

Abstract
Stereoselective formation of substituted 1,3-dioxolanes was achieved through an assembly of three components: alkene, carboxylic acid and silyl enol ether. The reaction proceeded via stereospecific generation of a 1,3-dioxolan-2-yl cation intermediate during oxidation of alkene substrates with hypervalent iodine. The stereoselective trapping of the cation intermediate with silyl enol ether completed the formation of the dioxolane product.
Keywords: hypervalent iodine, oxidation, dioxolane, dioxolanyl cation, stereoselective synthesis
1. Introduction
Oxidative difunctionalization of alkenes with hypervalent iodine reagents is one of the many attractive and powerful transformations, as well as halogenation, oxygenation, amidation and arylation (alkylation), for forming useful synthetic intermediates [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44]. During oxidation, two nucleophiles are attached to the carbon-carbon double bond (Figure 1a) [20,21,22,23,24,25,26]. This approach has also been applied to the regio- and stereoselective preparation of heterocyclic compounds. An alkene substrate containing a simple intramolecular nucleophile provides a heterocyclic product (Figure 1b) [27,28,29,30,31,32,33,34,35,36,37,38,39,40]. When a bidentate nucleophile such as urea is introduced to the alkene substrate, bicyclic products are obtained (Figure 1c) [41,42,43,44]. To expand upon the diverse structures of the resulting heterocyclic products, we postulated that intermolecular heterocyclic formation would be achieved using an external bidentate nucleophile (Figure 1d) [45,46,47]. To demonstrate this concept, we focused on the 1,3-dioxolan-2-yl cation intermediate [48,49] generated during hypervalent-iodine-mediated Prévost and Woodward reactions [50,51,52,53,54,55,56,57,58]. In these reactions, a carboxylic acid acts as a bidentate nucleophile; however, the heterocyclic intermediate changes to a ring-opening product. To maintain the heterocyclic structure of dioxolane, a subsequent nucleophilic attack should occur at the 2-position to give a dioxolane product in stable form [59,60]. Herein, we describe the stereoselective preparation of substituted dioxolanes through condensation of three components: alkene, carboxylic acid and carbon nucleophile during the hypervalent-iodine-mediated Prévost and Woodward reaction. The stereoselectivity must be controlled by two steps: the oxidative formation of a 1,3-dioxolan-2-yl cation intermediate and its nucleophilic trapping (Figure 1d).
Figure 1.

Difunctionalization of alkenes with hypervalent iodine(III) using (a) external nucleophiles; (b) intramolecular nucleophile; (c) intramolecular bidentate nucleophile; and (d) external bidentate nucleophile.
2. Results and Discussion
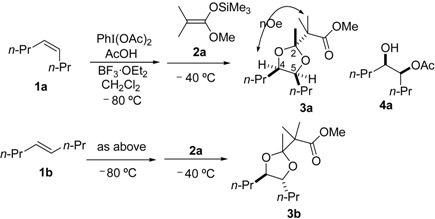
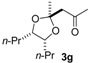
We initially selected cis-4-octene (1a) as a model substrate to test the feasibility of this transformation (Table 1). The reaction of 1a with (diacetoxyiodo)benzene was initiated by injection of boron trifluoride diethyl etherate at −80 °C into a dichloromethane solution containing AcOH. After warming to −40 °C, dimethyl ketene silyl acetal 2a was added to the reaction mixture to produce a dioxolane product 3a as a single diastereomer (Table 1, entries 1–3). The yield of 3a was affected by the amount of nucleophiles (acetic acid and 2a), as well as the temperature of reaction termination; the maximum yield was 62% (entry 3). In the 13C-NMR of 3a, ten signals were observed. This indicates that the adduct 3a has Cs symmetry (meso configuration). Moreover, the stereochemistry of 3a was determined to be the (2r)-configuration by nOe between H-4,5 and the dimethyl moiety (the illustration in Table 1), which was observed in an NMR NOESY experiment (Supplementary Materials (SM)). The corresponding chiral racemic isomer 3b was selectively obtained from the reaction with trans-4-octene 1b (entry 4). In the 13C-NMR spectrum of 3b, fifteen signals were detected.
Table 1.
Reaction of cis-4-octene 1a and trans-4-octene 1ba.
| Entry | 1 | AcOH (mmol) | 2a (mmol) | Quench Temp. | Yield (%) |
|---|---|---|---|---|---|
| 1 | 1a | 1.7 | 0.5 | rt | 3a, 2; 4a, 58 |
| 2 | 1a | 0.5 | 1.5 | rt | 3a, 46 |
| 3 | 1a | 0.5 | 1.5 | −30 °C | 3a, 62 |
| 4 | 1b | 0.5 | 1.5 | −30 °C | 3b, 48 |
a Reaction was initiated at −80 °C in the presence of 1 (0.32 mmol), PhI(OAc)2 (0.40 mmol) and BF3·OEt2 (0.8 mmol) in CH2Cl2 (4 mL) and AcOH. Then, 2a was added at −40 °C.
The stereospecific and stereoselective formation of the substituted dioxolane products can be explained by reaction pathways involving the 1,3-dioxolan-2-yl cation intermediate (Scheme 1). This intermediate was generated by participation of the neighboring acetoxy group in the oxidation of the alkene substrate with hypervalent iodine in the presence of AcOH. The neighboring group participation may cause stereospecific formation of the dioxolanyl cation intermediate, i.e., the cis alkene 1a forms the meso cation and the trans-alkene 1b forms the chiral cation.
Scheme 1.

Plausible pathway for stereoselective formation of dioxolane 3.
Stereoselectivity in nucleophilic trapping with 2a affects the stereochemical outcome of the dioxolane products. The meso cation generated from the cis alkene has diastereofaces for the nucleophilic addition. Thus, the diastereoselective formation of the trapping product requires stereoface control in the nucleophilic attack, as illustrated in Scheme 1. The actual diastereoselective formation of the (2r)-meso product starting from cis alkene may be attributed to the steric fence provided by the 4,5-substituents of the meso cation. Owing to the C2 symmetry of the chiral cation generated from the trans-octene, the nucleophilic trapping of the chiral cation led to the chiral dioxolane product 3b as a single diastereomer without any stereocontrol during the nucleophilic trapping.
The proposed reaction pathway (Scheme 1) was also supported by control experiments without the addition of the trapping reagent 2a (Scheme 2). The acetoxyhydroxy products 4a and 4b were stereospecifically obtained from cis-octene 1a and trans-octene 1b, respectively; their stereochemical configurations were confirmed after transformation to diacetoxy products: meso form 5a and racemic form 5b. The stereochemical outcome obeyed that of a typical Woodward reaction: trapping of 1,3-dioxolan-2-yl cation with water at the 2-position and subsequent ring-opening reaction result in retention of configuration of the cation intermediate.
Scheme 2.

Simple dioxygenation of 4-octenes 1a and 1b.
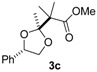
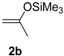
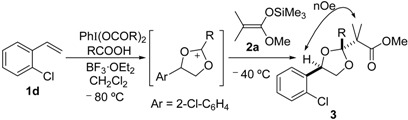
Under optimal conditions (Table 1, entries 3 and 4), the generality of the method was investigated (Table 2). Styrenes and cycloalkenes also successfully gave the dioxolane product 3 from reaction with dimethyl ketene silyl acetal 2a (entries 1–6). Isopropenyloxytrimethylsilane (2b), 1-ethoxy-1-trimethylsilyloxyethylene (2c) and 1-phenyl-1-trimethylsilyloxyethylene (2d) were also incorporated into the dioxolane product (entries 7–14) [61]. In the reaction of styrenes, quenching at rt led to a diastereomeric mixture of 3 and 3′ (entries 1 and 3). The diastereoselectivity was highly improved by quenching at a lower temperature (entries 2 and 4). The trans-configuration of the major product 3c or 3d was confirmed by the 1H-NMR NOESY experiment (SM). The stereochemical outcome may be explained by the steric fence owing to the aryl group during addition of the carbon nucleophile 2a to the 1,3-dioxolan-2-yl cation, similar to the case of the meso cation generated from cis-octene (Scheme 1). Thus, the aryl group at the 4-position of the dioxolanyl cation was able to control the diastereoface selectivity in the nucleophilic trapping. However, the diastereomeric ratio decreased at a higher quenching temperature (entries 1 and 3). To gain an insight into the effect of quenching temperature on diastereoselectivity, the following control experiment was performed. A sample of a single diastereomer of 3d with trans-configuration was treated with BF3·OEt2 in a dichloromethane solution at −40 °C and then warmed to rt. In the crude reaction mixture, isomerized cis dioxolane 3d′ was detected by 1H-NMR (3d(trans)/3d′(cis) = ca. 1/1). Thus, the lowering of the diastereomeric ratio may be due to a secondary reaction of the acetal product, i.e., isomerization mediated by a Lewis acid.
Table 2.
Stereoselective formation of dioxolane 3a.
| Entry | 1 | Nu-SiMe3 | Product | Yield (%) |
|---|---|---|---|---|
| 1 b |  |
 |
 |
15 (1.2:1) c |
| 2 | 56 d | |||
| 3 b | 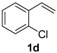 |
2a | 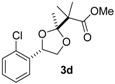 |
25 (1:1.2) c |
| 4 | 40 d | |||
| 5 |  |
2a | 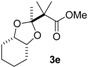 |
52 d |
| 6 |  |
2a | 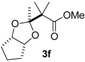 |
46 d |
| 7 | 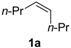 |
 |
 |
39 d |
| 8 | 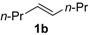 |
2b | 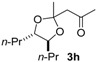 |
37 |
| 9 | 1c | 2b | 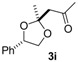 |
29 d |
| 10 | 1d | 2b | 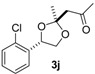 |
25 (8:1) c |
| 11 | 1a | 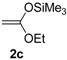 |
 |
34 d |
| 12 | 1b | 2c | 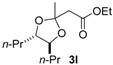 |
21 |
| 13 | 1d | 2c | 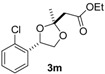 |
29 (7:1) c |
| 14 | 1b | 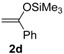 |
 |
27 |
a Reaction was initiated at −80 °C in the presence of 1 (0.32 mmol), PhI(OAc)2 (0.40 mmol), acetic acid (0.5 mmol) and BF3·OEt2 (0.8 mmol) in CH2Cl2 (4 mL). Then, 2 (1.5 mmol) was added at −40 °C and the reaction mixture was quenched at −30 °C by the addition of water; b The reaction was quenched after warming to rt; c The value in parentheses is the diastereomeric ratio of 3 and 3′ (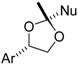 ); d Diastereomer was not detected by 1H-NMR.
); d Diastereomer was not detected by 1H-NMR.
Diastereoselectivity was also affected by the steric effect of the carbon nucleophile; β-unsubstituted silyl enol ethers 2b and 2c led to low diastereoselectivity in the reaction with 1d (entries 10 and 13, respectively). In contrast, a single diastereomer was formed by the reaction of cis alkene (entries 7 and 11), even with compact β-unsubstituted nucleophiles. The diastereoselectivity in the reaction of styrenes was affected by the steric effect of the carbon nucleophile 2. Thus, the stereocontrol by the aryl group in the 4-aryl-1,3-dioxolan-2-yl cation may be slightly reduced compared with the 4,5-dialkyl substituted dioxolanyl cation generated from cis alkene.
Next, a range of carboxylic acid nucleophiles was tested (Table 3). As a primitive examination, (diacetoxyiodo)benzene was used in the reaction of 1d with propanoic acid; however, the protocol resulted in a mixture of 3d and 3o. Therefore, a set of hypervalent iodine and carboxylic acid compounds was employed as reagents for the reaction. The dioxolane product was obtained from benzoic acid (entry 4), as well as primary and secondary aliphatic carboxylic acids (entries 1 and 2, respectively). NOESY analysis indicated that they have trans-configuration, similar to the acetate-incorporated dioxolane 3d. However, the corresponding dioxolane product was not obtained from the tertiary carboxylic acid (entry 3). In this case, 2-hydroxy-1-arylethyl pivalate 6 was produced as an ordinary oxidation product. The sterically bulky pivalic acid was incorporated into the dioxolane product 3r when the compact nucleophile 2c was used (Scheme 3). This reaction led to a 1:1 mixture of 3r and 6, and the dioxolane trapping product 3r was isolated in 10% yield as a single diastereomer with cis configuration. The relative stereochemistry was confirmed by the nOe observed in the NOESY spectrum (SM), in accordance with stereoface differentiation during nucleophilic addition to the dioxolanyl cation intermediate, as discussed above.
Table 3.
Scope of carboxylic acids a.
| Entry | R | Yield (%) |
|---|---|---|
| 1 | Et | 3o, 41 |
| 2 | i-Pr | 3p, 35 |
| 3 | t-Bu | b |
| 4 | Ph | 3q, 26 |
a Reaction was initiated in the presence of 1d (0.32 mmol), PhI(OCOR)2 (0.40 mmol), RCOOH (0.5 mmol) and BF3·OEt2 (0.8 mmol) in CH2Cl2 (4 mL). Then, 2a (1.5 mmol) was added at −40 °C; b No dioxolane 3 was observed and 2-hydroxy-1-arylethyl pivalate 6 was obtained in 19% yield.
Scheme 3.

Reaction with pivalic acid leading to 3r.
On the basis of the reaction pathway discussed above, we examined enantioselective preparation of the dioxolane product using a chiral hypervalent iodine reagent, as shown in Scheme 4. The lactate-based chiral hypervalent iodine reagent 7 [62,63,64,65,66,67,68,69,70,71] has been previously used for enantioselective oxidation of styrenes and has led to high enantioselectivity; a Woodward reaction of 1d with 7 gave the oxyacetylation product with 92% ee [57]. Comparable enantioselectivity was observed for the three-component assembly but the yield of 3d was low (20%). The yield was slightly improved to 36% when trimethylsilyl acetate was added instead of acetic acid (Scheme 4). An aliphatic alkene 1b was also examined for the enantioselective reaction, and enantioselectivity was slightly decreased in comparison with the styrene substrate. The ee value of the dioxolane 3h (77% ee) was comparable with that of the acetoxy product 4b (76% ee) obtained in a separate reaction without addition of the carbon nucleophile 2b.
Scheme 4.

Enantioselective dioxolane formation.
In conclusion, we achieved the stereoselective formation of 1,3-dioxolane compounds via a 1,3-dioxolan-2-yl cation intermediate, which is stereospecifically generated during the oxidation of alkenes with hypervalent iodine. Three components, alkene, carboxylic acid and carbon nucleophile, were stereoselectively assembled into the dioxolane product over the course of the reaction.
3. Experimental Section
3.1. General Information
Dichloromethane was purified by distillation over calcium hydride prior to use. Boron trifluoride diethyl etherate was distilled over calcium hydroxide, and kept under nitrogen. All commercial available reagents were used without further purification. (Diacyloxyiodo)benzenes were prepared according to the literature [24]. Reaction temperature was controlled using low temperature baths with magnetic stirrer PSL-1800 (EYELA, Tokyo, Japan) and PSL-1810 (EYELA). Proton and 13C-NMR spectra were measured on a JEOL ECA-600 spectrometer (JEOL, Tokyo, Japan) as solutions in CDCl3. Proton NMR spectra were recorded using the residual CHCl3 as an internal reference (7.24 ppm) and 13C-NMR using CDCl3 as an internal reference (77.00 ppm). For mass spectra measurements was used JEOL JMS-T100LC (JEOL). Optical rotations were measured on a Perkin-Elmer 241 polarimeter (Perkin-Elmer, Waltham, MA, USA). Gas chromatography was performed on Shimadzu GC-17A (Shimadzu, Kyoto, Japan) equipped with a chiral column (Chirasil-DEX-CB, 25 m × 0.25 mm × 0.25 µm film thickness, Agilent Technology, Santa Clara, CA, USA).
3.2. Typical Procedure for the Three-Component Assembly Reaction
(Diacetoxyiodo)benzene (130 mg, 0.40 mmol) and cis-4-octene 1a (50 µL, 0.32 mmol) were dissolved in dichloromethane (4 mL) in the presence of acetic acid (30 µL, 0.5 mmol). The solution was cooled at −80 °C using a low temperature bath with magnetic stirrer (EYELA, PSL-1800). Boron trifluoride diethyl etherate (0.1 mL, 0.8 mmol) was added to the solution at −80 °C. The solution was warmed up to −40 °C over 1 h. Dimethylketene methyl trimethylsilyl acetal 2a (0.3 mL, 1.5 mmol) was added to the solution at −40 °C and then the mixture was allowed to warm up to −30 °C over 1 h. The reaction mixture was quenched by the addition of water and extracted with dichloromethane. The organic phase was dried with MgSO4 and concentrated in vacuo. The crude mixture was then purified by column chromatography (SiO2, eluent: 10% ethyl acetate in hexane) to yield 3a (54.2 mg, 0.20 mmol, 62% yield).
Methyl 2-methyl-2-((2r,4R,5S)-2-methyl-4,5-dipropyl-1,3-dioxolan-2-yl)propanoate (3a). 1H-NMR (600 MHz, CDCl3) δ 4.05 (m, 2H), 3.65 (s, 3H), 1.54–1.46 (m, 4H), 1.38 (s, 3H), 1.35–1.27 (m, 4H), 1.22 (s, 6H), 0.92 (t, J = 6.9 Hz, 6H); 13C-NMR (150 MHz, CDCl3) δ 176.2, 110.8, 79.5, 52.0, 51.9, 32.4, 25.0, 21.8, 19.6, 14.1; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C15H28NaO4 295.1885; Found 295.1882.
Methyl 2-methyl-2-((4R*,5R*)-2-methyl-4,5-dipropyl-1,3-dioxolan-2-yl)propanoate (3b). 1H-NMR (600 MHz, CDCl3) δ 3.64 (s, 3H), 3.62 (m, 1H), 3.45 (m, 1H), 1.53–1.30 (m, 8H), 1.33 (s, 3H), 1.21 (s, 3H), 1.20 (s, 3H), 0.92 (t, J = 6.9 Hz, 3H), 0.91 (t, J = 6.9 Hz, 3H); 13C-NMR (150 MHz, CDCl3) δ 176.0, 110.5, 82.1, 80.2, 51.8, 51.1, 35.6, 34.0, 22.3, 21.4, 21.1, 19.3, 19.2, 14.23, 14.16; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C15H28NaO4 295.1885; Found 295.1874.
Methyl 2-methyl-2-((2R*,4R*)-2-methyl-4-phenyl-1,3-dioxolan-2-yl)propanoate (3c). 1H-NMR (600 MHz, CDCl3) δ 7.36–7.26 (m, 5H), 4.96 (dd, J = 9.6, 6.2 Hz, 1H), 4.36 (dd, J = 8.9, 6.2 Hz, 1H), 3.70 (s, 3H), 3.67 (dd, J = 9.6, 8.9 Hz, 1H), 1.54 (s, 3H), 1.33 (s, 3H), 1.32 (s, 3H); 13C-NMR (150 MHz, CDCl3) δ 175.7, 138.4, 128.6, 128.1, 126.2, 112.8, 79.6, 72.1, 52.0, 51.3, 21.7, 21.6, 20.7; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C15H20NaO4 287.1259; Found 287.1256. Selected 1H-NMR data for the diastereomeric isomer 3c′: 1H-NMR (600 MHz, CDCl3) δ 5.11 (dd, J = 8.9, 6.2 Hz, 1H), 4.28 (dd, J = 7.6, 6.2 Hz, 1H), 3.69 (s, 3H), 3.53 (dd, J = 8.9, 7.6 Hz, 1H).
Methyl 2-((2R*,4R*)-4-(2-chlorophenyl)-2-methyl-1,3-dioxolan-2-yl)-2-methylpropanoate (3d). 1H-NMR (600 MHz, CDCl3) δ 7.61 (d, J = 7.6 Hz, 1H), 7.31–7.26 (m, 2H), 7.21 (t, J = 7.6 Hz, 1H), 5.29 (dd, J = 9.6, 6.2 Hz, 1H), 4.64 (dd, J = 8.9, 6.2 Hz, 1H), 3.71 (s, 3H), 3.55 (dd, J = 9.6, 8.9 Hz, 1H), 1.51 (s, 3H), 1.34 (s, 3H), 1.33 (s, 3H); 13C-NMR (150 MHz, CDCl3) δ 175.7, 137.2, 131.6, 129.2, 128.8, 127.1, 126.7, 112.7, 76.5, 70.4, 52.1, 51.2, 21.5, 21.4, 20.9; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C15H1935ClNaO4 321.0870; Found 321.0873. Selected 1H-NMR data for the diastereomeric isomer 3d′: 1H-NMR (600 MHz, CDCl3) δ 7.71 (d, J = 7.6 Hz, 1H), 5.41 (dd, J = 8.9, 6.2 Hz, 1H), 4.55 (dd, J = 7.6, 6.2 Hz, 1H), 3.69 (s, 3H), 3.39 (dd, J = 8.9, 7.6 Hz, 1H), 1.48 (s, 3H), 1.35 (s, 3H), 1.35 (s, 3H).
Methyl 2-((2r,3aS,7aR)-hexahydro-2-methylbenzo[d][1,3]dioxol-2-yl)-2-methylpropanoate (3e). 1H-NMR (600 MHz, CDCl3) δ 4.16 (t, J = 4.1 Hz, 2H), 3.66 (s, 3H), 1.80–1.69 (m, 4H), 1.53–1.47 (m, 2H), 1.50 (s, 3H), 1.25–1.20 (m, 2H), 1.23 (s, 6H); 13C-NMR (150 MHz, CDCl3) δ 176.2, 111.4, 75.3, 52.4, 52.0, 28.5, 24.7, 21.9, 20.5; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C13H22NaO4 265.1416; Found 265.1419.
Methyl 2-((2r,3aS,6aR)-tetrahydro-2-methyl-3aH-cyclopenta[d][1,3]dioxol-2-yl)-2-methylpropanoate (3f). 1H-NMR (600 MHz, CDCl3) δ 4.67 (t, J = 4.8 Hz, 2H), 3.66 (s, 3H), 1.86 (dd, J = 14.4, 6.9 Hz, 2H), 1.70 (m, 1H), 1.55 (m, 1H), 1.41 (s, 3H), 1.39 (m, 2H), 1.21 (s, 6H); 13C-NMR (150 MHz, CDCl3) δ 176.1, 114.5, 84.1, 52.4, 52.0, 34.2, 23.4, 22.6, 21.7; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C12H20NaO4 251.1259; Found 251.1256.
1-((2r,4S,5R)-2-Methyl-4,5-dipropyl-1,3-dioxolan-2-yl)propan-2-one (3g). 1H-NMR (600 MHz, CDCl3) δ 4.06 (m, 2H), 2.70 (s, 2H), 2.20 (s, 3H), 1.57–1.44 (m, 4H), 1.43 (s, 3H), 1.38–1.26 (m, 4H), 0.93 (t, J = 6.9 Hz, 6H); 13C-NMR (150 MHz, CDCl3) δ 206.6, 106.4, 78.2, 53.1, 31.8, 31.5, 27.0, 19.4, 14.1; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C13H24NaO3 251.1623; Found 251.1619.
1-((4R*,5R*)-2-Methyl-4,5-dipropyl-1,3-dioxolan-2-yl)propan-2-one (3h). 1H-NMR (600 MHz, CDCl3) δ 3.64–3.53 (m, 2H), 2.72 (s, 2H), 2.20 (s, 3H), 1.52–1.43 (m, 6H), 1.42–1.30 (m, 2H), 1.37 (s, 3H), 0.92 (t, J = 6.9 Hz, 6H); 13C-NMR (150 MHz, CDCl3) δ 206.5, 107.0, 81.2, 80.8, 54.1, 34.9, 34.7, 31.8, 26.1, 19.40, 19.37, 14.2, 14.1; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C13H24NaO3 251.1623; Found 251.1626.
1-((2R*,4R*)-2-Methyl-4-phenyl-1,3-dioxolan-2-yl)propan-2-one (3i). 1H-NMR (600 MHz, CDCl3) δ 7.38–7.27 (m, 5H), 5.07 (dd, J = 8.9, 6.2 Hz, 1H), 4.33 (dd, J = 8.9, 6.2 Hz, 1H), 3.72 (t, J = 8.9 Hz, 1H), 2.87 (s, 2H), 2.25 (s, 3H), 1.57 (s, 3H); 13C-NMR (150 MHz, CDCl3) δ 205.9, 138.3, 128.6, 128.2, 126.2, 108.9, 78.4, 71.7, 52.8, 31.8, 25.3; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C13H16NaO3 243.0997; Found 243.0999.
1-((2R*,4R*)-4-(2-Chlorophenyl)-2-methyl-1,3-dioxolan-2-yl)propan-2-one (3j). 1H-NMR (600 MHz, CDCl3) δ 7.60 (d, J = 7.6 Hz, 1H), 7.32 (d, J = 7.6 Hz, 1H), 7.29 (t, J = 7.6 Hz, 1H), 7.22 (t, J = 7.6 Hz, 1H), 5.41 (dd, J = 7.6, 6.9 Hz, 1H), 4.58 (dd, J = 8.2, 6.9 Hz, 1H), 3.65 (dd, J = 8.2, 7.6 Hz, 1H), 2.89 (s, 2H), 2.25 (s, 3H), 1.58 (s, 3H); 13C-NMR (150 MHz, CDCl3) δ 205.8, 137.2, 131.6, 129.2, 128.9, 127.1, 126.6, 108.9, 75.3, 70.3, 52.5, 31.7, 24.9; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C13H1535ClNaO3 277.0607; Found 277.0613. Selected 1H-NMR data for the diastereomeric isomer 3j′: 1H-NMR (600 MHz, CDCl3) δ 7.58 (d, J = 7.6 Hz, 1H), 4.56 (dd, J = 8.2, 6.9 Hz, 1H), 3.61 (dd, J = 8.2, 7.6 Hz, 1H), 2.93 (s, 2H), 2.25 (s, 3H), 1.53 (s, 3H).
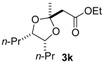
Ethyl 2-((2r,4S,5R)-2-methyl-4,5-dipropyl-1,3-dioxolan-2-yl)acetate (3k). 1H-NMR (600 MHz, CDCl3) δ 4.11 (q, J = 6.9 Hz, 2H), 4.06 (m, 2H), 2.62 (s, 2H), 1.53 (s, 3H), 1.52–1.40 (m, 4H), 1.37–1.25 (m, 4H), 1.23 (t, J = 6.9 Hz, 3H), 0.92 (t, J = 6.9 Hz, 6H); 13C-NMR (150 MHz, CDCl3) δ 169.7, 106.3, 78.1, 60.4, 44.6, 31.8, 27.0, 19.4, 14.2, 14.1; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C14H26NaO4 281.1729; Found 281.1716.
Ethyl 2-((4R*,5R*)-2-methyl-4,5-dipropyl-1,3-dioxolan-2-yl)acetate (3l). 1H-NMR (600 MHz, CDCl3) δ 4.12 (q, J = 6.9 Hz, 2H), 3.64 (td, J = 8.2, 4.1 Hz, 1H), 3.60 (td, J = 8.2, 4.1 Hz, 1H), 2.64 (d, J = 13.1 Hz, 1H), 2.62 (d, J = 13.1 Hz, 1H), 1.53–1.42 (m, 6H), 1.48 (s, 3H), 1.41–1.32 (m, 2H), 1.24 (t, J = 6.9 Hz, 3H), 0.92 (t, J = 6.9 Hz, 6H); 13C-NMR (150 MHz, CDCl3) δ 169.6, 106.8, 81.1, 81.0, 60.4, 45.7, 34.8, 34.7, 26.2, 19.30, 19.26, 14.19, 14.17, 14.1; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C14H26NaO4 281.1729; Found 281.1721.
Ethyl 2-((2R*,4R*)-4-(2-chlorophenyl)-2-methyl-1,3-dioxolan-2-yl)acetate (3m). 1H-NMR (600 MHz, CDCl3) δ 7.60 (d, J = 7.6 Hz, 1H), 7.31 (d, J = 7.6 Hz, 1H), 7.28 (t, J = 7.6 Hz, 1H), 7.21 (t, J = 7.6 Hz, 1H), 5.45 (dd, J = 7.6, 6.9 Hz, 1H), 4.57 (dd, J = 8.2, 6.9 Hz, 1H), 4.17 (q, J = 6.9 Hz, 2H), 3.66 (dd, J = 8.2, 7.6 Hz, 1H), 2.79 (s, 2H), 1.68 (s, 3H), 1.27 (t, J = 6.9 Hz, 3H); 13C-NMR (150 MHz, CDCl3) δ 169.3, 137.4, 131.6, 129.2, 128.8, 127.0, 126.7, 108.7, 75.3, 70.6, 60.7, 44.2, 25.0, 14.2; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C14H1735ClNaO4 307.0713; Found 307.0711. Selected 1H-NMR data for the diastereomeric isomer 3m′: 1H-NMR (600 MHz, CDCl3) δ 7.69 (d, J = 7.6 Hz, 1H), 5.42 (dd, J = 8.2, 6.9Hz, 1H), 4.17 (q, J = 6.9 Hz, 2H), 3.59 (t, J = 8.2 Hz, 1H), 2.82 (s, 2H), 1.62 (s, 3H).
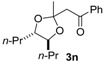
2-((4R*,5R*)-2-Methyl-4,5-dipropyl-1,3-dioxolan-2-yl)-1-phenylethanone (3n). 1H-NMR (600 MHz, CDCl3) δ 7.97 (d, J = 7.6 Hz, 2H), 7.52 (t, J = 7.6 Hz, 1H), 7.42 (t, J = 7.6 Hz, 2H), 3.56 (td, J = 7.6, 3.4 Hz, 1H), 3.42 (td, J = 7.6, 3.4 Hz, 1H), 3.30 (d, J = 13.7 Hz, 1H), 3.27 (d, J = 13.7 Hz, 1H), 1.48 (s, 3H), 1.45–1.18 (m, 8H), 0.85 (t, J = 6.9 Hz, 3H), 0.84 (t, J = 6.9 Hz, 3H); 13C-NMR (150 MHz, CDCl3) δ 197.6, 138.0, 132.8, 129.0, 128.2, 107.6, 81.2, 80.6, 48.9, 34.6, 34.3, 26.8, 19.3, 19.2, 14.11, 14.08; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C18H26NaO3 313.1780; Found 313.1777.
Methyl 2-((2R*,4R*)-4-(2-chlorophenyl)-2-ethyl-1,3-dioxolan-2-yl)-2-methylpropanoate (3o). 1H-NMR (600 MHz, CDCl3) δ 7.73 (d, J = 7.6 Hz, 1H), 7.31 (d, J = 7.6 Hz, 1H), 7.29 (t, J = 7.6 Hz, 1H), 7.21 (t, J = 7.6 Hz, 1H), 5.37 (dd, J = 9.6, 6.9 Hz, 1H), 4.70 (dd, J = 8.2, 6.9 Hz, 1H), 3.70 (s, 3H), 3.58 (dd, J = 9.6, 8.2 Hz, 1H), 1.96 (dq, J = 15.1, 7.6 Hz, 1H), 1.94 (dq, J = 15.1, 7.6 Hz, 1H), 1.33 (s, 3H), 1.32 (s, 3H), 0.92 (t, J = 7.6 Hz, 3H); 13C-NMR (150 MHz, CDCl3) δ 176.0, 136.8, 131.5, 129.2, 128.7, 127.0, 126.4, 114.5, 76.5, 72.1, 52.2, 52.1, 28.4, 21.9, 21.6, 8.1; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C16H2135ClNaO4 335.1026; Found 335.1014.
Methyl 2-((2R*,4R*)-4-(2-chlorophenyl)-2-isopropyl-1,3-dioxolan-2-yl)-2-methylpropanoate (3p). 1H-NMR (600 MHz, CDCl3) δ 7.75 (d, J = 7.6 Hz, 1H), 7.30 (t, J = 7.6 Hz, 1H), 7.28 (d, J = 7.6 Hz, 1H), 7.21 (t, J = 7.6 Hz, 1H), 5.44 (dd, J = 9.6, 6.9 Hz, 1H), 4.70 (t, J = 6.9 Hz, 1H), 3.69 (s, 3H), 3.53 (dd, J = 9.6, 6.9 Hz, 1H), 2.38 (sept, J = 6.9 Hz, 1H), 1.36 (s, 3H), 1.34 (s, 3H), 1.04 (d, J = 6.9 Hz, 3H), 0.94 (d, J = 6.9 Hz, 3H); 13C-NMR (150 MHz, CDCl3) δ 176.3, 137.0, 131.4, 129.3, 128.7, 127.0, 126.5, 115.2, 76.7, 71.6, 52.4, 52.0, 34.9, 22.6, 22.5, 19.1, 18.3; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C17H2335ClNaO4 349.1183; Found 349.1186.
Methyl 2-((2R*,4R*)-4-(2-chlorophenyl)-2-phenyl-1,3-dioxolan-2-yl)-2-methylpropanoate (3q). 1H-NMR (600 MHz, CDCl3) δ 7.54 (d, J = 7.6 Hz, 2H), 7.41 (t, J = 7.6 Hz, 1H), 7.35 (t, J = 7.6 Hz, 2H), 7.30 (t, J = 7.6 Hz, 1H), 7.25 (t, J = 7.6 Hz, 1H), 7.14 (d, J = 7.6 Hz, 1H), 7.13 (d, J = 7.6 Hz, 1H), 5.46 (dd, J = 9.6, 5.5 Hz, 1H), 4.61 (dd, J = 8.2, 5.5 Hz, 1H), 3.65 (s, 3H), 3.24 (dd, J = 9.6, 8.2 Hz, 1H), 1.33 (s, 3H), 1.27 (s, 3H); 13C-NMR (150 MHz, CDCl3) δ 175.0, 140.1, 135.9, 131.9, 129.1, 128.8, 128.2, 127.7, 127.5, 127.3, 127.0, 112.4, 77.6, 70.6, 51.9, 51.5, 21.5, 21.2; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C20H2135ClNaO4 383.1026; Found 383.1013.
Ethyl 2-((2R*,4S*)-2-tert-butyl-4-(2-chlorophenyl)-1,3-dioxolan-2-yl)acetate (3r). 1H-NMR (600 MHz, CDCl3) δ 7.70 (d, J = 7.6 Hz, 1H), 7.31 (d, J = 7.6 Hz, 1H), 7.28 (t, J = 7.6 Hz, 1H), 7.21 (t, J = 7.6 Hz, 1H), 5.52 (dd, J = 9.6, 6.9 Hz, 1H), 4.66 (t, J = 6.9 Hz, 1H), 4.19 (dq, J = 9.6, 6.9 Hz, 1H), 4.16 (dq, J = 9.6, 6.9 Hz, 1H), 3.47 (dd, J = 9.6, 6.9 Hz, 1H), 2.88 (d, J = 13.7 Hz, 1H), 2.82 (d, J = 13.7 Hz, 1H), 1.29 (t, J = 6.9 Hz, 3H), 1.06 (s, 9H); 13C-NMR (150 MHz, CDCl3) δ 170.7, 136.6, 131.5, 129.3, 128.7, 127.0, 126.4, 114.2, 75.8, 71.6, 60.7, 40.8, 40.6, 25.4, 14.2; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C17H2335ClNaO4 349.1183; Found 349.1182.
1-(2-Chlorophenyl)-2-hydroxyethyl pivalate (6). 1H-NMR (600 MHz, CDCl3) δ 7.59 (d, J = 7.6 Hz, 1H), 7.33 (d, J = 7.6 Hz, 1H), 7.29 (t, J = 7.6 Hz, 1H), 7.23 (t, J = 7.6 Hz, 1H), 5.36 (dd, J = 7.6, 2.7 Hz, 1H), 4.35 (dd, J = 11.7, 2.7 Hz, 1H), 4.20 (dd, J = 11.7, 7.6 Hz, 1H), 1.18 (s, 9H); 13C-NMR (150 MHz, CDCl3) δ 179.1, 137.3, 132.0, 129.4, 129.1, 127.8, 127.0, 69.6, 67.8, 38.9, 27.2; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C13H1735ClNaO3 279.0764; Found 279.0761.
3.3. Isomerization of 3d Mediated by a Lewis Acid
Boron trifluoride diethyl etherate (0.1 mL, 0.8 mmol) was added to a dichloromethane solution (4 mL) containing 3d (36.2 mg, 0.12 mmol) at −40 °C. The solution was gradually warmed up to 0 °C over 1 h. The reaction mixture was quenched by the addition of water and extracted with dichloromethane. The organic phase was dried with Na2SO4 and concentrated in vacuo. The crude mixture contained 3d and the diastereomer 3d′ in a 45 to 55 ratio, in addition to decomposed products. The crude mixture was purified by column chromatography (SiO2, eluent: 10% ethyl acetate in hexane) to give a diastereomeric mixture (3d/3d′ = 48/52, 8.4 mg, 23% yield).
3.4. Enantioselective Reaction of 1d with 7 and 2a
The lactate-based hypervalent iodine(III) reagent 7 (260 mg, 0.50 mmol) and 2-chlorostyrene 1d (55 μL, 0.43 mmol) were dissolved in dichloromethane (4 mL) in the presence of trimethylsilyl acetate (30 μL, 0.20 mmol). The solution was cooled at −80 °C using a low temperature bath with magnetic stirrer (EYELA, PSL-1800). Boron trifluoride diethyl etherate (0.1 mL, 0.8 mmol) was added to the solution at −80 °C and the mixture was stirred for 1 h at −80 °C. Dimethylketene methyl trimethylsilyl acetal 2a (0.3 mL, 1.5 mmol) was added to the solution at −80 °C and the mixture was stirred for 30 min at −80 °C. The reaction mixture was quenched by the addition of water and extracted with dichloromethane. The organic phase was dried with Na2SO4 and concentrated in vacuo. The crude mixture was purified by column chromatography (SiO2, eluent: 10% ethyl acetate in hexane) to give 3d (46.6 mg, 0.16 mmol, 36% yield). Selected data for (2S,4S)-3d: +33.5 (c 0.83 in CH2Cl2). Ee of 3d was determined to be 95% by GC on a chiral stationary phase (DEX-CB, i.d. 0.25 mm × 25 m). Retention times of (2R,4R)-3d and (2S,4S)-3d were 30.1 and 31.1 min, respectively, when the column temperature was maintained at 160 °C. The absolute stereochemistry was based on the analogy of the enantioselective Woodward reaction of 1d [57].
3.5. Enantioselective Reaction of 1b with 7 and 2b
Boron trifluoride diethyl etherate (0.1 mL, 0.8 mmol) was added to a dichloromethane solution (4 mL) containing 7 (260 mg, 0.50 mmol), 1b (68 μL, 0.43 mmol) and trimethylsilyl acetate (30 μL, 0.20 mmol) at −80 °C. The solution was warmed to −40 °C over 1 h and 2b (0.3 mL, 1.8 mmol) was subsequently added to the solution at −40 °C. The mixture was warmed to −30 °C over 1 h and quenched by the addition of water. Extracts with dichloromethane were dried with Na2SO4 and concentrated in vacuo. The crude mixture was purified by column chromatography (SiO2, eluent: 10% ethyl acetate in hexane) to give 3h (17.9 mg, 0.078 mmol, 18% yield). Selected data for (4S,5S)-3h: −18.9 (c 0.36 in CH2Cl2). Ee of 3h was determined to be 77% by GC on a chiral stationary phase (DEX-CB, i.d. 0.25 mm × 25 m). Retention times of (4R,5R)-3h and (4S,5S)-3h were 39.6 and 40.2 min, respectively, when the column temperature was maintained at 100 °C. The absolute stereochemistry was based on the analogy of the enantioselective Woodward reaction of 1b, which was described below.
(4R*,5S*)-5-Hydroxyoctan-4-yl acetate 4a: Boron trifluoride diethyl etherate (0.1 mL, 0.8 mmol) was added to a dichloromethane solution (4 mL) containing PhI(OAc)2 (130 mg, 0.40 mmol), 1a (50 μL, 0.32 mmol), acetic acid (0.2 mL, 3.5 mmol) and trimethylsilyl acetate (0.2 mL, 1.3 mmol) at −80 °C. The solution was gradually warmed to rt and stirred overnight. The reaction mixture was quenched by the addition of water and extracted with dichloromethane. The organic phase was dried with MgSO4 and concentrated in vacuo. The crude mixture was purified by column chromatography (SiO2, eluent: 20% ethyl acetate in hexane) to give 4a (51.8 mg, 0.275 mmol, 86% yield). 1H-NMR (600 MHz, CDCl3) δ 4.86 (dt, J = 10.3, 3.4 Hz, 1H), 3.68 (m, 1H), 2.07 (s, 3H), 1.75–1.23 (m, 9H), 0.92 (t, J = 6.9 Hz, 3H), 0.90 (t, J = 6.9 Hz, 3H); 13C-NMR (150 MHz, CDCl3) δ 171.3, 77.5, 73.0, 34.3, 30.8, 21.2, 19.1, 18.9, 14.0, 13.9; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C10H20NaO3 211.1310; Found 211.1309.
Acetylation of 4a with acetic anhydride gave meso-4,5-diacetoxyoctane (5a) in 87% yield. Selected data for 5a: 1H-NMR (600 MHz, CDCl3) δ 4.98 (d, J = 11.0 Hz, 2H), 2.02 (s, 6H), 1.54 (m, 2H), 1.46 (m, 2H), 1.36 (m, 2H), 1.25 (m, 2H), 0.89 (t, J = 7.6 Hz, 6H); 13C-NMR (150 MHz, CDCl3) δ 170.7, 74.0, 31.3, 21.0, 18.8, 13.8. The NMR data agreed well with the reported values [72].
(4S,5S)-5-Hydroxyoctan-4-yl acetate (4S,5S)-4b: A Woodward reaction of trans-4-octene (1b) was performed according to similar procedures described above and gave a racemic sample of 4b in 77% yield. 1H-NMR (600 MHz, CDCl3) δ 4.82 (dt, J = 7.6, 4.2 Hz, 1H), 3.57 (m, 1H), 2.07 (s, 3H), 1.63–1.27 (m, 8H), 0.90 (t, J = 6.9 Hz, 3H); 13C-NMR (150 MHz, CDCl3) δ 171.0, 76.4, 72.3, 35.9, 32.8, 21.1, 18.8, 18.7, 13.99, 13.95. The NMR data agreed well with the reported values [72]. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C10H20NaO3 211.1310; Found 211.1303.
Enantioselective reaction of 1b (0.32 mmol) was performed using 7 and gave optically active 4b in 53% yield (0.17 mmol). −22.2 (c 0.90 in CHCl3). Ee of the product was determined to be 76% ee of (4S,5S)-4b by GC on a chiral stationary phase (DEX-CB, i.d. 0.25 mm × 25 m). Retention times of (4S,5S)-4b and (4R,5R)-4b were 7.8 and 8.2 min, respectively, when the column temperature was maintained at 130 °C.
Acetylation of (4S,5S)-4b with acetic anhydride gave the corresponding diacetoxy compound (4S,5S)-5b in 87% yield. Selected data for (4S,5S)-5b: 1H-NMR (600 MHz, CDCl3) δ 4.99 (m, 2H), 2.06 (s, 6H), 1.53–1.24 (m, 8H), 0.88 (t, J = 6.9 Hz, 6H); 13C-NMR (150 MHz, CDCl3) δ 170.7, 73.7, 32.9, 20.9, 18.5, 13.9. The NMR data agreed well with the reported values [73]. −42.1 (c 0.96 in CHCl3). Ee of the product was determined to be 76% ee of (4S,5S)-5b by GC on a chiral stationary phase (DEX-CB, i.d. 0.25 mm × 25 m). Retention times of (4S,5S)-5b and (4R,5R)-5b were 7.7 and 7.9 min, respectively, when the column temperature was maintained at 130 °C.
The sample of (4S,5S)-5b was hydrolyzed under basic conditions and octane-4,5-diol was obtained. Optical rotation of the diol obtained ( −33 (c 0.54 in EtOH)) indicates that the sample has (4S,5S)-configuration; reported value for (4R,5R)-octane-4,5-diol: +44.4 (c 0.12 in EtOH) [74].
Acknowledgments
This research was partially supported by the Japan Society for the Promotion of Science (JSPS) through a Grant-in-Aid for Scientific Research (C) (23550059 and 26410057).
Supplementary Materials
Supplementary materials can be accessed at: http://www.mdpi.com/1420-3049/20/09/17041/s1.
Author Contributions
M.S. and M.F. designed the project, and performed experiments. M.F. wrote the manuscript. T.S. helped with evaluation of the project.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Not available.
References and Notes
- 1.Varvoglis A. The Organic Chemistry of Polycoordinated Iodine. VCH; New York, NY, USA: 1992. [Google Scholar]
- 2.Varvoglis A. Hypervalent Iodine in Organic Synthesis. Academic Press; San Diego, CA, USA: 1997. [Google Scholar]
- 3.Wirth T. Hypervalent Iodine Chemistry. Springer; Berlin, Germany: 2003. [Google Scholar]
- 4.Zhdankin V.V. Hypervalent Iodine Chemistry. John Wiley & Sons; Chichester, UK: 2014. [Google Scholar]
- 5.Moriarty R.M. Organohypervalent iodine: Development, applications, and future directions. J. Org. Chem. 2005;70:2893–2903. doi: 10.1021/jo050117b. [DOI] [PubMed] [Google Scholar]
- 6.Wirth T. Hypervalent iodine chemistry in synthesis: Scope and new directions. Angew. Chem. Int. Ed. 2005;44:3656–3665. doi: 10.1002/anie.200500115. [DOI] [PubMed] [Google Scholar]
- 7.Zhdankin V.V., Stang P.J. Chemistry of polyvalent iodine. Chem. Rev. 2008;108:5299–5358. doi: 10.1021/cr800332c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ochiai M., Miyamoto K. Catalytic version of and reuse in hypervalent organo-λ3- and -λ5-iodane oxidation. Eur. J. Org. Chem. 2008:4229–4239. doi: 10.1002/ejoc.200800416. [DOI] [Google Scholar]
- 9.Ngatimin M., Lupton D.W. The discovery of catalytic enantioselective polyvalent iodine mediated reactions. Aust. J. Chem. 2010;63:653–658. doi: 10.1071/CH09625. [DOI] [Google Scholar]
- 10.Liang H., Ciufolini M.A. Chiral hypervalent iodine reagents in asymmetric reactions. Angew. Chem. Int. Ed. 2011;50:11849–11851. doi: 10.1002/anie.201106127. [DOI] [PubMed] [Google Scholar]
- 11.Uyanik M., Ishihara K. Conformationally-flexible chiral hypervalent organoiodine catalysts for enantioselective oxidative transformations. J. Synth. Org. Chem. Jpn. 2012;70:1116–1122. doi: 10.5059/yukigoseikyokaishi.70.1116. [DOI] [Google Scholar]
- 12.Rawling M.J., Tomkinson N.C.O. Metal-free syn-dioxygenation of alkenes. Org. Biomol. Chem. 2013;11:1434–1440. doi: 10.1039/c3ob27387c. [DOI] [PubMed] [Google Scholar]
- 13.Parra A., Reboredo S. Chiral hypervalent iodine reagents: Synthesis and reactivity. Chem. Eur. J. 2013;19:17244–17260. doi: 10.1002/chem.201302220. [DOI] [PubMed] [Google Scholar]
- 14.Brown M., Farid U., Wirth T. Hypervalent iodine reagents as powerful electrophiles. Synlett. 2013;24:424–431. [Google Scholar]
- 15.Dohi T., Kita Y. New site-selective organoradical based on hypervalent iodine reagent for controlled alkane sp3 C–H oxidations. ChemCatChem. 2014;6:76–78. doi: 10.1002/cctc.201300666. [DOI] [Google Scholar]
- 16.Singh F.V., Wirth T. Hypervalent iodine-catalyzed oxidative functionalizations including stereoselective reactions. Chem. Asian J. 2014;9:950–971. doi: 10.1002/asia.201301582. [DOI] [PubMed] [Google Scholar]
- 17.Romero R.M., Wöste T.H., Muñiz K. Vicinal difunctionalization of alkenes with iodine(III) reagents and catalysts. Chem. Asian J. 2014;9:972–983. doi: 10.1002/asia.201301637. [DOI] [PubMed] [Google Scholar]
- 18.Harned A.M. Asymmetric oxidative dearomatizations promoted by hypervalent iodine(III) reagents: An opportunity for rational catalyst design? Tetrahedron Lett. 2014;55:4681–4689. doi: 10.1016/j.tetlet.2014.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berthiol F. Reagent and catalyst design for asymmetric hypervalent iodine oxidations. Synthesis. 2015;47:587–603. doi: 10.1055/s-0034-1379892. [DOI] [Google Scholar]
- 20.Kang Y.-B., Gade L.H. The nature of the catalytically active species in olefin dioxygenation with PhI(OAc)2: Metal or proton? J. Am. Chem. Soc. 2011;133:3658–3667. doi: 10.1021/ja110805b. [DOI] [PubMed] [Google Scholar]
- 21.Zhong W., Yang J., Meng X., Li Z. BF3·OEt2-Promoted diastereoselective diacetoxylation of alkenes by PhI(OAc)2. J. Org. Chem. 2011;76:9997–10004. doi: 10.1021/jo201752y. [DOI] [PubMed] [Google Scholar]
- 22.Li Y., Studer A. Transition-metal-free trifluoromethylaminoxylation of alkenes. Angew. Chem. Int. Ed. 2012;51:8221–8224. doi: 10.1002/anie.201202623. [DOI] [PubMed] [Google Scholar]
- 23.Nocquet-Thibault S., Retailleau P., Cariou K., Dodd R.H. Iodine(III)-mediated umpolung of bromide salts for the ethoxybromination of enamides. Org. Lett. 2013;15:1842–1845. doi: 10.1021/ol400453b. [DOI] [PubMed] [Google Scholar]
- 24.Wang Y., Zhang L., Yang Y., Zhang P., Du Z., Wang C. Alkene oxyalkylation enabled by merging rhenium catalysis with hypervalent iodine(III) reagents via decarboxylation. J. Am. Chem. Soc. 2013;135:18048–18051. doi: 10.1021/ja410195j. [DOI] [PubMed] [Google Scholar]
- 25.He Y.-T., Li L.-H., Yang Y.-F., Zhou Z.-Z., Hua H.-L., Liu X.-Y., Liang Y.-M. Copper-catalyzed intermolecular cyanotrifluoromethylation of alkenes. Org. Lett. 2014;16:270–273. doi: 10.1021/ol403263c. [DOI] [PubMed] [Google Scholar]
- 26.Danneman M.W., Hong K.B., Johnston J.N. Oxidative inter-/intermolecular alkene diamination of hydroxy styrenes with electron-rich amines. Org. Lett. 2015;17:2558–2561. doi: 10.1021/acs.orglett.5b01177. [DOI] [PubMed] [Google Scholar]
- 27.Lovick H.M., Michael F.E. Metal-free highly regioselective aminotrifluoroacetoxylation of alkenes. J. Am. Chem. Soc. 2010;132:1249–1251. doi: 10.1021/ja906648w. [DOI] [PubMed] [Google Scholar]
- 28.Wardrop D.J., Bowen E.G., Forslund R.E., Sussman A.D., Weerasekera S.L. Intramolecular oxamidation of unsaturated O-alkyl hydroxamates: A remarkably versatile entry to hydroxy lactams. J. Am. Chem. Soc. 2010;132:1188–1189. doi: 10.1021/ja9069997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karila D., Leman L., Dodd R.H. Copper-catalyzed iminoiodane-mediated aminolactonization of olefins: Application to the synthesis of 5,5-disubstituted butyrolactones. Org. Lett. 2011;13:5830–5833. doi: 10.1021/ol202436a. [DOI] [PubMed] [Google Scholar]
- 30.Wei H.-L., Piou T., Dufour J., Neuville L., Zhu J. Iodo-carbocyclization of electron-deficient alkenes: Synthesis of oxindoles and spirooxindoles. Org. Lett. 2011;13:2244–2247. doi: 10.1021/ol2005243. [DOI] [PubMed] [Google Scholar]
- 31.Zhu R., Buchwald S.L. Copper-catalyzed oxytrifluoromethylation of unactivated alkenes. J. Am. Chem. Soc. 2012;134:12462–12465. doi: 10.1021/ja305840g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tu D., Ma L., Tong X., Deng X., Xia C. Synthesis of pyrrolo[2,3-b]indole via iodine(III)-mediated intramolecular annulation. Org. Lett. 2012;14:4830–4833. doi: 10.1021/ol302158h. [DOI] [PubMed] [Google Scholar]
- 33.Zheng Y., Li X., Ren C., Zhang-Negrerie D., Du Y., Zhao K. Synthesis of oxazoles from enamides via phenyliodine diacetate-mediated intramolecular oxidative cyclization. J. Org. Chem. 2012;77:10353–10361. doi: 10.1021/jo302073e. [DOI] [PubMed] [Google Scholar]
- 34.Fujita M., Mori K., Shimogaki M., Sugimura T. Asymmetric synthesis of 4,8-dihydroxyisochroman-1-one polyketide metabolites using chiral hypervalent iodine(III) Org. Lett. 2012;14:1294–1297. doi: 10.1021/ol300185u. [DOI] [PubMed] [Google Scholar]
- 35.Zhu R., Buchwald S.L. Enantioselective functionalization of radical intermediates in redox catalysis: Copper-catalyzed asymmetric oxytrifluoromethylation of alkenes. Angew. Chem. Int. Ed. 2013;52:12655–12658. doi: 10.1002/anie.201307790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fujita M., Mori K., Shimogaki M., Sugimura T. Total synthesis of (12R)- and (12S)-12-hydroxymonocerins: Stereoselective oxylactonization using a chiral hypervalent iodine(III) species. RSC Adv. 2013;3:17717–17725. doi: 10.1039/c3ra43230k. [DOI] [Google Scholar]
- 37.Li L., Deng M., Zheng S.-C., Xiong Y.-P., Tan B., Liu X.-Y. Metal-free direct intramolecular carbotrifluoromethylation of alkenes to functionalized trifluoromethyl azaheterocycles. Org. Lett. 2014;16:504–507. doi: 10.1021/ol403391v. [DOI] [PubMed] [Google Scholar]
- 38.Chen H., Kaga A., Chiba S. Diastereoselective aminooxygenation and diamination of alkenes with amidines by hypervalent iodine(III) reagents. Org. Lett. 2014;16:6136–6139. doi: 10.1021/ol503000c. [DOI] [PubMed] [Google Scholar]
- 39.Takesue T., Fujita M., Sugimura T., Akutsu H. A series of two oxidation reactions of ortho-alkenylbenzamide with hypervalent iodine(III): A concise entry into (3R,4R)-4-hydroxymellein and (3R,4R)-4-hydroxy-6-methoxymellein. Org. Lett. 2014;16:4634–4637. doi: 10.1021/ol502225p. [DOI] [PubMed] [Google Scholar]
- 40.Alhalib A., Kamouka S., Moran W.J. Iodoarene-catalyzed cyclizations of unsaturated amides. Org. Lett. 2015;17:1453–1456. doi: 10.1021/acs.orglett.5b00333. [DOI] [PubMed] [Google Scholar]
- 41.Steuff J., Hövelmann C.H., Nieger M., Muñiz K. Palladium(II)-catalyzed intramolecular diamination of unfunctionalized alkenes. J. Am. Chem. Soc. 2005;127:14586–14587. doi: 10.1021/ja055190y. [DOI] [PubMed] [Google Scholar]
- 42.Muñiz K., Hövelmann C.H., Steuff J. Oxidative diamination of alkenes with ureas as nitrogen sources: Mechanistic pathways in the presence of a high oxidation state palladium catalyst. J. Am. Chem. Soc. 2008;130:763–773. doi: 10.1021/ja075041a. [DOI] [PubMed] [Google Scholar]
- 43.Cochran B.M., Michael F.E. Metal-free oxidative cyclization of urea-tethered alkenes with hypervalent iodine. Org. Lett. 2008;10:5039–5042. doi: 10.1021/ol8022165. [DOI] [PubMed] [Google Scholar]
- 44.Mizar P., Laverny A., El-Sherbini M., Farid U., Brown M., Malmedy F., Wirth T. Enantioselective diamination with novel chiral hypervalent iodine catalysts. Chem. Eur. J. 2014;20:9910–9913. doi: 10.1002/chem.201403891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anumandla D., Littlefield R., Jeffrey C.S. Oxidative 1,4-diamination of dienes using simple urea derivatives. Org. Lett. 2014;16:5112–5115. doi: 10.1021/ol502460j. [DOI] [PubMed] [Google Scholar]
- 46.Karade N.N., Shirodkar S.G., Patil M.N., Potrelar R.A., Larade H.N. Diacetoxyiodobenzene-mediated oxidative addition of 1,3-dicarbonyl compounds to olefins: An efficient one-pot synthesis of 2,3-dihydrofuran derivatives. Tetrahedron Lett. 2003;44:6729–6731. doi: 10.1016/S0040-4039(03)01644-7. [DOI] [Google Scholar]
- 47.Yoshimura A., Middleton K.R., Todora A.D., Kastern B.J., Koski S.R., Maskaev A.V., Zhdankin V.V. Hypervalent iodine catalyzed generation of nitrile oxides from oximes and their cycloaddition with alkenes or alkynes. Org. Lett. 2013;15:4010–4013. doi: 10.1021/ol401815n. [DOI] [PubMed] [Google Scholar]
- 48.Pittman C.U., Jr., McManus S.P., Larsen J.W. 1,3-Dioxolan-2-ylium and related heterocyclic cations. Chem. Rev. 1972;72:357–438. doi: 10.1021/cr60278a003. [DOI] [Google Scholar]
- 49.Lorenz W., Maas G. O-Acylation of α-diazo ketones. A novel route to alkenediazonium and 1,3-dioxolium salts. J. Org. Chem. 1987;52:375–381. [Google Scholar]
- 50.Prévost C. Sur un complexe iodo-argento-benzoïque et son application à l’oxydation des combinaisons éthyléniques en α-glycols. Comptes Rendus. 1933;196:1129–1131. [Google Scholar]
- 51.Woodward R.B., Brutcher F.V., Jr. cis-Hydroxylation of a synthetic steroid intermediate with iodine, silver acetate and wet acetic acid. J. Am. Chem. Soc. 1958;80:209–211. doi: 10.1021/ja01534a053. [DOI] [Google Scholar]
- 52.Zhdankin V.V., Tykwinski R., Berglund B., Mullikin M., Caple R., Zefirov N.S., Koz’min A.S. Iodosobenzene tetrafluoroborate, hexafluoroantimonate, and hexafluorophosphate: Stable electrophilic hypervalent iodine reagents without nucleophilic ligands. J. Org. Chem. 1989;54:2609–2612. doi: 10.1021/jo00272a029. [DOI] [Google Scholar]
- 53.Ochiai M., Miyamoto K., Shiro M., Ozawa T., Yamaguchi K. Isolation, characterization, and reaction of activated iodosylbenzene monomer hydroxy(phenyl)iodonium ion with hypervalent bonding: Supramolecular complex PhI+OH·18-crown-6 with secondary I···O interactions. J. Am. Chem. Soc. 2003;125:13006–13007. doi: 10.1021/ja0377899. [DOI] [PubMed] [Google Scholar]
- 54.Emmanuvel L., Shaikh T.M.A., Sudalai A. NaIO4/LiBr-mediated diastereoselective dihydroxylation of olefins: A catalytic approach to the Prevost−Woodward reaction. Org. Lett. 2005;7:5071–5074. doi: 10.1021/ol052080n. [DOI] [PubMed] [Google Scholar]
- 55.Li Y., Song D., Dong V.M. Palladium-catalyzed olefin dioxygenation. J. Am. Chem. Soc. 2008;130:2962–2964. doi: 10.1021/ja711029u. [DOI] [PubMed] [Google Scholar]
- 56.Seayad J., Seayad A.M., Chai C.L.L. Copper-catalyzed diacetoxylation of olefins using PhI(OAc)2 as oxidant. Org. Lett. 2010;12:1412–1415. doi: 10.1021/ol902813m. [DOI] [PubMed] [Google Scholar]
- 57.Fujita M., Wakita M., Sugimura T. Enantioselective Prévost and Woodward reactions using chiral hypervalent iodine(III): Switchover of stereochemical course of an optically active 1,3-dioxolan-2-yl cation. Chem. Commun. 2011;47:3983–3985. doi: 10.1039/c1cc10129c. [DOI] [PubMed] [Google Scholar]
- 58.Zhong W., Liu S., Yang J., Meng X., Li Z. Metal-free, organocatalytic syn diacetoxylation of alkenes. Org. Lett. 2012;14:3336–3339. doi: 10.1021/ol301311e. [DOI] [PubMed] [Google Scholar]
- 59.Mukaiyama T., Hayashi Y., Hashimoto Y. Regioselective alkylation of 1,3-dioxolan-2-ylium cation derived from α,β-unsaturated aldehyde ethylene acetal with lithium organo compounds. Chem. Lett. 1986:1627–1630. doi: 10.1246/cl.1986.1627. [DOI] [Google Scholar]
- 60.Hayashi Y., Wariishi K., Mukaiyama T. Oxidative carbon-carbon bond forming reaction via a 1,3-dioxolan-2-ylium cation. Chem. Lett. 1987:1243–1246. doi: 10.1246/cl.1987.1243. [DOI] [Google Scholar]
- 61.An enamine nucleophile, 4-(1-phenylvinyl)morpholine was used instead of 2d in the reaction of 1b. However, no 3n was observed.
- 62.Fujita M., Okuno S., Lee H.J., Sugimura T., Okuyama T. Enantiodifferentiating tetrahydrofuranylation of but-3-enyl carboxylates using optically active hypervalent iodine(III) reagents via a 1,3-dioxan-2-yl cation intermediate. Tetrahedron Lett. 2007;48:8691–8694. doi: 10.1016/j.tetlet.2007.10.015. [DOI] [Google Scholar]
- 63.Uyanik M., Yasui T., Ishihara K. Enantioselective Kita oxidative spirolactonization catalyzed by in situ generated chiral hypervalent iodine(III) species. Angew. Chem. Int. Ed. 2010;49:2175–2177. doi: 10.1002/anie.200907352. [DOI] [PubMed] [Google Scholar]
- 64.Fujita M., Yoshida Y., Miyata K., Wakisaka A., Sugimura T. Enantiodifferentiating endo-selective oxylactonization of ortho-alk-1-enylbenzoate with a lactate-derived aryl-λ3-iodane. Angew. Chem. Int. Ed. 2010;49:7068–7071. doi: 10.1002/anie.201003503. [DOI] [PubMed] [Google Scholar]
- 65.Uyanik M., Yasui T., Ishihara K. Chiral hypervalent iodine-catalyzed enantioselective oxidative Kita spirolactonization of 1-naphthol derivatives and one-pot diastereo-selective oxidation to epoxyspirolactones. Tetrahedron. 2010;66:5841–5851. doi: 10.1016/j.tet.2010.04.060. [DOI] [Google Scholar]
- 66.Röben C., Souto J.A., González Y., Lishchynskyi A., Muñiz K. Enantioselective metal-free diamination of styrenes. Angew. Chem. Int. Ed. 2011;50:9478–9482. doi: 10.1002/anie.201103077. [DOI] [PubMed] [Google Scholar]
- 67.Farid U., Wirth T. Highly stereoselective metal-free oxyaminations using chiral hypervalent iodine reagents. Angew. Chem. Int. Ed. 2012;51:3462–3465. doi: 10.1002/anie.201107703. [DOI] [PubMed] [Google Scholar]
- 68.Kong W., Feige P., de Haro T., Nevado C. Regio- and enantioselective aminofluorination of alkenes. Angew. Chem. Int. Ed. 2013;52:2469–2473. doi: 10.1002/anie.201208471. [DOI] [PubMed] [Google Scholar]
- 69.Farid U., Malmedy F., Claveau R., Albers L., Wirth T. Stereoselective rearrangements with chiral hypervalent iodine reagents. Angew. Chem. Int. Ed. 2013;52:7018–7022. doi: 10.1002/anie.201302358. [DOI] [PubMed] [Google Scholar]
- 70.Shimogaki M., Fujita M., Sugimura T. Enantioselective oxidation of alkenylbenzoates catalyzed by chiral hypervalent iodine(III) to yield 4-hydroxyisochroman-1-ones. Eur. J. Org. Chem. 2013;2013:7128–7138. doi: 10.1002/ejoc.201300959. [DOI] [Google Scholar]
- 71.Wu H., He Y.-P., Xu L., Zhang D.-Y., Gong L.-Z. Asymmetric organocatalytic direct C(sp2)–H/C(sp3)–H oxidative cross-coupling by chiral iodine reagents. Angew. Chem. Int. Ed. 2014;53:3466–3469. doi: 10.1002/anie.201309967. [DOI] [PubMed] [Google Scholar]
- 72.Lethbridge A., Norman R.O.C., Thomas C.B., Parr W.J.E. Oxidation of oct-1-ene and trans-oct-4-ene by lead(IV), thallium(III), and mercury(II) acetates. J. Chem. Soc. Perkin Trans. 1975;1:231–241. doi: 10.1039/p19750000231. [DOI] [Google Scholar]
- 73.Uemura S., Ohe K., Fukuzawa S.-I., Patil S.R., Sugita N. Dominant cis-diacetoxylation of alkenes with tellurium(IV) oxide and lithium bromide in acetic acid. J. Organomet. Chem. 1986;316:67–78. doi: 10.1016/0022-328X(86)82076-9. [DOI] [Google Scholar]
- 74.Fujioka H., Matsunaga N., Kitagawa H., Nagatomi Y., Kondo M., Kita Y. Asymmetric synthesis using C2-symmetric diols: Use of (5R,6R)-3-acetoxy-5,6-diphenyl-1,4-dioxan-2-one as a chiral synthetic equivalent of 1,2-ethanediol 1,2-dicarbocation. Tetrahedron Asymmetry. 1995;6:2117–2020. doi: 10.1016/0957-4166(95)00278-W. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.