

Abstract
Key points
Maternal high‐fat diet (MHF) consumption led to metabolic and liver disorders in male offspring, which are associated with reduced sirtuin (SIRT)1 expression and activity in the offspring liver
SIRT1 overexpression in MHF offspring reduced their body weight and adiposity and normalized lipid metabolic markers in epididymal and retroperitoneal adipose tissues
SIRT1 overexpression in MHF offspring improved glucose tolerance, as well as systemic and hepatic insulin sensitivity
SIRT1 overexpression ameliorated MHF‐induced lipogenesis, oxidative stress and fibrogenesis in the liver of offspring.
Abstract
Maternal obesity can increase the risk of metabolic disorders in the offspring. However, the underlying mechanism responsible for this is not clearly understood. Previous evidence implied that sirtuin (SIRT)1, a potent regulator of energy metabolism and stress responses, may play an important role. In the present study, we have shown, in C57BL/6 mice, that maternal high‐fat diet (HFD) consumption can induce a pre‐diabetic and non‐alcoholic fatty liver disease phenotype in the offspring, associated with reduced SIRT1 expression in the hypothalamus, white adipose tissues (WAT) and liver. Importantly, the overexpression of SIRT1 in these offspring significantly attenuated the excessive accumulation of epididymal (Epi) white adipose tissue (WAT) and retroperitoneal (Rp)WAT (P < 0.001), glucose intolerance and insulin resistance (both P < 0.05) at weaning age. These changes were associated with the suppression of peroxisome proliferator‐activated receptor gamma (PPAR)γ (P < 0.01), PPARγ‐coactivator 1‐alpha (P < 0.05) and sterol regulatory element‐binding protein‐1c in EpiWAT (P < 0.01), whereas there was increased expression of PPARγ in RpWAT (P < 0.05). In the liver, PPARγ mRNA expression, as well as Akt protein expression and activity, were increased (P < 0.05), whereas fatty acid synthase and carbohydrate response element binding protein were downregulated (P < 0.05), supporting increased insulin sensitivity and reduced lipogenesis in the liver. In addition, hepatic expression of endogenous anti‐oxidants, including glutathione peroxidase 1 and catalase, was increased (P < 0.01 and P < 0.05 respectively), whereas collagen and fibronectin deposition was suppressed (P < 0.01). Collectively, the present study provides direct evidence of the mechanistic significance of SIRT1 in maternal HFD‐induced metabolic dysfunction in offspring and suggests that SIRT1 is a promising target for fetal reprogramming.
Keywords: obesity, sirtuin, developmental programming, metabolism, liver
Key points
Maternal high‐fat diet (MHF) consumption led to metabolic and liver disorders in male offspring, which are associated with reduced sirtuin (SIRT)1 expression and activity in the offspring liver
SIRT1 overexpression in MHF offspring reduced their body weight and adiposity and normalized lipid metabolic markers in epididymal and retroperitoneal adipose tissues
SIRT1 overexpression in MHF offspring improved glucose tolerance, as well as systemic and hepatic insulin sensitivity
SIRT1 overexpression ameliorated MHF‐induced lipogenesis, oxidative stress and fibrogenesis in the liver of offspring.
Introduction
The term of developmental origin of health and diseases (DOHaD) or fetal programming refers to the effects of parental lifestyle and health conditions on subsequent generations (Armitage et al. 2008). Maternal obesity and high‐fat diet (HFD) consumption in particular have been shown to predispose offspring to obesity and related metabolic disorders (Howie et al. 2009; Oben et al. 2010; Glastras et al. 2015; Saben et al. 2016). Importantly, clinical trials of lifestyle intervention during pregnancy only marginally improve postnatal outcomes, partially as a result of poor compliance and late engagement in these programmes (Catalano, 2015). Clearly, understanding all of the contributing factors and transmission pathways in maternal obesity‐related DOHaD is crucial to the development of therapeutic strategies. Recent studies suggest that maternal obesity during pregnancy can cause the transmission of excessive nutrients to the fetus, which further induces oxidative stress and inflammatory responses, leading to epigenetic modifications in fetal tissues to reprogram energy metabolism (Heerwagen et al. 2010).
A particular protein has been suggested to play a central role in this multidimensional mechanism, namely sirtuin (SIRT)1 (Nguyen et al. 2016). As a histone deacetylase, SIRT1 has a direct regulatory effect on chromatin tertiary structure, thus playing an important role in DNA repair and replication, as well as genetic and epigenetic regulation. SIRT1 activity strictly depends on the cellular availability of NAD+, which fluctuates in accordancewith the circadian rhythm and energy status (Houtkooper et al. 2010). These characteristics make SIRT1 a potent regulator of lifespan, metabolism and stress responses (Haigis & Sinclair, 2010; Houtkooper et al. 2012). Both ageing and obesity negatively affect SIRT1 expression and activity (Pedersen et al. 2008; dos Santos Costa et al. 2010; Mariani et al. 2015). Conversely, overexpression of SIRT1 mimics caloric restriction (Wood et al. 2004), leading to a longer lifespan and resistance to obesity‐related disorders in various animal models (Wood et al. 2004; Milne et al. 2007; Pfluger et al. 2008). With respect to DOHaD, several studies have reported reduced SIRT1 expression/activity in the fetus and neonatal tissues as a result of maternal HFD feeding (MHF) (Suter et al. 2012; Nguyen et al. 2017).
In the present study, we induced a systemic overexpression of SIRT1 in the offspring to confirm the causative role of SIRT1 regulation in maternal obesity‐related DOHaD. The results of the present study suggest that SIRT1 is a promising target for fetal reprogramming of metabolic disorders as a result of maternal obesity.
Methods
Animals
The present study was approved by the Animal Care and Ethics Committee of the University of Sydney (RESP/15/22). All methods were performed in accordance with the relevant guidelines and regulations in the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes. Eight‐week old female wild‐type C57BL/6 mice (WT) were fed a HFD (20 kJ g–1, 43.5% calorie as fat; Specialty Feed, Glen Forest, WA, Australia) (MHF, n = 21) or standard rodent chow (11 kJ g–1, 14% calorie as fat; Gordon's Speciality Stockfeeds, Yanderra, NSW, Australia) (MC, n = 13) for 6 weeks before mating, throughout gestation and lactation (Glastras et al. 2015). To test the hypothesis that SIRT1 expression in the offspring per se can attenuate maternal obesity‐induced metabolic programming, these female mice were mated with hemizygous transgenic sires (Tg) to produce both WT and Tg offspring without maternal genotypic modification (resulting in the offspring groups: MC‐WT, n = 21; MC‐Tg, n = 8; MHF‐WT, n = 26; MHF‐Tg, n = 11). The strain of male breeders was B6.Cg‐Col1a1tm1(CAG‐Sirt1)Dsin/Mmjax, derived from C57BL/6. The original Tg colony was a generous gift from Dr Lindsay Wu (University of New South Wales, Sydney, NSW, Australia).
As we have previously shown sex‐specific downregulation of SIRT1 by MHF in male offspring (Nguyen et al. 2017), only male offspring were examined in the present study. To limit the difference in milk competition between the litters (Chen et al. 2008), the newborn litters were adjusted to four to six pups per litter. Because HFD‐fed dams tend to cannibalize their pups, the number of pups per litter was subject to how many male mice survived until weaning and how many of these were genotyped to be wild‐type and transgenic. The proportion of Tg offspring was ∼30% of the litter size and there was typically one Tg male mouse per litter. The offspring were genotyped at postnatal (P)14 in accordance to the Jackson Laboratory Genotyping Protocol for B6.Cg‐Col1a1tm1(CAG‐Sirt1)Dsin/Mmjax strain using crude tail DNA extracted with DirectPCR Lysis Reagent (Mouse Tail) (Viagen Biotech, Los Angeles, CA, USA).
At weaning (P20), all pups were deeply anaesthetized with 3% isoflurane and killed upon cardiac puncture for blood collection after 5 h of fasting. PBS (1%) was used for whole body perfusion. Tissues were snap frozen or embedded in optimal cutting temperature (OCT) compound and stored at −80°C or fixed in neutral buffered formalin (10%) for ∼36 h for later analyses.
Hypothalamus dissection
The first coronal cut was made at the rostral edge of the hypothalamus, using the sulcus as a guide, at Bregma +0.5 mm. A second coronal cut was made 3.2 mm caudal to the second cut. This section (Bregma +0.5 mm to –2.7 mm) contains the hypothalamus. It was then placed on its rostral surface and the hypothalamic area dissected by cutting at the level of the hypothalamic sulcus. The hypothalamus was then removed by cutting just above the top of the third ventricle.
Intraperitoneal glucose tolerance test (IPGTT)
At P18, the animals were weighed and fasted for 5 h prior to IPGTT (Chen et al. 2008) and then d‐glucose was injected (2 g kg–1 i.p.). Tail blood glucose level was recorded prior to glucose injection, and then at 15, 30, 60 and 90 min after injection using a glucometer (Accu‐Chek® glucose meter; Roche Diagnostics, Basel, Switzerland). The area under the curve was calculated for each animal.The Quantitative Insulin Sensitivity Check Index (QUICKI) was calculated as reported previously (Chen et al. 2005; Cacho et al. 2008).
Milk intake estimation
At P19, offspring were separated from their dams and fasted for 5 h. They were weighed before returning to their dams. After 2 h of feeding, the offspring were weighed again. The difference in the body weight during the 2 h period was used for milk intake estimation. No access to solid food was provided during the lactation phase to ensure no chow/HFD consumption by the pups. The same method has been described previously (Jara‐Almonte & White, 1972; Del Prado et al. 1997).
Protein and lipid extraction from tissues
The tissues were homogenized in Triton X‐100 lysis buffer (pH 7.4, 150 mmNaOH, 50 mmTris‐HCl, 1% Triton X‐100; Roche protease inhibitor) using TissueRuptor (Qiagen, Hilden, Germany). Lipid and protein were extracted and quantified in accordancewith previously reported protocols (Nguyen et al. 2017) using Roche triglyceride reagent GPO‐PAP (Roche Life Science, North Ryde, NSW, Australia) and the Pierce BCA Protein Assay Kit (Thermo Scientific, Scoresby, VIC, Australia) in accordancewith the manufacturer's instructions. Lipid concentrations were normalized to the weight of tissue homogenized. Protein concentrations were standardized to 5 μg μL–1.
Quantitative RT‐PCR
The Minimum Information for Publication of Quantitative Real‐Time PCR Experiments (MIQE) guidelines were followed (Bustin et al. 2009). Total RNA was extracted from liver tissues using an RNeasy Plus Mini Kit (Qiagen Pty Ltd, Valancia, CA, USA) in accordancewith the manufacturer's instructions, whereas RNA in fat tissues and the hypothalamus was extracted using Trizol Reagent (Sigma‐Aldrich, St Louis, MO, USA). The purified total RNA was used as a template to generate cDNA using the First Strand cDNA Synthesis Kit (Roche Life Science). The amplicons of target genes were amplified with SYBR Green probes. Primer sequences have been reported in ourprevious study (Nguyen et al. 2018) and are summarized in Table 1. Before acquiring the actual data, all of the new primers were tested for amplification efficiency (90–110%) and specificity (single peak in dissociation curve analysis). The final concentration for all primers in a quantitative PCR reaction was 200 nm. Several commonly used housekeeping genes in the literature, including 18s, α‐tubulin and β‐actin, were tested. Β‐actin showed the least variation in mRNA expression among the groups. Therefore, gene expression was standardized to β‐actin mRNA and log‐transformed.
Table 1.
RT‐PCR primers sequences
| Number | Gene | Forward primer sequence | Reverse primer sequence |
|---|---|---|---|
| 1 | IL1B | GGATGATGATGATAACCTGC | CATGGAGAATATCACTTGTTGG |
| 2 | Leptin | GAGACCCCTGTGTCGGTTC | GACTGCGTGTGTGAAATGTC |
| 3 | MC4R | GACGGAGGATGCTATGAG | GCAGGTTCTTGTTCTTGGC |
| 4 | MCP‐1 | GCCTGCTGTTCACAGTTGC | CAGGTGAGTGGGGCGTTA |
| 5 | NPY | GGCTGTGTGGACTGACCCT | GATGTAGTGTCGCAGAGCGG |
| 6 | NPY1R | TGATCTCCACCTGCGTCAAC | ATGGCTATGGTCTCGTAGTC |
| 7 | Ob‐Rb | CCAGGTGAGGAGCAAGAG | CTGCACAGTGCTTCCCAC |
| 8 | POMC | GAGATTCTGCTACAGTCGCTC | TTGATGATGGCGTTCTTGAA |
| 9 | TGFBR1 | CAGCTCCTCATCGTGTTGG | CAGAGGTGGCAGAAACACTG |
| 10 | TGFBR2 | TCCATCTGTGAGAAGCCACA | GGGTCATGGCAAACTGTCTC |
| 11 | GLUT1 | AGCCTGCAAACTCACTGCTC | CCTACCCTCAATCCCACAAGC |
| 12 | GLUT2 | TCGCCCTCTGCTTCCAGTAC | GAACACGTAAGGCCAAGGA |
Immunoblotting
The same amount of protein (20 μg) was electrophoresed and electroblotted onto the Hybond nitrocellulose membrane (Amersham Pharmacia Biotech, Amersham, UK), which was then incubated with a primary antibody at 4°C overnight. Antibody information and the applied concentrations are shown in Table 2. Two commonly used housekeeping genes in the literature including β‐actin and GAPDH were tested for loading normalization in a western blot. GAPDH showed less variation in expression among groups compared to β‐actin and hence was selected for the present study. Subsequently, the membrane was incubated with a horseradish peroxidase‐conjugated secondary antibodies. The immunoblots were developed by adding the Luminata Western HRP Substrates (Millipore, Burlington, MA, USA) to the membrane and imaged using ImageQuant LAS 4000 (Fujifilm, Tokyo, Japan). ImageJ (National Institutes of Health, Bethesda, MD, USA) was used for densitometric analyses.
Table 2.
Antibody information
| Target | Catalogue number | Size (kDa) | Dilution (WB) | Dilution (IHC) | Company | Address |
|---|---|---|---|---|---|---|
| GAPDH | sc‐47724 | 36 | 1:2000 | Santa Cruz Biotechnology | Santa Cruz, TX, USA | |
| SIRT1 | 07‐131 | 110 | 1:2000 | EMD Millipore | North Ryde, NSW, Australia | |
| PGC‐1α | NBP1‐04676 | 91 | 1:2000 | Novus Biologicals | Littleton, CO, USA | |
| SREBP1 | 04‐469 | 62 | 1:500 | EMD Millipore | NSW, Australia | |
| GLUT2 | 07‐1402 | 50 | 1:1000 | EMD Millipore | NSW, Australia | |
| p‐Akt (S473) | 13038 | 60 | 1:1000 | Cell Signaling | Danvers, MA, USA | |
| Akt | 9272 | 60 | 1:1000 | Cell Signaling | MA, USA | |
| p‐AMPKα (T172) | 2535 | 62 | 1:1000 | Cell Signaling | MA, USA | |
| AMPKα | 5831 | 62 | 1:1000 | Cell Signaling | MA, USA | |
| Collagen I | ab34710 | 1:750 | Abcam | Cambridge, UK | ||
| Collagen III | ab7778 | 1:750 | Abcam | Cambridge, UK |
WB, western blot.
SIRT1 activity assay
Nuclear protein was extracted in accordancewith the protocol ‘Nuclear protein extraction without the use of detergent’ (Sigma‐Aldrich, Dublin, Ireland) using a hypotonic lysis buffer (10 mmHepes, pH 7.9, with 1.5 mmMgCl2 and 10 mmKCl, 1 mmdithiothreitol), followed by centrifugation to separate nuclear and cytoplasmic fractions. The nuclear proteins were extracted using extraction buffer (20 mmHepes, pH 7.9, with 1.5 mmMgCl2, 0.42 mNaCl, 0.2 mmEDTA, 25% (v/v) glycerol, 1 mmdithiothreitol). No protease inhibitor was used in the extraction to avoid interference with SIRT1 activity measurement. SIRT1 activity was then measured using the SIRT1 activity assay kit (Abcam, Cambridge, UK)in accordance with the manufacturer's instructions.
Tissue morphology
Tissues were fixed in 10% formalin for 36 h and embedded in paraffin or frozen‐embedded in OCT solution (Tissue‐Tek; Sakura Finetek USA, Inc., Torrance, CA, USA). Paraffin sections and frozen sections were prepared at 4 and 12 μm thickness, respectively, and mounted on microscope slides (Trajan Scientific and Medical, Ringwood, VIC, Australia).
The sections were stained with haematoxylin and eosin (H&E) for general morphology. For adipocyte frequency analysis, H&E‐stained sections were analysed using a bright‐field microscope (Leica Microsystems, Wetzlar, Germany) and six random non‐overlapping fields were captured at 200× magnification. All slides were randomly coded by an independent person before analysis. Adipocyte size analysis was conducted using Adiposoft software (https://imagej.net/Adiposoft) (Galarraga et al. 2012). For collagen staining, paraffin sections were stained with Fast Green for 30 min followed by incubation in Picro‐Sirius Red (PSR) for another 30 min. Cytoplasm was stained in orange or light red while collagen was stained in dark red. For lipid droplet visualization, both H&E and Oil Red O (ORO) staining was used. In ORO staining, frozen tissues were sectioned ata thickness of 12 μm and stained with ORO (Sigma‐Aldrich, St Louis, MO, USA) for 15 min.
Immunohistochemistry (IHC)
IHC staining was performed as described previously (Nguyen et al. 2017). Antigen‐retrieval was performed at 99°C for 20 min in 0.01 m (pH 6.0) citric buffer. Endogenous peroxidase was deactivated with 3% H2O2 (Sigma‐Aldrich, Dublin, Ireland). The liver slices were then blocked (Protein Block Serum‐Free; Dako, Glostrup, Denmark) and incubated with primary antibodies overnight at 4°C, and then biotinylated with secondary anti‐rabbit IgG antibodies (Dako) and horseradish peroxidase‐conjugated streptavidin (Dako). All slides were randomly coded and then assessed by two independent investigators. Using bright field microscopy, six consecutive non‐overlapping fields from each liver section were photographed under high magnification (200×). ImageJ (National Institutes of Health) was used for estimation of the specific stained area. Briefly, colour deconvolution was performed on the image and the DAB channel was selected for analysis. The same threshold was applied to all images, above which positive staining area was measured. The whole process was automated usingthe Macro function in ImageJ. IHC score was determined by the stained area.
Statistical analysis
The data were analysed using two‐way ANOVA (two factors: maternal diet and offspring genotype) and planned pairwise comparisons (MC‐WT vs. MC‐Tg, MC‐WT vs.MHF‐WT and MHF‐WT vs.MHF‐Tg), which were performed independently of the results of the ANOVA. Because the former two comparisons were to confirm previous findings in the literature, and only the comparison between MHF‐WT and MHF‐Tg addresses the hypothesis, no correction for multiple comparison was performed. Data are presented as the mean ± SD unless otherwise specified. P< 0.05 was considered statistically significant.
Results
SIRT1 overexpression reduces body weight, fat and liver mass but not hyperlipidaemia in MHF offspring
Consistent with our previous studies, body weight was significantly increased in the MHF‐WT offspring compared to MC‐WT offspring (P< 0.001) (Table 3). The net weight and percentage of body weight of epididymal (Epi) and retroperitoneal (Rp) white adipose tissue (WAT) were also significantly higher in the MHF‐WT group (P< 0.001) (Table 3). Net liver weight was also increased (P< 0.001) proportionally to body weight. Plasma levels of non‐esterified fatty acid (NEFA) were elevated to a small extent as a result of maternal HFD consumption (two‐way ANOVA, P MHF< 0.05). Similarly, plasma triglyceride levels were increased in MHF‐WT offspring compared to the MC‐WT group (P< 0.01).
Table 3.
Anthropometric results
| MC‐WT (n = 21) (mean ± SD) | MC‐Tg (n = 8) (mean ± SD) | MHF‐WT (n = 26) (mean ± SD) | MHF‐Tg (n = 11) (mean ± SD) | Main effect | MC‐WT vs. MC‐Tg | MC‐WT vs. MHF‐WT | MHF‐WT vs. MHF‐Tg | |
|---|---|---|---|---|---|---|---|---|
| BW (g) | 7.95 ± 1.37 | 7.40 ± 0.99 | 10.44 ± 1.28 | 8.62 ± 1.85 | b, c | NS | *** | ††† |
| EpiWAT (mg) | 30.9 ± 9.1 | 15.8 ± 8.9 | 113.7 ± 31.8 | 76.8 ± 35.4 | b, c | NS | *** | ††† |
| EpiWAT (%BW) | 0.39 ± 0.07 | 0.21 ± 0.11 | 1.01 ± 0.23 | 0.83 ± 0.31 | b, c | NS | *** | † |
| RpWAT (mg) | 7.8 ± 2.1 | 4.2 ± 2.3 | 29.2 ± 7.8 | 21.4 ± 11.6 | b, c | NS | *** | †† |
| RpWAT (%BW) | 0.10 ± 0.03 | 0.05 ± 0.03 | 0.27 ± 0.08 | 0.21 ± 0.09 | b, c | NS | *** | †† |
| Liver (mg) | 425.4 ± 91.5 | 305.5 ± 70.1 | 597.9 ± 103.8 | 376.2 ± 113.4 | b, c | * | *** | ††† |
| Liver (%) | 4.74 ± 0.79 | 4.04 ± 0.75 | 5.16 ± 0.61 | 4.38 ± 0.70 | c | * | NS | † |
| NEFA (mm) | 0.36 ± 0.06 | 0.35 ± 0.08 | 0.44 ± 0.09 | 0.41 ± 0.09 | b | NS | NS | NS |
| Triglyceride (g/L) | 0.51 ± 0.16 | 0.52 ± 0.12 | 0.90 ± 0.34 | 0.79 ± 0.10 | b | NS | ** | NS |
BW, body weight. N = 6 for NEFA and triglyceride measurement. Compared to MC‐WT: * P < 0.05, ** P < 0.01, *** P < 0.001; vs. MHF‐WT: †P < 0.05, ††P < 0.01, †††P < 0.001, NS, non‐significant. a P interaction < 0.05. b P MHF < 0.05. c P SIRT1‐tg < 0.05.
MHF‐Tg offspring had a significantly lower body weight compared to MHF‐WT offspring (P< 0.001) (Table 3). The net weight and percentage of body weight of EpiWAT and RpWAT in these offspring was also significantly lower than in MHF‐WT offspring (P< 0.001, P< 0.05, P< 0.01 and P< 0.01, respectively). Liver weight (net and %) was also significantly lower in Tg offspring born to either chow‐ or HFD‐fed dams (all P< 0.05). Despite reduced adiposity, no changes were found in NEFA and triglyceride levels as a result of SIRT1 overexpression in the offspring (Table 3).
SIRT1 overexpression does not change milk intake in MHF offspring
As shown in Fig. 1 A and B, MHF‐WT offspring had a higher milk intake (P< 0.001 vs. MC‐WT) and plasma leptin level (P< 0.01 vs. MC‐WT) compared to the control group. Concomitantly, there were significant reductions of hypothalamic mRNA expression of SIRT1 (P< 0.001) (Fig. 1 C) and leptin receptor (Ob‐Rb) (P< 0.05 vs. MC‐WT) (Fig. 1 D). In addition, mRNA expression of the orexigenic neuropeptide Y (NPY) was significantly decreased (P< 0.05) in MHF‐WT animals, probably as result of the appetite suppressing effect of leptin. Maternal HFD consumption did not affect mRNA expression of signal transducer and activator of transcription (STAT)3, a downstream marker in the leptin signalling pathway, as well as neuropeptide Y1 receptor (NPY‐1R), pro‐opiomelanocortin (POMC) and melanocortin 4 receptor (MC4R).
Figure 1. The effects of perinatal SIRT1 overexpression on estimated milk intake and appetite regulators in MHF offspring.
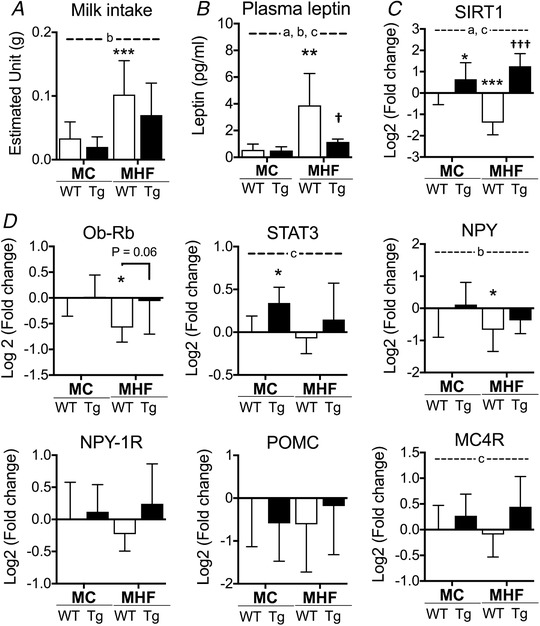
A, estimated appetite (n = 10, 5, 13 and 5 respectively). B, plasma leptin levels. mRNA expression of (C) SIRT1 and (D) appetite regulators (n = 8). Compared to MC‐WT: * P < 0.05, ** P < 0.01, *** P < 0.001; vs. MHF‐WT: †P < 0.05, †††P < 0.001. aTwo‐way ANOVA, P interaction < 0.05. bTwo‐way ANOVA, P MHF < 0.05. cTwo‐way ANOVA, P SIRT1‐tg < 0.05.
Plasma levels of leptin were significantly reduced in MHF‐Tg offspring (P< 0.05) (Fig. 1 B). However, despite the trend to normalized mRNA expression of leptin receptor (P = 0.06) and MC4R (two‐way ANOVA, P SIRT1‐Tg< 0.05), no reduction in milk intake was detected (Fig. 1 A). STAT3 was elevated by SIRT1 overexpression in MC offspring only (MC‐WT vs. MC‐Tg, P < 0.05) (Fig. 1 D). Similarly, there were no differences between groups in the mRNA expression of NPY1, NPY1R and POMC.
SIRT1 overexpression increases glucose tolerance and insulin sensitivity in MHF offspring
As expected, MHF‐WT offspring showed significantly higher levels of blood glucose during IPGTT (P < 0.01 vs. MC‐WT) (Fig. 2 A), suggesting glucose intolerance. This was associated with hyperinsulinaemia (P < 0.05) and significant reduction of QUICKI (P < 0.05) (Fig. 2 B), indicating impaired insulin sensitivity in the offspring as a result of maternal HFD consumption. SIRT1 overexpression prevented glucose intolerance (P < 0.01 vs. MC‐WT) (Fig. 1 A) and normalized plasma levels of insulin, as well as QUICKI.
Figure 2. Perinatal SIRT1 overexpression increases glucose tolerance and insulin sensitivityin MHF offspring.
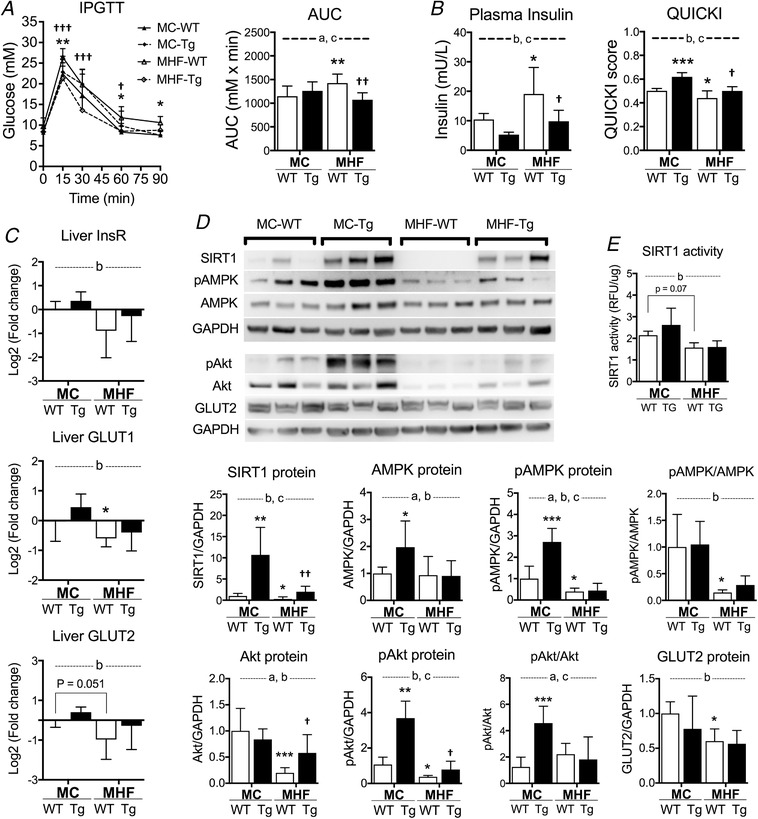
A, IPGTT and area under the curve (n = 11, 8, 13 and 8 respectively). B, plasma insulin levels and quantitative insulin sensitivity check index (QUICKI) (n = 6). C, liver mRNA expression of insulin receptor and glucose transporter 1 and 2 (n = 6). D, protein expression of SIRT1 and insulin signalling regulators (n = 6). E, SIRT1 activity (n = 6).Compared to MC‐WT: * P < 0.05, ** P < 0.01, *** P < 0.001; vs. MHF‐WT: †P < 0.05, ††P < 0.01, †††P < 0.001. aTwo‐way ANOVA, P interaction < 0.05. bTwo‐way ANOVA, P MHF < 0.05. cTwo‐way ANOVA, P SIRT1‐tg < 0.05.
In association with the reduction of systemic insulin sensitivity in MHF‐WT offspring, mRNA expression of insulin receptor and glucose transporters (GLUT)1 and 2 in the liver was also downregulated (two‐way ANOVA, P MHF < 0.05) (Fig. 2 C), suggesting impaired insulin signalling and glucose transport in offspring liver as a result of maternal HFD consumption. In line with the quantitative RT‐PCR result, the immunoblotting result also indicated reduced GLUT2 protein expression in MHF‐WT offspring (two‐way ANOVA, P MHF < 0.05) (Fig. 2 D). AMPK and Akt are both important regulators of insulin sensitivity, which are phosphorylated upon activation (pAMPK and pAkt) (Khamzina et al. 2005). Our results indicate that the protein levels of pAMPK, Akt and pAkt were significantly suppressed in MHF‐WT offspring (P < 0.05, P < 0.001 and P < 0.05, respectively) (Fig. 2 D). AMPK expression was unchanged and the pAMPK/AMPK ratio was significantly reduced (P < 0.05), suggesting reduced activity rather than expression. Because both pAkt and Akt levels were reduced, the relative ratio of pAkt/Akt was unchanged. Consistent with previous findings (Borengasser et al. 2014), liver expression of SIRT1 was reduced as a result of maternal HFD consumption (P < 0.05, MHF‐WT vs. MC‐WT). SIRT1 overexpression significantly reversed the expression of Akt and pAkt (P < 0.05) but not that of InsR, GLUT, AMPK or pAMPK (Fig. 2 C and D).
SIRT1 overexpression attenuates adipocyte hypertrophy in MHF offspring
H&E staining of EpiWAT revealed enlarged adipocytes in MHF offspring compared to the control (two‐way ANOVA, P MHF < 0.05) (Fig. 3 A), which was associated with a significant reduction in SIRT1 mRNA expression (two‐way ANOVA, P MHF < 0.05) (Fig. 3 B). Concomitantly, mRNA expression of peroxisome proliferator‐activated receptor gamma (PPAR) coactivator 1‐alpha (PGC‐1α) and sterol regulatory element‐binding protein (SREBP)1c in MHF‐WT offspring were both significantly upregulated (P < 0.05) (Fig. 3 B). No change was found in PPARγ mRNA levels. Similar to EpiWAT, RpWAT showed a significant reduction in SIRT1 mRNA levels in MHF offspring (two‐way ANOVA, P MHF < 0.05) (Fig. 3 B). However, in contrast to EpiWAT, PGC‐1α and PPARγ mRNA expression in RpWAT were significantly decreased in MHF‐WT offspring (P < 0.05 and P < 0.01, respectively) (Fig. 3 B), whereas SREBP1c levels were unchanged.
Figure 3. Perinatal SIRT1 overexpression reversed hypertrophy in white adipose tissue in MHF offspring.
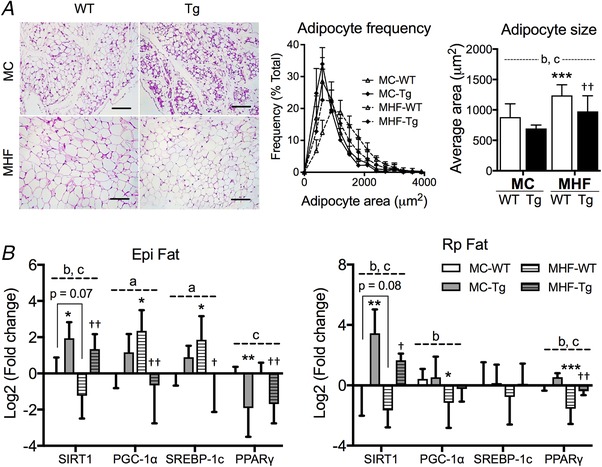
A, H&E staining and adipocyte frequency analysis. 200× magnification (n = 6). B, quantitative RT‐PCR analysis of offspring EpiWAT and RpWAT (n = 6). Compared to MC‐WT: * P < 0.05, ** P < 0.01, *** P < 0.001; vs. MHF‐WT: ††P < 0.01. aTwo‐way ANOVA, P interaction < 0.05. bTwo‐way ANOVA, P MHF < 0.05. cTwo‐way ANOVA, P SIRT1‐tg < 0.05. Scale bar = 200 μm. [Color figure can be viewed at wileyonlinelibrary.com]
SIRT1 overexpression significantly attenuated adipocyte hypertrophy in MHF‐Tg offspring EpiWAT (P < 0.01) (Fig. 3 A). The effect was associated with marked reductions in the mRNA expression of PGC‐1α, SREBP‐1c and PPARγ (P < 0.01, P < 0.05 and P < 0.01, respectively) (Fig. 3 B). There was a significant interaction between maternal diet and SIRT1 overexpression on mRNA expression of PGC‐1α and SREBP‐1c in EpiWAT (two‐way ANOVA, P interaction < 0.05). These two markers appear to be upregulated in MC‐Tg offspring but downregulated in MHF‐Tg offspring. In RpWAT, no significant change was found in PGC‐1α and SREBP‐1c mRNA expression as a result of SIRT1 overexpression. Conversely, PPARγ expression was significantly increased (P < 0.01) (Fig. 3 B).
SIRT1 overexpression attenuates liver lipogenesis and lipotoxicity in MHF offspring
In the present study, MHF offspring showed increased lipid accumulation as reflected by liver TG levels, ORO staining (both P < 0.001) (Fig. 4 A), as well as H&E staining (Fig. 4 B), in association with significantly reduced SIRT1 protein expression (Fig. 4 B). The mRNA expression of SREBP‐1c, fatty acid synthase (FASN) and fatty acid uptake fatty acid binding protein (FABP)1 was significantly increased in MHF offspring (P < 0.05, P < 0.05 and P < 0.01, respectively) (Fig. 4 D), suggesting lipotoxicity. Carbohydrate‐responsive element‐binding protein (ChREBP), a hepatic lipogenesis marker that acts in a glucose concentration‐dependent manner, was also slightly upregulated as a result of MHF (two‐way ANOVA, P MHF < 0.05). PPARα mRNA expression was also increased (two‐way ANOVA, P MHF < 0.05). By contrast, PGC‐1α and PPARγ mRNA expression were significantly downregulated (MHF‐WT vs. MC‐WT, P < 0.05) (Fig. 4 C). The results of SREBP1 and PGC‐1α were validated by protein expression analysis (MHF‐WT vs. MC‐WT, P < 0.05) (Fig. 4 D). mRNA expression of liver X receptor (LXR)β, a marker of cholesterol efflux, was unchanged as a result of maternal HFD consumption.
Figure 4. Perinatal SIRT1 overexpression reduced liver weight and lipotoxicity in MHF offspring.
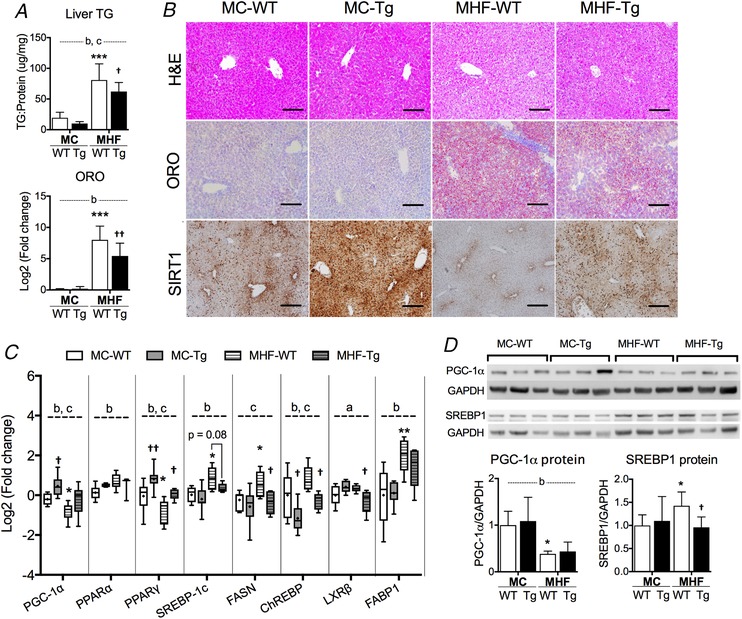
A, liver triglyceride (TG) and Oil Red O (ORO) staining quantitation (n = 6). B, representative images of H&E, ORO and IHC staining of SIRT1. C, box plots of RT‐PCR analysis of offspring liver. The boxes extend from the 25th to 75th percentiles, the central lines indicate the median. Whiskers extend from minimum to maximum values (n = 6). D, protein expression of genes in SIRT1 signalling network (n = 6). Compared to MC‐WT: * P < 0.05, ** P < 0.01, *** P < 0.001; vs. MHF‐WT: †P < 0.05, ††P < 0.01. aTwo‐way ANOVA, P interaction < 0.05. bTwo‐way ANOVA, P MHF < 0.05. cTwo‐way ANOVA, P SIRT1‐tg < 0.05. Scale bar = 100 μm. [Color figure can be viewed at wileyonlinelibrary.com]
SIRT1 overexpression significantly decreased TG and ORO staining levels in the liver of MHF offspring (P < 0.05 and P < 0.01) (Fig. 4 A and B). mRNA but not protein levels of PGC‐1α were upregulated in Tg offspring (two‐way ANOVA, P SIRT1‐tg < 0.05). By contrast, the mRNA level of SREBP‐1c was not significantly changed (P = 0.06) (Fig. 4 C), although its protein level was significantly suppressed (P < 0.05) (Fig. 4 D). Similar to the RpWAT, the PPARγ mRNA level in the liver was normalized by SIRT1 overexpression in MHF‐Tg offspring (P < 0.05) (Fig. 4 C). On the other hand, FASN and ChREBP were significantly suppressed (all P < 0.05) (Fig. 4 C). There was an interactive effect between maternal diet and SIRT1 overexpression on LXRβ expression (two‐way ANOVA, P interaction < 0.05), with a significant decrease in MHF‐Tg compared to MHF‐WT offspring (P < 0.05).
SIRT1 overexpression attenuates inflammatory disorder, oxidative stress and fibrogenesis in the liver of MHF offspring
In association with increased lipid accumulation and altered levels of lipotoxicity markers, MHF‐WT offspring also demonstrated increased mRNA expression of monocyte chemotactic protein (MCP)1, an inflammatory marker, and reduced expression of TGFβ receptor (TGFβR) types 1 and 2 in the liver (all P < 0.05) (Fig. 5 A). mRNA expression of anti‐oxidant enzymes, including superoxide dismutase (SOD)2, glutathione peroxidase (GPx)‐1 and catalase, in MHF‐WT offspring liver was significantly suppressed by maternal HFD consumption (all P < 0.01) (Fig. 5 B). Maternal HFD consumption also significantly increased protein expression of liver extracellular matrix (ECM) markers, including collagen (COL)1A, COL4 and fibronectin (FN) in offspring liver, as reflected by IHC staining (all P < 0.05) (Fig. 5 C). The results were confirmed by PSR staining for collagens (P < 0.001) (Fig. 5 C). No significant change in plasma alanine aminotransferase (ALT) was detected among these groups (data not shown), suggesting these early changes have not induced measurable liver cell injury.
Figure 5. Perinatal SIRT1 overexpression attenuates inflammatory dysregulation and oxidative stress as a result of MHF in offspring liver.
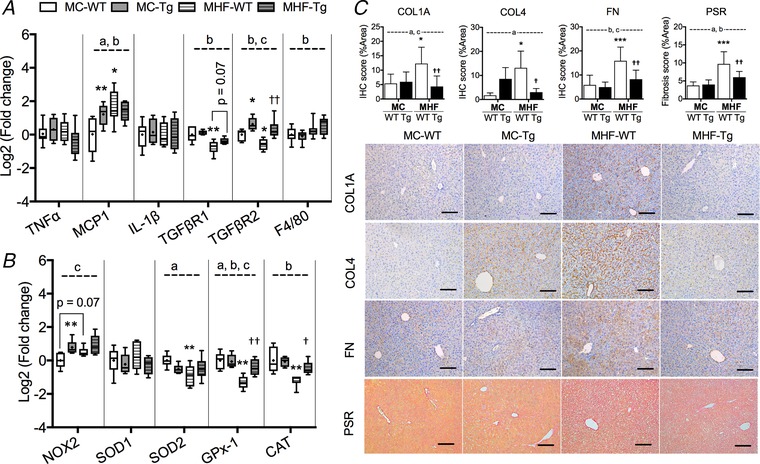
Box plots showing mRNA expression of (A) inflammatory markers and (B) oxidative stress regulators. The boxes extend from the 25th to 75th percentiles, the central lines indicate the median. Whiskers extend from minimumto maximum values (n = 6). C, IHC and PSR staining quantification (n = 6). Compared to MC‐WT: * P < 0.05, ** P < 0.01, *** P < 0.001; vs. MHF‐WT: †P < 0.05, ††P < 0.01. aTwo‐way ANOVA, P interaction < 0.05. bTwo‐way ANOVA, P MHF < 0.05. cTwo‐way ANOVA, P SIRT1‐tg < 0.05. Scale bar = 100 μm. [Color figure can be viewed at wileyonlinelibrary.com]
SIRT1 overexpression normalized hepatic levels of TGFβR2 (P < 0.01) but had little effect on the other inflammatory and macrophage markers in offspring liver (Fig. 5 A). It also increased mRNA expression of NAPDH oxidase 2 (two‐way ANOVA, P SIRT1‐tg < 0.05) but concomitantly improved the levels of GPx‐1 (P < 0.01) and catalase (P < 0.05) (Fig. 5 B), which may reflect increased reactive oxygen species production and appropriate anti‐oxidant defence. The Tg offspring also showed significantly attenuated levels of COL1A, COL4 and FN, suggesting reduced fibrogenesis (all P < 0.05), which is supported by the PSR staining (P < 0.01) (Fig. 5 C).
Discussion
In the present study, we confirm the negative effects of maternal HFD consumption on increased body weight and adiposity, reduced glucose tolerance and insulin sensitivity, as well as induced hepatic steatosis, inflammatory and oxidative stress responses and fibrogenesis, in offspring at weaning. Importantly, we demonstrate that SIRT1 is a key player in this process, the systemic overexpression of which resulted in the attenuation in all the above‐mentioned disorders in the offspring of HFD‐fed dams, suggesting improved metabolic homeostasis.
Although hypothalamic SIRT1 has been shown to play an important role in appetite regulation (Çakir et al. 2009; Sasaki et al. 2010), no difference in milk intake was found between WT and Tg offspring in the present study. Because both WT and Tg offspring were born to the same WT dams, offspring of both genotypes probably received similar nutrients during gestation and lactation. As such, the key difference that underlines the reduced body weight and adiposity in Tg offspring probably lies in the increased metabolic rate, which is a well‐established characteristic of SIRT1‐overexpressed mice (Pfluger et al. 2008).
SIRT1 has been shown to upregulate PGC‐1α and increase fatty acid oxidation in muscles (Gerhart‐Hines et al. 2007), with concordant suppression of PPARγ and SREBP1c expression in adipose tissue, leading to reduced adipogenesis (Picard et al. 2004; Ponugoti et al. 2010). We demonstrate that these markers are differentially regulated in two types of adipose tissues (i.e. EpiWAT and RpWAT) in the offspring by maternal HFD and SIRT1 overexpression, respectively. In EpiWAT, SREBP1c was increased in MHF offspring, which is consistent with the adipocyte hypertrophy phenotype. Interestingly, PGC‐1α was also increased, probably suggesting an adaptive mechanism. SIRT1 overexpression suppressed SREBP1c and PPARγ, which is in line with reduced EpiWAT mass and fat cell size in MHF‐Tg offspring. By contrast to EpiWAT, the RpWAT in MHF offspring showed a significant reduction of PGC‐1α and PPARγ, which is associated with smaller RpWAT mass and increased systemic insulin sensitivity. It has been shown that the expression of PPARγ and SREBP‐1c in s.c. WAT is suppressed in obese patients with insulin resistance (Kolehmainen et al. 2001; Dubois et al. 2006). Thus, PPARγ agonists are used clinically as insulin sensitizers (Lebovitz et al. 2001; Ahmadian et al. 2013) and MHF mouse offspring treated with pioglitazone, a PPARγ agonist, have reduced body weight, reduced visceral WAT gain, increased s.c. WAT mass in association with lower levels of fasting glucose and insulin resistance (Kalanderian et al. 2013). In the present study, IPGTT was reduced and QUICKI was improved in MHF‐Tg offspring, suggesting the positive effect of SIRT1 overexpression on systemic insulin sensitivity and glucose homeostasis. This may be partially attributed to the regulation of PPARγ in the offspring RpWAT.
The reduced expression of GLUTs, InsR, pAMPK and pAkt indicates impaired glucose transport and insulin signalling in the offspring liver as a result of maternal HFD consumption(Nawano et al. 1999; Iglesias et al. 2002), which may also underpin the dysregulation of systemic glucose. SIRT1 overexpression only reversed the expression and activity of Akt and not the other markers, suggesting that SIRT1 overexpression regulates liver insulin sensitivity via an Akt‐dependent mechanism in our model. To support this hypothesis, PGC‐1α, the common downstream marker of both SIRT1 and AMPK, was also not upregulated in MHF‐Tg offspring. An explanation for this is the lack of important cofactors NAD+ and AMP as a result of a positive energy balance that is required for SIRT1 and AMPK to become active. The unchanged activity of SIRT1 in the MHF‐Tg offspring liver is also in line with this hypothesis. By contrast, SIRT1 has been shown to promote phosphorylation of Akt via deacetylationindependent pathways (Wang et al. 2011; Ramakrishnan et al. 2014).
Apart from Akt, PPARγ expression was also upregulated in Tg offspring, which suggests another pathway for improved insulin sensitivity in the liver (Tiikkainen et al. 2004). Importantly, PPARγ is also involved in liver lipid metabolism, of which activation has been shown to reduce liver lipid contents (Tiikkainen et al. 2004).The reduced expression of de novolipogenesis markers including SREBP‐1c, ChREBP and FASN, probably also contributes to such an effect. LXRβ not only is the master regulator of cholesterol, but also is involved in lipogenesis by stimulating the expression of SREBP‐1c and FASN (Grefhorst et al. 2002). The downregulation ofLXRβ in MHF‐Tg offspring supports a reduction of hepatic steatosis (Patel et al. 2011). Conversely, no change in FABP1 expression was found, suggesting that lipid uptake in MHF‐Tg offspring liver was unchanged. As noted, AMPK/PGC‐1α signalling was still impaired despite SIRT1 overexpression, suggesting no improvement in lipolysis (Viollet et al. 2006). Considering that liver lipid content is the consequence of the balance between de novo lipogenesis, lipid uptake and lipolysis, it is understandable why, in the present study, SIRT1‐mediated suppression of de novo lipogenesis by itself can not fully normalize lipid accumulation in the liver as a result of maternal HFD consumption.
Maternal HFD consumption was associated with inflammatory dysregulation, as reflected by increased expression of MCP‐1 and reduced expression of TGFβ receptors 1 and 2. The result is consistent with our previous findings in the kidneys of MHF offspring in rats (Nguyen et al. 2017). Further investigation regarding TGFβ signalling pathways is required to determine whether these abnormalities reflect inflammatory disorders or compensatory responses. In the present study, maternal HFD also led to significant suppression of endogenous anti‐oxidants, including SOD2, GPx‐1 and CAT, suggesting increased oxidative damage in offspring liver. As such, the expression of fibrogenic markers COL1A, COL4 and FN was significantly elevated, which reflects increased susceptibility for liver fibrosis. Despite significant liver remodelling, there was no change in the plasma levels of ALT in MHF offspring, which suggests that no major liver damages had occurred. However, additional exposure to postnatal HFD can cause significant lipotoxicity, oxidative stress and inflammation later in life(McCurdy et al. 2009). SIRT1 overexpression in the offspring was able to enhance anti‐oxidant capacity and attenuate fibrogenesis in the liver as a result of maternal HFD consumption. On one hand, these positive effects can be partially attributed to the effects of SIRT1 to suppress glucotoxicity and lipotoxicity in Tg offspring. On the other hand, SIRT1 has been shown to have direct regulatory effects on inflammation, oxidative stress and fibrosis in models of acute tissue injuries (He et al. 2010; Wu et al. 2015).
In the present study, we did not control for single pup per litter, which can be a limitation in DOHaD research (Dickinson et al. 2016). However, given that there was typically one transgenic male mouse per litter, the possible bias in the analysis of the effects of SIRT1 overexpression is probably minimal.
Collectively, the present study provides direct evidence of the importance of SIRT1 in linking maternal HFD consumption to metabolic dysfunction in the offspring, and suggests that targeting SIRT1 in the offspring in early developmental periods may reprogram metabolic disorders as a result of maternal HFD feeding. Further studies are required to examine the long‐term effects of these approaches in adulthood and across generations. In addition, because SIRT1 has been shown to modulate zygotic histone code (Adamkova et al. 2017), examination of epigenetic modifications including DNA methylation and histone acetylation can provide additional understanding of the fetal reprogramming effects of SIRT1 in the setting of maternal obesity.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
LN designed and conducted all of the main experiments, performed the data analysis, and preparedthe figures and the manuscript. AZ assisted with the tissue processing for histology. HC, CP and SS co‐ordinated the execution of the project and were involved in the experimental design. HC, CP and SS reviewed the data analysis and the manuscript. All authors have critically revised and approved the final copy of the manuscript submitted for publication.
Funding
LN was supported by Sydney Medical School's ECR PhD Scholarship and Amgen research scholarship.
Acknowledgments
We thank Dr Lindsay Wu from The University of New South Wales, Australia, for kindly sharing the SIRT1‐transgenic colony.
Biography
Long The Nguyen recently completed his PhD programme and started his academic career as a junior postdoctoral associate. Throughout different stages of his education, he has been involved in multiple research projects in different fields, such as cancer therapy, fetal programming, smoking and metabolic disorders, which involves kidney, liver and brain research. His research productivity has been consistently outstanding, with 10 publications in the last 4 years, and with six these as the first author. His current research focuses on metabolic disorders and chronic kidney diseases.

Edited by: Laura Bennet & Janna Morrison
This is an Editor's Choice article from the 15 January 2019 issue.
Linked articles: This article is highlighted in a Perspectives article by Ong. To read this article, visit https://doi.org/10.1113/JP277280.
References
- Adamkova K, Yi Y‐J, Petr J, Zalmanova T, Hoskova K, Jelinkova P, Moravec J, Kralickova M, Sutovsky M, Sutovsky P & Nevoral J (2017). SIRT1‐dependent modulation of methylation and acetylation of histone H3 on lysine 9 (H3K9) in the zygotic pronuclei improves porcine embryo development. J Anim Sci Biotechnol 8, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M & Evans RM (2013). PPAR [gamma] signaling and metabolism: the good, the bad and the future. Nat Med 99, 557–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armitage JA, Poston L & Taylor PD (2008). Developmental origins of obesity and the metabolic syndrome: the role of maternal obesity. Front Horm Res 36, 73–84. [DOI] [PubMed] [Google Scholar]
- Borengasser SJ, Kang P, Faske J, Gomez‐Acevedo H, Blackburn ML, Badger TM & Shankar K (2014). High fat diet and in utero exposure to maternal obesity disrupts circadian rhythm and leads to metabolic programming of liver in rat offspring. PLoS ONE 9, e84209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW & Shipley GL (2009). The MIQE guidelines: minimum information for publication of quantitative real‐time PCR experiments. Clin Chem 55, 611–622. [DOI] [PubMed] [Google Scholar]
- Cacho J, Sevillano J, de Castro J, Herrera E & Ramos M (2008). Validation of simple indexes to assess insulin sensitivity during pregnancy in Wistar and Sprague‐Dawley rats. Am J Physiol Endocrinol Metab 295, E1269–E1276. [DOI] [PubMed] [Google Scholar]
- Çakir I, Perello M, Lansari O, Messier NJ, Vaslet CA & Nillni EA (2009). Hypothalamic Sirt1 regulates food intake in a rodent model system. PLoS ONE 4, e8322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalano P (2015). Maternal obesity and metabolic risk to the offspring: why lifestyle interventions may have not achieved the desired outcomes. Int J Obes 39, 642–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Simar D, Lambert K, Mercier J & Morris MJ (2008). Maternal and postnatal overnutrition differentially impact appetite regulators and fuel metabolism. Endocrinology 149, 5348–5356. [DOI] [PubMed] [Google Scholar]
- Chen H, Sullivan G & Quon MJ (2005). Assessing the predictive accuracy of QUICKI as a surrogate index for insulin sensitivity using a calibration model. Diabetes 54, 1914. [DOI] [PubMed] [Google Scholar]
- Del Prado M, Delgado G & Villalpando S (1997). Maternal lipid intake during pregnancy and lactation alters milk composition and production and litter growth in rats. J Nutr 127, 458–462. [DOI] [PubMed] [Google Scholar]
- Dickinson H, Moss T, Gatford K, Moritz K, Akison L, Fullston T, Hryciw D, Maloney C, Morris M & Wooldridge A (2016). A review of fundamental principles for animal models of DOHaD research: an Australian perspective. J Dev Orig Health Dis 7, 449–472. [DOI] [PubMed] [Google Scholar]
- dos Santos Costa C, Hammes TO, Rohden F, Margis R, Bortolotto JW, Padoin AV, Mottin CC & Guaragna RM (2010). SIRT1 transcription is decreased in visceral adipose tissue of morbidly obese patients with severe hepatic steatosis. Obes Surg 20, 633–639. [DOI] [PubMed] [Google Scholar]
- Dubois SG, Heilbronn LK, Smith SR, Albu JB, Kelley DE & Ravussin E (2006). Decreased expression of adipogenic genes in obese subjects with type 2 diabetes. Obesity 14, 1543–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galarraga M, Campión J, Muñoz‐Barrutia A, Boqué N, Moreno H, Martínez JA, Milagro F & Ortiz‐de‐Solórzano C (2012). Adiposoft: automated software for the analysis of white adipose tissue cellularity in histological sections. J Lipid Res 53, 2791–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhart‐Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R, Alt FW, Wu Z & Puigserver P (2007). Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC‐1α. EMBO J 26, 1913–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glastras S, Tsang M, Teh R, Chen H, McGrath R, Zaky A, Pollock C & Saad S (2015). Maternal obesity promotes diabetic nephropathy in rodent offspring. Sci Rep 6, 27769–27769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grefhorst A, Elzinga BM, Voshol PJ, Plösch T, Kok T, Bloks VW, van der Sluijs FH, Havekes LM, Romijn JA, Verkade HJ & Kuipers F (2002). Stimulation of Lipogenesis by pharmacological activation of the liver X receptor leads to production of large, triglyceride‐rich very low density lipoprotein particles. J Biol Chem 277, 34182–34190. [DOI] [PubMed] [Google Scholar]
- Haigis MC & Sinclair DA (2010). Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol 5, 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W, Wang Y, Zhang M‐Z, You L, Davis LS, Fan H, Yang H‐C, Fogo AB, Zent R & Harris RC (2010). Sirt1 activation protects the mouse renal medulla from oxidative injury. J Clin Invest 120, 1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heerwagen MJ, Miller MR, Barbour LA & Friedman JE (2010). Maternal obesity and fetal metabolic programming: a fertile epigenetic soil. Am J Physiol Regul Integr Comp Physiol 299, R711–R722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Cantó C, Wanders RJ & Auwerx J (2010). The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr Rev 31, 194–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Pirinen E & Auwerx J (2012). Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol 13, 225–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie G, Sloboda D, Kamal T & Vickers M (2009). Maternal nutritional history predicts obesity in adult offspring independent of postnatal diet. J Physiol 587, 905–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias MA, Ye J‐M, Frangioudakis G, Saha AK, Tomas E, Ruderman NB, Cooney GJ & Kraegen EW (2002). AICAR administration causes an apparent enhancement of muscle and liver insulin action in insulin‐resistant high‐fat‐fed rats. Diabetes 51, 2886–2894. [DOI] [PubMed] [Google Scholar]
- Jara‐Almonte M & White JM (1972). Milk production in laboratory mice. J Dairy Sci 55, 1502–1505. [DOI] [PubMed] [Google Scholar]
- Kalanderian A, Abate N, Patrikeev I, Wei J, Vincent KL, Motamedi M, Saade GR & Bytautiene E (2013). Pioglitazone therapy in mouse offspring exposed to maternal obesity. Am J Obstet Gynecol 208, e301–e308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khamzina L, Veilleux A, Sb B & Marette A (2005). Increased activation of the mammalian target of rapamycin pathway in liver and skeletal muscle of obese rats: possible involvement in obesity‐linked insulin resistance. Endocrinology 146, 1473–1481. [DOI] [PubMed] [Google Scholar]
- Kolehmainen M, Vidal H, Alhava E & Uusitupa MI (2001). Sterol regulatory element binding protein 1c (SREBP‐1c) expression in human obesity. Obesity 9, 706–712. [DOI] [PubMed] [Google Scholar]
- Lebovitz HE, Dole JF, Patwardhan R, Rappaport EB & Freed MI (2001). Rosiglitazone monotherapy is effective in patients with type 2 diabetes. J Clin Endocrinol Metab 86, 280–288. [DOI] [PubMed] [Google Scholar]
- Mariani S, Fiore D, Basciani S, Persichetti A, Contini S, Lubrano C, Salvatori L, Lenzi A & Gnessi L (2015). Plasma levels of SIRT1 associate with non‐alcoholic fatty liver disease in obese patients. Endocrine 49, 711–716. [DOI] [PubMed] [Google Scholar]
- McCurdy CE, Bishop JM, Williams SM, Grayson BE, Smith MS, Friedman JE & Grove KL (2009). Maternal high‐fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J Clin Invest 119, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB & Vu CB (2007). Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 450, 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawano M, Ueta K, Oku A, Arakawa K, Saito A, Funaki M, Anai M, Kikuchi M, Oka Y & Asano T (1999). Hyperglycemia impairs the insulin signaling step between PI 3‐kinase and Akt/PKB activations in ZDF rat liver. Biochem Biophys Res Commun 266, 252–256. [DOI] [PubMed] [Google Scholar]
- Nguyen LT, Chen H, Mak C, Zaky A, Pollock C & Saad S (2018). SRT1720 attenuates obesity and insulin resistance but not liver damage in the offspring due to maternal and postnatal high‐fat diet consumption. Am J Physiol Endocrinol Metab 315, E196–E203. [DOI] [PubMed] [Google Scholar]
- Nguyen LT, Chen H, Pollock C & Saad S (2017). SIRT1 reduction is associated with sex‐specific dysregulation of renal lipid metabolism and stress responses in offspring by maternal high‐fat diet. Sci Rep 7, 8982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LT, Chen H, Pollock CA & Saad S (2016). Sirtuins – mediators of maternal obesity‐induced complications in offspring? FASEB J 30, 1383–1390. [DOI] [PubMed] [Google Scholar]
- Oben JA, Mouralidarane A, Samuelsson A‐M, Matthews PJ, Morgan ML, Mckee C, Soeda J, Fernandez‐Twinn DS, Martin‐Gronert MS & Ozanne SE (2010). Maternal obesity during pregnancy and lactation programs the development of offspring non‐alcoholic fatty liver disease in mice. J Hepatol 52, 913–920. [DOI] [PubMed] [Google Scholar]
- Patel R, Patel M, Tsai R, Lin V, Bookout AL, Zhang Y, Magomedova L, Li T, Chan JF, Budd C, Mangelsdorf DJ & Cummins CL (2011). LXRβ is required for glucocorticoid‐induced hyperglycemia and hepatosteatosis in mice. J Clin Invest 121, 431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen SB, Ølholm J, Paulsen SK, Bennetzen MF & Richelsen B (2008). Low Sirt1 expression, which is upregulated by fasting, in human adipose tissue from obese women. Int J Obes 32, 1250–1255. [DOI] [PubMed] [Google Scholar]
- Pfluger PT, Herranz D, Velasco‐Miguel S, Serrano M & Tschöp MH (2008). Sirt1 protects against high‐fat diet‐induced metabolic damage. Proc Natl Acad Sci U S A 105, 9793–9798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard F, Kurtev M, Chung N, Topark‐Ngarm A, Senawong T, Machado de Oliveira R, Leid M, McBurney MW & Guarente L (2004). Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR‐γ. Nature 429, 771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponugoti B, Kim D‐H, Xiao Z, Smith Z, Miao J, Zang M, Wu S‐Y, Chiang C‐M, Veenstra TD & Kemper JK (2010). SIRT1 deacetylates and inhibits SREBP‐1C activity in regulation of hepatic lipid metabolism. J Biol Chem 285, 33959–33970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan G, Davaakhuu G, Kaplun L, Chung W‐C, Rana A, Atfi A, Miele L & Tzivion G (2014). Sirt2 deacetylase is a novel AKT binding partner critical for AKT activation by insulin. J Biol Chem 289, 6054–6066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saben JL, Boudoures AL, Asghar Z, Thompson A, Drury A, Zhang W, Chi M, Cusumano A, Scheaffer S & Moley KH (2016). Maternal metabolic syndrome programs mitochondrial dysfunction via germline changes across three generations. Cell Rep 16, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Kim H‐J, Kobayashi M, Kitamura Y‐I, Yokota‐Hashimoto H, Shiuchi T, Minokoshi Y & Kitamura T (2010). Induction of hypothalamic Sirt1 leads to cessation of feeding via agouti‐related peptide. Endocrinology 151, 2556–2566. [DOI] [PubMed] [Google Scholar]
- Suter MA, Chen A, Burdine MS, Choudhury M, Harris RA, Lane RH, Friedman JE, Grove KL, Tackett AJ & Aagaard KM (2012). A maternal high‐fat diet modulates fetal SIRT1 histone and protein deacetylase activity in nonhuman primates. FASEB J 26, 5106–5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiikkainen M, Häkkinen A‐M, Korsheninnikova E, Nyman T, Mäkimattila S & Yki‐Järvinen H (2004). Effects of rosiglitazone and metformin on liver fat content, hepatic insulin resistance, insulin clearance, and gene expression in adipose tissue in patients with type 2 diabetes. Diabetes 53, 2169–2176. [DOI] [PubMed] [Google Scholar]
- Viollet B, Foretz M, Guigas B, Horman S, Dentin R, Bertrand L, Hue L & Andreelli F (2006). Activation of AMP‐activated protein kinase in the liver: a new strategy for the management of metabolic hepatic disorders. J Physiol 574, 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R‐H, Kim H‐S, Xiao C, Xu X, Gavrilova O & Deng C‐X (2011). Hepatic Sirt1 deficiency in mice impairs mTorc2/Akt signaling and results in hyperglycemia, oxidative damage, and insulin resistance. J Clin Invest 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M & Sinclair D (2004). Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature 430, 686. [DOI] [PubMed] [Google Scholar]
- Wu Y, Liu X, Zhou Q, Huang C, Meng X, Xu F & Li J (2015). Silent information regulator 1 (SIRT1) ameliorates liver fibrosis via promoting activated stellate cell apoptosis and reversion. Toxicol Appl Pharmacol 289, 163–176. [DOI] [PubMed] [Google Scholar]