

Abstract
Thrombotic thrombocytopenic purpura (TTP) is a thrombotic microangiopathy characterized by severe congenital or immune‐mediated deficiency in ADAMTS13, the enzyme that cleaves von Willebrand factor multimers. This rare condition leads invariably and rapidly to a fatal outcome in the absence of treatment, and therefore raises multiple diagnostic and therapeutic challenges. The novel concepts and mechanisms identified in the laboratory for this disease have been rapidly and successfully translated into the clinic for the benefit of patients, making TTP an archetypal disease that has benefited from targeted therapies. After decades of empirical treatment with plasma exchange, identification of ADAMTS13 as the key enzyme involved in TTP pathophysiology provided an explanation for the remarkable efficacy of plasma administration, in which the missing enzyme is replenished, and paved the way for development of a recombinant form of the enzyme. Similarly, the demonstration of a major role of anti‐ADAMTS13 antibodies through models of passive transfer of autoimmunity spurred development of immunomodulatory strategies based on B‐cell depletion. More recently, an inhibitor of the platelet‐von Willebrand factor interaction demonstrated efficacy in large clinical trials through prevention of formation of further microthrombi and protection of organs from ischemia. These translational breakthroughs in TTP are described in our review.
Keywords: ADAMTS13, caplacizumab, precision medicine, rituximab, targeted therapies, thrombotic thrombocytopenic purpura
Essentials.
TTP is still under‐diagnosed. A delay in diagnosis remains a prognostic concern.
Death rate of acute TTP scarcely changed for > 20 years. Most deaths occur in the first days of the management; these patients need new strategies.
An increasing number of targeted therapies based on anti‐vWF agents and recombinant ADAMTS13 should help in decreasing early TTP mortality and relapse.
These new therapies were derived from a better understanding of TTP pathophysiology, reflecting a shift from empiricism to targeted therapies.
1. INTRODUCTION
Thrombotic thrombocytopenic purpura (TTP) is a devastating disease characterized by microangiopathic hemolytic anemia, thrombocytopenia, and organ failure of variable severity. Historically, TTP had a fatal prognosis. This was transformed with the use of intensive therapeutic plasma exchange (TPE) in 1991.1 In the following years, the rapid and systematic use of TPE in association with corticosteroids until durable remission emerged as the standard regimen for TTP treatment. In 1998, the identification of a dysfunction of the von Willebrand factor (vWF)‐cleaving protease ADAMTS13 (A Disintegrin And Metalloproteinase with ThromboSpondin‐1 motifs; 13th member of the family) as a result of the production of autoantibodies against the enzyme in autoimmune TTP (iTTP) provided a rationale for the evaluation of B‐cell depleting therapies.2, 3 The addition of rituximab to the standard regimen in the mid‐2000s represented the second breakthrough in the management of the disease.4 Indeed rituximab, formerly administered as adjuvant therapy in iTTP in patients with a suboptimal response to the standard regimen, is now increasingly used frontline, and still more recently as a preemptive strategy to prevent relapse.5 In the very near future, this therapeutic arsenal will be strengthened by more targeted therapies stemming directly from our understanding of disease pathophysiology, specifically recombinant ADAMTS13 and caplacizumab, an agent that inhibits platelets‐vWF adhesion. In this review, we discuss recent advances as well as future perspectives in the rapidly evolving therapeutic landscape of TTP.
2. PATHOPHYSIOLOGICAL BASIS OF TTP TREATMENT
iTTP episodes occur in patients with severe, autoantibody‐mediated ADAMTS13 deficiency. In this context, highly adhesive vWF multimers accumulate leading to excessive platelet clumping in the microvasculature under high shear stress conditions, with resultant multi‐organ failure and death if effective management is not instituted. Consequently, the basis of current treatment is to supply ADAMTS13 through the administration of very large volumes of plasma, in association with plasmapheresis to prevent fluid overload and possibly to remove unusually large vWF multimers and anti‐ADAMTS13 autoantibodies. Autoantibodies directed against ADAMTS13 block the proteolytic activity of ADAMTS13 and/or increase its clearance from the circulation by forming noncovalent circulating immune complexes. Models of passive transfer of autoimmunity in baboons and rodents have supported these concepts,4 and led to wider use of immunosuppressive treatments. However, the immune cells precisely involved in the production of anti‐ADAMTS13 autoantibodies and accounting for the remarkable efficacy of B‐cell depleting agents in this disease remain to be identified.
3. CLINICAL PRESENTATION
The epidemiology of iTTP and clinical features on presentation have now been well characterized from large national registries.4, 5 The incidence of TTP has been reported to be two to four cases per million people per year.6 iTTP occurs more frequently in females (3:2 female‐to‐male ratio) with a median age in the fourth decade. While the onset of disease is typically sudden, prodromic manifestations including fatigue, arthralgias, myalgias, and abdominal and/or lumbar pain suggestive of a flu‐like illness are frequent. iTTP occurs in association with predisposing conditions in 50% of cases that need to be identified for appropriate management: connective tissue diseases, pregnancy, or more rarely HIV infection, and cancer.6 Therefore, unexplained anemia and thrombocytopenia in such contexts should prompt investigation for possible underlying iTTP.
Microangiopathic hemolytic anemia and peripheral thrombocytopenia are constant clinical features of TTP and are variably associated with organ injury. Cerebral and digestive manifestations are the most usual. Renal failure is typically mild or absent7 (for a concise review, see Kremer Hovinga et al4). In recent years, clinicians have become increasingly aware of cardiac involvement including infarction, congestive heart failure, arrhythmias, cardiogenic shock, and sudden cardiac arrest. Importantly, an elevated serum troponin level upon presentation is a common event (observed in up to 60% of patients) and represents an independent predictor of death, treatment refractoriness, and subsequent acute myocardial infarction.8, 9
The above clinical features are not specific for TTP, and TTP presentation may overlap with that of other thrombotic microangiopathy (TMA) syndromes. However, there is now accumulating evidence from large series of patients that severe acquired ADAMTS13 deficiency is associated with specific clinical features, such as mild renal involvement and severe thrombocytopenia compared with other TMAs. Lastly, some patients with TMAs other than TTP may have a low but detectable level of ADAMTS13 activity, and it cannot be excluded that subtle dysfunction of ADAMTS13 caused by yet unknown mechanisms may have a role in the TMA process.10
4. IMPORTANCE OF RAPID, ACCURATE DIAGNOSIS
With the increasing availability of effective treatments, rapid diagnosis of TTP by sensitive criteria is mandatory, and this urgency led to a decrease in the stringency of the diagnostic criteria in the clinical trial that documented the effectiveness of TPE.1 Consequently, the association of microangiopathic hemolytic anemia with peripheral thrombocytopenia should be enough to strongly suggest a diagnosis of TTP, before organ failure occurs. In line with this view, patients with an apparent diagnosis of immune thrombocytopenia (ITP) or Evan's syndrome who are not responding to the usual therapies should be investigated for schistocytes on repeated blood smears to revisit the diagnosis.11
Although the identification of severe (activity <10%) autoimmune‐mediated ADAMTS13 deficiency is required to document the diagnosis of iTTP, results of ADAMTS13 activity measurements are rarely available in emergency situations. Moreover, commercial kits may provide discrepant results in 12% of cases.5 Until assays are able to provide accurate ADAMTS13 activity within several hours, clinical scores for rapid prediction of individuals with severe ADAMTS13 deficiency will continue to have an important role in diagnosis.12, 13, 14 Two scores (the French score, and more recently the PLASMIC score),12, 13 derived from standard parameters easily available on presentation, offer a comparable and reliable way to identify patients with severe ADAMTS13 deficiency. Both scores use absence of an associated condition (eg, cancer, transplant, and disseminated intravascular coagulation), severe thrombocytopenia (<30 G/L) and mild renal involvement (serum creatinine level < or 2.25 mg/dL) as criteria for identifying patients with probable TTP (Table 1). It is likely that platelet count and serum creatinine level are the most useful and reliable values to predict severe ADAMTS13 deficiency. These scores are not aimed at redefining TTP diagnostic criteria, which are based on severe ADAMTS13 deficiency,15, 16 but they can help to rapidly identify patients who are most likely to have iTTP and therefore most likely to benefit from emergency treatment including TPE.
Table 1.
Comparison of the two clinical scores (French score and PLASMIC score)12, 13, 57 predicting severe ADAMTS13 deficiency
| French score | PLASMIC score | |
|---|---|---|
| Platelet count | <30 G/L (+1) | <30 G/L (+1) |
| Serum creatinine level | <2.25 mg/dL (+1) | <2 mg/dL (+1) |
| Hemolysis | ||
| Indirect bilirubin >2 mg/dL | –a | +1 |
| Or reticulocyte count >2.5% | ||
| Or undetectable haptoglobin | ||
| No active cancer in previous year | –a | +1 |
| No history of solid organ or SCT | –a | +1 |
| INR < 1.5 | –a | +1 |
| MCV < 90 fLb | – | +1 |
| Prediction of severe | 0: 2% | 0‐4: 0%‐4% |
| ADAMTS13 deficiency | 1: 70% | 5: 5%‐24% |
| (Activity <10%)c | 2: 94% | 6‐7: 62%‐82% |
Each item is associated with one point (+1).
INR, international normalized ratio; MCV, mean corpuscular value; SCT, stem cell transplantation.
The French score considered patients with a thrombotic microangiopathy (TMA) syndrome (which includes hemolysis with schistocytes in the definition) and assumes that there is no history of or clinical evidence for associated cancer, transplantation or disseminated intravascular coagulopathy; so these items are intrinsic to the score.
MCV was not incorporated in the French score.
Results correspond to those of the derivation cohort and those of a validation by (French score) the bootstrap resampling technique (internal validation) (Coppo et al,12 Bendapudi et al,13 and manuscript in preparation), or (PLASMIC score) different samples of patients from the same institution (internal validation) or from a different institution (external validation).12, 13
5. THE CURRENT STANDARD OF CARE
TTP is a medical emergency and requires rapid diagnosis and urgent management. Older age, a very high LDH level (reflecting organ damage as well as hemolysis) and increased cardiac troponin level on diagnosis, are associated with death and treatment refractoriness. Cardiac troponin assessment at the time of initial diagnosis to identify the more severe patients should be part of standard evaluation.8, 9
TPE remains the cornerstone of current management of iTTP and should be started as soon as the diagnosis is strongly suspected. Given the autoimmune nature of iTTP, there is a rationale for the use of corticosteroids, although evidence is limited17, 18, 19, 20 (see Figure 1).
Figure 1.
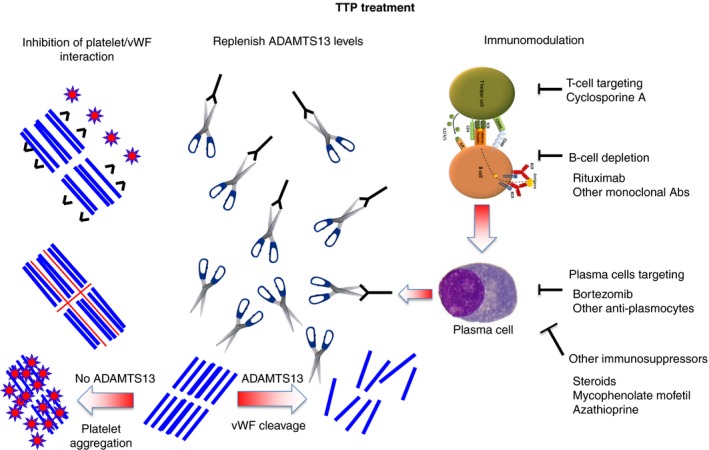
The three strategies of immune thrombotic thrombocytopenic purpura treatment. Abs, antibodies; vWF, von Willebrand factor.  , ADAMTS13;
, ADAMTS13;  , platelets;
, platelets;  , von Willebrand factor;
, von Willebrand factor; 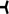 , anti‐ADAMTS13 antibodies;
, anti‐ADAMTS13 antibodies;  , anti‐vWF nanobody (caplacizumab);
, anti‐vWF nanobody (caplacizumab);  , N‐acetylcysteine
, N‐acetylcysteine
6. IMMUNOMODULATION THROUGH B‐CELL DEPLETION: WHERE DO WE STAND?
The introduction of the humanized anti‐CD20 monoclonal antibody rituximab has been the second major breakthrough in iTTP management. Clinical studies conducted to assess the role of rituximab in iTTP have faced the usual challenges that confront clinical trials in rare diseases such as difficulty with recruitment and cross‐over.21 These challenges are compounded in iTTP by the necessity to intervene rapidly, leaving little time for determination of eligibility and informed consent. As such, current knowledge about the use of rituximab in iTTP is based on observational studies that enrolled a limited number of patients and provide low to moderate levels of evidence.
Rituximab was first introduced in patients with a suboptimal response to conventional treatment (ie, disease exacerbation or refractoriness). From published studies of ≥10 iTTP patients treated with rituximab impressive remission rates of >90% occurring rapidly (typically in less than 4 weeks) were observed.4 In most instances, we administer four infusions of rituximab 375 mg/m2 once or twice weekly. The twice weekly administration schedule is justified by the significant clearance of rituximab by TPE (65% clearance per plasma volume22). Rituximab is started immediately after the end of a TPE session. When feasible, we wait a minimum of 18 hours after rituximab infusion until the next TPE to avoid excessive clearance of rituximab.23, 24 The response rates observed with rituximab exceed those of other historical salvage therapies, such as vincristine and cyclosporine A.4 No relapse was observed during the first year of follow‐up, but there were relapses beyond 1 year. Most significant side effects were mild infections. A rituximab regimen based on B‐cell depletion provided evidence that similar results could be obtained with only two to three rituximab infusions.25 Trials are ongoing to define more accurately the minimal effective dose of rituximab for this indication (ClinicalTrials.gov, NCT01554514).
The convincing results reported with rituximab in patients with a suboptimal response to standard therapy prompted investigators to evaluate its efficacy as frontline therapy in all patients with iTTP. In 2011, the UK group reported that frontline treatment with rituximab resulted in shorter hospitalization and fewer relapses that occurred later than in a historical group not treated with rituximab.26 Fewer and later relapses were also seen in rituximab treated patients by the French TMA Reference Center Network23 and the Oklahoma TTP registry.27
Although the time to response with rituximab is shorter in iTTP (median, 2 weeks between the first infusion and durable platelet count recovery) than for many other autoimmune diseases,23, 26 rituximab failed to prevent early deaths occurring in the first 10 days,23 leaving a crucial unmet need.
7. THE UNMET NEEDS WITH STANDARD TREATMENT
TPE transformed the historically fatal prognosis of iTTP to a treatable disease with an overall survival rate of 85%. Despite further improvements in the management of iTTP, including immunomodulation with rituximab, the last 20 years have seen only modest gains in survival.1, 23, 28 The two main causes of death are insufficient awareness of TTP diagnosis, leading to diagnostic delay,11 and the unmet need for new weapons for the management of the most severe cases, particularly early in the disease course. Another limitation of standard treatment is that TPE and catheter placement represent a cumbersome procedure associated with major complications including infection, bleeding, catheter‐associated thrombosis, toxicities of plasma infusion, and death.29 It is hoped that new drugs stemming from a better understanding of TTP pathophysiology will potentially bring about further reductions in mortality and healthcare burden.
8. THE FORTHCOMING THERAPEUTIC ARSENAL
8.1. Caplacizumab: inhibiting von Willebrand factor–platelet interaction
Caplacizumab (formerly ALX‐0081) is a nanobody (single‐domain antibody) derived from single‐chain antibodies naturally occurring in Camelidae. It was assessed in the TITAN and HERCULES trials, two multicenter randomized placebo‐controlled phase II and phase III studies, respectively, in patients with iTTP. Although the TITAN trial enrolled a lower number of patients than planned due to persistent recruitment challenges, some of whom did not have confirmed severe ADAMTS13 deficiency, both trials provided comparable conclusions. Time‐to‐platelet count recovery was significantly shorter and biomarkers reflecting ischemic organ damage normalized more rapidly in patients who received caplacizumab in addition to standard of care treatment.14, 30 The incidence of exacerbations was also reduced with caplacizumab, as the compound was continued for 30 days after TPE, thus covering the period for exacerbation. Because caplacizumab does not target the ongoing autoimmune response, increased relapses were observed in both trials shortly after withdrawal of caplacizumab. Relapsing patients had persistent severe ADAMTS13 deficiency. These observations raise two novel management considerations in iTTP. First, they provide further evidence that B‐cell depletion and corticosteroids should be initiated as soon as possible to hasten ADAMTS13 recovery. As caplacizumab provides a therapeutic bridge until ADAMTS13 improvement, it may be pursued over a longer period of time in patients with persistent severe ADAMTS13 deficiency, though this strategy must be balanced against the potential bleeding risk. Second, they suggest the potential of caplacizumab to prevent formation of further microthrombi and ischemic organ injury in the early, critical phase of the disease (ie, until rituximab and corticosteroids are effective).31 Caplacizumab substantially reduced the burden of care of iTTP patients by reducing the number of TPE sessions, length of hospital stay, and length of stay in the intensive care unit.14 Minor bleeding was more common in caplacizumab‐treated patients, but importantly there were no major or fatal bleeding cases.14 More safety data are needed from future studies on the risk of bleeding, especially in patients treated concomitantly with anticoagulants or when caplacizumab is administered over a period longer than 30 days (see Figure 1 and Algorithm A1).
Algorithm A1.
Initial evaluation and management of patients with acquired, autoimmune TTP. Because of the urgency to begin treatment for TTP, a clinical score (Table 1, 12, 13), which can be calculated from the initial clinical and laboratory data, is used to establish a probable diagnosis. When the clinical score suggests a high probability for ADAMTS13 activity <10% (French score 1‐2; PLASMIC score 6‐7), it is important to begin the full treatment for TTP immediately. *If the likelihood for ADAMTS13 activity <10% is intermediate (for example, PLASMIC score = 5; Table 1), we recommend empiric treatment for TTP because of the potential harms of withholding or delaying treatment, particularly TPE. This salvage treatment should be subsequently completed with immunosuppressive strategies and caplacizumab only when/if severe ADAMTS13 deficiency is confirmed. If ADAMTS13 activity is subsequently reported to be ≥10%, the diagnosis of TTP may still be correct and treatment should be continued unless an alternative diagnosis is established. It is important to recognize that some patients with ADAMTS13 ≥ 10% activity may have well documented TTP.7, 17
When the clinical score suggests a low probability for ADAMTS13 activity <10%, it may be appropriate to withhold treatment for TTP; however the diagnosis of TTP cannot be excluded. In these patients it is critical to search for an alternative etiology for the MAHA and thrombocytopenia, especially atypical (or complement‐mediated) hemolytic uremic syndrome (HUS) as well as a post‐infection HUS when renal involvement is predominant. If ADAMTS13 activity is subsequently reported to be <10%, treatment for TTP is indicated unless a clear alternative etiology for MAHA and thrombocytopenia has been established. Some patients in whom an alternative etiology for MAHA and thrombocytopenia is clearly established (eg, systemic infection or malignancy) may have ADAMTS13 activity <10%.7
The diagnosis of TTP requires clinical judgment as well as measurement of ADAMTS13 activity. Corticosteroids may be prednisone, 1 mg/kg/day, or for patients who are acutely ill, high doses of corticosteroids may be given initially (eg, intravenous methylprednisolone, 1000 mg/day for 3 days) before beginning oral prednisone. We use the conventional regimen for rituximab: 375 mg/m2/week, four doses once or twice weekly. Current studies are evaluating alternative regimens for rituximab, to determine if a lower dose or fewer infusions are equally efficacious. Caplacizumab is listed as initial treatment although it is not yet approved as treatment for TTP in the United States or Europe. Patients may continue to have deficient ADAMTS13 activity when their platelet count recovers to normal. Therefore weekly measurements of ADAMTS13 activity are important to guide the duration of corticosteroid and caplacizumab treatment. Persistent ADAMTS13 activity <20% requires continued treatment until ADAMTS13 improvement, because exacerbations of TTP may occur when plasma exchange is stopped. Weekly measurements should be continued until ADAMTS13 activity recovers to normal (≥50%). When ADAMTS13 activity returns to normal (≥50%) with effective immunosuppression, serial measurements continue at increasingly greater intervals. Because of the risk for relapse many years after the initial episode, indefinite follow‐up with annual measurement of ADAMTS13 activity is important.
In conclusion, caplacizumab may improve survival of patients with iTTP in the early phase of the disease until improvement of ADAMTS13 activity with B‐cell depleting therapies, which usually occurs within 30 days.23, 26 Consequently, ADAMTS13 activity should be monitored closely (for example, once per week) after platelet count recovery to determine the optimal time for stopping caplacizumab when ADAMTS13 is no longer severely deficient (for example, ADAMTS13 activity >20%). Forthcoming studies that focus specifically on the impact of caplacizumab on outcomes in the most severely affected patients are anxiously awaited.
9. RECOMBINANT ADAMTS13: THE NEXT STEP?
The identification of ADAMTS13 as the key missing component in TTP‐fueled development of a recombinant form of the protein (rADAMTS13) (SHP655; BAX930), for which tolerability and efficacy were recently evaluated in a phase 1 study of patients with congenital TTP. The therapeutic enzyme was reported to be safe and well tolerated, with a half‐life of 53 hours, which is comparable to the half‐life of wild‐type ADAMTS13.32 No significant adverse events were reported; specifically, no anti‐ADAMTS13 antibodies were detected. Finally, the study provided evidence of rADAMTS13 activity: platelet counts increased and the larger multimers of VWF decreased.33 Consequently, a phase 3 randomized controlled trial is planned to assess the efficacy of recombinant ADAMTS13 versus standard, plasma infusion‐based, treatment in congenital TTP (https://clinicaltrials.gov/ct2/show/NCT03393975). In the future, recombinant ADAMTS13 could be important not only for congenital TTP but also for the autoimmune‐mediated form. By saturating anti‐ADAMTS13 antibodies and cleaving large vWF multimers, recombinant ADAMTS13 could, together with immunomodulatory therapies and caplacizumab, decrease the burden of TPE treatment and hospitalization stay. The need to saturate anti‐ADAMTS13 antibodies in patients with iTTP suggests that higher doses of recombinant ADAMTS13 may be required for patients with iTTP than the replacement doses required for cTTP. Alternatively, recombinant forms of the enzyme engineered to be resistant to the autoantibodies could circumvent this limitation and be more cost‐effective.34 As reported for severe hemophilia A,35 the use of recombinant ADAMTS13 in cTTP could enhance the risk of developing inhibitors or boosting inhibitor titers compared with administration of plasma‐derived ADAMTS13 by the activation of ADAMTS13 specific memory B‐ and T‐cells.36
10. MANAGEMENT OF PATIENTS FOLLOWING THE ACUTE PHASE
10.1. Prevention of relapses
Historically, 40% of patients with iTTP experience one or multiple relapses.37 Relapses result from severe ADAMTS13 deficiency caused by the persistence or recurrence of anti‐ADAMTS13 autoantibodies. Whereas persistent severe ADAMTS13 deficiency during remission has been consistently associated with clinical relapse, the predictive value of anti‐ADAMTS13 antibodies for iTTP relapse remains controversial.38, 39 Younger age was also associated with an increased risk of relapse in one study.39 Each relapse exposes the patient to risk of death and to complications related to TPE or to intensive care unit hospitalization. Therefore, the prevention of relapse in TTP represents a major goal. As detailed above, the use of rituximab in the acute phase of the disease dramatically decreases the relapse rate at 1 year. However, beyond this period, anti‐ADAMTS13 autoantibodies may recur along with peripheral B‐cell reconstitution, exposing patients to risk of clinical relapse. These observations provided a rationale to evaluate the efficacy of rituximab in iTTP as preemptive therapy for patients in clinical remission, but with persistent or recurrent severe ADAMTS13 deficiency. In this context, rituximab remarkably reduces the incidence of iTTP relapse by diminishing the production of anti‐ADAMTS13 antibodies and rapidly restoring ADAMTS13 activity, which parallels peripheral B cell depletion.40, 41, 42 In our practice, we assess ADAMTS13 regularly during follow‐up (typically every 3 months). After serial ADAMTS13 assessments have remained normal (>50%) durably (typically 3 years), measurements are spaced out to twice a year for 2 years and then yearly. When ADAMTS13 activity becomes undetectable (activity <10% or even <20%), a single infusion of rituximab 375 mg/m2 is administered. In >80% of patients, ADAMTS13 activity post‐rituximab is detectable or even normalizes (20% to >50%) as early as 4‐6 weeks after infusion. However, in up to 50% of patients, ADAMTS13 recovery is transient and drops again with peripheral B‐cell reconstitution, typically 12 months later. Consequently, further rituximab infusions may be required to maintain a detectable ADAMTS13 activity and prevent clinical relapse.40, 41, 42 Patients with persistent severe (activity <10%‐20%) ADAMTS13 deficiency are exposed to a high risk of relapse, with a 7‐year cumulative incidence of relapse of 74%.42 Moreover, 10%‐15% of patients are primarily unresponsive to rituximab or experience refractoriness after an initial response.40 In these cases, a more intensive regimen inspired from those used in lymphoid malignancies and consisting of 4‐6 weekly infusions and/or maintenance treatment (4 infusions per year for 2 years) may overcome rituximab refractoriness42 (R. Saleem, Z. R. Rogers, C. Neunert, & J. N. George, Submitted).
Multiple infusions of rituximab may expose patients to infections or other long‐term complications, although treatment is generally well tolerated. Based on estimates that preemptive treatment with rituximab improves ADAMTS13 activity in 85% of cases and prevents a clinical relapse that may occur in 74% of cases, we can estimate that the number of patients needed to treat to prevent one relapse is only 1/(0.85 × 0.74), ie, 1.6. Moreover, if the risk of death with relapse is 5%,38 the number of patients needed to treat to prevent one death is 1/(0.85 × 0.74 × 0.05), ie, 32.42, 43 The most serious infectious complication associated with rituximab, progressive multifocal leukoencephalopathy, is only observed in one in 25 000 patients, and one in 500 000 if one only considers patients without AIDS or cancer. Therefore, persistent or recurrent severe autoimmune ADAMTS13 deficiency should be considered as a reasonable indication for preemptive treatment with rituximab (See Algorithm A2).40, 44
Algorithm A2.
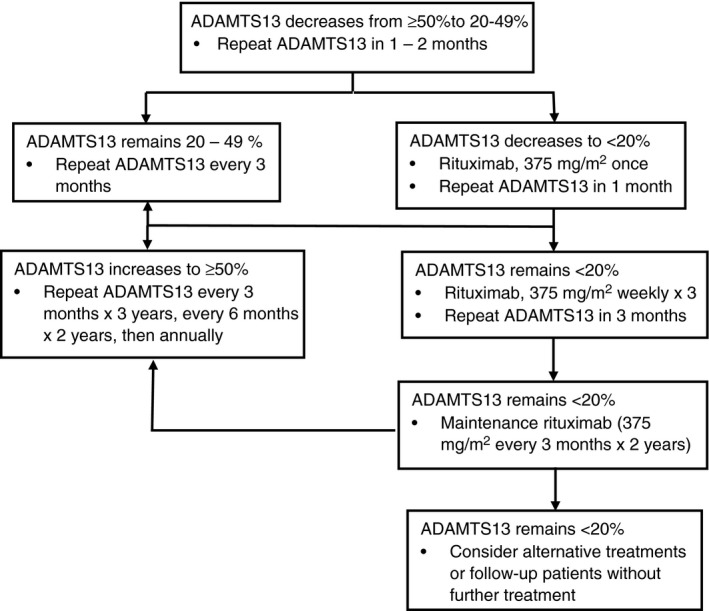
Management of patients with acquired, autoimmune TTP during remission. If ADAMTS13 activity decreases to 20%‐49%, more frequent measurements of ADAMTS13 activity are required. If ADAMTS13 activity decreases to <20%, rituximab is recommended to allow recovery of ADAMTS13 activity to normal (≥50%). In approximately 80% of patients in whom ADAMTS13 activity decreases to <20% during remission, activity will return to normal with rituximab, often with only a single administration. If only partial recovery occurs (ADAMTS13 activity 20%‐50%), then no further rituximab is required, but more frequent measurements of ADAMTS13 activity are recommended. If maintenance rituximab for 2 y does not cause recovery of ADAMTS13 activity to <20%, alternative treatments (eg cyclosporine, splenectomy) may be considered or the patient may be followed without further treatment.
The pathophysiological mechanisms underlying the different scenarios in iTTP observed after treatment with rituximab (ie, durable response, temporary response with subsequent recurrence of anti‐ADAMTS13 autoantibodies, refractoriness) as well as the specific B‐ and T‐cell and plasma cell subpopulations involved still remain to be elucidated.45 Future studies should also assess whether rituximab selects for survival of long‐lived plasma cells involved in the production of anti‐ADAMTS13 autoantibodies, which would raise new therapeutic challenges.46, 47 Splenectomy has also been reported to decrease relapses with an acceptable safety profile and may therefore represent an alternative to rituximab.4, 48, 49 A comparison of the efficacy of splenectomy versus rituximab to prevent relapse deserves further investigation, possibly through a large international registry.
10.2. Long‐term follow‐up: connective tissue diseases, cognitive disturbances, and greater risk of death
iTTP is associated with an increased risk for other autoimmune diseases (mostly systemic lupus erythematosus and Sjögren's syndrome), which may present before, concomitant with, or after diagnosis of iTTP. The cumulative incidence of autoimmune disorders after iTTP diagnosis is 9.9% after 5 years, 13.5% after 10 years, and 25.9% after 12 years.50, 51 The incidence is higher after rather than before the first iTTP episode and increases over time, suggesting a role for increasing age.
Recent data have emphasized that after recovery from acute episodes of iTTP, patients report minor cognitive abnormalities as well as problems with concentration and endurance, possibly resulting from posttraumatic stress disorder. Such complications need to be recognized and appropriately managed. Of note, neither depression nor cognitive impairment was significantly associated with the occurrence of relapses or ADAMTS13 activity <10%‐20% during remission.52 Moreover, prevalence of hypertension and major depression is greater as is mortality among iTTP survivors in remission compared with a reference population.51 The excess of early death in patients with a history of iTTP remains unexplained and could result from sudden undiagnosed relapses, increased cardiovascular risk related to persistent severe ADAMTS13 deficiency, organ damage following the acute episode, or other causes.50
The long‐term complications of iTTP were described and recognized only recently and emphasize the need to consider iTTP as a chronic disease. Accordingly, patients should receive long‐term follow‐up and monitoring for associated autoimmune diseases and manifestations of post‐traumatic stress disorder. Future studies should assess whether new targeted therapies lower the burden of long‐term complications.
11. HOW TO IMPROVE TTP MANAGEMENT?
11.1. Make clinicians more aware of TTP diagnosis
The rapid diagnosis and early initiation of treatment are clearly related to a favorable outcome. Diagnosis of iTTP in an emergency setting is challenged by the rarity of the disease and nonspecificity of presenting signs and symptoms, which may lead to a delay in initiation of treatment.11, 21 Although the diagnosis of iTTP is still delayed in some patients, clinicians are becoming increasingly aware of this condition. In line with this statement, the frequency of organ failure (especially cerebral involvement) has decreased in the last 10 years.12, 53 In an attempt to further improve management of patients with iTTP, various measures are being developed in a growing number of countries. These include educational programs for generalists, emergency department physicians, and other specialists aimed at enhancing recognition and improving management of the disease. Importantly, there should also be educational programs for patients about the typical features suggestive of a relapse.
11.2. More rapidly identify the most severely affected patients
The other important cause of death that must be considered is primary refractory disease. Primary refractory disease is characterized by the absence of platelet count improvement under standard treatment by day 4 and organ involvement, often manifesting as an increased cardiac troponin. These patients require earlier intensified treatment.9, 31, 54, 55 The prompt availability of ADAMTS13 activity or the use of surrogate markers12, 13, 56 to predict severe enzyme deficiency, coupled with early prognostic markers such as cardiac troponin and ADAMTS13 antigen/anti‐ADAMTS13 autoantibody titers,9, 55 could facilitate adjustment of initial treatment to the severity of the disease and improve the historical 10%‐15% death rate in the acute phase.
11.3. Reference centers with experienced practitioners
TTP and other TMAs represent rare diseases requiring a high level of skill for their management. These disorders require dedicated national or regional reference centers which have been established in an increasing number of countries.57, 58 In France, the Rare Diseases National Plan provides expertise for management of rare disorders, a platform for conducting studies, and resources and information for patients, their families, and their physicians. Telemedicine allows reference centers to provide services to remote locations. TTP and the other TMA syndromes may be particularly amenable to telemedicine approaches because of the need to answer reports promptly at all hours of the day.58 Future work should evaluate the effectiveness of telemedicine activity from reference centers on outcomes in patients with rare diseases including iTTP.
11.4. Improve the level of evidence of therapeutic studies in TTP
Clinical studies in iTTP have been limited by the low incidence of this disease. In particular, conventional methods (eg, randomized controlled trials) are challenging in TTP,21 except for extremely expensive and lengthy pharmaceutical trials.14 However, over the past few years, several groups have established large registries that include hundreds of patients. These registries have shed light on the epidemiology, clinical presentation, prognosis, and long‐term outcomes of the disease.12, 28, 55, 56, 59, 60, 61, 62 Those efforts also provide evidence that collaborations at the national and international level remain key to the continued advancement of knowledge and treatment of rare diseases.14 Collaborative efforts have led to proposal of consensus treatment modalities and definitions of treatment responses based on large series of patients.15 Though arbitrary and based only on clinical experience, these consensuses foster a common language and study designs that will facilitate meta‐analyses and data synthesis in the future. There is no doubt that the understanding of iTTP will require close collaboration between multiples disciplines, including hematology, transfusion medicine, nephrology, internal medicine, immunology, and intensive care medicine.
12. UNANSWERED QUESTIONS IN TTP MANAGEMENT
As new standards in the management of iTTP are defined15 and new treatments are implemented, new questions will need to be addressed, ideally through international collaborative studies:
Are diagnostic algorithms, (particularly platelet count and serum creatinine level),4, 13, 14 which are easily available in real‐time, sufficiently accurate to begin treatments while awaiting the report of ADAMTS13 activity? Such an approach would allow the immediate use of targeted therapies (B‐cell depleting therapies, caplacizumab, and possibly recombinant ADAMTS13) in addition to TPE and corticosteroids;
Just as rituximab is being progressively incorporated into standard initial therapy along with TPE and corticosteroids,4, 5 should we also consider caplacizumab as a new standard of care for all iTTP patients from diagnosis? Such a strategy would be expected to facilitate faster platelet count and organ damage recovery and protect patients from exacerbations until ADAMTS13 recovery14;
Should preemptive treatment be recommended for all patients with severe (activity <10%‐20%) immune‐mediated ADAMTS13 deficiency in clinical remission? On one hand, this strategy significantly decreases the incidence of relapse and is well tolerated. On the other hand, 50% of patients need multiple courses of rituximab over time to maintain a detectable ADAMTS13 activity, exposing them to possible side effects,40, 42 and some patients with severe ADAMTS13 deficiency do not relapse;
Could a recombinant form of ADAMTS13 improve outcomes in patients with iTTP by saturating anti‐ADAMTS13 antibodies and cleaving ultra‐large vWF multimers? One could speculate that recombinant ADAMTS13 may reduce the number of TPE sessions or even replace TPE;
Future studies are needed to define the efficacy of additional therapeutic agents. In patients who are refractory to rituximab, it has been hypothesized that long‐lived plasma cells could be the source of anti‐ADAMTS13 antibodies, suggesting a potential role for anti‐plasma cell strategies. So far, promising results were reported with the proteasome inhibitor bortezomib,37, 63 which suggests that monoclonal antibodies targeting the CD38 antigen on plasma cells could also be of interest. N‐acetylcysteine (NAC), an FDA‐approved mucolytic agent, was demonstrated to reduce disulfide bonds in vWF, thereby decreasing VWF multimer size.64 Although animal models suggested efficacy of NAC as a prophylactic agent, the administration of NAC was not effective in treating TTP even though a reduction in vWF multimers was observed.65 A Phase I trial is in progress to assess the possible efficacy of this compound in clinical practice (ClinicalTrials.gov NCT018008521).
13. CONCLUSION: TTP AS A SUCCESSFUL EXAMPLE FOR TRANSLATIONAL MEDICINE
The prospect of targeted therapies promises to alter the landscape of TTP therapy in the coming years. In particular, the combination of recombinant ADAMTS13 in association with immunomodulation (steroids and B‐cell depletion) and caplacizumab may constitute a therapeutic triplet that reduces the number of TPE sessions, shortens hospital stay, and improves survival. TTP has entered the era of targeted therapies and the history of this disease can be considered as a convincing example of the success of translational medicine.
RELATIONSHIP DISCLOSURES
PC is member of the Advisory Boards of Ablynx for the development of caplacizumab, Alexion for the development of eculizumab and Shire for the development of Bax930. He has received funds from Ablynx, Alexion, Octapharma, and Roche. The French Reference Center for Thrombotic Microangiopathies (www.cnr-mat.fr) is in part supported by the French Ministry of Health (Plan National Maladies Rares, Direction Générale de l'Offre de Soin) and the Programme Hospitalier de Recherche Clinique (PHRC 2012 P120118, Clinicaltrials.gov identifier NCT02134171). AC has served as a consultant or advisory board member for Bioverativ, Genzyme, Kedrion, Stago, and Synergy and his institution has received research support on his behalf from Alexion, Bayer, Bioverativ, Novo Nordisk, Pfizer, Shire, and Spark. JG has no competing interests.
AUTHOR CONTRIBUTIONS
PC, AC, and JNG wrote the manuscript. All authors agreed the submitted version of the manuscript.
Coppo P, Cuker A, George JN. Thrombotic thrombocytopenic purpura: Toward targeted therapy and precision medicine. Res Pract Thromb Haemost. 2019;3:26–37. 10.1002/rth2.12160
Contributor Information
Paul Coppo, Email: paul.coppo@aphp.fr, @Filiere_MaRIH.
Adam Cuker, @CukerMd.
REFERENCES
- 1. Rock GA, Shumak KH, Buskard NA, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. Canadian Apheresis Study Group. N Engl J Med. 1991;325:393–7. [DOI] [PubMed] [Google Scholar]
- 2. Furlan M, Robles R, Galbusera M, et al. von Willebrand factor‐cleaving protease in thrombotic thrombocytopenic purpura and the hemolytic‐uremic syndrome. N Engl J Med. 1998;339:1578–84. [DOI] [PubMed] [Google Scholar]
- 3. Tsai HM, Lian EC. Antibodies to von Willebrand factor‐cleaving protease in acute thrombotic thrombocytopenic purpura. N Engl J Med. 1998;339:1585–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kremer Hovinga JA, Coppo P, Lammle B, Moake JL, Miyata T, Vanhoorelbeke K. Thrombotic thrombocytopenic purpura. Nat Rev Dis Primers. 2017;3:17020. [DOI] [PubMed] [Google Scholar]
- 5. Joly BS, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura. Blood. 2017;129:2836–46. [DOI] [PubMed] [Google Scholar]
- 6. Mariotte E, Azoulay E, Galicier L, et al. Epidemiology and pathophysiology of adulthood‐onset thrombotic microangiopathy with severe ADAMTS13 deficiency (thrombotic thrombocytopenic purpura): a cross‐sectional analysis of the French national registry for thrombotic microangiopathy. Lancet Haematol. 2016;3:e237–45. [DOI] [PubMed] [Google Scholar]
- 7. Page EE, Kremer Hovinga JA, Terrell DR, Vesely SK, George JN. Thrombotic thrombocytopenic purpura: diagnostic criteria, clinical features, and long‐term outcomes from 1995 through 2015. Blood Adv. 2017;1:590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hughes C, McEwan JR, Longair I, et al. Cardiac involvement in acute thrombotic thrombocytopenic purpura: association with troponin T and IgG antibodies to ADAMTS 13. J Thromb Haemost. 2009;7:529–36. [DOI] [PubMed] [Google Scholar]
- 9. Benhamou Y, Boelle PY, Baudin B, et al. Cardiac troponin‐I on diagnosis predicts early death and refractoriness in acquired thrombotic thrombocytopenic purpura. Experience of the French Thrombotic Microangiopathies Reference Center. J Thromb Haemost. 2015;13:293–302. [DOI] [PubMed] [Google Scholar]
- 10. Jamme M, Raimbourg Q, Chauveau D, et al. Predictive features of chronic kidney disease in atypical haemolytic uremic syndrome. PLoS One. 2017;12:e0177894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Grall M, Azoulay E, Galicier L, et al. Thrombotic thrombocytopenic purpura misdiagnosed as autoimmune cytopenia: causes of diagnostic errors and consequence on outcome. Experience of the French thrombotic microangiopathies reference centre. Am J Hematol. 2017;92:381–7. [DOI] [PubMed] [Google Scholar]
- 12. Coppo P, Schwarzinger M, Buffet M, et al. Predictive features of severe acquired ADAMTS13 deficiency in idiopathic thrombotic microangiopathies: the French TMA reference center experience. PLoS One. 2010;5:e10208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bendapudi PK, Hurwitz S, Fry A, et al. Derivation and external validation of the PLASMIC score for rapid assessment of adults with thrombotic microangiopathies: a cohort study. Lancet Haematol. 2017;4:e157–64. [DOI] [PubMed] [Google Scholar]
- 14. Scully M, Cataland S, Peyvandi F, et al. Results of the randomized, double‐blind, placebo‐controlled, phase 3 HERCULES study of caplacizumab in patients with acquired thrombotic thrombocytopenic purpura. Blood. 2017;130(Suppl 1):LBA–1. [Google Scholar]
- 15. Scully M, Cataland S, Coppo P, et al. Consensus on the standardization of terminology in thrombotic thrombocytopenic purpura and related thrombotic microangiopathies. J Thromb Haemost. 2016;15(2):312–22. [DOI] [PubMed] [Google Scholar]
- 16. Ayanambakkam A, Kremer Hovinga JA, Vesely SK, George JN. Diagnosis of thrombotic thrombocytopenic purpura among patients with ADAMTS13 Activity 10%‐20. Am J Hematol. 2017;92:E644–6. [DOI] [PubMed] [Google Scholar]
- 17. Bell WR, Braine HG, Ness PM, Kickler TS. Improved survival in thrombotic thrombocytopenic purpura‐hemolytic uremic syndrome. Clinical experience in 108 patients. N Engl J Med. 1991;325:398–403. [DOI] [PubMed] [Google Scholar]
- 18. McClain RS, Terrell DR, Vesely SK, George JN. Plasma exchange complications in patients treated for thrombotic thrombocytopenia purpura‐hemolytic uremic syndrome: 2011 to 2014. Transfusion. 2014;54:3257–9. [DOI] [PubMed] [Google Scholar]
- 19. Cataland SR, Kourlas PJ, Yang S, et al. Cyclosporine or steroids as an adjunct to plasma exchange in the treatment of immune‐mediated thrombotic thrombocytopenic purpura. Blood Adv. 2017;1:2075–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Balduini CL, Gugliotta L, Luppi M, et al. High versus standard dose methylprednisolone in the acute phase of idiopathic thrombotic thrombocytopenic purpura: a randomized study. Ann Hematol. 2010;89:591–6. [DOI] [PubMed] [Google Scholar]
- 21. Uhl L, Kiss JE, Malynn E, Terrell DR, Vesely SK, George JN. Rituximab for thrombotic thrombocytopenic purpura: lessons from the STAR trial. Transfusion. 2017;57:2532–8. [DOI] [PubMed] [Google Scholar]
- 22. McDonald V, Manns K, Mackie IJ, Machin SJ, Scully MA. Rituximab pharmacokinetics during the management of acute idiopathic thrombotic thrombocytopenic purpura. J Thromb Haemost. 2010;8:1201–8. [DOI] [PubMed] [Google Scholar]
- 23. Froissart A, Buffet M, Veyradier A, et al. Efficacy and safety of first‐line rituximab in severe, acquired thrombotic thrombocytopenic purpura with a suboptimal response to plasma exchange. Experience of the French Thrombotic Microangiopathies Reference Center. Crit Care Med. 2012;40:104–11. [DOI] [PubMed] [Google Scholar]
- 24. Westwood JP, Webster H, Guckin SM, McDonald V, Machin SJ, Scully M. Rituximab for thrombotic thrombocytopenic purpura (TTP): benefit of early administration during acute episodes and use of prophylaxis to prevent relapse. J Thromb Haemost. 2013;11(3):481–90. [DOI] [PubMed] [Google Scholar]
- 25. Benhamou Y, Paintaud G, Azoulay E, et al. Efficacy of a rituximab regimen based on B cell depletion in thrombotic thrombocytopenic purpura with suboptimal response to standard treatment: results of a phase II, multicenter noncomparative study. Am J Hematol. 2016;91:1246–51. [DOI] [PubMed] [Google Scholar]
- 26. Scully M, McDonald V, Cavenagh J, et al. A phase 2 study of the safety and efficacy of rituximab with plasma exchange in acute acquired thrombotic thrombocytopenic purpura. Blood. 2011;118:1746–53. [DOI] [PubMed] [Google Scholar]
- 27. Page EE, Kremer Hovinga JA, Terrell DR, Vesely SK, George JN. Rituximab reduces risk for relapse in patients with thrombotic thrombocytopenic purpura. Blood. 2016;127(24):3092–4. [DOI] [PubMed] [Google Scholar]
- 28. Hovinga JA, Vesely SK, Terrell DR, Lammle B, George JN. Survival and relapse in patients with thrombotic thrombocytopenic purpura. Blood. 2010;115:1500–11; quiz 1662. [DOI] [PubMed] [Google Scholar]
- 29. Rizvi MA, Vesely SK, George JN, et al. Complications of plasma exchange in 71 consecutive patients treated for clinically suspected thrombotic thrombocytopenic purpura‐hemolytic‐uremic syndrome. Transfusion. 2000;40:896–901. [DOI] [PubMed] [Google Scholar]
- 30. Peyvandi F, Scully M, Kremer Hovinga JA, et al. Caplacizumab for acquired thrombotic thrombocytopenic purpura. N Engl J Med. 2016;374:511–22. [DOI] [PubMed] [Google Scholar]
- 31. Benhamou Y, Assie C, Boelle PY, et al. Development and validation of a predictive model for death in acquired severe ADAMTS13 deficiency‐associated idiopathic thrombotic thrombocytopenic purpura: the French TMA Reference Center experience. Haematologica. 2012;97:1181–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Furlan M, Robles R, Morselli B, Sandoz P, Lammle B. Recovery and half‐life of von Willebrand factor‐cleaving protease after plasma therapy in patients with thrombotic thrombocytopenic purpura. Thromb Haemost. 1999;81:8–13. [PubMed] [Google Scholar]
- 33. Scully M, Knobl P, Kentouche K, et al. Recombinant ADAMTS‐13: first‐in‐human pharmacokinetics and safety in congenital thrombotic thrombocytopenic purpura. Blood. 2017;130:2055–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jian C, Xiao J, Gong L, et al. Gain‐of‐function ADAMTS13 variants that are resistant to autoantibodies against ADAMTS13 in patients with acquired thrombotic thrombocytopenic purpura. Blood. 2012;119:3836–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cannavo A, Valsecchi C, Garagiola I, et al. Nonneutralizing antibodies against factor VIII and risk of inhibitor development in severe hemophilia A. Blood. 2017;129:1245–50. [DOI] [PubMed] [Google Scholar]
- 36. Kremer Hovinga JA, Voorberg J. Improving on nature: redesigning ADAMTS13. Blood. 2012;119:3654–5. [DOI] [PubMed] [Google Scholar]
- 37. Patriquin CJ, Thomas MR, Dutt T, et al. Bortezomib in the treatment of refractory thrombotic thrombocytopenic purpura. Br J Haematol. 2016;173:779–85. [DOI] [PubMed] [Google Scholar]
- 38. Peyvandi F, Lavoretano S, Palla R, et al. ADAMTS13 and anti‐ADAMTS13 antibodies as markers for recurrence of acquired thrombotic thrombocytopenic purpura during remission. Haematologica. 2008;93:232–9. [DOI] [PubMed] [Google Scholar]
- 39. Jin M, Casper TC, Cataland SR, et al. Relationship between ADAMTS13 activity in clinical remission and the risk of TTP relapse. Br J Haematol. 2008;141:651–8. [DOI] [PubMed] [Google Scholar]
- 40. Hie M, Gay J, Galicier L, et al. Preemptive rituximab infusions after remission efficiently prevent relapses in acquired thrombotic thrombocytopenic purpura: experience of the French Thrombotic Microangiopathies Reference Center. Blood. 2014;124(2):204–10. [DOI] [PubMed] [Google Scholar]
- 41. Westwood JP, Thomas M, Alwan F, et al. Rituximab prophylaxis to prevent thrombotic thrombocytopenic purpura relapse: outcome and evaluation of dosing regimens. Blood Adv. 2017;1:1159–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jestin M, Benhamou Y, Schelpe AS, et al. Preemptive rituximab prevents long‐term relapses in immune‐mediated thrombotic thrombocytopenic purpura. Blood. 2018;132(20):2143–53. [DOI] [PubMed] [Google Scholar]
- 43. Sadler JE. Pathophysiology of thrombotic thrombocytopenic purpura. Blood. 2017;130:1181–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. van Vollenhoven RF, Fleischmann RM, Furst DE, Lacey S, Lehane PB. Longterm safety of rituximab: final report of the Rheumatoid Arthritis Global Clinical Trial Program over 11 years. J Rheumatol. 2015;42:1761–6. [DOI] [PubMed] [Google Scholar]
- 45. Hrdinova J, D'Angelo S, Graca NAG, et al. Dissecting the pathophysiology of immune thrombotic thrombocytopenic purpura: interplay between genes and environmental triggers. Haematologica. 2018;103(7):1099–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mahevas M, Patin P, Huetz F, et al. B cell depletion in immune thrombocytopenia reveals splenic long‐lived plasma cells. J Clin Invest. 2013;123:432–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Thai LH, Le Gallou S, Robbins A, et al. BAFF and CD4(+) T cells are major survival factors for long‐lived splenic plasma cells in a B‐cell‐depletion context. Blood. 2018;131:1545–55. [DOI] [PubMed] [Google Scholar]
- 48. Kremer Hovinga JA, Studt JD, Demarmels Biasiutti F, et al. Splenectomy in relapsing and plasma‐refractory acquired thrombotic thrombocytopenic purpura. Haematologica. 2004;89:320–4. [PubMed] [Google Scholar]
- 49. Kappers‐Klunne MC, Wijermans P, Fijnheer R, et al. Splenectomy for the treatment of thrombotic thrombocytopenic purpura. Br J Haematol. 2005;130:768–76. [DOI] [PubMed] [Google Scholar]
- 50. Deford CC, Reese JA, Schwartz LH, et al. Multiple major morbidities and increased mortality during long‐term follow‐up after recovery from thrombotic thrombocytopenic purpura. Blood. 2013;122:2023–9; quiz 2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Roriz M, Landais M, Desprez J, et al. Risk factors for autoimmune diseases development after thrombotic thrombocytopenic purpura. Medicine (Baltimore). 2015;94:e1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Han B, Page EE, Stewart LM, et al. Depression and cognitive impairment following recovery from thrombotic thrombocytopenic purpura. Am J Hematol. 2015;90:709–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Veyradier A, Obert B, Houllier A, Meyer D, Girma JP. Specific von Willebrand factor‐cleaving protease in thrombotic microangiopathies: a study of 111 cases. Blood. 2001;98:1765–72. [DOI] [PubMed] [Google Scholar]
- 54. Soucemarianadin M, Benhamou Y, Delmas Y, et al. Twice‐daily therapeutical plasma exchange‐based salvage therapy in severe autoimmune thrombotic thrombocytopenic purpura: the French TMA Reference Center experience. Eur J Haematol. 2016;97:183–91. [DOI] [PubMed] [Google Scholar]
- 55. Alwan F, Vendramin C, Vanhoorelbeke K, et al. Presenting ADAMTS13 antibody and antigen levels predict prognosis in immune‐mediated thrombotic thrombocytopenic purpura. Blood. 2017;130:466–71. [DOI] [PubMed] [Google Scholar]
- 56. Bentley MJ, Lehman CM, Blaylock RC, Wilson AR, Rodgers GM. The utility of patient characteristics in predicting severe ADAMTS13 deficiency and response to plasma exchange. Transfusion. 2010;50:1654–64. [DOI] [PubMed] [Google Scholar]
- 57. Dutt T, Scully M. A proposal: the need for thrombotic thrombocytopenic purpura Specialist Centres—providing better outcomes. Br J Haematol. 2015;170:737–42. [DOI] [PubMed] [Google Scholar]
- 58. Coppo P, Corre E, Rondeau E, et al. Telemedicine in thrombotic microangiopathies: a way forward in rare diseases requiring emergency care. Rev Med Interne. 2016;37:514–20. [DOI] [PubMed] [Google Scholar]
- 59. Raife T, Atkinson B, Montgomery R, Vesely S, Friedman K. Severe deficiency of VWF‐cleaving protease (ADAMTS13) activity defines a distinct population of thrombotic microangiopathy patients. Transfusion. 2004;44:146–50. [DOI] [PubMed] [Google Scholar]
- 60. Cataland SR, Yang SB, Witkoff L, et al. Demographic and ADAMTS13 biomarker data as predictors of early recurrences of idiopathic thrombotic thrombocytopenic purpura. Eur J Haematol. 2009;83:559–64. [DOI] [PubMed] [Google Scholar]
- 61. Lotta LA, Mariani M, Consonni D, et al. Different clinical severity of first episodes and recurrences of thrombotic thrombocytopenic purpura. Br J Haematol. 2010;151(5):488–94. [DOI] [PubMed] [Google Scholar]
- 62. Fujimura Y, Matsumoto M. Registry of 919 patients with thrombotic microangiopathies across Japan: database of Nara Medical University during 1998‐2008. Intern Med. 2010;49:7–15. [DOI] [PubMed] [Google Scholar]
- 63. Shortt J, Oh DH, Opat SS. ADAMTS13 antibody depletion by bortezomib in thrombotic thrombocytopenic purpura. N Engl J Med. 2013;368:90–2. [DOI] [PubMed] [Google Scholar]
- 64. Chen J, Reheman A, Gushiken FC, et al. N‐acetylcysteine reduces the size and activity of von Willebrand factor in human plasma and mice. J Clin Invest. 2011;121:593–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tersteeg C, Roodt J, Van Rensburg WJ, et al. N‐acetylcysteine in preclinical mouse and baboon models of thrombotic thrombocytopenic purpura. Blood. 2017;129:1030–8. [DOI] [PubMed] [Google Scholar]