

Abstract
Key points
Significant and selective up‐regulation of the Na+/H+ exchanger NHA2 (SLC9B2) was observed in cysts of patients with autosomal dominant polycystic kidney disease.
Using the MDCK cell model of cystogenesis, it was found that NHA2 increases cyst size. Silencing or pharmacological inhibition of NHA2 inhibits cyst formation in vitro.
Polycystin‐1 represses NHA2 expression via Ca2+/NFAT signalling whereas the dominant negative membrane‐anchored C‐terminal fragment (PC1‐MAT) increased NHA2 levels.
Drugs (caffeine, theophylline) and hormones (vasopressin, aldosterone) known to exacerbate cysts elicit NHA2 expression.
Taken together, the findings reveal NHA2 as a potential new player in salt and water homeostasis in the kidney and in the pathogenesis of polycystic kidney disease.
Abstract
Autosomal dominant polycystic kidney disease (ADPKD) is caused by mutations in PKD1 and PKD2 encoding polycystin‐1 (PC1) and polycystin‐2 (PC2), respectively. The molecular pathways linking polycystins to cyst development in ADPKD are still unclear. Intracystic fluid secretion via ion transporters and channels plays a crucial role in cyst expansion in ADPKD. Unexpectedly, we observed significant and selective up‐regulation of NHA2, a member of the SLC9B family of Na+/H+ exchangers, that correlated with cyst size and disease severity in ADPKD patients. Using three‐dimensional cultures of MDCK cells to model cystogenesis in vitro, we showed that ectopic expression of NHA2 is causal to increased cyst size. Induction of PC1 in MDCK cells inhibited NHA2 expression with concordant inhibition of Ca2+ influx through store‐dependent and ‐independent pathways, whereas reciprocal activation of Ca2+ influx by the dominant negative membrane‐anchored C‐terminal tail fragment of PC1 elevated NHA2. We showed that NHA2 is a target of Ca2+/NFAT signalling and is transcriptionally induced by methylxanthine drugs such as caffeine and theophylline, which are contraindicated in ADPKD patients. Finally, we observed robust induction of NHA2 by vasopressin, which is physiologically consistent with increased levels of circulating vasopressin and up‐regulation of vasopressin V2 receptors in ADPKD. Our findings have mechanistic implications on the emerging use of vasopressin V2 receptor antagonists such as tolvaptan as safe and effective therapy for polycystic kidney disease and reveal a potential new regulator of transepithelial salt and water transport in the kidney.
Keywords: Na+/H+ exchanger, polycystin, vasopressin, Ca2+ signaling, NFAT
Key points
Significant and selective up‐regulation of the Na+/H+ exchanger NHA2 (SLC9B2) was observed in cysts of patients with autosomal dominant polycystic kidney disease.
Using the MDCK cell model of cystogenesis, it was found that NHA2 increases cyst size. Silencing or pharmacological inhibition of NHA2 inhibits cyst formation in vitro.
Polycystin‐1 represses NHA2 expression via Ca2+/NFAT signalling whereas the dominant negative membrane‐anchored C‐terminal fragment (PC1‐MAT) increased NHA2 levels.
Drugs (caffeine, theophylline) and hormones (vasopressin, aldosterone) known to exacerbate cysts elicit NHA2 expression.
Taken together, the findings reveal NHA2 as a potential new player in salt and water homeostasis in the kidney and in the pathogenesis of polycystic kidney disease.
Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is a highly prevalent hereditary disease, affecting one in 400–1000 humans (Harris & Rossetti, 2010; Ong et al. 2015). ADPKD accounts for up to 10% of end‐stage renal disease cases, making it one of the leading causes of kidney failure (Ong et al. 2015). A better understanding of the underlying pathophysiology is key to management of ADPKD and related disorders. ADPKD is caused by mutations in PKD1 and PKD2, encoding polycystin‐1 (PC1) and polycystin‐2 (PC2), respectively. PC1 is a transmembrane protein with a large extra‐cytoplasmic N‐terminal domain, which is thought to function as a receptor, and a C‐terminal cytoplasmic tail (Hughes et al. 1995; Nims et al. 2003). PC2 is a transmembrane protein that functions as a non‐selective Ca2+ channel. PC1 interacts with PC2 via C‐terminal coil‐coiled domains that are required for proper function and trafficking (Qian et al. 1997; Kim et al. 2014). The C‐terminal cytoplasmic tail of PC1 can undergo proteolytic cleavage and nuclear translocation leading to activation of signal transducer and activator of transcription (STAT) signalling (Talbot et al. 2011). In renal tubular cells, the polycystins localize to the primary cilium where they regulate intracellular Ca2+ and cAMP levels in response to mechano‐stimulating urinary flow. A disruption of PC1–PC2 interaction is thought to lead to cyst formation due to abnormal cellular proliferation and fluid secretion, although the specific molecular pathways that connect the underlying genetic defects to disease pathogenesis and cyst development are poorly understood (Harris & Rossetti, 2010; Ong et al. 2015).
In this context, increased plasma membrane Na+/H+ exchange (NHE) activity has been reported in cilium‐deficient collecting duct cells from the Oak Ridge mouse model of polycystic kidney disease (Avner et al. 2012; Olteanu et al. 2012). Apical mislocalization of NHE1 in these cells leads to hyperabsorption of Na+ and epidermal growth factor‐induced cell proliferation (Olteanu et al. 2012; Coaxum et al. 2014). The potential role of other Na+/H+ exchanger isoforms in polycystic kidney disease (PKD) pathophysiology has not been explored. Na+/H+ exchangers belong to the superfamily of monovalent cation/proton antiporters (CPA) that share a common transmembrane organization of 12–14 hydropathic helices with detectable sequence similarity. The CPA1 subgroup includes the electroneutral NHE family of Na+(K+)/H+ exchangers represented by the well‐characterized plasma membrane transporter NHE1 (Donowitz et al. 2013; Fuster & Alexander, 2014; Kondapalli et al. 2014). More recently, a new clade of CPA2 genes was discovered in animals, sharing ancestry with electrogenic bacterial NapA and NhaA antiporters (Xiang et al. 2007; Fuster et al. 2008). Of the two human CPA2 gene products, expression of NHA2 has ubiquitous tissue distribution, whereas the closely related NHA1 isoform is restricted to testis (Xiang et al. 2007; Fuster et al. 2008; Chen et al. 2016). Despite the multiplicity of genes encoding Na+/H+ exchangers in mammals, emerging evidence indicates that CPA1 and CPA2 subtypes have distinct, non‐redundant transport roles based on differences in chemiosmotic coupling, inhibitor sensitivity and pH activation (Kondapalli et al. 2012). At the plasma membrane, NHE1 couples proton extrusion to the inwardly directed Na+ electrochemical gradient, established by the Na+/K+‐ATPase, to mediate salt reabsorption in epithelia and recovery from acid load. Consistent with these functions, NHE isoforms are activated by low cytoplasmic pH. In contrast, NHA2 has an alkaline pH optimum and appears to couple inward transport of protons to mediate sodium (or lithium) efflux at the cell membrane, recapitulating the function of the bacterial CPA2 orthologue NhaA (Fuster et al. 2008; Schushan et al. 2010; Kondapalli et al. 2012; Donowitz et al. 2013).
Human NHA2 has been implicated as a marker of essential hypertension, with potential roles in kidney diseases relating to salt and water balance (Xiang et al. 2007; Schushan et al. 2010; Kondapalli et al. 2017). NHA2 expression in kidney is confined to the distal nephron and renal collecting duct, regions that play critical roles in salt, pH and volume homeostasis (Fuster et al. 2008). NHA2 is localized to both principal cells and intercalated cells of the distal tubule in vivo. Principal cells maintain sodium and water balance and the intercalated cells control acid–base homeostasis (Kondapalli et al. 2017). Although the physiological function of NHA2 is largely obscure, defining the pathways regulating NHA2 expression is an important first step towards deciphering its potential role in hypertension and kidney disease.
The nuclear factor of activated T‐cells (NFAT) family of transcription factors has been implicated in the regulation of genes that control numerous aspects of normal physiology including cell cycle progression and differentiation, and in pathological conditions such as inflammation and tumorigenesis (Pan et al. 2013). The TNF family member receptor activator of NF‐κB ligand (RANKL) elicits Ca2+ oscillations in differentiating osteoclasts that converge on the calcineurin/NFAT pathway (Kim & Kim, 2014). During RANKL‐induced osteoclast differentiation, NHA2 ranked among the top five NFATc1‐dependent transcripts, although the physiological role of this up‐regulation is yet to be determined (Charles et al. 2012). Consistent with these results, an independent study revealed strong down‐regulation of NFATc1 expression and consequently, >30‐fold lower NHA2 levels in macrophage colony stimulation factor (M‐CSF)‐ and RANKL‐primed osteoclast precursor myeloid blasts seeded on plastic, relative to cells seeded on bone (De Vries et al. 2015). Similarly, independent studies have reported NFAT‐mediated regulation of NHA2 expression in T cells (Blomberg et al. 2009; Martinez et al. 2015). Furthermore, NFAT activation might also underlie recent reports of TNF‐α‐induced up‐regulation of NHA2 expression in human endothelial cells (Knyazev et al. 2018). In this study, we extend the link between NFAT and NHA2 expression to an in vitro model of PKD.
A key aspect of precision medicine is to harness patient databases to uncover novel risk factors and potential therapeutic targets. This approach led to our unexpected discovery of selective and significant up‐regulation of NHA2 expression in PKD. Using an established Madin–Darby canine kidney (MDCK) cell model of in vitro cystogenesis we show that NHA2 expression correlates with cyst size, replicating patient data. PC1‐mediated Ca2+ transients and NFAT signalling regulate NHA2 expression, providing a mechanistic link to PKD pathogenesis. To extend physiological relevance, we demonstrate robust vasopressin‐mediated NHA2 expression, which is consistent with increased levels of circulating vasopressin and up‐regulation of vasopressin V2 receptors in PKD. Importantly, pharmacological inhibition of the NHA2 transporter drastically attenuated cyst size. Taken together, our findings reveal a hitherto under‐recognized role for Na+/H+ exchange activity in PKD progression and allow us to propose NHA2 as a potential chemotherapeutic target.
Methods
Cell culture
A stable MDCK cell line with inducible PC1 expression was generated using the Flp‐In System (Thermo Fisher Scientific, Waltham, MA, USA) by transfecting a pcDNA5/FRT/TO‐based wild‐type mPkd1 expression plasmid according to the manufacturer's protocol (Cebotaru et al. 2014). These cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 150 μg ml−1 hygromycin and 5 μg ml−1 blasticidin, with PC1 expression induced with tetracycline (2 μg ml−1) for 17 h. A stable MDCK cell line with NHA2 expression was generated by transfecting the pEGFPC2 vector carrying human NHA2 tagged to green fluorescent protein (GFP) at the N terminus and selecting for G418 antibiotic resistant clones (Kondapalli et al. 2012). MDCK cells (control and NHA2+) were cultured in minimum essential medium (MEM) supplemented with 10% fetal bovine serum. HEK293 cells were cultured in DMEM supplemented with 10% fetal bovine serum. Culture was in a 5% CO2 incubator at 37°C. Three‐dimensional cysts were cultured in 10% Matrigel with a 100% Matrigel basement membrane and grown in full medium, which was changed every 2 days (Yang et al. 2008, Sun et al. 2011). MDCK hemicysts in monolayer cultures were generated as previously described (Lever, 1979; Su et al. 2007). Hormonal treatment protocols (1 μM vasopressin or 100 nM aldosterone) were previously reported to regulate transporter expression and function in MDCK cells (Stewart et al. 2009; Carmosino et al. 2011). Lithium sensitivity was determined by measuring cell growth in the presence of medium containing 90–100 mm LiCl. Cell growth was quantified using a 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyl‐tetrazolium bromide (MTT) assay (Thermo Fisher Scientific), following the manufacturer's instructions (Kondapalli et al. 2012).
Plasmids and transfection
PC1 full‐length plasmid was a gift from Dr Gregory Germino (Addgene plasmid, cat. no. 21369). Membrane anchored C‐terminal tail fragment of PC1 (PC1‐MAT) plasmid was a gift from Dr Thomas Weimbs (Addgene plasmid, cat. no. 41567). GFP‐tagged NFAT1 (Ong et al. 2015) plasmid was described previously (Feng et al. 2010). Constitutively active NFAT (CA‐NFAT) was a gift from Dr Anjana Rao (Addgene plasmid, cat. no. 11102). pGL3‐NFAT‐luc was a gift from Dr Jerry Crabtree (Addgene plasmid, cat. no. 17870). pSV40‐RL was a gift from Dr Jennifer L. Pluznick (Johns Hopkins University). HsNHA2 short hairpin RNA (shRNA) sequence (5′‐CACTGTAGGCCTTTGTGTTGTTCAAGAGACAACACAAAGGCCTACAGTTTTTTCAATT‐3′) was designed to knockdown NHA2 (ShRNA 1). An additional knockdown construct (ShRNA 2) targeting NHA2 with sequence (5′‐CCGGGCATTGCAGTATTGATACGAACTCGAGTTCGTATCAATACTGCAATGCTTTTTTG‐3′) was purchased from Sigma‐Aldrich (St Louis, MO, USA; cat. no. TRCN0000130075). MDCK cells were transfected using lentiviral packaging and expression. HEK293 cells were transfected using Lipofectamine 2000 reagent, as per the manufacturer's instructions (Thermo Fisher Scientific).
Antibodies and reagents
Specific rabbit polyclonal NHA2 antibody was raised against a 15‐amino‐acid peptide of NHA2 (Xiang et al. 2007; Kondapalli et al. 2012). Mouse monoclonal antibodies used were anti‐PC1 (7E12) (cat. no. sc‐130554, Santa Cruz Biotechnology, Dallas, TX, USA), anti‐α‐tubulin (cat. no. T9026, Sigma‐Aldrich), anti‐β‐actin (cat. no. A5441, Sigma‐Aldrich) and anti‐E‐cadherin (cat. no. 610181, BD Transduction Laboratories, San Jose, CA, USA). Aldosterone (A9477), blasticidin (cat. no. 15205), caffeine (cat. no. C0750), ionomycin (cat. no. I3909), lithium chloride (cat. no. L9650), phloretin (cat. no. P7912), thapsigargin (cat. no. T9033), theophylline (cat. no. T1633), and vasopressin (cat. no. V9879) were obtained from Sigma‐Aldrich. Tetracycline hydrochloride (A1004‐5) and hygromycin B (cat. no. 10843555001) were purchased from Zymo Research (Irvine, CA, USA) and Roche (Basel, Switzerland), respectively.
Bioinformatics
Public datasets were mined to uncover novel mechanisms of gene regulation, as described earlier (Prasad & Rao, 2018). We analysed drug‐induced gene expression signatures from 1078 microarray studies in human cells to identify drugs that significantly altered NHA2 expression. Experimental conditions eliciting a minimum of ±2‐fold change in gene expression were selected for further pathway analysis. Normalized gene expression data were obtained from Genevestigator (Nebion AG, Zürich, Switzerland). Other mammalian gene expression datasets included in the study are GSE7869, GSE37219, GSE50971 and GSE57468.
Cytoplasmic pH measurement
Cytoplasmic pH was measured using pHrodo Red AM Intracellular pH Indicator (cat. no. P35372, Thermo Fisher Scientific), following the manufacturer's instructions. Briefly, cells were rinsed with serum‐free medium and then incubated with 1 μl ml−1 of pHrodo Red AM at 37°C for 30 min. Cells were washed, trypsinized and pH was determined by flow cytometry analysis of ∼10,000 cells in biological triplicates using the FACSCalibur instrument (BD Biosciences, San Jose, CA, USA). Cells were gated on a forward scatter and side scatter to obtain live single cells. A four‐point calibration curve with different pH values (4.5, 5.5, 6.5 and 7.5) was generated using an Intracellular pH Calibration Buffer Kit (cat. no. P35379, Thermo Fisher Scientific) in the presence of 10 μM K+/H+ ionophore nigericin and 10 μM K+ ionophore valinomycin.
Quantitative real‐time PCR
mRNA was isolated using RNeasy Mini kit (cat. no. 74104, Qiagen, Hilden, Germany) with DNaseI (cat. no. 10104159001, Roche) treatment and complementary DNA was synthesized using the high‐Capacity RNA‐to‐cDNA Kit (cat. no. 4387406, Thermo Fisher Scientific), following the manufacturer's instructions. Gene expression was assessed by quantitative real‐time PCR using the following TaqMan gene expression assays: canine: NHA2, Cf02729492_m1; PC2, Cf02690612_m1; glyceraldehyde 3‐phosphate dehydrogenase (GAPDH), Cf04419463_gH; human: NHA2, Hs01104990_m1; GAPDH, Hs02786624_g1.
Western blot
Cells were harvested and lysed with 1% Nonidet P‐40 (Sigma‐Aldrich) supplemented with protease inhibitor cocktail (Roche). Following sonication, cell lysates were centrifuged for 15 min at 16,100 g at 4°C. The protein concentrations were measured using the BCA assay (Thermo Fisher Scientific). Equal amounts of total protein were electrophoretically separated by polyacrylamide gel electrophoresis (NuPAGE) before transferring onto nitrocellulose membranes (Bio‐Rad, Hercules, CA, USA). The membranes were blocked with 5% milk, followed by overnight incubation with primary antibodies and 1h incubation with HRP‐conjugated secondary antibodies (GE Healthcare, Chicago, IL, USA). Amersham 600 chemiluminescence imager system (GE Healthcare) was used to capture images.
Confocal microscopy
To determine colocalization of NHA2–GFP with E‐cadherin, cultured MDCK NHA2+ cells on polyornithine coverslips were pre‐extracted with PHEM buffer (60 mm PIPES, 25 mm HEPES, 10 mm EGTA and 2 mm MgCl2, pH 6.8) containing 0.025% saponin for 2 min, then washed twice for 2 min with PHEM buffer containing 0.025% saponin and 8% sucrose. Following fixation (4% paraformaldehyde and 8% sucrose in phosphate‐buffered saline (PBS)) for 30 min, cells were blocked with 1% BSA and 0.025% saponin in PBS for 1 h. Cells were stained with primary anti‐E‐cadherin antibody (overnight) and Alexa Fluor 568‐conjugated secondary antibody (1 h; Thermo Fisher Scientific). On the other hand, to monitor nuclear translocation of NFAT, cultured HEK293 cells with NFAT–GFP transfection were fixed with a solution of 4% paraformaldehyde in PBS. Following 4′,6‐diamidino‐2‐phenylindole (DAPI) staining, coverslips from both experiments were mounted onto slides using Dako fluorescence mounting medium (Santa Clara, CA, USA) and imaged using an LSM 700 Confocal microscope (Zeiss, Oberkochen, Germany). Confocal imaging was performed with a ×63 oil immersion objective and the fractional colocalization was determined using the JACoP ImageJ plugin.
Calcium imaging
Calcium imaging was performed as we previously described (Feng et al. 2010); 5 × 105 cells were cultured on 25 mm circular coverslips. Cells were briefly washed with PBS and loaded with Fura‐2 AM (Thermo Fisher Scientific, cat. no. F1201) at 1 mg ml−1 in calcium imaging buffer (2 mm CaCl2, 126 mm NaCl, 4.5 mm KCl, 2 mm MgCl2, 10 mm glucose, 20 mm HEPES, pH 7.4). After incubation at room temperature for 30 min, cells were washed 3 times for 5 min in the calcium imaging buffer without Fura‐2 AM. The coverslips were transferred to an imaging chamber in calcium‐free imaging buffer (126 mm NaCl, 4.5 mm KCl, 2 mm MgCl2, 10 mm glucose, 20 mm HEPES, pH 7.4). Cells were imaged with the Metafluor imaging software using a ×40 oil immersion objective on a Zeiss Axio Observer A.1 inverted microscope, and baseline recordings were established prior to further treatment with Ca2+, ionomycin (Sigma‐Aldrich cat. no. I3909) or thapsigargan (Sigma‐Aldrich cat. no. T9033). Cells were excited at 340 nm and 380 nm for 100 ms each, and Fura‐2 AM emission at 505 nm was monitored. Ratiometric recordings were calculated and plotted.
Luciferase assay
Luciferase assay was performed as we previously described (Prasad & Rao, 2018). Briefly, pGL3‐NFAT‐luc (encoding a 3× NFAT binding sequence and firefly luciferase) and pSV40‐RL (encoding for a constitutively active SV40 promoter and Renilla luciferase) plasmids were transiently transfected into HEK293 cells using Lipofectamine 2000 reagent, as per the manufacturer's instructions. The ratio of firefly luciferase and Renilla luciferase, a measure of NFAT activity, was determined using the Dual Luciferase Assay System (cat. no. E1910, Promega, Madison, WI, USA). Data were collected using a FLUOstar Omega automated plate reader (BMG LabTech, Ortenberg, Germany).
Results
NHA2 is up‐regulated in PKD and promotes cyst development in vitro
We sought to investigate differences in transcript levels of plasma membrane isoforms of the SLC9 superfamily of Na+/H+ exchangers in cysts obtained from patients with ADPKD (Song et al. 2009). Transcriptome data from renal cysts of different sizes were obtained from five polycystic kidneys removed for medical reasons, as reported by Song et al. (2009). Transcripts of SLC9A (NHE) subfamily genes remained unchanged (NHE1 and NHE3) or showed modest repression (NHE2) (Fig. 1 A–C). Unexpectedly, we observed significant up‐regulation of a member of the SLC9B subfamily, NHA2, relative to normal kidney tissue that correlated with cyst size and disease severity (Fig. 1 D): 4.6‐fold in large cysts (** P = 0.002), 3.4‐fold in medium cysts (** P = 0.001), 3.1‐fold in small cysts (*** P = 0.0004) and 1.2‐fold in minimally cystic tissue (NS P = 0.49). Based on this observation, we hypothesized that increased expression of NHA2 could contribute to pathophysiology of PKD. To test this hypothesis, we used an established in vitro MDCK cell model of cystogenesis (Yang et al. 2008; Sun et al. 2011). To model NHA2 up‐regulation, we used MDCK cells stably expressing GFP‐tagged NHA2, which colocalizes with the basolateral membrane marker E‐cadherin (Kondapalli et al. 2012; Fig. 1 E–F; Pearson's correlation coefficient = 0.66 ± 0.11; Manders’ coefficient = 0.64 ± 0.17; n = 25).
Figure 1. NHA2 is up‐regulated in polycystic kidney disease.
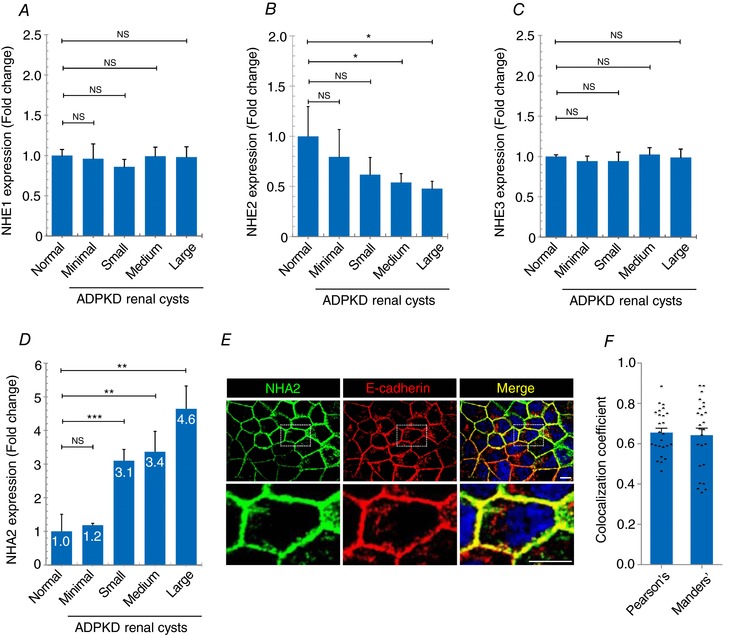
A–C, expression profiling of plasma membrane SLC9A (NHE) genes in ADPKD patient cysts of different sizes showed no change (NHE1 and NHE3) or modest repression (NHE2). D, NHA2 (SLC9B2) expression was increased relative to normal kidney tissue and correlated with cyst size and disease severity. (normal: n = 3; minimal cyst: n = 5; small cyst: n = 5; medium cyst: n = 5; large cyst: n = 3.) Student's t test: NS P > 0.05, * P < 0.05, ** P < 0.01, *** P < 0.001. E, representative confocal microscopic images of MDCK cells stably expressing NHA2–GFP (green) showing colocalization with basolateral marker E‐cadherin (red) in DAPI (blue) stained cells, as seen in the merged image (yellow). Bottom row is higher magnification of boxed region from top row. F, quantification of colocalization. Pearson's correlation coefficient = 0.66 ± 0.11; Manders’ coefficient = 0.64 ± 0.17; n = 25. Scale bar: 10 μm. Error bars are S.E.
Three‐dimensional culturing of MDCK cells in Matrigel produces cysts with polarized, single‐layer, thinned epithelium surrounding a fluid‐filled lumen (Fig. 2 A and B). Similar to PKD kidneys, MDCK cells in cysts undergo proliferation, fluid transport and matrix remodelling. Cysts showed surface expression of NHA2–GFP (Fig. 2 B), similar to monolayer cultures. Consistent with our hypothesis linking NHA2 up‐regulation to cyst development, we observed significant expansion in cyst size with NHA2 expression relative to non‐transfected controls (Fig. 2 C and D). Notably, we documented a dramatic difference in the relative cyst diameter on day 10 of culture (Control: 100 ± 66.4, n = 363; NHA2 up‐regulation: 232.3 ± 213.7, n = 367; **** P < 0.0001). We classified cysts derived from control cells based on their relative diameter: small (<50 percentile), medium (50–90 percentile) and large (>90 percentile). We observed a significantly higher percentage of larger cysts in NHA2+ cells (control: 10% vs. NHA2 up‐regulation: 44%; **** P < 0.0001, chi‐square test; Fig. 2 E). Intriguingly, we also observed several multilayered and multiloculated cysts in NHA2+ MDCK cells that were not seen in cysts derived from control MDCK cells (Fig. 2 F and G). Taken together, these findings led us to hypothesize that NHA2 up‐regulation in polycystic kidney disease promotes cyst development.
Figure 2. NHA2 promotes cyst development in vitro .
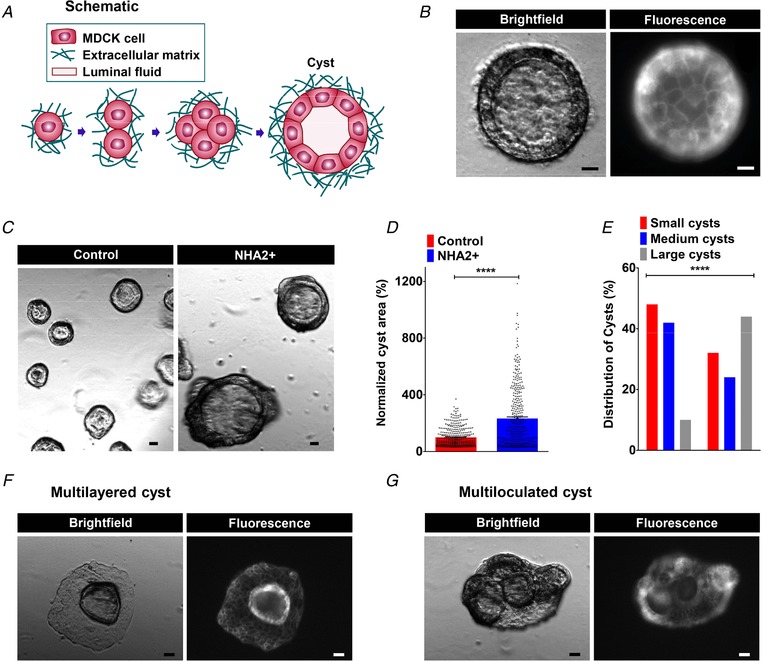
A, schematic depiction of in vitro MDCK cell model of cystogenesis in extracellular matrix (Matrigel). B, representative image of a MDCK cyst with polarized, single‐layer, thinned epithelium surrounding a fluid‐filled lumen (left; brightfield) with surface expression of NHA2–GFP (right; fluorescence). C, NHA2+ MDCK cells formed larger cysts (right) relative to control (left). D, quantification of cyst size on day 10 of culture showed significant cyst expansion from NHA2+ cells relative to control (Control: 100 ± 66.4, n = 363; NHA2+: 232.3 ± 213.7, n = 367; Student's t test, **** P < 0.0001). E, percentage of large cysts is significantly higher from NHA2+ cells as compared to control. Classification of cysts in three subclasses according to their relative diameter (small/medium/large) showed significantly elevated percentage of cysts with large size in NHA2+ and reduced percentage of cysts with small and medium sizes (χ2 test, **** P < 0.0001). F and G, representative images of MDCK cysts documenting mutilayered and multiloculated cysts in NHA2+ MDCK cells (left; brightfield) with surface expression of NHA2–GFP (right; fluorescence) that was not seen in cysts derived from control MDCK cells. Scale bars: 10 μm. All error bars are S.E.
Polycystin 1 downregulates Ca2+ influx and NHA2 expression
Previously, using a well‐characterized MDCK cell model of inducible PC1 expression, we showed that PC1 negatively regulates PC2 levels post‐translationally, by enhancing its degradation via the aggresome–autophagosome pathway (Cebotaru et al. 2014). Using this model, we sought to determine if PC1 induction also alters expression of NHA2 (Fig. 3 A). Tetracycline‐inducible PC1 expression (for 17 h) resulted in significant down‐regulation of NHA2 protein levels by ∼2.9‐fold, relative to non‐induced control (* P = 0.0353; Fig. 3 B and C), similar to our previous findings with PC2 (Cebotaru et al. 2014). Whereas transcript levels of PC2 remained unchanged upon PC1 induction (NS P = 0.663; Fig. 3 D), we observed significantly lower NHA2 mRNA levels, consistent with PC1‐mediated transcriptional down‐regulation of NHA2 (** P = 0.0029; Fig. 3 D). Given that PC2 is a known calcium channel, we next determined if calcium influx is altered by PC1 induction in MDCK cells. We tested two modes of Ca2+ entry: store‐independent Ca2+ entry (SICE) and store‐operated Ca2+ entry (SOCE), both mechanisms that are implicated in normal physiology and pathological conditions (Feng et al. 2010; Cross et al. 2014). In the absence of PC1 induction, MDCK cells showed robust SICE that was significantly and proportionately attenuated upon induction of PC1 for 24 and 48 h (Fig. 3 E and F). Similarly, PC1 induction for 48 h also reduced SOCE following thapsigargin‐mediated release of store Ca2+ (Fig. 3 G), consistent with previous reports of PC1‐mediated inhibition of STIM1 translocation during store depletion (Woodward et al. 2010). Given these concordant observations on NHA2 expression and Ca2+ influx, we investigated whether reduced Ca2+ influx in PC1‐induced cells is causal to transcriptional down‐regulation of NHA2. Treatment with the Ca2+ ionophore ionomycin increased cytoplasmic Ca2+ in PC1‐induced MDCK cells (Fig. 3 H). Importantly, ionomycin resulted in significant and dose‐dependent increase in NHA2 expression to levels similar to MDCK cells without PC1 induction (Fig. 3 I). These observations were independently validated by bioinformatic analysis of a publicly available microarray dataset describing Ca2+‐induced activation of primary T lymphoblasts following treatment with ionomycin and phorbol myristate (Villarroya‐Beltri et al. 2013). Intriguingly, we observed SLC9 gene expression changes similar to ADPKD cysts, including ∼2.4‐fold increase in NHA2 levels, no change in NHE1, and lower, but non‐significant levels of NHE2 in response to Ca2+ influx in these cells (Fig. 4 A–C). Taken together these findings are consistent with Ca2+‐mediated regulation of NHA2 expression by PC1.
Figure 3. Polycystin 1 downregulates Ca2+ influx and NHA2 expression.
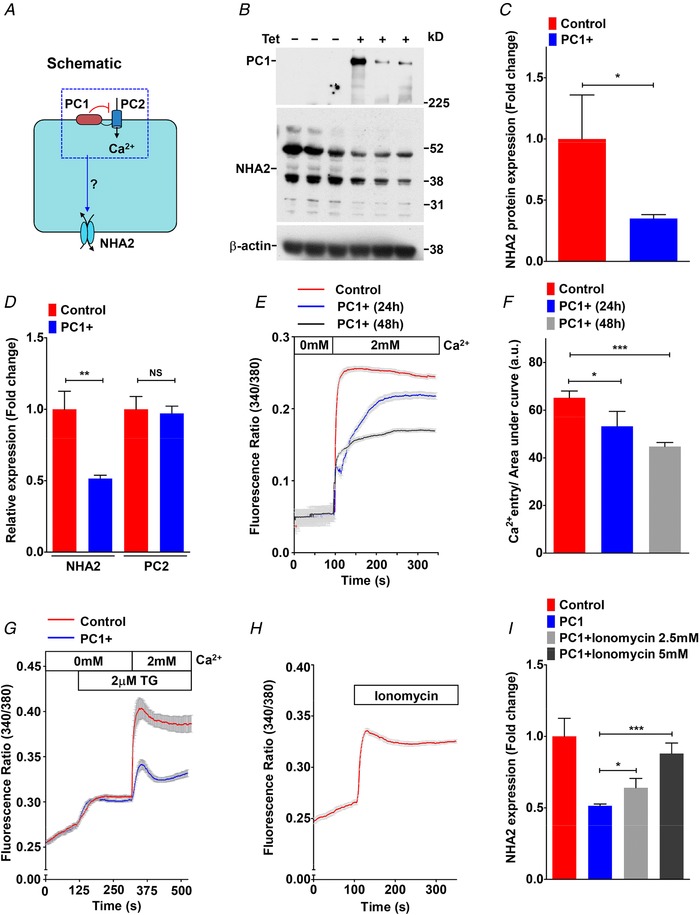
A, hypothesis for PC1‐mediated inhibition of Ca2+ influx through PC2 and NHA2 expression in MDCK cells. B, western blotting of MDCK cell lysates in the absence or presence of tetracycline for 17 h to induce PC1 expression, in biological triplicates, probed with antibody to PC1 (top), NHA2 (middle) and β‐actin (bottom). C, densitometric quantification of western blot shown in B. NHA2 protein expression was normalized to actin controls and was significantly down‐regulated by ∼2.9‐fold (n = 3; Student's t test, * P < 0.05) in MDCK cells with PC1 induction. D, quantitative PCR (qPCR) showing significant down‐regulation of NHA2 mRNA (n = 3; Student's t test, ** P < 0.01) and no change in PC2 mRNA (n = 3; Student's t test, NS P > 0.05) with PC1 induction. E and F, representative Fura‐2 fluorescence ratio traces (E) and quantitation (F) showing significant and proportionate reduction in store‐independent calcium entry (SICE) upon induction of PC1 for 24 h (n = 3; Student's t test, * P < 0.05) and 48 h (n = 3; Student's t test, *** P < 0.001) in MDCK cells. G, representative Fura‐2 fluorescence ratio traces showing reduction in store‐operated calcium entry (SOCE) following thapsigargin‐mediated release of store Ca2+ in MDCK cells with PC1 induction for 48 h. H, representative Fura‐2 fluorescence ratio traces showing efficacy of Ca2+ ionophore ionomycin to enhance cytosolic Ca2+ levels. I, qPCR showing significant and dose‐dependent increase in NHA2 expression with ionomycin treatment to levels similar to MDCK cells without PC1 induction. Error bars are S.D.
Figure 4. NFAT and Ca2+‐mediated regulation of NHA2 expression.
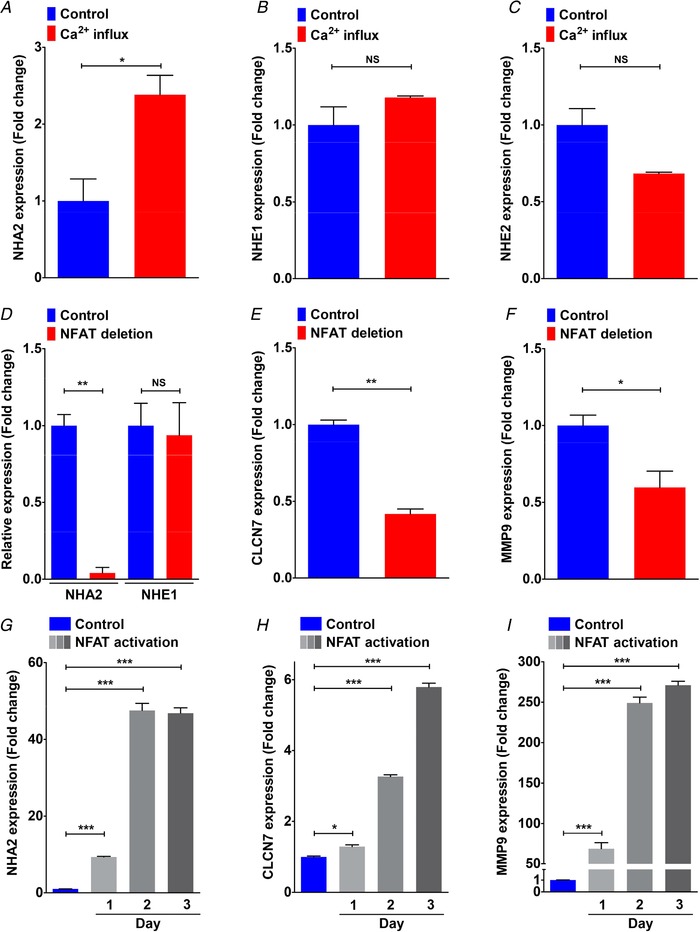
A–C, gene expression changes in response to Ca2+ influx following treatment with ionomycin and phorbol myristate in human primary T lymphoblasts obtained from the GSE50971 dataset. A, NHA2 showed significant up‐regulation (∼2.4‐fold higher; Student's t test, * P < 0.05). B, NHE1 expression showed no change (Student's t test, NS P > 0.05). C, NHE2 expression was lower, but non‐significant (Student's t test, NS P > 0.05). D, NFATc1 deletion resulted in profound, >95% down‐regulation of NHA2 expression (Student's t test, ** P < 0.01) and no change in related NHE1 isoform (Student's t test, NS P > 0.05) during osteoclast differentiation in vitro. E and F, down‐regulation of well‐known NFAT target genes, CLCN7 and MMP9, is shown for comparison. G–I, gene expression changes were determined from the GSE57468 dataset. In this experiment, mouse bone marrow‐derived osteoclast precursors were treated with RANKL for up to 3 days to differentiate into osteoclasts. G, robust, time‐dependent up‐regulation of NHA2 levels in response to NFAT‐mediated osteoclast differentiation in vitro (∼10‐fold on day 1; ∼50‐fold on day 2–3; Student's t test, *** P < 0.001). H and I, significant up‐regulation of well‐known NFAT target genes, CLCN7 (∼1.3‐fold on day 1, ∼3.3‐fold on day 2, ∼5.8‐fold on day 3; Student's t test, * P < 0.05, *** P < 0.001) and MMP9 (∼69‐fold on day 1, ∼249‐fold on day 2, ∼270‐fold on day 3; Student's t test, *** P < 0.001). Error bars are S.D.
NFAT mediates Ca2+‐dependent NHA2 expression
Studies have established the importance of NFAT signalling in normal kidney development and in pathological conditions including polycystic kidney disease (Puri et al. 2004; Aguiari et al. 2008; Burn et al. 2011). Analysis of microarray data on NFAT‐mediated osteoclast differentiation in vitro (Charles et al. 2012; An et al. 2014) revealed profound, >95% down‐regulation of NHA2 levels upon NFAT deletion (Fig. 4 D). There was no change in expression of the NHE1 isoform (Fig. 4 D). Expression patterns of well‐known NFAT target genes, CLCN7 and MMP9 (Aliprantis et al. 2008), are shown for comparison (Fig. 4 E and F). In contrast, time‐dependent up‐regulation of NHA2 transcript levels (∼50‐fold on day 2–3), as well as CLCN7 and MMP9, occurred upon NFAT activation (Fig. 4 G–I). Our analysis of NFATc1 expression in ADPKD patient‐derived cysts revealed significant up‐regulation relative to normal kidney tissue that correlated with cyst size and disease severity (Fig. 5 A): large cysts (3.5‐fold, ** P = 0.005), medium cysts (3.5‐fold, NS P = 0.235), small cysts (2.2‐fold, ** P = 0.003) and minimally cystic tissue (1.5‐fold, ** P = 0.003). Based on findings from the literature and our data so far, we propose a model in which PC1‐mediated modulation of Ca2+ homeostasis regulates NHA2 expression through NFAT signalling (Fig. 5 B). To test this hypothesis, we used a membrane anchored C‐terminal tail fragment of PC1 (PC1‐MAT) that was previously shown to function as a dominant negative effector and mimic cellular pathologies associated with patient mutations in PC1, including activation of downstream AP‐1, WNT, STAT3 and NFAT signalling (Arnould et al. 1998; Kim et al. 1999; Puri et al. 2004; Talbot et al. 2011; Fig. 5 C). We therefore asked if exogenous expression of the PC1‐MAT fragment could induce NHA2 expression in HEK293 cells. Using the firefly and Renilla luciferase reporter gene system, we first confirmed that ectopic expression of PC1‐MAT resulted in significant, ∼5‐fold increase in NFAT reporter activity, relative to empty vector transfection control (**** P < 0.0001; Fig. 5 D). These results were independently validated by visualizing localization of GFP‐tagged NFAT: in vector transfected cells, NFAT–GFP is predominantly localized in the cytoplasm, whereas increased nuclear translocation of NFAT–GFP was documented in cells expressing PC1‐MAT, similar to cells expressing constitutively active NFAT–GFP (CA‐NFAT) (Fig. 5 E and F). In striking contrast, expression of full‐length PC1 showed significant, ∼4‐fold down‐regulation of NFAT reporter activity, relative to empty vector transfection control (**** P < 0.0001; Fig. 5 D). These results are consistent with down‐regulation of Ca2+ influx (Fig. 3 E and F) and NHA2 expression (Fig. 3 B and C) by induction of full length PC1 in MDCK cells. We have previously shown that store‐independent Ca2+ entry (SICE) regulates NFAT nuclear translocation and promotes cell proliferation (Feng et al. 2010). We therefore analysed SICE in cells transfected with PC1‐MAT. In contrast to our SICE data in MDCK cells expressing full‐length PC1 (Fig. 3 E and F), we observed robust ∼2.4‐fold increase in Ca2+ entry, compared to empty vector control (* P = 0.02; Fig. 5 G and H). We also observed a higher baseline with PC1‐MAT, relative to the empty vector control, suggesting higher basal Ca2+ levels with PC1‐MAT expression (Fig. 5 G). Taken together, these data provide strong evidence for reciprocal down‐ and up‐regulation of SICE/NFAT signalling by full length PC1 and PC1‐MAT, respectively. Importantly, consistent with our proposed model, we observed ∼25% lower NHA2 levels in cells expressing full‐length PC1 (** P = 0.0016; Fig. 5 I) and on the other hand, ∼2.6‐fold higher NHA2 expression in cells expressing PC1‐MAT (*** P = 0.0002; Fig. 5 J), relative to empty vector transfection. These findings are also consistent with our data from MDCK cells expressing full‐length PC1 (Fig. 3 B–D), and evidence from the literature showing significant up‐regulation of NHA2 upon ectopic expression of engineered constitutively active NFAT1 in T cells (Martinez et al. 2015). These findings, taken together, suggest that polycystin 1‐mediated modulation of Ca2+ influx regulates NHA2 expression through the calcineurin/NFAT pathway.
Figure 5. NFAT mediates Ca2+‐dependent NHA2 expression.
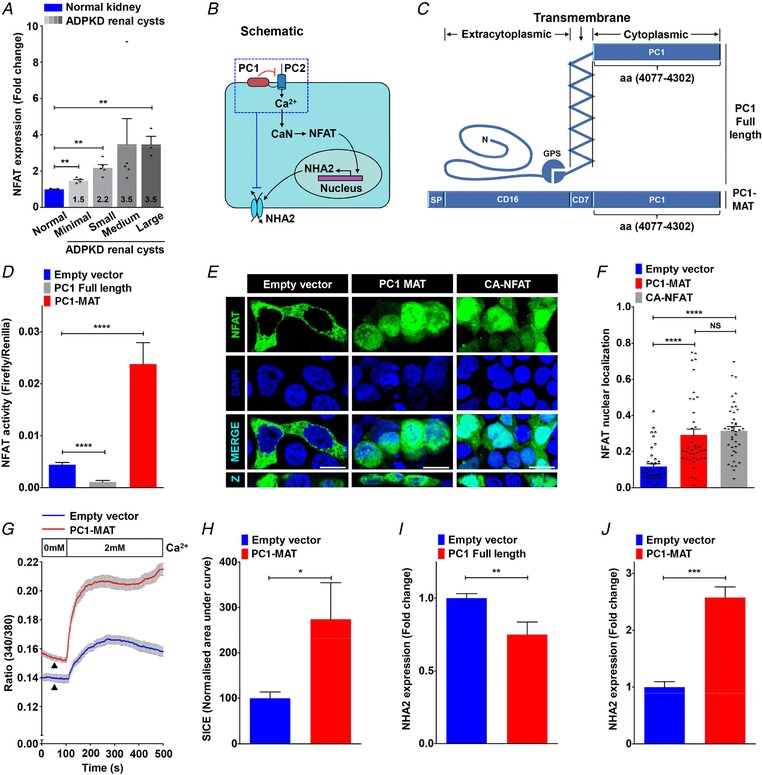
A, NFATc1 expression profiling of ADPKD patient cysts of different sizes revealed up‐regulation of NFAT expression relative to normal kidney tissue that correlated with cyst size and disease severity. B, proposed model for PC1‐mediated modulation of Ca2+ homeostasis and regulation of NHA2 expression through calcineurin (CaN) and NFAT signalling. C, schematic representation of full‐length PC1 (top) and membrane‐anchored C‐terminal tail fragment of PC1 (PC1‐MAT) (bottom) containing the C‐terminal tail fragment of PC1 linked to the signal peptide (SP) of CD16 and transmembrane domain of CD7. D, Luciferase assay in HEK293 cells to determine NFAT activation using a 3× NFAT binding sequence that drives a firefly luciferase reporter gene and is measured luminometrically. Renilla luciferase driven by a constitutively active SV40 promoter was used to normalize for variations in both cell number and the transfection efficiency. Expression of full‐length PC1 resulted in reduction (∼4‐fold lower; n = 3; Student's t test, **** P < 0.0001) and of PC1‐MAT resulted in increase (∼5‐fold higher; n = 3; Student's t test, **** P < 0.0001) in NFAT reporter activity, relative to empty vector transfection control. E and F, representative micrographs (E, scale bar= 10 μm) and quantification using Manders’ coefficient (F) determining fractional colocalization of NFAT–GFP (green) with DAPI (blue). Colocalization is evident in the merge and orthogonal slices (Z) as cyan puncta. In vector transfected cells, NFAT–GFP is predominantly localized in the cytoplasm. Note prominent overlap between NFAT–GFP and DAPI, consistent with increased nuclear translocation, in cells expressing PC1‐MAT, similar to cells expressing constitutively active NFAT–GFP (CA‐NFAT) (n = 40/each condition; Student's t test, **** P < 0.0001). G and H, representative Fura‐2 fluorescence ratio traces (G) and quantification (H) showing ∼2.4‐fold increase in store‐independent calcium entry (SICE) in cells transfected with PC1‐MAT (n = 3; Student's t test, * P < 0.05), relative to empty vector transfection. Note a higher baseline with PC1‐MAT, relative to the empty vector control, suggesting higher basal Ca2+ levels with PC1‐MAT expression. I and J, qPCR showing ∼2.6‐fold higher NHA2 expression in cells expressing PC1‐MAT (n = 3; Student's t test, *** P < 0.001) and ∼25% lower NHA2 levels in cells expressing full‐length PC1 (n = 3; Student's t test, ** P < 0.01), relative to empty vector transfection. Error bars S.D.
Potential modulation of kidney function by drug and hormonal regulation of NHA2
Public datasets can be leveraged to identify novel mechanisms of gene regulation, as described earlier (Prasad & Rao, 2018). To gain new insights into the regulation of NHA2 expression and to predict a functional role of NHA2 in PKD, we performed an unbiased bioinformatics approach to analyse drug‐induced gene expression signatures from 1078 microarray studies in human cells. We identified experimental conditions eliciting a minimum of ±2‐fold change in NHA2 gene expression (Fig. 6 A). Notably, the highest up‐regulation of NHA2 (≥3‐fold) was observed in response to methyl xanthine drugs: caffeine (7.5 mm; 24 h) and theophylline (10 mm; 24 h). Further analysis of these individual microarray studies showed that the ability of these drugs to induce NHA2 expression is dependent on dosage and duration of treatment (Fig. 6 B and C). Interestingly, these methyl xanthine drugs are suggested to promote cyst development in PKD patients (Belibi et al. 2002). Furthermore, both caffeine and theophylline are known to activate ryanodine‐sensitive receptors (RyR) and release Ca2+ stores in response to an initial Ca2+ entry through PC2 and other plasma membrane Ca2+ channels, thereby effectively amplifying cytoplasmic Ca2+ (Kong et al. 2008).
Figure 6. Drug and hormonal regulation of NHA2.
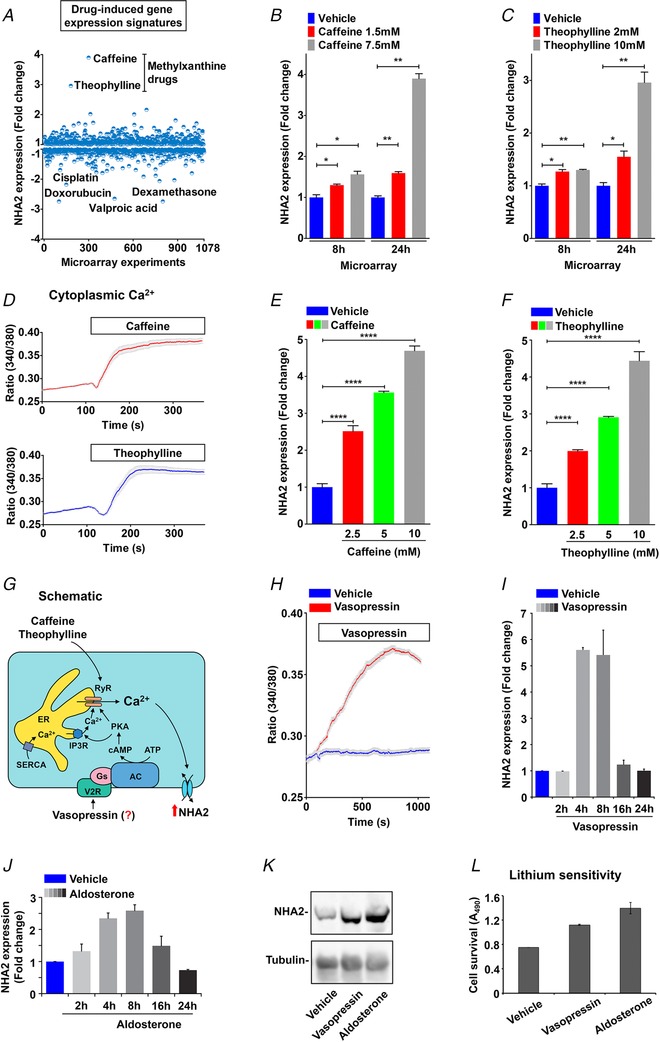
A, expression profiling of NHA2 expression (y‐axis) obtained from an unbiased bioinformatics analysis of 1078 microarray studies (x‐axis), as described in Methods. Note that highest up‐regulation of NHA2 (≥3‐fold) was observed in response to methyl xanthine drugs: caffeine (7.5 mm; 24 h) and theophylline (10 mm; 24 h). Four drugs that resulted in maximal down‐regulation of NHA2 are valproic acid, dexamethasone, cisplatin and doxorubicin. B and C, bar graphs of NHA2 expression derived from microarray analysis of caffeine (1.5 and 7.5 mm; B) and theophylline (2 and 10 mm; C) treatments showing dose‐ and duration‐ (8 h and 24 h) dependent effects. D, representative Fura‐2 fluorescence ratio traces showing significant increase in cytoplasmic Ca2+ with caffeine (top) and theophylline (bottom) treatment in MDCK cells. E and F, qPCR analysis to validate bioinformatic studies documenting significant, dose‐dependent increase in NHA2 expression with caffeine (E) and theophylline (F), relative to vehicle controls in HEK293 cells. G, hypothesis for vasopressin‐mediated regulation of NHA2 expression in renal epithelial cells. Vasopressin stimulation of the V2R receptors results in accumulation of cAMP and activation of PKA. Like caffeine and theophylline, vasopressin‐driven PKA activation stimulates Ca2+ release via the ryanodine receptor channel from the endoplasmic reticulum (ER). Increased cytosolic Ca2+ would in turn increase NHA2 expression. H, representative Fura‐2 fluorescence ratio traces showing significant increase in cytoplasmic Ca2+ with vasopressin treatment in MDCK cells, relative to vehicle control. I, qPCR data showing fold change in NHA2 transcript levels following treatment with vasopressin for indicated time periods ranging from 2 to 24 h. Note significant and phasic increase in NHA2 expression with vasopressin treatment that peaked (∼6‐fold) at 4–8 h and reached baseline by 24 h. J, qPCR data showing fold change in NHA2 transcript levels following treatment of MDCK cells with aldosterone for indicated time periods ranging from 2 to 24 h. Note significant and phasic increase in NHA2 expression with aldosterone treatment that peaked (∼2.6‐fold) at 8 h and reached baseline by 24 h. K, western blot showing NHA2 expression levels following a 6 h treatment of MDCK cells with vehicle (lane 1), vasopressin (lane 2) or aldosterone (lane 3), with tubulin serving as a loading control (bottom panel). L, lithium sensitivity assay to evaluate functional consequences of the hormonal induction of NHA2. Consistent with increased NHA2 mRNA and protein expression, treatment with either vasopressin or aldosterone for 8 h resulted in increased cell survival in medium supplemented with 90 mm LiCl, relative to untreated control. Cell survival in the presence of LiCl was measured using an MTT assay. Error bars are S.D.
On the other hand, at least two drugs that resulted in maximal down‐regulation of NHA2, namely valproic acid and dexamethasone, are known to reduce cytoplasmic Ca2+ concentration (Nilsson et al. 1992; Suwanjang et al. 2013). Interestingly, the therapeutic potential of valproic acid has been shown in zebra fish and mouse models of PKD (Cao et al. 2009). Taken together, top hits in NHA2 gene expression (both up‐ and down‐regulation) funnelled into pathways that modulate cytoplasmic Ca2+, further bolstering the credibility of our model of Ca2+‐mediated regulation of NHA2 expression. Our previous studies show that NHA2 is an important cellular lithium efflux transporter that might be involved in Li+ clearance in kidney (Kondapalli et al. 2012). In this context it is important to note that the methylxanthine drugs caffeine and theophylline that increase NHA2 levels are well known to promote Li+ clearance via kidney. These drugs enhance renal salt and water transport and are contraindicated for Li+ therapy (Perry et al. 1984; Grandjean & Aubry, 2009). Using ratiometric Fura 2 Ca2+ imaging, we first confirmed significant increase in cytoplasmic Ca2+ with caffeine and theophylline treatment in MDCK cells (Fig. 6 D). Furthermore, caffeine and theophylline treatment elicit significant, dose‐dependent increase in NHA2 expression (Fig. 6 E and F), relative to vehicle controls, validating the findings from bioinformatic studies (Fig. 6 A–C).
Given that physiological concentrations of vasopressin, a hormone strongly implicated in PKD pathogenesis and a promising drug target, also function via activation of RyR (Chebib et al. 2015), we hypothesized vasopressin mediated regulation of NHA2 expression in renal epithelial cells (Fig. 6 G). The MDCK cell line is a well‐established model to study transport function changes in the distal renal tubule in response to hormones, including vasopressin and aldosterone (Stewart et al. 2009; Carmosino et al. 2011). To test our hypothesis, we used the MDCK cell model and first documented robust increase in cytoplasmic Ca2+ following vasopressin treatment, relative to vehicle control (Fig. 6 H). Compared with the quick (∼50–100 s) initial slope of the Ca2+ rise in caffeine and theophylline treatment, the slope of the Ca2+ rise was more gradual (∼500–1000 s) consistent with indirect activation of downstream signalling cascades by vasopressin (Fig. 6 G). Of note, the peak heights of cytosolic [Ca2+] were comparable between caffeine, theophylline and vasopressin. It is worth noting that both caffeine and vasopressin have been shown to activate NFAT nuclear translocation and signalling (Scicchitano et al. 2005; Stiber et al. 2005). Therefore, to determine if a rise in cytosolic Ca2+ with vasopressin is translated to increase in NHA2 expression, we treated MDCK cells with vasopressin for different intervals, ranging from 2 to 24 h. We observed significant and phasic increase in NHA2 expression with vasopressin treatment that peaked (∼6‐fold) at 4–8 h and reached baseline by 24 h (Fig. 6 I). Using western blotting we confirmed robust elevation of NHA2 protein with vasopressin treatment for 6 h (Fig. 6 K).
These findings are consistent with our recent in vivo studies showing that high salt diet in mice significantly increased transcript and protein expression of NHA2 in renal tubules (Kondapalli et al. 2017). High salt diet significantly raises circulating levels of vasopressin to retain water and help sustain normal plasma osmolarity (Kjeldsen et al. 1985). Likewise, increased vasopressin concentrations have been associated with PKD severity and disease progression and in experimental studies vasopressin has been shown to directly modulate transepithelial fluid transport and regulate cyst growth (Chebib et al. 2015). Intriguingly, studies have documented that the NHA2 inhibitor phloretin attenuates vasopressin‐stimulated solute movement (Levine et al. 1973). Thus, NHA2 might regulate salt and water transport across the epithelium and help contribute to the functions of vasopressin in PKD and high salt diet, both of which are associated with hypertension.
For comparison, we tested the effect of another hormone, aldosterone, on NHA2 expression in MDCK cells. Basic and clinical research has shown increased activation of the renin–angiotensin–aldosterone system and associations with cyst enlargement and hypertension in ADPKD (Chapman et al. 1990; Schrier, 2009; Tkachenko et al. 2013). Intriguingly, like vasopressin, aldosterone also increased NHA2 mRNA (Fig. 6 J) and protein levels (Fig. 6 K). Similarly, induction with both vasopressin and aldosterone expression has been reported in the literature for aquaporin‐2 and the γ subunit of the epithelial sodium channel (ENaC) (Hasler et al. 2003; Perlewitz et al. 2010). Moreover, a synergistic effect of vasopressin and aldosterone has been described for Na+ channel and Na+/K+‐ATPase activities (Schafer & Hawk, 1992; Coutry et al. 1995). It is worth noting that aldosterone has been shown to enhance global Ca2+ transients, through activation of protein kinase A (PKA) (de Almeida et al. 2013). To evaluate functional consequences of the hormonal induction of NHA2, we monitored growth sensitivity to lithium in MDCK cells. Previously, we showed that NHA2 is functionally coupled to plasma membrane V‐ATPase in MDCK cells to mediate robust H+‐driven efflux of Li+, resulting in selective survival and growth advantage in media with a high concentration of lithium (Kondapalli et al. 2012). Consistent with increased NHA2 mRNA and protein expression, treatment with either vasopressin or aldosterone for 8 h resulted in increased cell survival in medium supplemented with 90 mm LiCl, relative to untreated control (Fig. 6 L).
Vasopressin and aldosterone play key roles in the fine adjustment of transport of salt and water in the nephron (Schafer & Hawk, 1992; Coutry et al. 1995; Perlewitz et al. 2010). In order to test the potential role of NHA2 in regulating transepithelial fluid transport, we generated fluid‐filled hemicysts or domes, a simple, yet powerful assay widely used over the last 40 years to determine vectorial salt and water transport in vitro (Misfeldt et al. 1976; Lever, 1979). MDCK cells grown on impermeable substratum, such as plastic, spontaneously form hemicysts due to transepithelial transport and accumulation of fluid in focal regions, beneath the cell layer (Fig. 7 A and B). Studies have established that cyclic AMP levels, Na+/H+ exchange activity and PC1 function regulate hemicyst formation (Lever, 1979; Bukanov et al. 2002; Su et al. 2007). Importantly, vasopressin and aldosterone treatment promote transepithelial transport and robustly stimulate hemicyst formation in MDCK cells (Rodrig et al. 1988; Oberleithner et al. 1990). Consistent with these findings, we showed that ectopic expression of NHA2 in MDCK cells resulted in a ∼2.8‐fold increase in area of hemicysts (Fig. 7 C and D), suggesting an increase in vectorial transport of salt and water with NHA2 expression that might, in part, contribute to the downstream effects of vasopressin and aldosterone in PKD.
Figure 7. NHA2 promotes hemicyst formation.
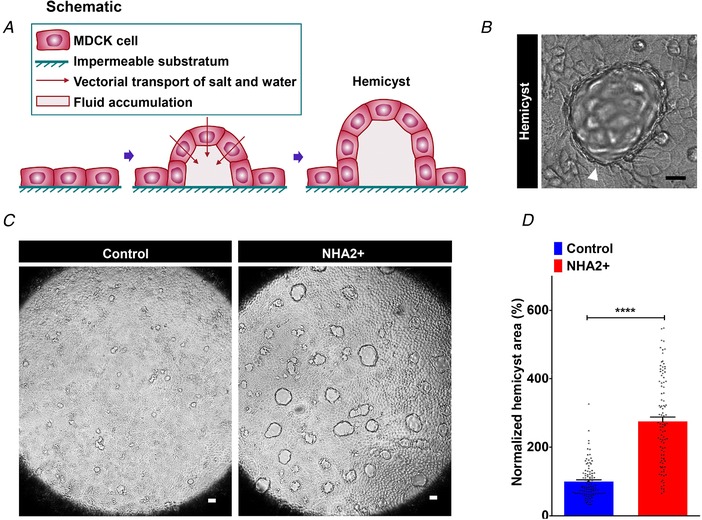
A, schematic depiction of generation of fluid‐filled hemicysts or domes with MDCK cells. B, representative image of a MDCK hemicyst with fluid accumulation focally beneath the epithelium (white arrow). C, NHA2+ MDCK cells formed larger hemicysts (right) relative to control (left). D, quantification of hemicyst development showed significant expansion in hemicyst area with NHA2 expression relative to control (Control: 100 ± 5.01, n = 100; NHA2+: 275.1 ± 12.75, n = 100; Student's t test, **** P < 0.0001), suggesting an increase in vectorial transport of salt and water with NHA2 expression. Scale bar: 10 μm. Error bars are S.E.
Knockdown or inhibition of NHA2 attenuates cyst development in vitro
Given that fluid secretion and cyst expansion are major factors for the progressive and irreversible decline and renal failure in PKD, therapies targeting salt and water transport are of major interest as an alternative to, or to complement, anti‐proliferative therapies in PKD (Terryn et al. 2011). To evaluate the therapeutic potential of targeting NHA2 in PKD, we used two complementary approaches: shRNA‐mediated knockdown of NHA2 and pharmacological inhibition of NHA2 using phloretin. MDCK cells have low endogenous NHA2 expression (Kondapalli et al. 2012), and therefore we knocked down NHA2 in NHA2+ MDCK cells (Fig. 8 A). We have previously shown that overexpression of NHA2 does not alter growth in MDCK cells (Kondapalli et al. 2012). Consistent with these findings, we now show that lentivirally mediated knockdown of NHA2 also did not alter growth of MDCK cells (Fig. 8 B). Pharmacological inhibition offered an alternative approach to NHA2 knockdown. NHA2 is insensitive to classical NHE inhibitors such as amiloride and its derivatives, but is sensitive to phloretin (Kondapalli et al. 2012). In contrast to the effect of pharmacological inhibition of NHE1, inhibition of NHA2 with phloretin did not significantly alter cytoplasmic pH (Fig. 8 C and D). Therefore, to functionally validate the effect of phloretin, we turned to lithium sensitivity as a defining phenotype of NHA2 transport function. Treatment of NHA2+ MDCK cells with phloretin conferred growth sensitivity to lithium (Fig. 8 E). Notably, lithium sensitivity was not significantly altered by phloretin treatment following NHA2 knockdown indicating the specificity of the lithium sensitivity assay as a marker of NHA2 activity (Fig. 8 E).
Figure 8. Knockdown or inhibition of NHA2 attenuates cyst development in vitro .
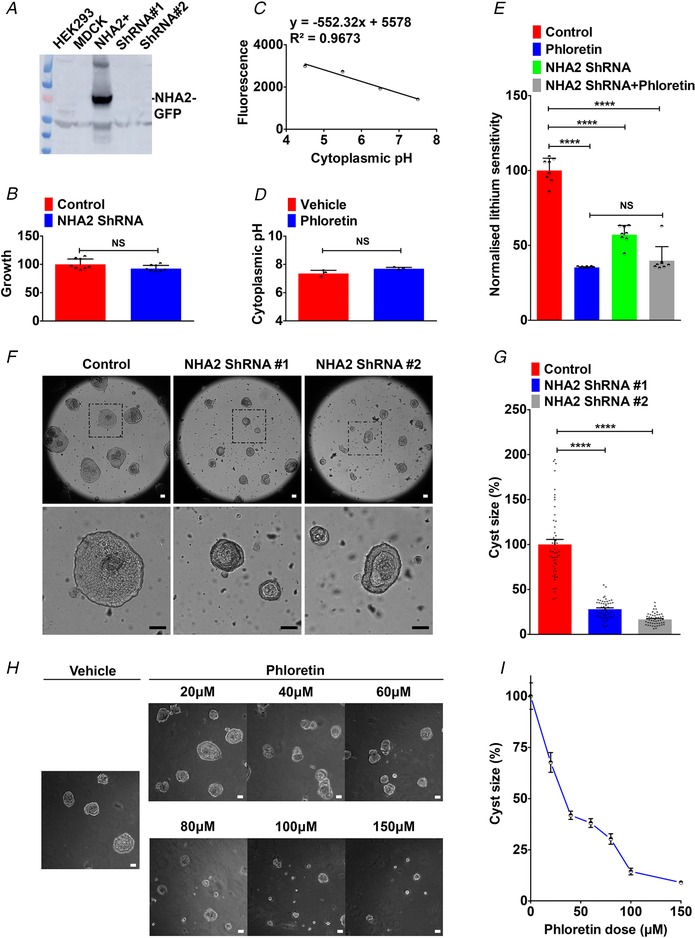
A, lentivirally mediated knockdown of NHA2 in NHA2+ MDCK cells, using two different lentiviral shRNA constructs (ShRNA 1 and ShRNA 2) targeting human NHA2, resulted in robust knockdown of GFP‐tagged NHA2. Western blot of total protein was probed using anti‐NHA2 antibody. B, lentivirally mediated knockdown of NHA2 does not alter growth of MDCK cells. Cell growth activity was measured by an MTT assay, normalized to the scrambled control (n = 8; Student's t test, NS P > 0.05). C and D, cytoplasmic pH in NHA2+ cells with vehicle or phloretin treatment was determined, as described under Methods. Calibration curve is shown in C. Inhibition of NHA2 with phloretin did not significantly alter cytoplasmic pH (D) (n = 3; Student's t test, NS P > 0.05). E, lithium sensitivity assay to validate the effect of phloretin. Cell survival in the presence of 100 mm LiCl was measured using MTT. Phloretin treatment and lentiviral NHA2 knockdown conferred significant growth sensitivity to lithium (n = 8; Student's t test, **** P < 0.0001). Note no difference in lithium sensitivity between phloretin treatment with or without NHA2 knockdown (n = 8; Student's t test, NS P > 0.05). F, lentivirally mediated knockdown of NHA2 shown in A resulted in formation of significantly smaller cysts, relative to control. Representative images are shown, and bottom row is higher magnification of boxed region from top row. G, cyst size is significantly reduced following NHA2 depletion relative to control on day 10 of culture (Control: 100.0 ± 5.6; n = 50, NHA2 ShRNA 1: 28.0 ± 1.46; n = 50, NHA2 ShRNA 2: 16.7 ± 0.91; n = 50, Student's t test, **** P < 0.0001). H, phloretin (20–150 μm) causes a dose‐dependent reduction in cyst size. Representative images are shown, with the vehicle treatment on the left. I, quantification of cyst size on day 10 of culture (n = 50/each condition). Scale bar: 25 μm. Error bars are S.E.
Next, we tested the effect of NHA2 knockdown on cyst formation by MDCK cells. Specific knockdown of NHA2, using two different lentiviral shRNA constructs, resulted in formation of significantly smaller (∼70–80% lower diameter) cysts (Fig. 8 F and G). We also analysed the effect of phloretin (20–150 μm) on cyst formation. Consistent with lower cyst size resulting from NHA2 knockdown (Fig. 8 F and G), we observed dose‐dependent attenuation of cyst size with NHA2 inhibition with phloretin (Fig. 8 H and I). Taken together, these findings suggest that NHA2 may be a potential drug target for attenuating cyst development in PKD.
Discussion
The three key components of cystogenesis in PKD are abnormalities in tubular cell proliferation, matrix remodelling and fluid secretion (Terryn et al. 2011). Cysts arise from the renal tubule as saccular expansions, and eventually become pinched off and anatomically separated from the tubule. Thus, the intracystic fluid is derived from transepithelial fluid secretion due to abnormal reversal of normally absorptive epithelium to a secretory one. This leads to a dramatic expansion of cystic volume, which is the single most important predictor of progressive deterioration and renal failure in PKD (Grantham et al. 2006; Terryn et al. 2011). Therefore, a better understanding of pathways driving salt and water transport in PKD models could lead to novel strategies to halt disease progression.
Na+/H+ exchangers are known to be of vital physiological importance for fluid and salt homeostasis in the kidney (Donowitz et al. 2013; Fuster & Alexander, 2014), and their contribution to the pathology of PKD has been recognized. NHE1 is mislocalized to the apical membrane in PKD cell models where it absorbs Na+ ions, in exchange for H+ (Avner et al. 2012; Olteanu et al. 2012). In contrast, we previously showed that NHA2 co‐localizes with the V‐ATPase at the basolateral domain of MDCK cells (Kondapalli et al. 2012). An unconventional coupling to the proton gradient allows NHA2 to extrude Na+ ions across the basal membrane in exchange for H+. Because H+ can be readily buffered at both extra‐ and intracellular milieus, we suggest that the net apical‐to‐basal transepithelial movement of Na+ ions could set up an osmotic potential for water movement, driving cyst formation. A direct testing of this model awaits a mechanistic study of transport in cysts. It is as yet unclear whether the normal physiological role of NHA2 in the renal tubular cell is in salt and water reabsorption or secretion or both depending on the coupling ion (sodium or protons) and localization (apical or basolateral) (Kondapalli et al. 2017). Of note, down‐regulation of related NHE2 isoform observed in our analysis of patient cysts could indicate renal inflammation driven by proinflammatory cytokines (Soleiman et al. 2017).
Although far from conclusive, there are tantalizing hints pointing to a role for NHA2 in renal function and hypertension that may be relevant to pathophysiology of PKD. First, SLC9B2, the gene encoding NHA2, lies within a limited (∼300 genes) region of human chromosome 4q24 genetically linked to phloretin‐sensitive and amiloride‐insensitive sodium–lithium countertransport (SLC), an activity linked to essential hypertension (Xiang et al. 2007, Schushan et al. 2010). Indeed, NHA2 mediates phloretin‐sensitive and amiloride‐insensitive SLC activity in stably transfected MDCK cells (Kondapalli et al. 2012). SLC activity has been associated with PKD and there is an intriguing, yet unexplained link between lithium treatment and formation of renal cysts in human and rodents (Kling et al. 1984; Vareesangthip et al. 1995; Shah et al. 2010). We suggest that lithium‐induced NFAT activation may provide a molecular mechanism for lithium and NHA2‐mediated cytogenesis in PKD (Gomez‐Sintes & Lucas, 2010). Second, a recent genome‐wide association study of renal function identified NHA2 as a genetic risk locus for estimated glomerular filtration rate by serum creatinine. Glomerular filtration rate, which shows heritability in the range of 36–75%, is commonly used to monitor kidney function decline in PKD (Schrier, 2009; Liu et al. 2018). Third, NHA2 expression is predominant in the distal nephron where sodium reabsorption is under hormonal and dietary control, consistent with the induction of renal NHA2 expression by a high salt diet in mouse (Fuster et al. 2008, Kondapalli et al. 2017). In addition to these findings, we now show that NHA2 is highly elevated in PKD, a disease, with a predisposition to hypertension (Chapman et al. 1990).
The dysregulation of two second messengers, Ca2+ and cAMP, is central to the disease mechanism in PKD and there is evidence connecting NHA2 to both. Here we show that methylxanthine drugs (caffeine and theophylline), which activate Ca2+ release via the ryanodine receptor channel (RyR), increase NHA2 expression, which is associated with cyst growth (Fig. 9). Furthermore, we show that NHA2 is up‐regulated by vasopressin, a hormone that in physiological concentrations functions through activation of RyR and increases cytoplasmic Ca2+ levels. Thus, NHA2 is a novel vasopressin effector in renal epithelial cells with potential implications for PKD.
Figure 9. Proposed role for NHA2 in PKD.
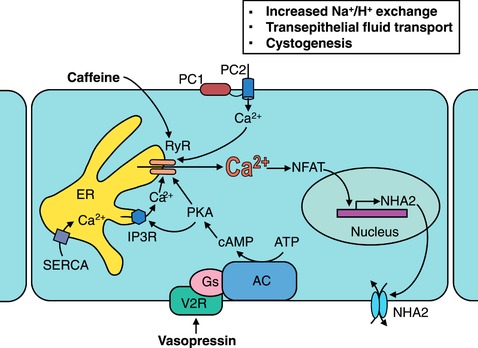
Polycystins 1 and 2 (PC1 and PC2) regulate Ca2+ influx, NFAT signalling and NHA2 expression. Methylxanthine drugs (caffeine and theophylline) activate ryanodine‐sensitive receptor channel (RyR) and release Ca2+ stores from the endoplasmic reticulum (ER) in response to an initial Ca2+ entry through PC2 and other plasma membrane Ca2+ channels, thereby effectively amplifying cytoplasmic Ca2+ and increasing NHA2 expression. Vasopressin stimulation of the V2R receptors results in accumulation of cyclic AMP (cAMP) and activation of protein kinase A (PKA). Like caffeine and theophylline, vasopressin‐driven PKA activation stimulates Ca2+ release via the RyR, leading to an increase NHA2 expression which is associated with cyst expansion.
The precise NHA2‐mediated regulation of transepithelial fluid transport and cystogenesis remains to be determined. Vasopressin is secreted in response to increased plasma osmolality resulting from high salt intake, consistent with our previous observation that high sodium diet in mice significantly increases expression of NHA2 in renal tubules (Kjeldsen et al. 1985; Kondapalli et al. 2017). Previous studies have shown that PC1‐MAT enhances caffeine‐induced intracellular Ca2+ levels (Puri et al. 2004). Similarly, caffeine has been reported to synergize the effects of desmopressin, a synthetic analogue of vasopressin (Belibi et al. 2002). Caffeine is also thought to promote cyst development in PKD patients (Belibi et al. 2002). Furthermore, caffeine and theophylline are well known to promote clearance of lithium through kidney (Perry et al. 1984; Grandjean & Aubry, 2009). Given our previous findings that NHA2 is an important cellular lithium efflux transporter (Kondapalli et al. 2012), we suggest that induction of NHA2 expression might, in part, contribute to the downstream effects of caffeine in PKD and lithium clearance.
NHA2 may mediate cross‐talk between cAMP and Ca2+ signalling. In spermatozoa, loss of NHA2, or the closely related NHA1 isoform, led to decreased soluble adenylyl cyclase and cAMP levels, and conversely, high levels of NHA antiporters stabilized soluble adenylyl cyclase expression (Chen et al. 2016). Given the central role of cAMP in cyst formation and growth, elevated NHA2 could exacerbate cAMP signalling in ADPKD patients, which might in turn modulate the activity of other transport proteins. PKD patients have high circulating levels of vasopressin. Treatment with the vasopressin V2‐receptor antagonist tolvaptan has been shown to inhibit cyst progression in preclinical studies and large clinical trials (Chebib et al. 2015). The potential synergistic or additive effects of NHA2 inhibition with tolvaptan merit further study. In summary, our findings warrant a thorough in vivo investigation of NHA2 as a potential new player in cellular salt and pH regulation in the kidney and in the pathogenesis of PKD and hypertension.
Additional information
Authors’ present addresses
H. Prasad: Department of Molecular Reproduction, Development and Genetics, Indian Institute of Science, Bangalore 560012, India.
K. C. Kondapalli: Department of Natural Sciences, University of Michigan‐Dearborn, MI 48128, USA.
N. Natarajan: Department of Stem Cell and Regeneration, Harvard University, MA 02138, USA.
Competing interests
None declared.
Author contributions
H.P., D.K.D, K.C.K, N.N. and V.C. designed, conducted and interpreted experiments. R.R. designed and interpreted experiments. H.P, D.K.D., V.C. and R.R. wrote and edited the paper. All authors have read and approved the final version of this manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by grants from the National Institutes of Health to R.R. (DK108304) and to V.C. (DK103078). H.P. was a Fulbright Fellow supported by the International Fulbright Science and Technology Award. K.C.K. was a postdoctoral fellow of the American Heart Association.
Acknowledgements
We gratefully acknowledge the Baltimore Polycystic Kidney Disease (PKD) Research and Clinical Core Centre for stable MDCK cells with inducible PC1 expression.
Biography
Donna K. Dang and Hari Prasad are newly minted PhD graduates of the Cellular and Molecular Medicine training program at the Johns Hopkins School of Medicine, under the mentorship of R.R. D.K.D.’s thesis work focused on the role of the Golgi Ca2+ pump, SPCA2, in E‐cadherin biogenesis and breast microcalcifications in the evolution of breast cancer; H.P.’s described the importance of the endosomal Na+/H+ exchanger NHE6 in the production and clearance of amyloid β in Alzheimer's disease. Together, they contributed their individual expertise to make sense of data left by previous lab members on NHA2, a mysterious and poorly described member of the Na+/H+ exchanger superfamily. Their collaboration has uncovered a potential role for NHA2 in polycystic kidney disease that illustrates the importance of salt and fluid transport in cystogenesis. Together, they exemplify the scholarship and collegiality that is critical for the advancement of science.

Edited by: Peying Fong & Dennis Brown
H. Prasad and D. K. Dang contributed equally.
Preprint publication: Prasad H, Dang DK, Kondapalli KC, Natarajan N, Cebotaru V & Rao R (2018). NHA2 promotes cyst development in an in vitro model of polycystic kidney disease. bioRxiv. http://doi.org/10.1101/364679.
Linked articles: This article is highlighted in a Perspectives article by Bevensee. To read this article, visit https://doi.org/10.1113/JP276727.
References
- Aguiari G, Trimi V, Bogo M, Mangolini A, Szabadkai G, Pinton P, Witzgall R, Harris PC, Borea PA, Rizzuto R & del Senno L (2008). Novel role for polycystin‐1 in modulating cell proliferation through calcium oscillations in kidney cells. Cell Prolif 41, 554–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aliprantis AO, Ueki Y, Sulyanto R, Park A, Sigrist KS, Sharma SM, Ostrowski MC, Olsen BR & Glimcher LH (2008). NFATc1 in mice represses osteoprotegerin during osteoclastogenesis and dissociates systemic osteopenia from inflammation in cherubism. J Clin Invest 118, 3775–3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An D, Kim K & Lu W (2014). Defective entry into mitosis 1 (Dim1) negatively regulates osteoclastogenesis by inhibiting the expression of nuclear factor of activated T‐cells, cytoplasmic, calcineurin‐dependent 1 (NFATc1). J Biol Chem 289, 24366–24373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnould T, Kim E, Tsiokas L, Jochimsen F, Gruning W, Chang JD & Walz G (1998). The polycystic kidney disease 1 gene product mediates protein kinase C alpha‐dependent and c‐Jun N‐terminal kinase‐dependent activation of the transcription factor AP‐1. J Biol Chem 273, 6013–6018. [DOI] [PubMed] [Google Scholar]
- Avner ED, McDonough AA & Sweeney WE Jr (2012). Transport, cilia, and PKD: must we in (cyst) on interrelationships? Focus on “Increased Na+/H+ exchanger activity on the apical surface of a cilium‐deficient cortical collecting duct principal cell model of polycystic kidney disease”. Am J Physiol Cell Physiol 302, C1434–C1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belibi FA, Wallace DP, Yamaguchi T, Christensen M, Reif G & Grantham JJ (2002). The effect of caffeine on renal epithelial cells from patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 13, 2723–2729. [DOI] [PubMed] [Google Scholar]
- Blomberg KE, Boucheron N, Lindvall JM, Yu L, Raberger J, Berglof A, Ellmeier W & Smith CE (2009). Transcriptional signatures of Itk‐deficient CD3+, CD4+ and CD8+ T‐cells. BMC Genomics 10, 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukanov NO, Husson H, Dackowski WR, Lawrence BD, Clow PA, Roberts BL, Klinger KW & Ibraghimov‐Beskrovnaya O (2002). Functional polycystin‐1 expression is developmentally regulated during epithelial morphogenesis in vitro: downregulation and loss of membrane localization during cystogenesis. Hum Mol Genet 11, 923–936. [DOI] [PubMed] [Google Scholar]
- Burn SF, Webb A, Berry RL, Davies JA, Ferrer‐Vaquer A, Hadjantonakis AK, Hastie ND & Hohenstein P (2011). Calcium/NFAT signalling promotes early nephrogenesis. Dev Biol 352, 288–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Semanchik N, Lee SH, Somlo S, Barbano PE, Coifman R & Sun Z (2009). Chemical modifier screen identifies HDAC inhibitors as suppressors of PKD models. Proc Natl Acad Sci U S A 106, 21819–21824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmosino M, Rizzo F, Ferrari P, Torielli L, Ferrandi M, Bianchi G, Svelto M & Valenti G (2011). NKCC2 is activated in Milan hypertensive rats contributing to the maintenance of salt‐sensitive hypertension. Pflugers Arch 462, 281–291. [DOI] [PubMed] [Google Scholar]
- Cebotaru V, Cebotaru L, Kim H, Chiaravalli M, Boletta A, Qian F & Guggino WB (2014). Polycystin‐1 negatively regulates Polycystin‐2 expression via the aggresome/autophagosome pathway. J Biol Chem 289, 6404–6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman AB, Johnson A, Gabow PA & Schrier RW (1990). The renin‐angiotensin‐aldosterone system and autosomal dominant polycystic kidney disease. N Engl J Med 323, 1091–1096. [DOI] [PubMed] [Google Scholar]
- Charles JF, Coury F, Sulyanto R, Sitara D, Wu J, Brady N, Tsang K, Sigrist K, Tollefsen DM, He L, Storm D & Aliprantis AO (2012). The collection of NFATc1‐dependent transcripts in the osteoclast includes numerous genes non‐essential to physiologic bone resorption. Bone 51, 902–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chebib FT, Sussman CR, Wang X, Harris PC & Torres VE (2015). Vasopressin and disruption of calcium signalling in polycystic kidney disease. Nat Rev Nephrol 11, 451–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SR, Chen M, Deng SL, Hao XX, Wang XX & Liu YX (2016). Sodium‐hydrogen exchanger NHA1 and NHA2 control sperm motility and male fertility. Cell Death Dis 7, e2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coaxum SD, Blanton MG, Joyner A, Akter T, Bell PD, Luttrell LM, Raymond JR Sr, Lee MH, Blichmann PA, Garnovskaya MN & Saigusa T (2014). Epidermal growth factor‐induced proliferation of collecting duct cells from Oak Ridge polycystic kidney mice involves activation of Na+/H+ exchanger. Am J Physiol Cell Physiol 307, C554–C560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutry N, Farman N, Bonvalet JP & Blot‐Chabaud M (1995). Synergistic action of vasopressin and aldosterone on basolateral Na+‐K+‐ATPase in the cortical collecting duct. J Membr Biol 145, 99–106. [DOI] [PubMed] [Google Scholar]
- Cross BM, Breitwieser GE, Reinhardt TA & Rao R (2014). Cellular calcium dynamics in lactation and breast cancer: from physiology to pathology. Am J Physiol Cell Physiol 306, C515–C526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida PW, de Freitas Lima R, de Morais Gomes ER, Rocha‐Resende C, Roman‐Campos D, Gondim AN, Gavioli M, Lara A, Parreira A, de Azevedo Nunes SL, Alves MN, Santos SL, Alenina N, Bader M, Resende RR, dos Santos Cruz J, Souza dos Santos RA & Guatimosim S (2013). Functional cross‐talk between aldosterone and angiotensin‐(1‐7) in ventricular myocytes. Hypertension 61, 425–430. [DOI] [PubMed] [Google Scholar]
- De Vries TJ, Schoenmaker T, Aerts D, Grevers LC, Souza PP, Nazmi K, van de Wiel M, Ylstra B, Lent PL, Leenen PJ & Everts V (2015). M‐CSF priming of osteoclast precursors can cause osteoclastogenesis‐insensitivity, which can be prevented and overcome on bone. J Cell Physiol 230, 210–225. [DOI] [PubMed] [Google Scholar]
- Donowitz M, Ming Tse C & Fuster D (2013). SLC9/NHE gene family, a plasma membrane and organellar family of Na+/H+ exchangers. Mol Aspects Med 34, 236–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng M, Grice DM, Faddy HM, Nguyen N, Leitch S, Wang Y, Muend S, Kenny PA, Sukumar S, Roberts‐Thomson SJ, Monteith GR & Rao R (2010). Store‐independent activation of Orai1 by SPCA2 in mammary tumors. Cell 143, 84–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuster DG & Alexander RT (2014). Traditional and emerging roles for the SLC9 Na+/H+ exchangers. Pflugers Arch 466, 61–76. [DOI] [PubMed] [Google Scholar]
- Fuster DG, Zhang J, Shi M, Bobulescu IA, Andersson S & Moe OW (2008). Characterization of the sodium/hydrogen exchanger NHA2. J Am Soc Nephrol 19, 1547–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez‐Sintes R & Lucas JJ (2010). NFAT/Fas signaling mediates the neuronal apoptosis and motor side effects of GSK‐3 inhibition in a mouse model of lithium therapy. J Clin Invest 120, 2432–2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandjean EM & Aubry JM (2009). Lithium: updated human knowledge using an evidence‐based approach: part III: clinical safety. CNS Drugs 23, 397–418. [DOI] [PubMed] [Google Scholar]
- Grantham JJ, Chapman AB & Torres VE (2006). Volume progression in autosomal dominant polycystic kidney disease: the major factor determining clinical outcomes. Clin J Am Soc Nephrol 1, 148–157. [DOI] [PubMed] [Google Scholar]
- Harris PC & Rossetti S (2010). Molecular diagnostics for autosomal dominant polycystic kidney disease. Nat Rev Nephrol 6, 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasler U, Mordasini D, Bianchi M, Vandewalle A, Feraille E & Martin PY (2003). Dual influence of aldosterone on AQP2 expression in cultured renal collecting duct principal cells. J Biol Chem 278, 21639–21648. [DOI] [PubMed] [Google Scholar]
- Hughes J, Ward CJ, Peral B, Aspinwall R, Clark K, San Millan JL, Gamble V & Harris PC (1995). The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet 10, 151–160. [DOI] [PubMed] [Google Scholar]
- Kim E, Arnould T, Sellin LK, Benzing T, Fan MJ, Gruning W, Sokol SY, Drummond I & Walz G (1999). The polycystic kidney disease 1 gene product modulates Wnt signaling. J Biol Chem 274, 4947–4953. [DOI] [PubMed] [Google Scholar]
- Kim H, Xu H, Yao Q, Li W, Huang Q, Outeda P, Cebotaru V, Chiaravalli M, Boletta A, Piontek K, Germino GG, Weinman EJ, Watnick T & Qian F (2014). Ciliary membrane proteins traffic through the Golgi via a Rabep1/GGA1/Arl3‐dependent mechanism. Nat Commun 5, 5482.25405894 [Google Scholar]
- Kim JH & Kim N (2014). Regulation of NFATc1 in osteoclast differentiation. J Bone Metab 21, 233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjeldsen SE, Os I, Forsberg G, Aakesson I, Skjoto J, Frederichsen P, Fonstelien E & Eide I (1985). Dietary sodium intake increases vasopressin secretion in man. J Clin Hypertens 1, 123–131. [PubMed] [Google Scholar]
- Kling MA, Fox JG, Johnston SM, Tolkoff‐Rubin NE, Rubin RH & Colvin RB (1984). Effects of long‐term lithium administration on renal structure and function in rats. A distinctive tubular lesion. Lab Invest 50, 526–535. [PubMed] [Google Scholar]
- Knyazev EN, Mal'tseva DV, Zacharyants AA, Zakharova GS, Zhidkova OV & Poloznikov AA (2018). TNFα‐induced expression of transport protein genes in HUVEC cells is associated with enhanced expression of transcription factor genes RELB and NFKB2 of the non‐canonical NF‐κB pathway. Bull Exp Biol Med 164, 757–761. [DOI] [PubMed] [Google Scholar]
- Kondapalli KC, Kallay LM, Muszelik M & Rao R (2012). Unconventional chemiosmotic coupling of NHA2, a mammalian Na+/H+ antiporter, to a plasma membrane H+ gradient. J Biol Chem 287, 36239–36250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondapalli KC, Prasad H & Rao R (2014). An inside job: how endosomal Na+/H+ exchangers link to autism and neurological disease. Front Cell Neurosci 8, 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondapalli KC, Todd Alexander R, Pluznick JL & Rao R (2017). NHA2 is expressed in distal nephron and regulated by dietary sodium. J Physiol Biochem 73, 199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong H, Jones PP, Koop A, Zhang L, Duff HJ & Chen SR (2008). Caffeine induces Ca2+ release by reducing the threshold for luminal Ca2+ activation of the ryanodine receptor. Biochem J 414, 441–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lever JE (1979). Inducers of mammalian cell differentiation stimulate dome formation in a differentiated kidney epithelial cell line (MDCK). Proc Natl Acad Sci U S A 76, 1323–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine S, Franki N & Hays RM (1973). Effect of phloretin on water and solute movement in the toad bladder. J Clin Invest 52, 1435–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HM, He JY, Zhang Q, Lv WQ, Xia X, Sun CQ, Zhang WD & Deng HW (2018). Improved detection of genetic loci in estimated glomerular filtration rate and type 2 diabetes using a pleiotropic cFDR method. Mol Genet Genomics 293, 225–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez GJ, Pereira RM, Aijo T, Kim EY, Marangoni F, Pipkin ME, Togher S, Heissmeyer V, Zhang YC, Crotty S, Lamperti ED, Ansel KM, Mempel TR, Lahdesmaki H, Hogan PG & Rao A (2015). The transcription factor NFAT promotes exhaustion of activated CD8+ T cells. Immunity 42, 265–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misfeldt DS, Hamamoto ST & Pitelka DR (1976). Transepithelial transport in cell culture. Proc Natl Acad Sci U S A 73, 1212–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson M, Hansson E & Ronnback L (1992). Agonist‐evoked Ca2+ transients in primary astroglial cultures—modulatory effects of valproic acid. Glia 5, 201–209. [DOI] [PubMed] [Google Scholar]
- Nims N, Vassmer D & Maser RL (2003). Transmembrane domain analysis of polycystin‐1, the product of the polycystic kidney disease‐1 (PKD1) gene: evidence for 11 membrane‐spanning domains. Biochemistry 42, 13035–13048. [DOI] [PubMed] [Google Scholar]
- Oberleithner H, Vogel U & Kersting U (1990). Madin‐Darby canine kidney cells. I. Aldosterone‐induced domes and their evaluation as a model system. Pflugers Arch 416, 526–532. [DOI] [PubMed] [Google Scholar]
- Olteanu D, Liu X, Liu W, Roper VC, Sharma N, Yoder BK, Satlin LM, Schwiebert EM & Bevensee MO (2012). Increased Na+/H+ exchanger activity on the apical surface of a cilium‐deficient cortical collecting duct principal cell model of polycystic kidney disease. Am J Physiol Cell Physiol 302, C1436–C1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong AC, Devuyst O, Knebelmann B, Walz G; ERA‐EDTA Working Group for Inherited Kidney Diseases (2015). Autosomal dominant polycystic kidney disease: the changing face of clinical management. Lancet 385, 1993–2002. [DOI] [PubMed] [Google Scholar]
- Pan MG, Xiong Y & Chen F (2013). NFAT gene family in inflammation and cancer. Curr Mol Med 13, 543–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlewitz A, Nafz B, Skalweit A, Fahling M, Persson PB & Thiele BJ (2010). Aldosterone and vasopressin affect α‐ and γ‐ENaC mRNA translation. Nucleic Acids Res 38, 5746–5760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry PJ, Calloway RA, Cook BL & Smith RE (1984). Theophylline precipitated alterations of lithium clearance. Acta Psychiatr Scand 69, 528–537. [DOI] [PubMed] [Google Scholar]
- Prasad H & Rao R (2018). Histone deacetylase‐mediated regulation of endolysosomal pH. J Biol Chem 293, 6721–6735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri S, Magenheimer BS, Maser RL, Ryan EM, Zien CA, Walker DD, Wallace DP, Hempson SJ & Calvet JP (2004). Polycystin‐1 activates the calcineurin/NFAT (nuclear factor of activated T‐cells) signaling pathway. J Biol Chem 279, 55455–55464. [DOI] [PubMed] [Google Scholar]
- Qian F, Germino FJ, Cai Y, Zhang X, Somlo S & Germino GG (1997). PKD1 interacts with PKD2 through a probable coiled‐coil domain. Nat Genet 16, 179–183. [DOI] [PubMed] [Google Scholar]
- Rodrig N, Osanai T, Iwamori M & Nagai Y (1988). Uncoupling of intracellular cyclic AMP and dome formation in cultured canine kidney epithelial cells: effects of gangliosides and vasopressin. J Biochem (Tokyo) 104, 215–219. [DOI] [PubMed] [Google Scholar]
- Schafer JA & Hawk CT (1992). Regulation of Na+ channels in the cortical collecting duct by AVP and mineralocorticoids. Kidney Int 41, 255–268. [DOI] [PubMed] [Google Scholar]
- Schrier RW (2009). Renal volume, renin‐angiotensin‐aldosterone system, hypertension, and left ventricular hypertrophy in patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 20, 1888–1893. [DOI] [PubMed] [Google Scholar]
- Schushan M, Xiang M, Bogomiakov P, Padan E, Rao R & Ben‐Tal N (2010). Model‐guided mutagenesis drives functional studies of human NHA2, implicated in hypertension. J Mol Biol 396, 1181–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scicchitano BM, Spath L, Musaro A, Molinaro M, Rosenthal N, Nervi C & Adamo S (2005). Vasopressin‐dependent myogenic cell differentiation is mediated by both Ca2+/calmodulin‐dependent kinase and calcineurin pathways. Mol Biol Cell 16, 3632–3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S, Watnick T & Atta MG (2010). Not all renal cysts are created equal. Lancet 376, 1024. [DOI] [PubMed] [Google Scholar]
- Soleiman AA, Thameem F & Khan I (2017). Mechanism of down regulation of Na‐H exchanger‐2 in experimental colitis. PLoS One 12, e0176767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Di Giovanni V, He N, Wang K, Ingram A, Rosenblum ND & Pei Y (2009). Systems biology of autosomal dominant polycystic kidney disease (ADPKD): computational identification of gene expression pathways and integrated regulatory networks. Hum Mol Genet 18, 2328–2343. [DOI] [PubMed] [Google Scholar]
- Stewart GS, Thistlethwaite A, Lees H, Cooper GJ & Smith C (2009). Vasopressin regulation of the renal UT‐A3 urea transporter. Am J Physiol Renal Physiol 296, F642–F648. [DOI] [PubMed] [Google Scholar]
- Stiber JA, Tabatabaei N, Hawkins AF, Hawke T, Worley PF, Williams RS & Rosenberg P (2005). Homer modulates NFAT‐dependent signaling during muscle differentiation. Dev Biol 287, 213–224. [DOI] [PubMed] [Google Scholar]
- Su HW, Yeh HH, Wang SW, Shen MR, Chen TL, Kiela PR, Ghishan FK & Tang MJ (2007). Cell confluence‐induced activation of signal transducer and activator of transcription‐3 (Stat3) triggers epithelial dome formation via augmentation of sodium hydrogen exchanger‐3 (NHE3) expression. J Biol Chem 282, 9883–9894. [DOI] [PubMed] [Google Scholar]
- Sun Y, Zhou H & Yang BX (2011). Drug discovery for polycystic kidney disease. Acta Pharmacol Sin 32, 805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suwanjang W, Holmstrom KM, Chetsawang B & Abramov AY (2013). Glucocorticoids reduce intracellular calcium concentration and protects neurons against glutamate toxicity. Cell Calcium 53, 256–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot JJ, Shillingford JM, Vasanth S, Doerr N, Mukherjee S, Kinter MT, Watnick T & Weimbs T (2011). Polycystin‐1 regulates STAT activity by a dual mechanism. Proc Natl Acad Sci U S A 108, 7985–7990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terryn S, Ho A, Beauwens R & Devuyst O (2011). Fluid transport and cystogenesis in autosomal dominant polycystic kidney disease. Biochim Biophys Acta 1812, 1314–1321. [DOI] [PubMed] [Google Scholar]
- Tkachenko O, Helal I, Shchekochikhin D & Schrier RW (2013). Renin‐angiotensin‐aldosterone system in autosomal dominant polycystic kidney disease. Curr Hypertens Rev 9, 12–20. [DOI] [PubMed] [Google Scholar]
- Vareesangthip K, Thomas TH & Wilkinson R (1995). Abnormal effect of thiol groups on erythrocyte Na/Li countertransport kinetics in adult polycystic kidney disease. Nephrol Dial Transplant 10, 2219–2223. [DOI] [PubMed] [Google Scholar]
- Villarroya‐Beltri C, Gutierrez‐Vazquez C, Sanchez‐Cabo F, Perez‐Hernandez D, Vazquez J, Martin‐Cofreces N, Martinez‐Herrera DJ, Pascual‐Montano A, Mittelbrunn M & Sanchez‐Madrid F (2013). Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat Commun 4, 2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward OM, Li Y, Yu S, Greenwell P, Wodarczyk C, Boletta A, Guggino WB & Qian F (2010). Identification of a polycystin‐1 cleavage product, P100, that regulates store operated Ca entry through interactions with STIM1. PLoS One 5, e12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang M, Feng M, Muend S & Rao R (2007). A human Na+/H+ antiporter sharing evolutionary origins with bacterial NhaA may be a candidate gene for essential hypertension. Proc Natl Acad Sci U S A 104, 18677–18681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Sonawane ND, Zhao D, Somlo S & Verkman AS (2008). Small‐molecule CFTR inhibitors slow cyst growth in polycystic kidney disease. J Am Soc Nephrol 19, 1300–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]