, 167.7, 168.4 (Ar-C) ppm; MS, m/z (%) 464 (M+ + 2, 7), 462 (M+, 28), 447 (64), 427 (80), 385 (75), 256 (52), 179 (92), 77 (100). Anal. calcd for C27H19ClN6 (462.14): C, 70.05; H, 4.14; N, 18.15; found: C, 70.15; H, 4.19; N, 18.26.
3-[(4-Bromophenyl)diazenyl]-2-methyl-7,9-diphenyl-1H-imidazo[1′,2′:1,5]pyrazolo[3,4-b]pyridine (5i). Brown solid, 73% yield; m.p. 234–236 °C; IR (KBr) υ 3418 (NH), 1606 (C=N) cm−1; 1H-NMR (300 MHz, DMSO-d6): δ 2.41 (s, 3H, CH3), 7.45–8.26 (m, 15H, Ar-H and pyridine H), 11.35 (s, 1H, NH); MS, m/z (%) 509 (M+ + 2, 15), 507(M+, 18), 430 (44), 428 (64), 352 (85), 275 (72), 198 (77), 77 (100). Anal. calcd for C27H19BrN6 (507.38): C, 63.91; H, 3.77; N, 16.56; found: C, 63.81; H, 3.71; N, 16.47.
2-Methyl-3-[(4-nitrophenyl)diazenyl]-7,9-diphenyl-1H-imidazo[1′,2′:1,5]pyrazolo[3,4-b]pyridine (5j). Brown solid, 68% yield; m.p. 249–251 °C; IR (KBr) υ 3425 (NH), 1602 (C=N) cm−1; 1H-NMR (300 MHz, DMSO-d6): δ 2.32 (s, 3H, CH3), 7.13 s, 1H, pyridine H), 7.15–8.10 (m, 14H, Ar-H), 11.23 (s, 1H, NH); MS, m/z (%) 473 (M+, 68), 427 (49), 351 (100), 274 (66), 178 (60), 77 (95). Anal. calcd for C27H19N7O2 (473.49): C, 68.49; H, 4.04; N, 20.71; found: C, 68.61; H, 4.15; N, 20.83.
2,7,9-Triphenyl-3-(phenyldiazenyl)-1H-imidazo[1′,2′:1,5]pyrazolo[3,4-b]pyridine (5k). Brown solid, 72% yield; m.p. 264–265 °C; IR (KBr) υ 3427 (NH), 1600 (C=N) cm−1; 1H NMR (300 MHz, DMSO-d6): δ 6.98 (s, 1H, pyridine H), 7.01–7.38 (m, 20H, Ar-H), 11.43 (s, 1H, NH); MS, m/z (%) 490 (M+, 71), 413 (92), 336 (68), 259 (82), 159 (60), 77 (100). Anal. calcd for C32H22N6 (490.56): C, 78.35; H, 4.52; N, 17.13; found: C, 78.55; H, 4.32; N, 17.30.
7,9-Diphenyl-3-(phenyldiazenyl)-2-(thiophen-2-yl)-1H-imidazo[1′,2′:1,5]pyrazolo[3,4-b]pyridine (5l). Brown solid, 74% yield; m.p. 222–224 °C; IR (KBr) υ 3394 (NH), 1597 (C=N) cm−1; 1H-NMR (300 MHz, DMSO-d6): δ 7.11–8.42 (m, 19H, Ar-H and pyridine H), 11.39 (s, 1H, NH); MS, m/z (%) 496 (M+, 87), 419 (77), 413 (59), 336 (85), 259 (52), 187 (70), 77 (100). Anal. calcd for C30H20N6S (496.58): C, 72.56; H, 4.06; N, 16.92; found: C, 72.32; H, 4.19; N, 16.79.
3.1.3. Synthesis of 2-Substituted-7,9-diphenyl-3H-imidazo[1′,2′:1,5]pyrazolo[3,4-b]pyridine (7a,b)
A mixture of 3-aminopyrazolopyridine 1b (2.86 g, 10 mmol) and haloketone 6a,b (10 mmol) in absolute EtOH (30 mL) was refluxed for 4 h. The product started to separate out during the course of reaction. The crystalline solid was filtered, washed with water, dried, and recrystallized from EtOH to give pure products 7a,b respectively.
2-Methyl-7,9-diphenyl-3H-imidazo[1′,2′:1,5]pyrazolo[3,4-b]pyridine (7a). Brown solid, 78% yield; m.p. 227–229 °C; IR (KBr) υ 1608 (C=N) cm−1; 1H-NMR (300 MHz, DMSO-d6): δ 2.37 (s, 3H, CH3), 6.55 (s, 1H, pyridine H), 7.14–8.25 (m, 11H, Ar-H), 11.71 (s, 1H, NH); 13C-NMR (DMSO-d6): δ 13.7 (CH3), 122.0, 126.0, 126.5, 126.8, 127.9, 128.1, 128.2, 128.7, 129.5, 130.7, 132.1, 135.1, 137.0, 140.0, 146.9, 155.2 (Ar-C) ppm; MS, m/z (%) 324 (M+, 100), 247 (44), 232 (82), 155 (64), 77 (73). Anal. calcd for C21H16N4 (324.38): C, 77.76; H, 4.97; N, 17.27; found: C, 77.52; H, 4.85; N, 17.39.
2,7,9-Triphenyl-1H-imidazo[1′,2′:1,5]pyrazolo[3,4-b]pyridine (7b). Brown solid, 78% yield; m.p. 178–180 °C; IR (KBr) υ 1604 (C=N) cm−1; 1H-NMR (300 MHz, DMSO-d6): δ 6.92 (s, 1H, pyridine H), 7.18–8.25 (m, 16H, Ar-H), 11.07 (s, 1H, NH); MS, m/z (%) 386 (M+, 20), 309 (34), 232 (79), 155 (34), 77 (100). Anal. calcd for C26H18N4 (386.45): C, 80.81; H, 4.69; N, 14.50; found: C, 80.50; H, 4.55; N, 14.32.
3.1.4. Coupling of 7a,b with Benzenediazonium Chloride
To a solution of 7a,b (1 mmol) in EtOH (20 mL) sodium acetate trihydrate (0.138 g, 1 mmol) was added, and the mixture was cooled to 0–5 °C in an ice bath. The resulting cold solution was added portion wise to a cold solution of benzenediazonium chloride (prepared by diazotizing aniline) (1 mmol) dissolved in hydrochloric acid (6 M, 1 mL) with a solution of sodium nitrite (0.07 g, 1 mmol) in water (2 mL). After complete addition of the diazonium salt, the reaction mixture was stirred for a further 30 min in an ice bath. The solid that separated was filtered off, washed with water, and finally recrystallized from DMF to give product proved to be identical in all respects (m.p., mixed m.p. and IR spectra) with compounds 5f and 5k respectively.
3.2. Pharmacology
3.2.1. Cyclooxygenase Inhibition Activity
The colorimetric COX (ovine) Inhibitor Screening Assay utilizes the peroxidase component of cyclooxygenase. The peroxidase activity is assayed colorimetrically by monitoring the appearance of oxidized N,N,N,N-tetramethyl-p-phenylenediamine (TMPD) at 590 nm. The estimation of COX-1 and COX-2 enzyme inhibitor activity was done using the kit supplied by Cayman Chemical (Ann Arbor, MI, USA). The kit contained Assay buffer (10×), Heme, COX-1 (Ovine), COX-2 (Ovine), Arachidonic acid, Potassium hydroxide, Colorimetric substrate, 96 well plate.
3.2.2. Anti-Inflammatory Activity: Carrageenan-Induced Edema (Rats Paw Test)
Groups of adult male albino rats (150–180 g), each of eight animals were orally dosed with tested compounds at a dose level of 2.5–5 mg/kg, as a suspension (50% w/w) in saline and 1% tween 80, one hour before the carrageenan challenge. Foot paw edema was induced by subplantar injection of 0.05 cm3 of a 1% suspension of carrageenan in saline into the plantar tissue of one hind paw. An equal volume of saline was injected to the other hind paw and served as control. Four hours after drug administration, the animals were decapitated, blood was collected, and the paws were rapidly excised. The average weight of edema was examined for the treated as well as for the control group, and the percentage inhibition of weight of edema was evaluated. Diclofenac potassium (5 mg/kg) was employed as standard reference to which the tested compounds were compared.
3.3. Statistical Analysis
Results are expressed as mean ± S.E.M. Differences between vehicle control and treatment groups were tested using one-way ANOVA, followed by multiple comparisons by the Dunnett’s test. A value of p ≤ 0.005 was considered statistically significant. Dose-response curves for percent inhibition were fitted by a four-parameter logistic function using a nonlinear least-squares regression.
3.4. Molecular Modeling
The structures of newly synthesized compounds were built with Sybyl-X software and minimized to 0.01 kcal/mol by the Powell method, using Gasteiger-Hückel charges and the Tripos force field [36]. The minimized molecules underwent 10 rounds of simulated annealing to search for the optimized conformation. During the simulation process, the starting conformation in each round was heated to 700 K within 1000 fs and then cooled to 200 K in the same period. Conformations were recorded at each temperature level (700 K and 200 K). The conformers located at the starting point at the each round of simulation were selected for the further energy refinement using the same parameter set as the ones in molecular construction. The minimized conformer with the lowest energy was selected as the optimized conformation molecular docking step. The proteins coordinates have been downloaded from Protein Data Bank website (PDB IDs:3ln1). The water molecules, and all other substructures including celecoxib were removed. The hydrogen atoms were added and the energy of the protein was minimized using the Amber force field with Amber charges. The energy-optimized lignads were docked into the binding site in the protein using GOLD. The parameters were set as the default values for GOLD. The maximum distance between hydrogen bond donors and acceptors for hydrogen bonding was set to 3.5 Å. After docking, the top three poses conformations of each lignad were merged into the ligands-free protein. The new ligand–protein complexes were subsequently subjected to energy minimization using the Amber force field with Amber charges. The energy minimization, in all cases, was performed using the Powell method with a 0.05 kcal/(mol Å) energy gradient convergence criterion and a distance dependent dielectric function.
4. Conclusions
The field of pain-control and anti-inflammation research is still suffering from several drawbacks arising from rapidly developed undesirable side effects. The clinical applications of non-steroidal anti-inflammatory drugs (NSAIDs) are hampered by suppression of COX-1 that mediates cytoprotection of gastric mucosa. Nevertheless, COX-2 is undetectable in healthy tissues and induced by inflammatory stimuli at site of inflammation. Therefore, the therapeutic benefits increase by selectively inhibiting COX-2 only. In the present study, we explored imidazopyrazolopyridine scaffold as a new potent and selective COX-2 inhibitor. Calculating the hypothetical binding mode of the lead compound 5a, obtained by virtual screening, within COX-2 active site showed that the phenyl group connected with diaza moiety is docked in the vicinity of Arg513. Since targeting such residue is a key step in designing selective COX-2 inhibitors, several analogues with hydrogen-bond acceptors para to the phenyl ring were designed and built. Interestingly, all of them revealed better COX-2 inhibitory activity and selectivity as well. Compound 5e that carries a para nitrophenyl group was the best COX-2 inhibitor in our series. It is 20 times more selective towards COX-2 and provided almost full edema protection in carrageenan-induced edema assay. Our attempts to improve the selectivity via increasing bulkiness around imidazopyrazolopyridine nucleus was achieved by less active compounds.
Abbreviations
COX, cyclooxygenase; COX-1, cyclooxygenase-1; COX-2, cyclooxygenase-2; NSAIDs, non-steroidal anti-inflammatory drugs; PGs, prostaglandins.
Author Contributions
Mohamed Badrey, Hassan Abdel-Aziz, and Sobhi Gomha conceived the study, designed the work, contributed in the strategy of the chemistry part, performed interpretation of the analytical data of the prepared compounds, prepared the manuscript and revised it for publication. Mohamed Abdalla designed the pharmacological part, contributed in performing pharmacology. Abdelrahman Mayhoub designed the molecular modeling part.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the synthesized compounds are available from the authors.
References
- 1.Misko T.P., Trotter J.L., Cross A.H. Mediation of inflammation by encephalitogenic cells: Interferon gamma induction of nitric oxide synthase and cyclooxygenase 2. J. Neuroimmunol. 1995;61:195–204. doi: 10.1016/0165-5728(95)00091-F. [DOI] [PubMed] [Google Scholar]
- 2.Seibert K., Masferrer J.L. Role of inducible cyclooxygenase (COX-2) in inflammation. Receptor. 1994;4:17–23. [PubMed] [Google Scholar]
- 3.Seed M.P., Willoughby D.A. COX-2, HO NO! Cyclooxygenase-2, heme oxygenase and nitric oxide synthase: Their role and interactions in inflammation. BIRAs Symposium, Saint Bartholomew’s Hospital, London, 26 April 1996. Inflamm. Res. 1997;46:279–281. doi: 10.1007/s000110050187. [DOI] [PubMed] [Google Scholar]
- 4.Kogiso M., Shinohara T., Dorey C.K., Shibata Y. Role of PPARgamma in COX-2 activation in mycobacterial pulmonary inflammation. Inflammation. 2012;35:1685–1695. doi: 10.1007/s10753-012-9486-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Renna N.F., Diez E.R., Lembo C., Miatello R.M. Role of Cox-2 in vascular inflammation: An experimental model of metabolic syndrome. Mediat. Inflamm. 2013 doi: 10.1155/2013/513251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin C.C., Lee I.T., Yang Y.L., Lee C.W., Kou Y.R., Yang C.M. Induction of COX-2/PGE(2)/IL-6 is crucial for cigarette smoke extract-induced airway inflammation: Role of TLR4-dependent NADPH oxidase activation. Free Radic. Biol. Med. 2010;48:240–254. doi: 10.1016/j.freeradbiomed.2009.10.047. [DOI] [PubMed] [Google Scholar]
- 7.Zarrilli R., Tuccillo C., Santangelo M., Nardone G., Romano M. Increased COX-2, but not COX-1, mRNA expression in Helicobacter pylori gastritis. Am. J. Gastroenterol. 1999;94:3376–3378. doi: 10.1111/j.1572-0241.1999.03376.x. [DOI] [PubMed] [Google Scholar]
- 8.Wang D., Dubois R. N. The role of COX-2 in intestinal inflammation and colorectal cancer. Oncogene. 2010;29:781–788. doi: 10.1038/onc.2009.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Agarwal S., Reddy G.V., Reddanna P. Eicosanoids in inflammation and cancer: The role of COX-2. Expert Rev. Clin. Immunol. 2009;5:145–165. doi: 10.1586/1744666X.5.2.145. [DOI] [PubMed] [Google Scholar]
- 10.Hahm K.B., Lim H.Y., Sohn S., Kwon H.J., Lee K.M., Lee J.S., Surh Y.J., Kim Y.B., Joo H.J., Kim W.S., et al. In vitro evidence of the role of COX-2 in attenuating gastric inflammation and promoting gastric carcinogenesis. J. Environ. Pathol. Toxicol. Oncol. 2002;21:165–176. doi: 10.1615/JEnvironPatholToxicolOncol.v21.i2.100. [DOI] [PubMed] [Google Scholar]
- 11.Misra U.K., Pizzo S.V. Evidence for a pro-proliferative feedback loop in prostate cancer: The role of Epac1 and COX-2-dependent pathways. PLoS ONE. 2013;8 doi: 10.1371/journal.pone.0063150. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.Clemente M., Sanchez-Archidona A.R., Sardon D., Diez L., Martin-Ruiz A., Caceres S., Sassi F., Pérez-Alenza M.D., Lllera J.C., Dunner S., et al. Different role of COX-2 and angiogenesis in canine inflammatory and non-inflammatory mammary cancer. Vet. J. 2013;197:427–432. doi: 10.1016/j.tvjl.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 13.Serna-Marquez N., Villegas-Comonfort S., Galindo-Hernandez O., Navarro-Tito N., Millan A., Salazar E.P. Role of LOXs and COX-2 on FAK activation and cell migration induced by linoleic acid in MDA-MB-231 breast cancer cells. Cell. Oncol. 2013;36:65–77. doi: 10.1007/s13402-012-0114-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nadda N., Setia S., Vaish V., Sanyal S.N. Role of cytokines in experimentally induced lung cancer and chemoprevention by COX-2 selective inhibitor, Etoricoxib. Mol. Cell. Biochem. 2013;372:101–112. doi: 10.1007/s11010-012-1451-3. [DOI] [PubMed] [Google Scholar]
- 15.Geis G.S. Update on clinical developments with celecoxib, a new specific COX-2 inhibitor: what can we expect. J. Rheumatol. 1999;56:31–36. doi: 10.1080/030097499750042407. [DOI] [PubMed] [Google Scholar]
- 16.Folco G.C. New NSAIDs and gastroduodenal damage. Ital. J. Gastroenterol. 1996;28:28–29. [PubMed] [Google Scholar]
- 17.Dammann H.G. Preferential COX-2 inhibition: Its clinical relevance for gastrointestinal non-steroidal anti-inflammatory rheumatic drug toxicity. Z. Gastroenterol. 1999;37:45–58. [PubMed] [Google Scholar]
- 18.Dannhardt G., Laufer S. Structural approaches to explain the selectivity of COX-2 inhibitors: Is there a common pharmacophore. Curr. Med. Chem. 2000;7:1101–1112. doi: 10.2174/0929867003374237. [DOI] [PubMed] [Google Scholar]
- 19.Zarghi A., Arfaei S. Selective COX-2 Inhibitors: A Review of Their Structure-Activity Relationships. Iran. J. Pharm. Res. 2011;10:655–683. [PMC free article] [PubMed] [Google Scholar]
- 20.Mason R.P., Walter M.F., McNulty H.P., Lockwood S.F., Byun J., Day C.A., Charles A.B.S., Jacob R.F. Rofecoxib increases susceptibility of human LDL and membrane lipids to oxidative damage: A mechanism of cardiotoxicity. J. Cardiovasc. Pharmacol. 2006;47:7–14. doi: 10.1097/00005344-200605001-00003. [DOI] [PubMed] [Google Scholar]
- 21.Park S.J., Buschmann H., Bolm C. Bioactive sulfoximines: Syntheses and properties of Vioxx analogs. Bioorg. Med. Chem. Lett. 2011;21:4888–4890. doi: 10.1016/j.bmcl.2011.06.029. [DOI] [PubMed] [Google Scholar]
- 22.Baxter I. Pyrazolopyridine Derivatives as Selective COX-2 Inhibitors. Pyrazolopyridine Derivatives as Selective COX; E.P. 1127058 A1. 2001 Sep 1;
- 23.Campbell I.B., Naylor A. Cyclooxygenase-2 Inhibitors (COX-2); 4-[2-(3-Fluoro-phenyl)-6-trifluoromethyl-pyrazolo[1,5-a]pyridin-3-yl]benzenesulfonamide for Example; Fever, Pain, Inflammation, Neurodegenerative Disorders, Oral Diseases, Influenza, Arthritis, Allergies. 7223772 B1. U.S. Patent. 2007 May 29;
- 24.Alberti M.J. Antiinflammatory Agents; Autoimmune Disease Treatments; Pyrazolo[1,5-a] Pyridine Derivatives. 7166597 B2. U.S. Patent. 2007 Jan 23;
- 25.Cavalli A., Poluzzi E., de Ponti F., Recanatini M. Toward a Pharmacophore for Drugs Inducing the Long Qt Syndrome: Insights from a Comfa Study of Herg K(+) Channel Blockers. J. Med. Chem. 2002;45:3844–3853. doi: 10.1021/jm0208875. [DOI] [PubMed] [Google Scholar]
- 26.Wang J.L., Limburg D., Graneto M.J., Springer J., Hamper J.R., Liao S., Pawlitz J.L., Kurumbail R.G., Maziasz T., Talley J.J., et al. The novel benzopyran class of selective cyclooxygenase-2 inhibitors. Part 2: The second clinical candidate having a shorter and favorable human half-life. Bioorg. Med. Chem. Lett. 2010;20:7159–7163. doi: 10.1016/j.bmcl.2010.07.054. [DOI] [PubMed] [Google Scholar]
- 27.Michaux C., Charlier C. Structural approach for COX-2 inhibition. Mini-Rev. Med. Chem. 2004;4:603–615. doi: 10.2174/1389557043403756. [DOI] [PubMed] [Google Scholar]
- 28.Marnett L.J., Kalgutkar A.S. Cyclooxygenase 2 inhibitors: Discovery, selectivity and the future. Trends Pharmacol. Sci. 1999;20:465–469. doi: 10.1016/S0165-6147(99)01385-1. [DOI] [PubMed] [Google Scholar]
- 29.Shi D.Q., Shi J.W., Yao H., Jiang H., Wang X.S. An Efficient Synthesis of Pyrazolo[3,4-b] pyridine Derivatives in Aqueous Media. J. Chin. Chem. Soc. 2007;54:1341–1345. doi: 10.1002/jccs.200700190. [DOI] [Google Scholar]
- 30.El-Gendy M.S., Abdel-Aziem A., Abdelhamid A.O. Reactions and Antimicrobial activity of (3-(3-(4-Methoxyphenyl)acryloyl)-2H-Chromen-2-one. Int. J. Adv. Res. 2013;1:557–568. [Google Scholar]
- 31.Copeland R.A., Williams J.M., Giannaras J., Nurnberg S., Covington M., Pinto D., Pick S., Trzaskos J.M. Mechanism of selective inhibition of the inducible isoform or prostaglandin G/H Synthase. Proc. Natl. Acad. Sci. USA. 1994;91:11202–11206. doi: 10.1073/pnas.91.23.11202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gouda A.M. Utility of 3-amino-4,6-dimethyl-1H-pyrazolo[3,4-b]pyridine in heterocyclic synthesis. J. Heterocycl. Chem. 2011;48:1–10. doi: 10.1002/jhet.481. [DOI] [Google Scholar]
- 33.Deeb A., Essawy A., El-Gendy A.M., Shaban A. Heterocyclic synthesis with 3-cyano-2(1H) pyridinethione: Synthesis of 3-oxo-2,3-dihydroisothiazolo[5,4-b]pyridine and related compounds. Monatsh. Chem. 1990;121:281–287. doi: 10.1007/BF00808929. [DOI] [Google Scholar]
- 34.Eweiss N.F., Osman A. Synthesis of heterocycles. Part II new routes to acetylthiadiazolines and alkylazothiazoles. J. Heterocycl. Chem. 1980;17:1713–1717. doi: 10.1002/jhet.5570170814. [DOI] [Google Scholar]
- 35.Shawali A.S., Abdelhamid A.O. Reaction of dimethylphenacylsulfonium bromide with N-nitrosoacetarylamides and reactions of the products with nucleophiles. Bull. Chem. Soc. Jpn. 1976;49:321–327. doi: 10.1246/bcsj.49.321. [DOI] [Google Scholar]
- 36.Wang J., Wolf R.M., Caldwell J.W., Kollman P.A., Case D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]