

Abstract
Eight novel N′-(2-oxoindolin-3-ylidene)-2-propylpentane hydrazide-hydrazone derivatives 4a–h were synthesized and fully characterized by IR, NMR (1H-NMR and 13C-NMR), elemental analysis, and X-ray crystallography. The cyto-toxicity and in vitro anti-cancer evaluation of the prepared compounds have been assessed against two different human tumour cell lines including human liver (HepG2) and leukaemia (Jurkat), as well as in normal cell lines derived from human embryonic kidney (HEK293) using MTT assay. The compounds 3e, 3f, 4a, 4c, and 4e revealed promising anti-cancer activities in tested human tumour cells lines (IC50 values between 3 and 7 μM) as compared to the known anti-cancer drug 5-Fluorouracil (IC50 32–50 μM). Among the tested compounds, 4a showed specificity against leukaemia (Jurkat) cells, with an IC50 value of 3.14 μM, but this compound was inactive in liver cancer and normal cell lines.
Keywords: valproic acid, isatin, hydrazide-hydrazone, anti-cancer activity
1. Introduction
Hydrazide-hydrazone derivatives are molecules containing a highly reactive group (CO-NH-N=CH) and considered to be a good candidate for development of a new drug [1]. Recently, hydrazide-hydrazones have been considered to be of great interest in medicinal chemistry due to their diverse biological properties, including anti-microbial [2,3,4], anti-mycobacterial [5,6], anti-convulsant [7], analgesic [8], anti-inflammatory [9], anti-platelet [10], anti-tubercular [10,11,12,13], and anti-tumoral activities [14,15,16,17,18,19]. In addition, hydrazide-hydrazones have been reported to elicit anti-cancer [18,19,20,21] and anti-HIV properties [22] and they are therefore increasingly considered to be of great value in medicinal chemistry [16,23,24,25,26,27].
Because of the remarkable biological value of isatin as an important constituent of bioactive compounds exhibiting caspase inhibitory [28,29], antibacterial, and antiproliferative activity [30], Schiff bases of isatin derivatives have antismallpox [31] and GAL3 receptor antagonist capabilities [32]. In addition, the analogous of isatin derivatives displayed inhibitory activity against eLF2 kinase activator [33], TNF-α, CDK2 [34] and SARS protease [35]. Isatin also displays antiviral [36], anti-inflammatory, analgesic [37], and anticonvulsant activities [38].
Valproic acid (VPA, 1) has an important biological value as a potent antiepileptic molecule [39,40,41], as well as showing inhibition of angiogenesis both in vitro and in vivo [42,43,44,45]. VPA also acts as a powerful histone deacetylase inhibitor [46,47] and induces differentiation and apoptosis in a variety of malignant cells in vitro [48]. Clinical trials with VPA have focused on acute myeloid leukaemia and the myelodysplastic syndromes. When it was used as mono therapy or in combination with all-trans retinoic acid, which synergizes in vitro, VPA achieved hematologic improvement in a subset of patients [48].
As a continuation to our previously reported data [49,50,51], herein we designed eight isatin hydrazide-hydrazone derivatives, considering some of the factors responsible for such activity, including (i) the presence of isatin moiety; (ii) the presence of the hydrazide-hydrazone functionality; and (iii) valproic acid moiety (Figure 1).
Figure 1.

Structure of the target product.
All the prepared compounds were assessed against two different human tumour cell lines, including human liver (HepG2) and leukaemia (Jurkat), as well as in normal cell lines derived from human embryonic kidney (HEK293) using MTT assay.
2. Results and Discussion
2.1. Chemistry
Compound 2 was prepared following the reported method [49] and condensed with isatin derivatives 3a–h in the presence of 2–3 drops of glacial HOAc and ethanol as the solvent to afford products 4a–h (Scheme 1). The structures of all synthesized compounds were in a good agreement with their spectral data.
Scheme 1.

Synthesis of N′-(2-oxoindolin-3-ylidene)-2-propylpentanehydrazide Derivatives.
The IR spectra of the compounds 4a–g reveal absorption bands in the region 3195–3217 cm−1 corresponding to the (NH), a band at 1720 and 1688 cm−1 corresponding to the C=O group, and a peak around 1596 cm−1 related to the C=N bond. The NMR of all the products 4a–g are in good agreement with their structures (Figures S1–S7).
As a prototype 1H-NMR of 4g showed multiple peaks at δ 0.86–0.89, 1.27–1.29, 1.40–1.44, 1.56–1.60 ppm, and a broad singlet at δ 2.51 ppm corresponding to the valporic acid moiety (2 CH3, 4 CH2, and CH, respectively). Also, two triplet peaks were observed at δ 3.77 and 4.20 ppm corresponding to the two methylene group (CH2-CH2Br), respectively. The observed peaks in the aromatic region at δ 7.18 (t, 1H, Ar-H), 7.32 (d, 1H, Ar-H), 7.53 (t, 1H, Ar-H), and 7.88 (brs, 1H, ArH) are related to the isatin moiety, while the broad singlet peak at δ 12.28 ppm is related to the NH. The 13C-NMR of 4g showed peaks at δ 14.5, 20.6, 29.9, 35.3, 39.7, 42.2, and 46.5 ppm, corresponding to the valporic acid moiety and the two methylene groups, in addition to seven peaks related to the aromatic carbons and imino function group. The two peaks at δ 169.8 and 176.0 ppm corresponded to the carbonyl groups of the isatin moiety and the hydrazide group, respectively.
It is expected that compound 4g could adopt two different geometrical isomers (Z and E) as shown in Figure 2A. Therefore, it is considered worthwhile to model the compounds using molecular mechanics MM2 calculations. In addition, quantum chemical calculations were carried out with the GAUSSIAN 98 suite of programs. Geometry optimizations were carried out using the DFT level (B3LYP/6-31G **) of theory to assess the relative stability of the Z and E isomeric species. Calculated relative energies of 4g Z and E isomers are −3585.4860873 au and 3585.4635100 au, respectively. Computed energies indicate the stability of the Z isomer over the E one by 0.0225773 au (14.1675 kcal/mol) (Figure 2B).
Figure 2.

(A) The expected geometrical isomers, Z and E forms, of N′-(1-(2-bromoethyl)-2-oxoindolin-3-ylidene)-2-propylpentane hydrazide; (B) The expected geometrical isomers, the 3D structure of Z and E forms, of N′-(1-(2-bromoethyl)-2-oxoindolin-3-ylidene)-2-propyl pentane hydrazide.
The X-ray single crystal structure determination of compound 4g (CCDC: 996592) confirmed that the structure exists in the Z-conformer rather than the E-conformer (Figure 3). The details of data collection and structure refinement are listed in Table 1, and the X-ray structures are shown in Figure 3. Compound 4g crystallized from ethanol in the triclinic space group P-1. All bond lengths and angles are in normal ranges [52].
Figure 3.

ORTEP diagram of the asymmetric unit consisting of two symmetry-independent molecules. The ellipsoids are drawn at the 50% probability level with hydrogen atoms being shown as spheres of arbitrary radii. One of the molecules has disorder in the side chain C11B—C13B.
Table 1.
Details of data collection and structure refinement for compound 4g.
| Compound | 4g |
|---|---|
| Empirical formula | C18H24BrN3O2 |
| Formula weight | 349.21 |
| Temperature (K) | 100 |
| Wavelength (A) | Mo Kα radiation, λ = 0.71073 Å |
| Crystal system, Space group | Triclinic, P-1 |
| Crystal | Plate, yellow |
| a (Å) | 8.8126 (11) |
| b (Å) | 14.4104 (18) |
| c (Å) | 14.479 (2) |
| α (°) | 92.002 (4) |
| β (°) | 97.498 (4) |
| γ (°) | 92.598 (4) |
| Volume (Å3) | 1819.6 (4) |
| Z, Dcalc (Mg·m−3) | 4, 1.275 |
| F (000) | 816 |
| Crystal size (mm) | 0.58 × 0.41 × 0.12 |
| θ Range for data (°) | 2.33–28.11 |
| Collection Limiting indices | −12 ≤ h ≤ 12, −20 ≤ k ≤ 20, −20 ≤ k ≤ 20 |
| Reflections collected/unique | 81,300/6824 [Rint = 0.136] |
| restraints/parameters | 112/469 |
| Goodness-of-fit on F2 | 1.009 |
| Final R indices[I > 2s(I)] | R1 = 0.053, wR2 = 0.135 |
| Absorption correction | Multi-scan, SADABS V2012/1 (Bruker AXS Inc., Madison, WI, USA) |
| Instrument | Bruker APEX-II D8 venture diffractometer |
In the crystal packing (Figure 4), the molecules are linked by intermolecular C17A—H17B···Br1B, C17B—H17D···O2B and C18A—H18B···O2A hydrogen bonds (Table 2) into two-dimensional networks parallel to the bc plane (Figure 4 and Table 2). The asymmetric unit contains two molecules of the compound with disorder in one of the ethylene arms. The molecular structure of compound 4g is composed of a 2-oxoindoline ring (C1/N1/C2-C8) which is linked with two side chains at N1 and C8. The two molecules in the asymmetric unit are with Z configuration about the C8=N2 double bond (Figure 3). The Z configuration of 4a is stabilized with intramolecular hydrogen N3—H3N···O2 (Table 2). The single bond N2—N3 is clearly characterized by the distances of 1.355 (3) Å and 1.361 (4) Å, respectively, for molecules A and B. The double bond of C8=N2 is characterized by the distances of 1.298 (3) Å and 1.283 (4) Å, for molecules A and B, respectively.
Figure 4.

Crystal packing showing intermolecular hydrogen bonds as dashed lines.
Table 2.
Hydrogen-bond geometry (Å, °).
| D—H···A | D—H | H···A | D···A | D—H···A |
|---|---|---|---|---|
| N3B−H3NB···O1B | 0.81 (3) | 2.10 (3) | 2.779 (3) | 141 (3) |
| N3A−H3NA···O1A | 0.86 (3) | 2.11 (3) | 2.799 (3) | 138 (3) |
| C10A−H10A···N2A | 0.9800 | 2.3700 | 2.815 (4) | 107.00 |
| C10B−H10B···N2B | 0.9800 | 2.3600 | 2.833 (4) | 109.00 |
| C17A−H17A···O1A | 0.9700 | 2.5700 | 2.930 (3) | 102.00 |
| C17A−H17B···Br1B i | 0.9700 | 2.9100 | 3.779 (3) | 149.00 |
| C17B−H17D···O2B ii | 0.9700 | 2.4100 | 3.139 (4) | 131.00 |
| C18A−H18B···O2A iii | 0.9700 | 2.2900 | 3.096 (3) | 140.00 |
Symmetry codes: (i) x − 1, y, z; (ii) −x + 1, −y + 1, −z; (iii) −x + 1, −y + 1, −z + 1.
Reaction of valporic acid hydrazide 2 with N-acetylisatin 3h proceeds in a different way, where the ring opening occurred to afford the product 4h. The IR and NMR data proved its structure in the open form, due to the attack of the hydrazide group on C2 instead of C3 [53]. The IR spectrum of 4h reveals absorption bands in the region 3310 and 3223 cm−1 corresponding to the NH, a band at 1720, 1709, 1688, and 1608 cm−1 corresponding to the C=O and C=N group, respectively. 1H-NMR of 4h showed multiple peaks at δ 0.86–0.89, 1.27–1.29, 1.40–1.44, 1.56–1.60, and a broad singlet peak at δ 2.21 ppm corresponding to the valporic acid moiety (2 CH3, 2 CH2, and 2CH2, CH, respectively). The observed peak at δ 2.16 ppm is corresponding to the N-acetyl group. The observed peaks in the aromatic region at δ 7.28 (t, 1H), 7.68 (t, 1H), 7.99 (d, 1H), and 8.17(d, 1H) ppm, are related to the phenyl ring, while the two singlet peaks at δ 10.03 and 10.72 ppm corresponded to the two NH. The 13C-NMR of 4h showed peaks at δ 14.6, 20.6, 29.9, 35.3, 43.8, and 46.5 ppm corresponding to the valporic acid moiety group, while the acetyl group was observed at δ 25.0 ppm, in addition to six peaks related to the aromatic carbons at δ 121.5, 121.8, 123.8, 133.6, 135.9, and 140.3 ppm. The three peaks observed at δ 164.1, 169.6, and 174.7 ppm related to the three CONH groups, while the peak observed at δ 193.0 ppm is related to the α-ketoamide group.
2.2. Anti-Cancer Activity
The cyto-toxicity activity was measured in vitro in HepG2, Jurkat, and HEK 293 cells using the MTT 1-(4,5-dimethylthiazol-2-yl)-3,5-diphenylformazan colorimetric assay. For comparison, 5-Fluorouracil (5-FU) was used as a standard anti-cancer drug. Treatment with dimethylsulfoxide (DMSO) was used as a control for the cancer cells.
The anti-cancer activity of the new prepared compounds against the HepG2 cell line revealed that compounds 3a, 3h, 4a, 4b, 4f, 4g, and 4h showed no anti-cancer activity. Similarly, the starting compounds 1 and 2 did not show promising anti-cancer activity in HepG2 cells. Compound 3e exhibited higher potency against HepG2 cell line with IC50 = 2.82 ± 40.7 μM, which is much lower than the IC50 value of 5-FU (32.15 ± 48.1 μM). Moreover, the results showed that compounds 3f, 4c, and 4e were also found to be potent and selective against HepG2 with IC50 values of 3.13 ± 42.0, 4.39 ± 43.2, and 7.61 ± 44.6 μM, respectively, which are also much lower than 5-FU (Figure 5 and Table 3).
Figure 5.


Dose-dependent responses of hydrazide-hydrazone derivatives in three cell lines. Each line represents the mean of three different experiments representing the % of mortality as compared to mock treated control cells.
Table 3.
In vitro cytotoxicity activity of the synthesized compounds in two cancer and one normal cell line(s) as measured with MTT assay *.
| Compd. No. | IC50 (μM) | ||
|---|---|---|---|
| HepG2 | Jurkat | HEK293 | |
| 1 | 69.74 ± 40.8 | 79.35 ± 40.7 | 62.98 ± 38.2 |
| 2 | 68.50 ± 37.1 | N.A. | N.A. |
| 3a | N.A. | N.A. | 16.73 ± 50.0 |
| 3e | 2.82 ± 40.7 | 7.13 ± 46.0 | N.A. |
| 3f | 3.13 ± 42.0 | 3.13 ± 42.0 | N.A. |
| 3h | N.A. | N.A. | N.A. |
| 4a | N.A. | 3.15 ± 43.3 | N.A. |
| 4b | N.A. | N.A. | 26.08 ± 31.3 |
| 4c | 4.39 ± 43.2 | 2.90 ± 36.3 | 4.77 ± 42.7 |
| 4d | N.A. | N.A. | N.A. |
| 4e | 7.61 ± 44.6 | 3.19 ± 46.9 | N.A. |
| 4f | N.A. | N.A. | N.A. |
| 4g | N.A. | N.A. | 45.43 ± 48.9 |
| 4h | N.A. | N.A. | N.A. |
| 5-FU | 32.15 ± 48.1 | 51.16 ± 47.4 | N.A. |
| DMSO | N.A. | N.A. | N.A. |
* Data were expressed as the mean ± standard deviation (SD) of six independent experiments; N.A.: IC50 values more than 100 μM was considered as No Activity.
The anti-cancer profile of newly synthesized compounds was also tested against leukaemia cells (Jurkat cells, originated from human T lymphocytes). The compound 4c was most active, with an IC50 value of just 2.90 ± 36.3 μM, which was much lower than the positive control (5-FU IC50 = 51.16 ± 47.4 μM). Similarly, the compounds 3f, 4a, and 4c showed strong activity and IC50 values were much lower as compared to 5-FU (Table 3 and Figure 5). The order of activity was 3f, 4a, 4e, and 3e in ascending order (Table 3 and Figure 5). The compound 1 showed a weaker level of activity against Jurkat cells (IC50 79.35 ± 40.7 μM) and, indeed, this was much weaker than 5-FU (IC50 51.16 ± 47.4 μM).
Finally the cyto-toxicities of these compounds were screened in the normal human embryonic kidney (HEK293) cell line, in order to assess whether these compounds are active only in cancer cells or whether they possess toxicity against normal cells as well. The results revealed that the compound 4c turned out to be most toxic by disrupting the cell survival of normal cells (HEK 293) with IC50 4.77 ± 42.7 μM. The compounds 3a, 4b, and 4g also possessed some level of cyto-toxicity against HEK 293 cells with IC50 values 16.73 ± 50.0, 26.08 ± 31.3, and 45.43 ± 48.9 μM, respectively. The IC50 values of these compounds in HEK cells, however, are much higher compared to their activity in cancer cells (Table 3 and Figure 5). The compounds 3e, 3f, 4a, and 4e showed no activity in HEK 293 cells, but strong activity in liver (HepG2) and leukaemia (Jurkat) cancer cells, suggesting that these compounds are potent and potentially viable anti-cancer molecules.
3. Experimental Section
3.1. Chemistry
3.1.1. Materials
The solvents used were of HPLC reagent grade. Melting points were determined with Melting points were obtained in open capillary tubes using a MEL-Temp melting point apparatus (Sigma-Aldrich Chemie GmbH, 82024 Taufkirchen, Germany) and are uncorrected and are uncorrected. Infrared (IR) spectra were recorded on a Perkin-Elmer 1600 series Fourier transform instrument (PerkinElmer Life and Analytical Sciences, Shelton, CT, USA) as KBr pellets. Nuclear Magnetic resonance spectra (1H-NMR and 13C-NMR spectra) were recorded on 400 MHz JEOL spectrometer (JEOL Ltd., Tokyo, Japan) at room temperature. Chemical shifts are reported in parts per million (ppm) and are referenced relative to residual solvent (e.g., CHCl3 at δ 7.26 ppm for CDCl3, DMSO at δ 2.50 ppm for DMSO-d6). Spin multiplicities are represented by the following signals: singlet (s), broad singlet (br s), doublet (d), broad doublet (br d), doublet of doublets (dd), triplet (t), doublet of triplets (dt), and multiplet (m). Elemental analyses were performed on a Perkin-Elmer 2400 elemental analyzer (PerkinElmer Inc., Waltham, MA USA), and the values found were within ±0.3% of the theoretical values. Follow-up of the reactions and checks of the purity of the compounds was done by TLC on silica gel-protected aluminum sheets (Type 60 GF254, Merck Millipore, Billerica, MA, USA) and the spots were detected by exposure to UV-lamp (Company Seven, Montpelier, MD, USA) at λ 254 nm for a few seconds. The compounds were named using Chem. Draw Ultra version 11, Cambridge soft Corporation. Crystallographic data for the structure reported in this paper have been deposited at the Cambridge Crystallographic Data Center and allocated with the deposition numbers: CCDC 996592 for compound 4g. CCDC 996592 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-mail: deposit@ccdc.cam.ac.uk).
3.1.2. Synthesis of Valproic Hydrazide 2
 |
The product was prepared according to the reported procedure [50] and obtained as white needle crystals, in 85%, yield; mp 123–124 °C; IR (KBr): 3284 (NH), 1631 (CO, amide) cm−1; 1H-NMR (CDCl3) δ (ppm): 0.86 (t, J = 4.4 Hz, 6H, 2CH3), 1.14–1.39 (m, 6H, 3CH2), 1.53–1.604 (m, 2H, CH2), 1.95–2.01 (m, 1H, CH), 3.95 (brs, 2H, NH2), 7.05 (s, 1H, NH). 13C-NMR (CDCl3) δ (ppm): 14.11, 20.86, 35.05, 45.61, 177.06.
3.1.3. Synthesis of Compounds 3e–h
Compounds 3e–h were prepared following the reported procedure [54]. The entire prepared compounds were in a good agreement with the reported data.
3.1.4. General Method for Preparation of N′-(2-Oxoindolin-3-ylidene)-2-propylpentane Hydrazide Derivatives 4a–h
A solution of valproic hydrazide 2 (316 mg, 2 mmol) in ethanol (20 mL) was added to a solution of substituted isatin 3a–h (1 mmol) in ethanol (20 mL), and glacial acetic acid (2 drops); the reaction mixture was refluxed for 3–4 h. The product was separated out on cooling, filtered, and recrystallized from ethanol or ethylacetate to afford N′-(2-oxoindolin-3-ylidene)-2-propylpentanehydrazide derivatives 4a–h.
(Z) N′-(2-Oxoindolin-3-ylidene)-2-propylpentane Hydrazide 4a
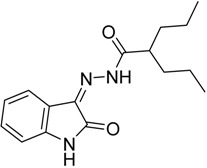 |
The product was obtained as yellow solid, in 89% yield; mp 175–176 °C. IR (KBr): 3194 (NH), 1720 (CO), 1663 (C=N), 1602 (CO) cm−1; 1H-NMR (DMSO-d6) δ (ppm): 0.85-0.86 (m, 6H, 2 CH3), 1.26–1.27 (m, 4H, 2 CH2), 1.40–1.42 (m, 2H, CH2), 1.56–1.58 (m, 2H, CH2), 2.51 (brs, 1H, CH), 6.90 (d, J = 4.6 Hz, 1H, Ar-H), 7.06 (brs, 1H, Ar-H), 7.38 (brs, 1H, ArH), 8.02 (brs,1H, ArH), 10.80 (s, 1H, NH), 11.08 (s, 1H, NH); 13C-NMR (DMSO-d6) δ (ppm): 14.5, 20.6, 35.1, 39.8,111.2, 115.9, 122.2, 126.6, 133.1, 144.3, 165.3, 169.80, 172.5. Anal. Calcd for C16H21N3O2: C, 66.88; H, 7.37; N, 14.62; found: C, 66.70; H, 7.44; N, 14.78.
(Z) N′-(5-Bromo-2-oxoindolin-3-ylidene)-2-propylpentane Hydrazide 4b
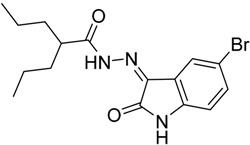 |
The product was obtained as yellow solid, in 86% yield; mp 172–173 °C; IR (KBr): 3222 (NH), 1729 (CO), 1663 (C=N), 1609 (CO) cm−1; 1H-NMR (DMSO-d6): δ (ppm): 0.85–0.89 (m, 6H, 2 CH3), 1.27–1.29 (m, 4H, 2 CH2), 1.40–1.43 (m, 2H, CH2), 1.57–1.59 (m, 2H, CH2), 2.50 (brs, 1H, CH), 6.90 (brs, 1H, Ar-H), 7.54 (brs, 1H, ArH), 8.02 (brs, 1H, ArH), 10.91 (s, 1H, NH), 11.31 (s, 1H, NH); 13C-NMR (DMSO-d6) δ (ppm): 14.6, 20.6, 35.1, 39.8, 112.9, 117.5, 122.2, 128.7, 135.1, 144.3, 166.3, 170.80, 172.5. Anal. Calcd for C16H20BrN3O2: C, 52.47; H, 5.50; N, 11.47; found: C, 52.66; H, 5.61; N, 11.23.
(Z) N′-(5-Chloro-2-oxoindolin-3-ylidene)-2-propylpentane Hydrazide 4c
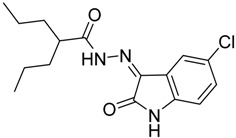 |
The product was obtained as yellow solid, in 88% yield; mp 154–156 °C; IR (KBr): 3233 (NH), 1720 (CO), 1689 (C=N), 1620 (CO) cm−1; 1H-NMR (DMSO-d6) δ (ppm): 0.87–0.93 (m, 6H, 2 CH3), 1.26–1.29 (m, 4H, 2 CH2), 1.40–1.43 (m, 2H, CH2), 1.57–1.59 (m, 2H, CH2), 2.51 (brs, 1H, CH), 6.91 (m, 1H, Ar-H), 7.42 (m, 1H, ArH), 8.2 (brs, 1H, ArH), 10.91 (s, 1H, NH), 11.35 (brs, 1H, NH); 13C-NMR (DMSO-d6) δ (ppm): 14.6, 20.6, 35.3, 39.7, 112.8, 118.5, 122.0, 126.2, 134.1, 145.3, 165.3, 170.80, 172.5. Anal. Calcd for C16H20ClN3O2: C, 59.72; H, 6.26; N, 13.06; found: C, 59.55; H, 6.18; N, 13.33.
(Z) N′-(5-Fluoro-2-oxoindolin-3-ylidene)-2-propylpentane Hydrazide 4d
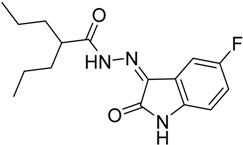 |
The product was obtained as yellow solid in 89% yield; mp 180–182 °C; IR (KBr): 3181 (NH), 1721 (CO), 1686 (C=N), 1630 (CO) cm−1; 1H-NMR (DMSO-d6) δ (ppm): 0.86–0.89 (m, 6H, 2 CH3), 1.26–1.27 (m, 4H, 2 CH2), 1.40–1.43 (m, 2H, CH2), 1.57–1.59 (m, 2H, CH2), 2.50 (brs, 1H, CH), 6.88 (m, 1H, Ar-H), 7.24 (m, 1H, ArH), 8.2 (brd, 1H, ArH), 10.81 (s, 1H, NH), 11.23 (brs, 1H, NH); 13C-NMR (DMSO-d6) δ (ppm): 14.6, 20.6, 35.3, 39.7, 112.8, 118.5, 119.8, 124.2, 134.1, 140.3, 162.3, 169.80, 172.5. Anal. Calcd for C16H20FN3O2: C, 62.94; H, 6.60; N, 13.76; found: C, 63.13; H, 6.77; N, 13.52.
(Z) N′-(1-Methyl-2-oxoindolin-3-ylidene)-2-propylpentane Hydrazide 4e
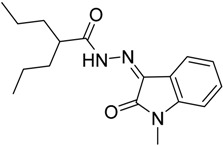 |
The product was obtained as yellow solid, in 89% yield; mp 154–156 °C; IR (KBr): 1720 (CO), 1689 (C=N), 1620 (CO) cm−1; 1H-NMR (DMSO-d6) δ (ppm): 0.85–0.87 (m, 6H, 2 CH3), 1.24–1.29 (m, 4H, 2 CH2), 1.40–1.43 (m, 2H, CH2), 1.56–1.59 (m, 2H, CH2), 2.50 (brs, 1H, CH), 3.18 (s, 3H, CH3), 7.08–7.14 (m, 2H, Ar-H), 7.46 (m, 1H, ArH), 8.20 (brs, 1H, ArH), 11.23 (brs, 1H, NH); 13C-NMR (DMSO-d6) δ (ppm): 14.5, 20.6, 27.6, 35.3, 39.7, 115.3, 119.6, 122.7, 129.2, 131.1, 134.8, 164.0, 169.80, 171.5. Anal. Calcd for C17H23N3O2: C, 67.75; H, 7.69; N, 13.94; found: C, 67.63; H, 7.78; N, 14.21.
(Z) N′-(1-Benzyl-2-oxoindolin-3-ylidene)-2-propylpentane Hydrazide 4f
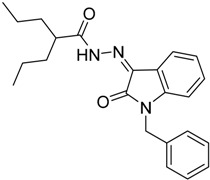 |
The product was obtained as yellow solid, in 83% yield; mp 170–172 °C; IR (KBr): 1718 (CO), 1686 (C=N), 1620 (CO) cm−1; 1H-NMR (DMSO-d6) δ (ppm): 0.86-0.89 (m, 6H, 2 CH3), 1.27–1.29 (m, 4H, 2 CH2), 1.40–1.44 (m, 2H, CH2), 1.56–1.60 (m, 2H, CH2), 2.51 (brs, 1H, CH), 4.96 (s, 2H, CH2), 7.00–7.20 (m, 2H, Ar-H), 7.33–7.39 (m, 6H, ArH), 8.20 (brs, 1H, ArH), 11.28 (brs, 1H, NH); 13C-NMR (DMSO-d6) δ (ppm): 14.6, 20.6, 35.3, 39.7, 43.2, 115.4, 122.8, 122.9, 126.8, 127.8, 128.1, 129.2, 129.3, 136.8, 144.2, 164.2, 170.5. Anal. Calcd for C23H27N3O2: C, 73.18; H, 7.21; N, 11.13; found: C, 73.41; H, 7.40; N, 10.94.
(Z)-N′-(1-(2-Bromoethyl)-2-oxoindolin-3-ylidene)-2-propylpentane Hydrazide 4g
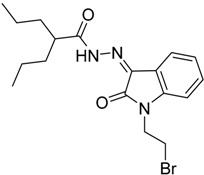 |
The product was obtained as yellow crystals, in 83% yield; mp 90–91 °C; IR (KBr): 1700 (CO), 1686 (C=N), 1608 (CO) cm−1; 1H-NMR (DMSO-d6) δ (ppm): 0.86–0.89 (m, 6H, 2 CH3), 1.27–1.29 (m, 4H, 2 CH2), 1.40–1.44 (m, 2H, CH2), 1.56–1.60 (m, 2H, CH2), 2.51 (brs, 1H, CH), 3.77 (t, J = 6.8 Hz, 2H, CH2-CH2Br), 4.20 (t, J = 6.4 Hz, 2H, CH2-CH2Br), 7.18 (t, J = 8.4 Hz, 1H, Ar-H), 7.32 (d, J = 8.0 Hz, 1H, Ar-H), 7.53 (t, J = 8.0 Hz, 1H, Ar-H), 7.88 (brs, 1H, ArH), 12.28 (brs, 1H, NH); 13C-NMR (DMSO-d6) δ (ppm): 14.5, 20.6, 29.9, 35.3, 39.7, 42.2, 46.5, 110.9, 119.7, 122.7, 123.2, 134.1, 142.9, 164.0, 169.80, 176.0. Anal. Calcd for C18H24BrN3O2: C, 54.83; H, 6.13; N, 10.66; found: C, 55.12; H, 6.23; N, 10.90.
N-(2-(2-Oxo-2-(2-(2-propylpentanoyl)hydrazinyl)acetyl)phenyl) Acetamide 4h
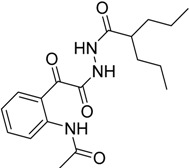 |
The product was obtained as yellowish white solid, in 83% yield; mp 180–181 °C; IR (KBr): 3310 (NH), 3223(NH), 1720 (CO), 1709 (CO), 1686 (C=N), 1608 (CO) cm−1; 1H-NMR (DMSO-d6) δ (ppm): 0.86–0.89 (m, 6H, 2 CH3), 1.27–1.29 (m, 4H, 2 CH2), 1.40–1.44 (m, 2H, CH2), 1.56–1.60 (m, 2H, CH2), 2.16 (s, 3H, CH3), 2.21 (brs, 1H, CH), 7.28 (t, J = 8.0 Hz, 1H, Ar-H), 7.68 (t, J = 8.0 Hz, 1H, Ar-H), 7.99 (d, J = 7.3 Hz, 1H, Ar-H), 8.17(d, J = 8.1 Hz, 1H, ArH), 10.03 (s, 1H, NH), 10.64 (s, 1H, NH), 10.72 (s, 1H, NH);13C-NMR (DMSO-d6) δ (ppm): 14.6, 20.6, 25.0, 29.9, 35.3, 43.8, 46.5, 121.5, 121.8, 123.8, 133.6, 135.9, 140.3, 164.1, 169.6, 174.7, 193.0. Anal. Calcd for C18H25N3O4: C, 62.23; H, 7.25; N, 12.10; found: C, 62.55; H, 7.42; N, 12.38.
3.2. X-ray Crystallography
Single crystals were obtained by slow evaporation from ethanol. A good crystal with a suitable size was selected for X-ray diffraction analysis. Data were collected on a D8 Venture area diffractometer equipped with graphite monochromatic MoK/α radiation (λ = 0.71073 Å) at 100 K. Cell refinement and data reduction were done by Bruker SAINT (Bruker, Madison, WI USA); the program used to solve structure and refine structure is SHELXS-97 [55]. The final refinement was performed by full-matrix least-squares techniques with anisotropic thermal data for non-hydrogen atoms on F2. All the hydrogen atoms were placed in calculated positions and constrained to ride on their parent atoms. Multi-scan absorption correction was applied by use of SADABS software [56].
3.3. Biology
3.3.1. Cell Culture and Cell Viability Assay
The stock concentration of the entire compound in DMSO was 10 mM and this concentration was used to prepare the working dilution. The final DMSO concentration used in the experiments was ≤0.5% as the working concentration. Human liver cancer cell lines (HepG2), human leukemia cell line (Jurkat), and human embryonic kidney cell line (HEK293) cells were cultured in high glucose Dulbecco’s Modified Eagle Medium (DMEM: Life technologies cat #11995073) supplemented with 10% Fetal bovine serum (FBS: Life technologies cat #16000044) in a humidified incubator with 5% CO2 at 37 °C. Around 2 ×103 cells were seeded in each well of a 24-well cell culture plate and were allowed to adhere and grow for 24 h. The Jurkat cells grow in suspension so they were cultured and allowed to grow overnight before the addition of compounds. The compounds in serial dilutions (1, 3, 15, 45, 100, and 300 μM) were added after 24 h of culture and the cells were cultured for another 24 h at 37 °C. The cell viability was determined in each experiment using MTT 1-(4,5-dimethylthiazol-2-yl)-3,5-diphenylformazan colorimetric assay. Briefly, the treated or untreated cells were trypsenized and centrifuged, and the resulting pellet was re-suspended in 100 μL of DMEM serum-free medium and incubated at 37 °C for 2 h. After incubation, 20 μL of MTT solution (5 mg/mL in PBS: Sigma Aldrich cat #M2003) was added to each well and further incubated for 2 h. The plate was centrifuged at 40,000 rpm for 10 min then the medium was removed from each well and 2-propanol containing 0.04 M HCl was added to dissolve the formazan produced in the cells. The optical density of the formazan product in solution was measured with a microplate reader at 540 nm. The experiment was conducted in triplicate. Data were calculated as percent of cell viability by the following formula:
| % cell viability = (Mean absorbance in test wells/Mean absorbance in control wells) × 100 | (1) |
The IC50 values were calculated from the means of six different concentrations by Bio Data Fit 1.02 using the software Bio Tool Kit (Version 300; Chang Bioscience Inc., Castro Valley, CA, USA) and online program http://ic50.tk/index.html.
3.3.2. Statistics
All data were analyzed using Origin (Version 6.1052; Origin Lab Corp Northampton, MA, USA). One-way ANOVA analysis of variance and student T TEST was used to compare different experimental groups, and data were considered statistically significant for p values less than 0.05.
4. Conclusions
In conclusion, the screening results from human cancer and normal cell lines suggested that isatin derivatives were remarkably influenced by various substituents on the isatin ring and at the N-terminal of the hydrazide-hydrazone moiety. The data revealed that replacement of N-hydrogen at position 1 of the isatin moiety 3a by methyl and benzyl of the targeted compound (3e and 3f) noticeably enhanced the activity against the selected two cell lines, HepG2 and Jurkat, for compound 3f. The presence of the valproic acid moiety as hydrazide-hydrazone derivatives 4a make the compound more targeted towards Jurakt cells in vitro. Introducing the chlorine atom to the molecule 4c, meanwhile, increased the reactivity more significantly than with 4a and 3a towards the three cell lines HepG2, Jurkat, and HEK293. Furthermore, the methyl group in the same analogous compound 4e increased the reactivity towards the two cell lines HepG2 and Jurkat more than the benzyl group 4f. The rest of the compounds had no effect on the three cell lines. Further studies to assess the effect of more derivatives on various cancer cell biomarkers are currently underway in our lab.
Acknowledgments
The authors extend their sincere appreciation to the Deanship of Scientific Research at King Saud University for funding this work through research group project no. RGP-234 (Saudi Arabia).
Supplementary Materials
Supplementary materials can be accessed at: http://www.mdpi.com/1420-3049/20/08/14638/s1.
Author Contributions
Ayman El-Faham and Sherine Khattab carried out the synthesis and designed the proposed methods and analyzed the data statistically together. Hazem Ghabbour and Hoong-Kun Fun carried out X-ray method and the characterization; Muhammad Farooq, Nael Abutaha, and Mohammad Wadaan carried out all the biological activities studies. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds 4 a–h are available from the authors.
References
- 1.Olayinka O.A., Craig A.O., Obinna C.N., David A.A. Microwave assisted synthesis and antimicrobial activity of 2-quinoxalinone-3-hydrazone derivatives. Bioorg. Med. Chem. 2010;18:214–221. doi: 10.1016/j.bmc.2009.10.064. [DOI] [PubMed] [Google Scholar]
- 2.Rollas S., Gulerman N., Edeniz H. Synthesis and antimicrobial activity of some new hydrazones of 4-fluorobenzoic acid hydrazide and 3-acetyl-2,5-disubstituted-1,3,4-oxadiazolines. Farmaco Prat. 2002;57:171–174. doi: 10.1016/S0014-827X(01)01192-2. [DOI] [PubMed] [Google Scholar]
- 3.Cukurovali A., Yilmaz B., Gur S., Kazaz C. Synthesis, antibacterial and antifungal activity of some new thiazolylhydrazone derivatives containing 3-substituted cyclobutane ring. Eur. J. Med. Chem. 2006;41:201–207. doi: 10.1016/j.ejmech.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 4.Capilla J., Serena C., Javier F., Ortoneda T., Guarro J. Efficacy of voriconazole in treatment of systemic scedosporiosis in neutropenic mice. Antimicrob. Agents Chemother. 2003;47:3976–3978. doi: 10.1128/AAC.47.12.3976-3978.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mamolo M.G., Falagiani V., Zampieri D., Vio U., Banfi E., Scialino G. Synthesis and antimycobacterial activity of (3,4-diaryl-3H-thiazol-2-ylidene)-hydrazide derivatives. Farmaco Prat. 2003;58:631–637. doi: 10.1016/S0014-827X(03)00103-4. [DOI] [PubMed] [Google Scholar]
- 6.Govindasami1 T., Pandey A., Palanivelu N., Pandey1 A. Synthesis, characterization and antibacterial activity of biologically important vanillin related hydrazone derivatives. Int. J. Org. Chem. 2011;1:71–77. doi: 10.4236/ijoc.2011.13012. [DOI] [Google Scholar]
- 7.Dimmock J.R., Vasishtha S.C., Stables J.P. Anticonvulsant properties of various acetylhydrazones, oxamoylhydrazones and semicarbazones derived from aromatic and unsaturated carbonyl compounds. Eur. J. Med. Chem. 2000;35:241–248. doi: 10.1016/S0223-5234(00)00123-9. [DOI] [PubMed] [Google Scholar]
- 8.Lima P.C., Lima L.M., Silva K.C., Leda P.H., Miranda A.L.P., Fraga C.A.M., Barreiro E.J. Synthesis and analgesic activity of novel N-acylarylhydrazones and isosters, derived from natural safrole. Eur. J. Med. Chem. 2000;35:187–203. doi: 10.1016/S0223-5234(00)00120-3. [DOI] [PubMed] [Google Scholar]
- 9.Salgin G.U., Gokham K.N., Gostal O., Koysal Y., Kilici E., Isik S., Aktay G., Ozalp M. 1-Acylthiose-micarbazides, 1,2,4-triazole-5(4H)-thiones, 1,3,4-thia-diazoles and hydrazones containing 5-methyl-2-benzoxazolinones: Synthesis, analgesic-anti-inflammatory and antimicrobial activities. Bioorg. Med. Chem. 2007;15:5738–5751. doi: 10.1016/j.bmc.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 10.Silva G.A., Costa L.M.M., Brito F.C.B., Miranda A.L.P., Barreiro E.J., Fraga C.A.M. New class of potent antinociceptive and antiplatelet 10H-phenothiazine-1-acylhydrazone derivatives. Bioorg. Med. Chem. 2004;12:3149–3158. doi: 10.1016/j.bmc.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 11.Imramovsky A., Polanc S., Vinsova J., Kocevar M., Jampitek J., Reckova Z., Kaustova J.A. A new modification of anti-tubercular active molecules. Bioorg. Med. Chem. 2007;15:2551–2559. doi: 10.1016/j.bmc.2007.01.051. [DOI] [PubMed] [Google Scholar]
- 12.Janin Y. Antituberculosis drugs: Ten years of research. Bioorg. Med. Chem. 2007;15:2479–2513. doi: 10.1016/j.bmc.2007.01.030. [DOI] [PubMed] [Google Scholar]
- 13.Du Toit L.C., Pillay V., Danckwerts M.P. Tuberculosis chemotherapy: Current drug delivery approaches. Respir. Res. 2006;7:118–136. doi: 10.1186/1465-9921-7-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Savini L., Chiasserini L., Travagli V., Pellerano C., Novellino E., Consentino S., Pisano M.B. New α-(N)-heterocyclichydrazones: Evaluation of anti-cancer, anti-HIV and antimicrobial activity. Eur. J. Med. Chem. 2004;39:113–122. doi: 10.1016/j.ejmech.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 15.El-Hawash A.M., Abdel W.A.E., El-Dewellawy M.A. Cyanoacetic acid hydrazones of 3-(and 4-) acetyl-pyridine and some derived ring systems as potential antitumor and anti-HCV agents. Arch. Pharm. 2006;339:14–23. doi: 10.1002/ardp.200500161. [DOI] [PubMed] [Google Scholar]
- 16.Wahba W., Nahed W., El-Sayed N.E., Mohareb R.M. Synthesis and anti-tumor evaluation of novel hydrazide and hydrazide-hydrazone derivatives. Acta Pharm. 2013;63:45–57. doi: 10.2478/acph-2013-0004. [DOI] [PubMed] [Google Scholar]
- 17.Mohareb R.M., Fleita D.H., Sakka O.K. Novel synthesis of hydrazide-hydrazone derivatives and their utilization in the synthesis of coumarin, pyridine, thiazole and thiophene derivatives with antitumor activity. Molecules. 2011;16:16–27. doi: 10.3390/molecules16010016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Terzioglu N., Gursoy A. Synthesis and anti-cancer evaluation of some new hydrazone derivatives of 2,6-dimethylimidazo[2,1-b][1,3,4]thiadiazole-5-carbohydrazide. Eur. J. Med. Chem. 2003;38:781–786. doi: 10.1016/S0223-5234(03)00138-7. [DOI] [PubMed] [Google Scholar]
- 19.Boga C., Fiume L., Baglioni M., Bertucci C., Farina C., Kratz F., Manerba M., Naldiand M., Stefano G. Characterization of the conjugate of the (6-maleimidocaproyl) hydrazone derivative of doxorubicin with lactosaminated human albumin by 13C-NMR spectroscopy. Eur. J. Pharm. Sci. 2009;38:262–269. doi: 10.1016/j.ejps.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 20.El-Sabbagh O.I., Rady H.M. Synthesis of new acridines and hydrazones derived from cyclic α-diketone for cytotoxic and antiviral evaluation. Eur. J. Med. Chem. 2009;44:3680–3686. doi: 10.1016/j.ejmech.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 21.Zhang H.Z., Drewe J., Tseng B., Kasibhatla S., Cai S.X. Discovery and SAR of indole-2-carboxylic acid benzylidene-hydrazides as a new series of potent apoptosis inducers using a cell-based HTS assay. Bioorg. Med. Chem. 2004;12:3649–3655. doi: 10.1016/j.bmc.2004.04.017. [DOI] [PubMed] [Google Scholar]
- 22.Vicini P., Incerti M.I., Collaand P.L., Loddo R. Anti HIV evaluation of benzo[d]isothiazole hydrazones. Eur. J. Med. Chem. 2009;44:1801–1807. doi: 10.1016/j.ejmech.2008.05.030. [DOI] [PubMed] [Google Scholar]
- 23.Rahman V.M., Mukhtar S., Ansari W.H., Lemiere G. Synthesis, stereochemistry and biological activity of some novel long alkyl chain substituted thiazolidin-4-ones and thiazan-4-one from 10-undecenoic acid hydrazide. Eur. J. Med. Chem. 2005;40:173–184. doi: 10.1016/j.ejmech.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 24.Kaushik D., Khan S.A., Chawla G., Kumar S. N′-[(5-chloro-3-methyl-1-phenyl-1H-pyrazol-4-yl)methylene] 2/4-substituted hydrazides: Synthesis and anticonvulsant activity. Eur. J. Med. Chem. 2010;45:3943–3949. doi: 10.1016/j.ejmech.2010.05.049. [DOI] [PubMed] [Google Scholar]
- 25.Xia Y.L., Chuan-Dong F., Zhao B.X., Zhao J., Shin D.S., Miaom J.Y. Synthesis and structure–Activity relationships of novel 1-arylmethyl-3-aryl-1H-pyrazole-5-carbohydrazide hydrazone derivatives as potential agents against A549 lung cancer cells. Eur. J. Med. Chem. 2008;43:2347–2353. doi: 10.1016/j.ejmech.2008.01.021. [DOI] [PubMed] [Google Scholar]
- 26.Menendez C., Chollet A., Rodriguez F., Inard C., Pasca M.R., Lherbet C., Baltas M. Chemical synthesis and biological evaluation of triazole derivatives as inhibitors of InhA and antituberculosis agents. Eur. J. Med. Chem. 2012;52:275–283. doi: 10.1016/j.ejmech.2012.03.029. [DOI] [PubMed] [Google Scholar]
- 27.Nerkar A.G., Saxena A.K., Ghone S.A., Thaker A.K. In Silico Screening, synthesis and in vitro evaluation of some quinazolinone and pyridine derivatives as dihydrofolate reductase inhibitors for anti-cancer activity. J. Chem. 2009;6:97–102. [Google Scholar]
- 28.Chu W., Zhang J., Zeng C., Rothfuss J., Tu Z., Chu Y., Reichert D.E., Welch M.J., Mach R.H. N-Benzylisation sulfonamide analogues as potent caspase-3 inhibitors: Synthesis, in vitro activity, and molecular modeling studies. J. Med. Chem. 2005;48:7637–7647. doi: 10.1021/jm0506625. [DOI] [PubMed] [Google Scholar]
- 29.Chu W., Rothfuss J., Chu Y., Zhou D., Mach R.H. Synthesis and in vitro evaluation of sulfonamide isatin Micheal acceptors as small molecule inhibitors of caspase-6. J. Med. Chem. 2009;52:2188–2191. doi: 10.1021/jm900135r. [DOI] [PubMed] [Google Scholar]
- 30.Chohan Z.H., Pervez H., Rauf A., Khan K.M., Supuran C.T. Isatin-derived antibacterial and antifungal compounds and their transition metal complexes. J. Enzyme Inhib. Med. Chem. 2004;19:417–423. doi: 10.1080/14756360410001710383. [DOI] [PubMed] [Google Scholar]
- 31.Pirrung M.C., Pansare S.V., das Sarma K., Keith K.A., Kern E.R. Combinatorial optimization of isatin-β-thiosemicarbazones as anti-poxvirus agents. J. Med. Chem. 2005;48:3045–3050. doi: 10.1021/jm049147h. [DOI] [PubMed] [Google Scholar]
- 32.Konkel M.J., Lagu B., Boteju L.W., Jimenez H., Noble S., Walker M.W., Chandrasena G., Blackburn T.P., Nikam S.S., Wright J.L., et al. 3-Arylimino-2-indolones are potent and selective galanin GAL3 receptor antagonists. J. Med. Chem. 2006;49:3757–3758. doi: 10.1021/jm060001n. [DOI] [PubMed] [Google Scholar]
- 33.Natarajan A., Fan Y.H., Chen H., Guo Y., Iysere J., Harbinski F., Christ H., Aktas W.J., Halperin Y.A. 3,3-Diaryl-1,3-dihydroindol-2-ones as antiproliferatives mediated by translation initiation inhibition. J. Med. Chem. 2004;47:1882–1885. doi: 10.1021/jm0499716. [DOI] [PubMed] [Google Scholar]
- 34.Matheus M.E., Violante F.D.A., Garden S.J., Pinto A.C., Fernandes P.D. Isatins inhibit cyclooxygenase-2- and inducible nitric oxide synthesis in a mouse macrophage cell line. Eur. J. Pharmacol. 2007;556:300–306. doi: 10.1016/j.ejphar.2006.10.057. [DOI] [PubMed] [Google Scholar]
- 35.Zhou L., Liu Y., Zhang W., Wei P., Huang C., Pei J., Yuan Y., Lai L. Isatin compounds as noncovalent SARS coronavirus 3C-like protease inhibitors. J. Med. Chem. 2006;49:3440–3443. doi: 10.1021/jm0602357. [DOI] [PubMed] [Google Scholar]
- 36.Jarrahpour A., Khalili D., de Clercq E., Salmi C., Brunel J.M. Synthesis, antibacterial, antifungal and antiviral activity evaluation of some new bis-Schiff bases of isatin and their derivatives. Molecules. 2007;12:1720–1730. doi: 10.3390/12081720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharaf O.A. Some pharmacological activities of new substituted pyrolloindoles, Indolothiazepine and azole derivatives. Bull. Fac. Pharm. 1997;35:79–82. [Google Scholar]
- 38.Verma M., Pandeya S.N., Singh K.N., Stables J.P. Anticonvulsant activity of Schiff bases of Isatin derivatives. Acta Pharm. 2004;54:49–56. [PubMed] [Google Scholar]
- 39.Bersudsky Y., Applebaum J., Gaiduk Y., Sharony L., Mishory A., Podberezsky A., Agam G., Belmaker R.H. Valnoctamide as a valproate substitute with low teratogenic potential in mania: A double-blind, controlled, add-on clinical trial. Bipolar Disord. 2010;12:376–382. doi: 10.1111/j.1399-5618.2010.00828.x. [DOI] [PubMed] [Google Scholar]
- 40.Bialer M. New antiepileptic drugs that are second generation to existing antiepileptic drugs. Expert Opin. Investig. Drugs. 2006;15:637–647. doi: 10.1517/13543784.15.6.637. [DOI] [PubMed] [Google Scholar]
- 41.Bialer M., Yagen B. Valproic acid: Second generation. Neurotherapeutics. 2007;4:130–137. doi: 10.1016/j.nurt.2006.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Michaelis M., Michaelis U.R., Fleming I., Suhan T., Cinatl J., Blaheta R.A., Hoffmann K., Kotchetkov R., Busse R., Nau H., et al. Valproic acid inhibits angiogenesis in vitro and in vivo. Mol. Pharm. 2004;65:520–527. doi: 10.1124/mol.65.3.520. [DOI] [PubMed] [Google Scholar]
- 43.Osuka S., Takano S., Watanabe S., Yamamoto T., Matsumura A. Valproic acid inhibits angiogenesis in vitro and glioma angiogenesis in vivo in the brain. Neurol. Med. Chir. (Tokyo) 2012;52:186–193. doi: 10.2176/nmc.52.186. [DOI] [PubMed] [Google Scholar]
- 44.Zgouras D., Becker U., Loitsch S., Stein J. Modulation of angiogenesis-related protein synthesis by valproic acid. Biochem. Biophys. Res. Commun. 2004;316:693–697. doi: 10.1016/j.bbrc.2004.02.105. [DOI] [PubMed] [Google Scholar]
- 45.Kitazoe K., Abe M., Hiasa M., Oda A., Amou H., Harada T., Nakano A., Takeuchi K., Hashimoto T., Ozaki S., et al. Valproic acid exerts anti-tumor as well as anti-angiogenic effects on myeloma. Int. J. Hematol. 2009;89:45–57. doi: 10.1007/s12185-008-0226-9. [DOI] [PubMed] [Google Scholar]
- 46.Farooq M., Sulochana K.N., Pan X., To J., Sheng D., Gong Z., Ge R. Histone deacetylase 3 (hdac3) is specifically required for liver development in zebrafish. J. Dev. Biol. 2008;317:336–353. doi: 10.1016/j.ydbio.2008.02.034. [DOI] [PubMed] [Google Scholar]
- 47.Gottlicher M., Minucci S., Zhu P., Kramer O.H., Schimpf A., Giavara S., Sleeman J.P., lo Coco F., Nervi C., Pelicci P.G., et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. Eur. Mol. Biol. Org. J. 2001;20:6969–6978. doi: 10.1093/emboj/20.24.6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kuendgen A., Gattermann N. Valproic acid for the treatment of myeloid malignancies. Cancer. 2007;110:943–954. doi: 10.1002/cncr.22891. [DOI] [PubMed] [Google Scholar]
- 49.Farooq M., El-Faham A., Ahmad M., Sami A.S., Wadaan M.A. Teratogenic profile of valproic acid and newly synthesized derivatives in zebrafish embryos. Int. J. Agric. Biol. 2012;14:894–900. [Google Scholar]
- 50.El-Faham A., Farooq M., Khattab S.N., Elkayal A.M., Ibrahim M.F., Abutaha N., Wadaan M.A.M., Hamed E.A. Synthesis and biological activity of Schiff base series of valproyl, N-valproyl glycinyl, and N-valproyl-4-aminobenzoyl hydrazide derivatives. Chem. Pharm. Bull. 2014;62:591–599. doi: 10.1248/cpb.c14-00143. [DOI] [PubMed] [Google Scholar]
- 51.Farooq M., El-Faham A., Khattab S.N., Elkayal A.M., Ibrahim M.F., Abutaha N., Baabbad A., Wadaan M.A.M., Hamed E.A. Biological screening of novel derivatives of valproic acid for anti-cancer and antiangiogenic properties. Asian Pac. J. Cancer Prev. 2014;15:7785–7792. doi: 10.7314/APJCP.2014.15.18.7785. [DOI] [PubMed] [Google Scholar]
- 52.Allen F.H., Kennard O., Watson D.G., Brammer L., Orpen A.G., Taylor R. Tables of bond lengths determined by X-ray and neutron diffraction. Part 1. Bond lengths in organic compounds. J. Chem. Soc. Perkin Trans. 1987;2:S1–S19. doi: 10.1039/p298700000s1. [DOI] [Google Scholar]
- 53.El-Faham A., Khattab S.N., Ghabbour H.A., Fun H.K., Siddiqui M.R. Microwave irradiation: Synthesis and characterization of α-ketoamide and bis (α-ketoamide) derivatives via the ring opening of N-acetylisatin. Chem. Cent. J. 2014;8:1–10. doi: 10.1186/1752-153X-8-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.El-Faham A., Elzatahry A.A., Al Othman Z.A., Elsayed E.A. Facile method for the synthesis of silver nanoparticles using 3-hydrazino-isatin derivatives in aqueous methanol and their antibacterial activity. Int. J. Nanomedicine. 2014;9:1167–1174. doi: 10.2147/IJN.S58571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sheldrick G.M. A short history of SHELX. Acta Crystallogr. A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 56.Bruker, APEX2, SAINT, SADABS. Bruker AXS Inc.; Madison, WI, USA: 2009. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.