

Abstract
Background
Knowledge on the prevalence of sex chromosome abnormalities (SCAs) is limited, and delayed diagnosis or non-diagnosis of SCAs are a continuous concern. We aimed to investigate change over time in incidence, prevalence and age at diagnosis among Turner syndrome (TS), Klinefelter syndrome (KS), Triple X syndrome (Triple X) and Double Y syndrome (Double Y).
Methods
This study is a nationwide cohort study in a public health care system. The Danish Cytogenetic Central Registry (DCCR) holds information on all karyotypes performed in Denmark since 1961. We identified all individuals in the DCCR with a relevant SCA during 1961–2014; TS: n = 1156; KS: n = 1235; Triple X: n = 197; and Double Y: n = 287. From Statistics Denmark, which holds an extensive collection of data on the Danish population, complete data concerning dates of death and migrations in and out of Denmark were retrieved for all individuals.
Results
The prevalence among newborns was as follows: TS: 59 per 100,000 females; KS: 57 per 100,000 males; Triple X: 11 per 100,000 females; and Double Y: 18 per 100,000 males. Compared with the expected number among newborns, all TS, 38% of KS, 13% of Triple X, and 18% of Double Y did eventually receive a diagnosis. The incidence of TS with other karyotypes than 45,X (P < 0.0001), KS (P = 0.02), and Double Y (P = 0.03) increased during the study period whereas the incidence of 45,X TS decreased (P = 0.0006). The incidence of Triple X was stable (P = 0.22).
Conclusions
The prevalence of TS is higher than previously identified, and the karyotypic composition of the TS population is changing. Non-diagnosis is extensive among KS, Triple X and Double Y, whereas all TS seem to become diagnosed. The diagnostic activity has increased among TS with other karyotypes than 45,X as well as among KS and Double Y.
Keywords: Turner syndrome, Klinefelter syndrome, Triple X syndrome, Double Y syndrome, Prevalence, Incidence, Age at diagnosis
Introduction
Sex chromosome abnormalities (SCAs) - Turner syndrome (TS [45,X]), Klinefelter syndrome (KS [47,XXY]), Triple X syndrome (Triple X, [47,XXX]) and Double Y syndrome (Double Y,[47,XYY]) - are estimated to affect approximately 1 per 400 births [1]. A number of representative cytogenetic surveys were conducted years ago on newborns in various countries. Previously, we pooled data from these surveys in order to estimate the prevalence of TS, KS, Triple X and Double Y [2–5] which were; TS: 50 per 100,000 newborn females [24 TS among 48,744 newborn females] [6–10]; KS: 152 per 100,000 newborn males [84 KS among 55,212 newborn males] [6, 7, 10–13]; Triple X: 84 per 100,000 newborn females [62 Triple X among 73,990 newborn females] [4]; 4) Double Y: 98 per 100,000 newborn males [51 Double Y among 52,004 newborn males] [7, 10, 12, 14, 15]. Estimates are, however, subject to much uncertainty.
The presence of a SCA may affect individuals at multiple organ levels, but the range of affection is very wide. Abnormal and delayed puberty as well as infertility are, however, distinctive features in individuals affected by TS or KS [16, 17]. Previously, we and others, have reported an almost four-fold increased mortality [3–5, 18, 19] as well as increased morbidity associated to a wide range of diseases [18, 20] in individuals affected by SCAs compared to age and sex-matched controls. Further, we have reported a reduced socioeconomic status in individuals affected by a SCA with lower education (except for TS), increased risk of being retired from the labor market, lower income, and reduced likelihood of living in a relationship [21–24].
Delayed or even non-diagnosis of SCAs is common. Individuals not diagnosed in infancy often do not receive a diagnosis until years after relevant medical therapy should have been initiated to alleviate symptoms [4, 5, 25–27]. Further, delayed diagnosis prevents timely screening and intervention for common associated health problems as well as learning and behavioral disabilities [28, 29]. Thus, health related and social consequences of non-diagnosis and delayed diagnosis are a continuous concern.
In the present study we aimed to investigate change over time in incidence, prevalence, and age at diagnosis in a national cohorts of SCAs.
Material and methods
Registries and cases
The Danish Civil Registration System was established in 1968 and since then all persons residing in Denmark have been assigned a unique 10-digit civil personal registration (CPR) number. The last digit in the CPR number allows identification of the persons’ officially registered sex (odd = male; even = female). Further, the CPR number allows accurate matching of data from different data sources [30]. The Danish health care system is a public tax funded system ensuring all citizens free and equal health care access.
The Danish Cytogenetic Central Registry (DCCR) was established in 1968, and holds data on all pre-and postnatal karyotypes performed in Denmark since 1961. Only postnatally diagnosed individuals or prenatally diagnosed individuals, subsequently postnatally confirmed, are included in the present study. Data in the DCCR includes: 1) CPR number; 2) karyotype; 3) date of birth; and 4) date of karyotyping. The DCCR holds no information on phenotype or reasons for performing a karyotype. Information concerning degrees of mosaicism is not available.
The DCCR was searched for all cases diagnosed with a karyotype compatible with TS, KS, Triple X or Double Y during 1961–2014. The distribution of accepted karyotypes for each syndrome are presented in Table 1. We considered females with a 45,X mosaic composition to have a phenotype most consistent with TS and these cases were thus included in the TS group. Females with more than three X chromosomes (e.g. 48,XXXX) and males with more than two Y chromosomes (e.g. 48,XYYY) were included in the Triple X and Double Y group, respectively. Cases with an autosomal aneuploidy were included in their respective group of SCA. Data were retrieved from the DCCR in October 2015.
Table 1.
Distribution of karyotypes among Turner syndrome, Klinefelter syndrome, Triple X and Double Y syndrome
| Karyotypes | Number |
|---|---|
| Turner syndrome | 1156 |
| 45,X | 422 |
| 45,X/46,XX | 287 |
| Karyotypes containing an isochromosome: 45,X/46,I(X) and equivalents | 117 |
| Karyotypes containing Y chromosome material: 45,X/46,XY; and equivalents | 47 |
| Other | 283 |
| Klinefelter syndrome | 1235 |
| 47,XXY | 1080 |
| 47,XXY/46,XY | 78 |
| 47,XXY/46,XX/46,XY; 46,XY/47,XXY/48,XXXY; and equivalents | 32 |
| Karyotypes with an autosomal aneuplodi: 48,XXY,+ 18; 48,XXY,+ 21 | 5 |
| Other | 40 |
| Triple X syndrome | 197 |
| 47,XXX | 104 |
| 47,XXX/46,XX | 58 |
| 48,XXXX; 49,XXXXX | 10 |
| Karyotypes with an autosomal aneuplodi: 48,XXX + 18; 48,XXX + 21; and quivalents | 10 |
| Other | 15 |
| Double Y syndrome | 287 |
| 47,XYY | 206 |
| 47,XYY/46,XY | 26 |
| Karyotypes containing an isochromosome: 46,XY/47,XY,+I(Yq); 45,X/47,XY,+I(Y)/46,XY; and equivalents | 6 |
| 48,XXYY; 48,XYYY; and equivalents | 35 |
| Other | 14 |
The Causes of Death Registry holds information on all deaths since 1970 including date of death and primary and auxiliary cause of death [31]. We retrieved data on all deceased cases during 1970 to December 31, 2014. Further, from the Civil Registration system complete data on migrations in and out of Denmark were retrieved.
Statistics Denmark is a state institution under the Ministry of Economic Affairs and the Interior and collects data on the Danish population. From Statistics Denmark (https://www.dst.dk/en), annually data concerning the number of females and males living in Denmark were retrieved. Likewise was annual data on the Danish birth cohorts.
Statistics
We distinguish between population-based prevalence and prevalence among newborns. In the following, we solely use the term “prevalence” when describing the prevalence of SCAs among newborns. All cases were incident the year they were diagnosed and prevalent the year they were born.
The population-based prevalence was estimated as the annual number of cases being alive in Denmark each year during the study period (1970–2014). 1970 was chosen as start of the observation period to avoid confounding from a run-in phase of the DCCR. We defined cases diagnosed prior to 1970 to be prevalent in 1970. Deceased cases were excluded the first year following death. Emigrated cases were excluded the first year following emigration, if emigration was not followed by subsequent immigration. A population-based prevalence was also estimated using the expected prevalence of the individual SCAs. Linear extrapolation were used to estimate when the observed and the expected population-based prevalence equals each other.
Incidence was estimated as the average number of diagnosed cases per million females (TS and Triple X) or males (KS and Double Y) in the background population each year during the study period. Cases diagnosed prior to 1970 were not included in this analysis.
Prevalence was estimated as the average number of cases being born per 100,000 newborn females (TS and Triple X) or males (KS and Double Y) in the background population. Cases were clustered according to 5-year calendar time periods, and the number of diagnosed cases per 5 years was divided by the sum of their five respective birth cohorts. Data on the Danish birth cohorts are available since 1901, thus observation periods started in 1901, and cases born prior to 1901 (TS: n = 5; KS: n = 11; Triple X: n = 2; and Double Y: n = 1) were thus not included in this analysis. All observation periods ended in 2014.
Time trend in incidence was analyzed using Poisson regression. Time trend in age at diagnosis during the study period was analyzed using linear regression. Differences in age at diagnosis among subgroups within one syndrome or among different syndromes were analyzed using Kruskal-Wallis test. P-values < 0.05 was considered statistically significant. Analyzes were made in StataCorp 13.1 and 15.1 for Windows.
Results
During 1961–2014 a total of 2875 individuals were diagnosed and recorded in the DCCR with a karyotype compatible with TS (n = 1156); KS (n = 1235); Triple X (n = 197); or Double Y (n = 287) (Table 1). Year of diagnosis did not differ among the syndromes (P = 0.07).
Population-based prevalence
The population-based prevalence of TS, KS, Triple X and Double Y increased during the study period (Fig. 1). Compared to the expected population-based prevalence, 70% of TS (980 out of 1418); 23% of KS (962 out of 4244); 7% of Triple X (165 out of 2381); and 9% of Double Y (239 out of 2736) were diagnosed and alive by the end of the study.
Fig. 1.

Absolute prevalence of Turner syndrome, Klinefelter syndrome, Triple X syndrome and Double Y syndrome in the Danish population. The observed number of a Turner syndrome (TS) and Triple X syndrome and b Klinefelter syndrome (KS) and Double Y syndrome (Double Y) being alive during 1970–2014 are illustrated by solid and dotted lines. Deceased or emigrated individuals were subtracted. Dashed lines indicate the expected number assuming a true prevalence of 1) 50 TS per 100,000 at birth; 2) 84 Triple X per 100,000 at birth; 3) 152 KS per 100,000 at birth; and 4) 98 Double Y per 100,000 at birth as well as a similar mortality as in the general population
Linear extrapolation of the observed and expected population-based prevalence of TS show that these theoretically equals each other in 2039 – in other words, all live TS will be diagnosed in 2039, given the current diagnostic practices continue unaltered, given the expected population-based prevalence is correct, and given the mortality rate is identical in TS and in the background population. For KS, this point in time was estimated to the year of 2471. Theoretically, the expected and observed prevalence of Triple X and Double Y will continue to diverge owing to a greater increase in the expected prevalence than in the observed prevalence.
Incidence
During the study period an average of 2,648,000 females in Denmark were at risk of being diagnosed with a SCA. The average annual number of diagnosed TS was 24. Thus, the average incidence of TS was 9.0 per million females. The average number of 45,X TS and TS with other karyotypes than 45,X (“other” TS) diagnosed annually was 8 and 16, respectively, leading to an average incidence of 3.1 45,X TS and 5.9 “other” TS per million females. The incidence among all TS was stable during the study period (P = 0.10), whereas the incidence was decreasing for 45,X TS (P = 0.0006) and increasing for “other” TS (P < 0.0001) (Fig. 2).
Fig. 2.
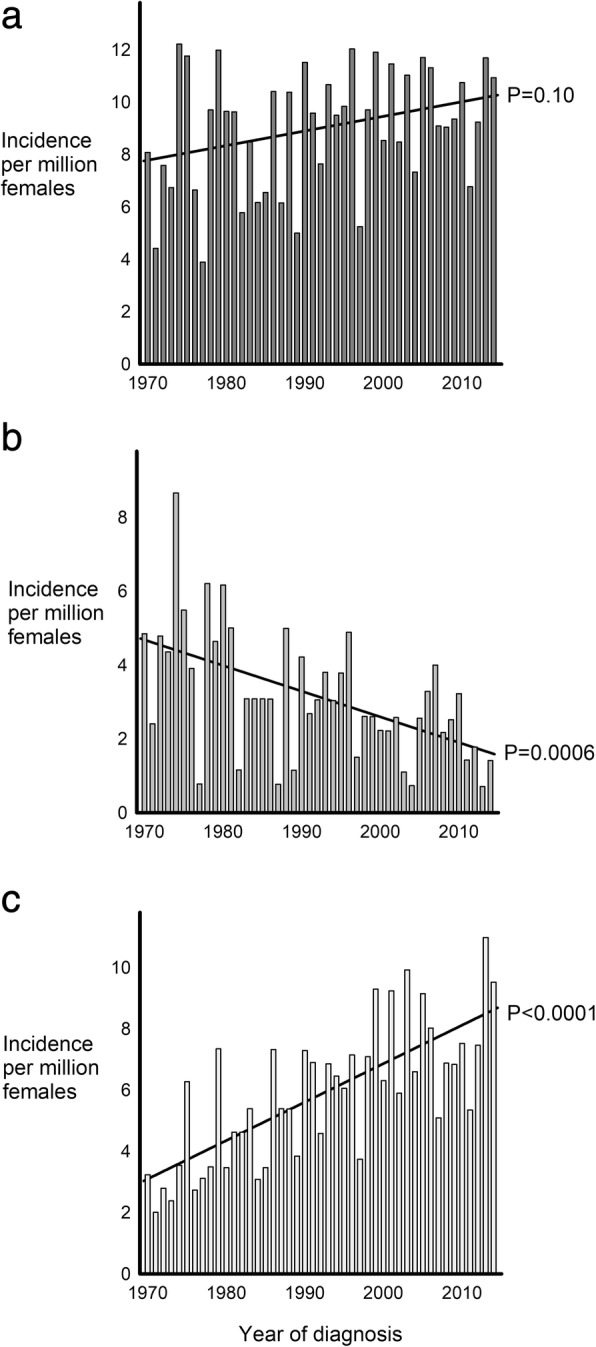
Incidence of Turner syndrome according to karyotype. Incidence of a all Turner syndrome (TS); b 45,X TS; and c TS with other karyotypes per million females during 1970–2014. Solid lines illustrate time trend in incidence during the 1970–2014. P-values indicate the significance level of time trend in incidence
An average of four Triple X was diagnosed each year during the study period leading to an incidence of 1.6 Triple X per million females. No change in incidence among Triple X was observed during the study period (P = 0.22) (Fig. 3a).
Fig. 3.

Incidence of Triple X syndrome, Klinefelter syndrome and Double Y syndrome. Incidence of a Triple X syndrome; b Klinefelter syndrome; and c Double Y syndrome per million females or males during 1970–2014. Solid lines illustrate time trend in incidence during the 1970–2014. P-values indicate the significance level of time trend in incidence
The average number of males in Denmark being at risk of being diagnosed with a SCA each year during 1970–2014 was 2,589,000, and annually an average of 24 KS and 6 Double Y were diagnosed. Thus, the average incidence of KS and Double Y were 9.4 and 2.2 per million males during the study period. The incidence was increasing for KS (P = 0.02) as well as for Double Y (P = 0.03) during the study period (Fig. 3b and c).
Prevalence among newborns
Median year of birth among the SCAs was as follows: 1) TS: 1971 (range: 1885–2014); 2) Triple X: 1975 (range: 1895–2014); 3) KS: 1965 (range: 1882–2013); and 4) Double Y: 1977 (range: 1899–2013). During 1901 to 2014, prevalence was divided into sub-periods according to differences in average prevalence. During 1961–1985 the maximum average prevalence of all TS was 59 per 100,000 newborn females (45,X TS: 21 per 100,000 newborn females and “other” TS: 38 per 100,000 newborn females), thus higher than expected. During 1971–1990 the maximum average prevalence of Triple X was 11 per 100,000 newborn females, corresponding to 13% of the expected prevalence. The maximum average prevalence of KS was observed during 1961–1990 being 57 per 100,000 newborn males, corresponding to 38% of the expected prevalence. Double Y had a maximum average prevalence of 18 per 100,000 newborn males during 1976–1990, corresponding to 18% of the expected prevalence (Fig. 4).
Fig. 4.

Prevalence of sex chromosome abnormalities among newborns. a Prevalence of Turner syndrome (TS). Black bars indicates the prevalence of 45,X TS among all TS; b Triple X syndrome (Triple X); c Klinefelter syndrome (KS); and d Double Y syndrome (Double Y) during 1901–2014. Dashed lines indicate the expected prevalence and dotted lines indicate the observed maximum average prevalence of TS, Triple X, KS and Double Y per 100,000 newborn females or males
Age at diagnosis
The median age at diagnosis was for TS 15.1 (range: 0.0–85.4) years, for Triple X 17.9 (0.0–73.2) years, for KS 27.5 (0–82.8) years, and for Double Y 15.1 (0–70.7) years (Fig. 5a and b). 45,X TS was diagnosed with significantly less delay than TS with other karyotypes (median age: 45,X TS = 11.4 years versus “other” TS = 19.0 years) (P < 0.0001) (Fig. 5b). There was no change in age at diagnosis during 1970–2014 among all TS (P = 0.17), whereas age at diagnosis was decreasing among 45,X TS (P = 0.005). Age at diagnosis among TS with other karyotypes was stable (P = 0.99) during the study period, as well as among the other SCAs (Triple X: P = 0.28; KS: P = 0.72; and Double Y: P = 0.33). It is clear from the violin plots that the pattern of age at diagnosis is very different among different groups of SCAs (Fig. 5), with most KS being diagnosed much later than most TS, and with Triple X and Double Y being intermediate.
Fig. 5.
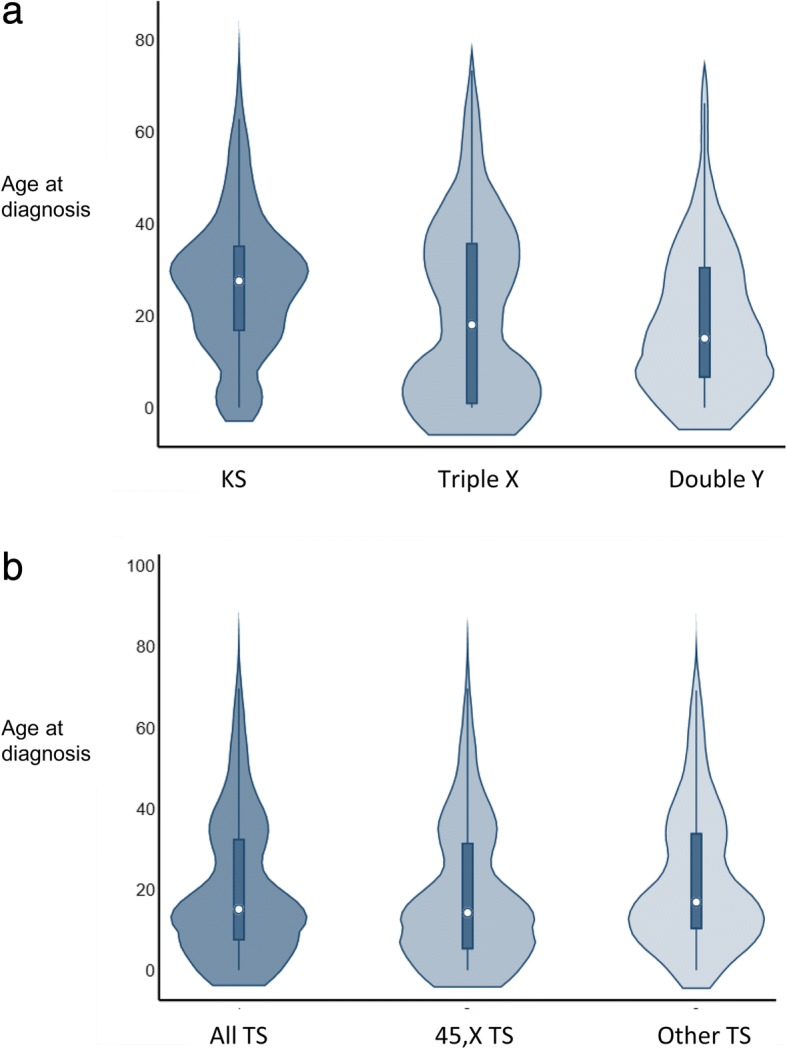
Age at diagnosis among sex chromosome abnormalities. Violin plots of age at diagnosis among a Klinefelter syndrome (KS), Triple X syndrome (Triple X) and Double Y syndrome (Double Y) and among b all Turner syndrome (TS), TS with a 45,X karyotype and TS with other karyotypes, diagnosed during 1970–2014. The small circle in the middle of the plot is median age, the dark rectangle depicts interquartile range, the thin dark lines depicts 95% confidence interval, and the density plot width equals frequency of age at diagnosis
Discussion
This population based study shows that the previously estimated prevalence of 50 TS per 100,000 may be an underestimate as we here present data showing a prevalence of 59 TS per 100,000 newborn females, corresponding to 1 per 1700. The karyotypic composition of the TS population is seemingly changing as the incidence of 45,X TS is decreasing and the incidence of TS with other karyotypes is increasing. Among KS and Double Y incidence is increasing as well, whereas it is stable among Triple X. Non-diagnosis remains extensive among KS, Triple X and Double Y. All TS eventually become diagnosed, although with a considerable delay.
The prevalence of the four forms of SCAs varied with a similar pattern during the study period. The marked increase in prevalence observed during the 1960’ties to the 1990’ties, possibly reflects that affected individuals from these birth cohorts were either born or reached puberty or fertil age at a time when karyotyping had become an established diagnostic procedure. However, comparing the maxiumum average prevalence with the expected prevalence of SCAs, the majority of KS (62%), Triple X (87%) and Double Y (82%) remain undiagnosed. In 2003, we reported that approximately 75% of expected KS was undiagnosed [2]. Although more with KS are diagnosed presently, we consider this far from satisfactory. In contrast, the average prevalence of TS exceeded the expected prevalence.
The population-based prevalence was steadily increasing for all SCAs during the study period caused by the build-up of the DCCR with continuing recruitment exceeding the rate of exit, a phenomenon known from other rare conditions as well [32]. The population-based prevalence will be stable when recruitment equals exit (death or emigration). Owing to increased mortality as well as increased rates of induced abortions among SCAs [33, 34] it will though remain lower than expected even with complete diagnosis of SCAs. However, it is difficult to ascertain how much prevalence is affected hereby. Only approximately 5% of pregnant women in Denmark have a prenatal karyotype performed [35], but they are of course selected as high-risk patients based on triple screening and non-invasive prenatal testing.
The incidence among all TS was stable during the study period. Interestingly, the incidence was steeply decreasing for 45,X TS and increasing for TS with other karyotypes. If this development continues it seems likely that 45,X TS are becoming extinct. Between 2004 and 2006, Denmark instituted a free prenatal screening program for Down syndrome (DS), in which over 90% of pregnant women in Denmark participate, and previously we have reported that approximately 40% of expected TS fetuses are detected by the DS screening [34]. The high induced abortion rate among prenatally detected 45,X TS fetuses likely contribute to the decreasing incidence of 45,X TS. Induced abortion among TS fetuses with other karyotypes are less pronounced [34], and a generally more favorable phenotype among these TS [36, 37] may prevent early diagnosis. A likely explanation for the increase in incidence among TS with other karyotypes may be that fertility treatment has become more common and thus more are diagnosed owing to fertility problems.
Late diagnosis as well as non-diagnosis of SCAs have been a continuous concern [25–27, 38] since SCAs are associated with increased morbidity and mortality [3, 5, 20, 23, 39], learning and/or behavioral disabilities [40–45] as well as reduced socioeconomic outcomes. Although there is lack of evidence regarding the influence of age at diagnosis on long-term outcomes, we believe that early diagnosis will provide better overall long-term outcome in SCAs by providing an opportunity for timely intervention against associated health problems as well as against learning and behavioral problems. Regrettably, the present study shows that delay in diagnosis remains a major problem (Fig. 5). Increased vigilance among health care professionals as well as a more liberal access to diagnostic tools was however hypothesized as having contributed to a decreasing diagnostic delay. As previously suggested for both TS and KS, neonatal screening programs would allow complete diagnosis of SCAs without diagnostic delay. Population-based, neonatal screening can be considered for conditions which have: 1) the magnitude to be an important health problem with a latent and early asymptomatic stage; and 2) has a well-understood natural history for which there are accepted treatments with associated facilities for diagnoses and treatment [46]. We consider these requirements fulfilled for SCAs. Due to the rarity of the syndromes it will, however, take a long time to demonstrate associations between early diagnosis, continuous specialized care and improved long-term outcomes.
The broad clinical spectrum observed among SCAs, ranging from overt to minimal or no apparent clinical features and only subtle symptoms [17, 47, 48] likely relates to the non-diagnosis and the delayed diagnosis. The fate of individuals with SCAs escaping diagnosis remains an enigma. Possibly, undiagnosed individuals experience a spectrum of similar challenges as those diagnosed, yet they might remain undiagnosed due to reluctance of seeking medical advice or due to lack of attention from health professionals. Based on our clinical experience, we believe that individuals diagnosed late in life more or less struggle with similar problems as those being diagnosed early in life. Recent data from UK among TS and Triple X support this hypothesis [36]. Here a large number of presumably undiagnosed 45,X and 47,XXX females were detected approximately about 250,000 examined females, presenting with typical features for both of these syndromes.
Conclusions
The karyotypic composition of TS is changing with less 45,X and more mosaic TS being diagnosed, possibly due to increased frequency of legal abortions among 45,X TS. TS have a higher prevalence than previously anticipated as observed in 1 per 1700 newborn females. The majority of KS, Triple X and Double Y remain undiagnosed despite an increase in diagnostic activity among KS and Double Y. Delayed diagnosis is a continuous problem among all SCAs, and no change over time has been observed. We consider a newborn screening program as the only opportunity to reduce both diagnostic delay as well as non-diagnosis and with a potential to improve health and socioeconomics among individuals affected by SCAs.
Acknowledgments
Data manager, Jan Hansen, from the DCCR is sincerely thanked for identifying patients in the DCCR.
Funding
We received grants from the Lundbeck Foundation, Novo Nordisk Foundation (grant agreement NNF13OC0003234 andNNF15OC0016474), “Fonden til lægevidenskabens fremme” and the Familien Hede Nielsen foundation. The sponsors had no influence on the preparation, design, analysis, and reporting of this study.
Availability of data and materials
The data that support the findings of this study are available from the DCCR and Statistics Denmark but restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available. Data are however available from the DCCR and Statistics Denmark upon request if permission from relevant authorities in Denmark has been obtained.
Abbreviations
- CPR number
Civil personal registration number
- DCCR
Danish Cytogenetic Central Registry
- Double Y
Double Y syndrome
- KS
Klinefelter syndrome
- SCA
Sex chromosome abnormalilty
- Triple X
Triple X syndrome
- TS
Turner syndrome
Authors’ contributions
MHV retrieved data from the DCCR and Statistics Denmark. AB did the statistical analyses and drafted the manuscript. MHV, KS and CHG contributed to interpretation of data. All authors contributed with critical revision of the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The study was approved by the Danish Data Protection Agency (journal number: 2013-41-2017). According to Danish law no approval was obtained by the National Committee on Health Research Ethics since the study solely includes registry data.
Consent for publication
The manuscript contains no individual person’s data in any form.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Agnethe Berglund, Email: agnethe.berglund@clin.au.dk.
Mette Hansen Viuff, Email: metteviuff@clin.au.dk.
Anne Skakkebæk, Email: asj@clin.au.dk.
Simon Chang, Email: simon.chang@rsyd.dk.
Kirstine Stochholm, Email: stochholm@dadlnet.dk.
Claus Højbjerg Gravholt, Email: ch.gravholt@dadlnet.dk.
References
- 1.Linden MG, Bender BG, Robinson A. Sex chromosome tetrasomy and pentasomy. Pediatrics. 1995;96:672–682. [PubMed] [Google Scholar]
- 2.Bojesen A, Juul S, Gravholt CH. Prenatal and postnatal prevalence of Klinefelter syndrome: a national registry study. J Clin Endocrinol Metab. 2003;88:622–626. doi: 10.1210/jc.2002-021491. [DOI] [PubMed] [Google Scholar]
- 3.Stochholm K, Juul S, Juel K, Naeraa RW, Gravholt CH. Prevalence, incidence, diagnostic delay, and mortality in turner syndrome. J Clin Endocrinol Metab. 2006;91:3897–3902. doi: 10.1210/jc.2006-0558. [DOI] [PubMed] [Google Scholar]
- 4.Kirstine S, Svend J, Højbjerg GC. Mortality and incidence in women with 47,XXX and variants. Am J Med Genet A. 2010;152A:367–372. doi: 10.1002/ajmg.a.33214. [DOI] [PubMed] [Google Scholar]
- 5.Stochholm K, Juul S, Gravholt CH. Diagnosis and mortality in 47,XYY persons: a registry study. Orphanet J Rare Dis. 2010;5:15. doi: 10.1186/1750-1172-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bochkov NP, Kuleshov NP, Chebotarev AN, Alekhin VI, Midian SA. Population cytogenetic investigation of newborns in Moscow. Hum Genet. 1974;22:139–152. doi: 10.1007/BF00278453. [DOI] [PubMed] [Google Scholar]
- 7.Hamerton JL, Canning N, Ray M, Smith S. A cytogenetic survey of 14,069 newborn infants. I. Incidence of chromosome abnormalities. Clin Genet. 1975;8:223–243. doi: 10.1111/j.1399-0004.1975.tb01498.x. [DOI] [PubMed] [Google Scholar]
- 8.Imaizumi KKY. Prevalence of turner syndrome in Japan. In: Hibi I, Takano K, editors. Basic and clinical approach to turner syndrome. Amsterdam: Excerpta Medica; 1993. pp. 3–6. [Google Scholar]
- 9.Jacobs PA, Melville M, Ratcliffe S, Keay AJ, Syme J. A cytogenetic survey of 11,680 newborn infants. Ann Hum Genet. 1974;37:359–376. doi: 10.1111/j.1469-1809.1974.tb01843.x. [DOI] [PubMed] [Google Scholar]
- 10.Nielsen J, Wohlert M. Sex chromosome abnormalities found among 34,910 newborn children: results from a 13-year incidence study in Arhus, Denmark. Birth Defects Orig Artic Ser. 1990;26:209–223. [PubMed] [Google Scholar]
- 11.Higurashi M, Iijima K, Ishikawa N, Hoshina H, Watanabe N. Incidence of major chromosome aberrations in 12,319 newborn infants in Tokyo. Hum Genet. 1979;46:163–172. doi: 10.1007/BF00291918. [DOI] [PubMed] [Google Scholar]
- 12.Ratcliffe SH. Development of children with sex chromosome abnormalities. Proc R Soc Med. 1976;69:189–191. [PMC free article] [PubMed] [Google Scholar]
- 13.Taylor AI, Moores EC. A sex chromatin survey of newborn children in two London hospitals. J Med Genet. 1967;4:258–259. doi: 10.1136/jmg.4.4.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goad WB, Robinson A, Puck TT. Incidence of aneuploidy in a human population. Am J Hum Genet. 1976;28:62–68. [PMC free article] [PubMed] [Google Scholar]
- 15.Maeda T, Ohno M, Matsunobu A, Yoshihara K, Yabe N. A cytogenetic survey of 14,835 consecutive liveborns. Jinrui Idengaku Zasshi. 1991;36:117–129. doi: 10.1007/BF01876812. [DOI] [PubMed] [Google Scholar]
- 16.Elsheikh M, Dunger DB, Conway GS, Wass JA. Turner’s syndrome in adulthood. Endocr Rev. 2002;23:120–140. doi: 10.1210/edrv.23.1.0457. [DOI] [PubMed] [Google Scholar]
- 17.Lanfranco F, Kamischke A, Zitzmann M, Nieschlag E. Klinefelter’s syndrome. Lancet. 2004;364:273–283. doi: 10.1016/S0140-6736(04)16678-6. [DOI] [PubMed] [Google Scholar]
- 18.Bojesen A, Gravholt CH. Morbidity and mortality in Klinefelter syndrome (47,XXY) Acta Paediatr. 2011;100:807–813. doi: 10.1111/j.1651-2227.2011.02274.x. [DOI] [PubMed] [Google Scholar]
- 19.Swerdlow AJ, Schoemaker MJ, Higgins CD, Wright AF, Jacobs PA. Cancer incidence and mortality in men with Klinefelter syndrome: a cohort study. J Natl Cancer Inst. 2005;97:1204–1210. doi: 10.1093/jnci/dji240. [DOI] [PubMed] [Google Scholar]
- 20.Gravholt CH, Juul S, Naeraa RW, Hansen J. Morbidity in turner syndrome. J Clin Epidemiol. 1998;51:147–158. doi: 10.1016/S0895-4356(97)00237-0. [DOI] [PubMed] [Google Scholar]
- 21.Bojesen A, Stochholm K, Juul S, Gravholt CH. Socioeconomic trajectories affect mortality in Klinefelter syndrome. J Clin Endocrinol Metab. 2011;96:2098–2104. doi: 10.1210/jc.2011-0367. [DOI] [PubMed] [Google Scholar]
- 22.Stochholm K, Hjerrild B, Mortensen KH, Juul S, Frydenberg M, Gravholt CH. Socioeconomic parameters and mortality in turner syndrome. Eur J Endocrinol. 2012;166:1013–1019. doi: 10.1530/EJE-11-1066. [DOI] [PubMed] [Google Scholar]
- 23.Stochholm K, Juul S, Gravholt CH. Socio-economic factors affect mortality in 47,XYY syndrome-a comparison with the background population and Klinefelter syndrome. Am J Med Genet A. 2012;158a:2421–2429. doi: 10.1002/ajmg.a.35539. [DOI] [PubMed] [Google Scholar]
- 24.Stochholm K, Juul S, Gravholt CH. Poor socio-economic status in 47,XXX --an unexpected effect of an extra X chromosome. Eur J Med Genet. 2013;56:286–291. doi: 10.1016/j.ejmg.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 25.Sävendahl L, Davenport ML. Delayed diagnoses of Turner’s syndrome: proposed guidelines for change. J Pediatr. 2000;137:455–459. doi: 10.1067/mpd.2000.107390. [DOI] [PubMed] [Google Scholar]
- 26.Herlihy AS, Gillam L, Halliday JL, McLachlan RI. Postnatal screening for Klinefelter syndrome: is there a rationale? Acta Paediatr. 2011;100:923–933. doi: 10.1111/j.1651-2227.2011.02151.x. [DOI] [PubMed] [Google Scholar]
- 27.Lee MC, Conway GS. Turner's syndrome: challenges of late diagnosis. Lancet Diabetes Endocrinol. 2014;2:333–338. doi: 10.1016/S2213-8587(13)70153-0. [DOI] [PubMed] [Google Scholar]
- 28.Gravholt CH, Andersen NH, Conway GS, Dekkers OM, Geffner ME, Klein KO, et al. Clinical practice guidelines for the care of girls and women with turner syndrome: proceedings from the 2016 Cincinnati international turner syndrome meeting. Eur J Endocrinol. 2017;177:G1–g70. doi: 10.1530/EJE-17-0430. [DOI] [PubMed] [Google Scholar]
- 29.Gravholt CH, Chang S, Wallentin M, Fedder J, Moore P, Skakkebaek A. Klinefelter syndrome - integrating genetics, neuropsychology and endocrinology. Endocr Rev. 2018;39(4):389–423. doi: 10.1210/er.2017-00212. [DOI] [PubMed] [Google Scholar]
- 30.Schmidt M, Schmidt SA, Sandegaard JL, Ehrenstein V, Pedersen L, Sorensen HT. The Danish National Patient Registry: a review of content, data quality, and research potential. Clin Epidemiol. 2015;7:449–490. doi: 10.2147/CLEP.S91125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Helweg-Larsen K. The Danish register of causes of death. Scand J Public Health. 2011;39:26–29. doi: 10.1177/1403494811399958. [DOI] [PubMed] [Google Scholar]
- 32.Groth KA, Hove H, Kyhl K, Folkestad L, Gaustadnes M, Vejlstrup N, et al. Prevalence, incidence, and age at diagnosis in Marfan syndrome. Orphanet J Rare Dis. 2015;10:153. doi: 10.1186/s13023-015-0369-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jeon KC, Chen LS, Goodson P. Decision to abort after a prenatal diagnosis of sex chromosome abnormality: a systematic review of the literature. Genet Med. 2012;14:27–38. doi: 10.1038/gim.0b013e31822e57a7. [DOI] [PubMed] [Google Scholar]
- 34.Viuff MH, Stochholm K, Uldbjerg N, Nielsen BB, Gravholt CH. Only a minority of sex chromosome abnormalities are detected by a national prenatal screening program for Down syndrome. Hum Reprod. 2015;30:2419–2426. doi: 10.1093/humrep/dev192. [DOI] [PubMed] [Google Scholar]
- 35.Petersen OB, Vogel I, Ekelund C, Hyett J, Tabor A. Potential diagnostic consequences of applying non-invasive prenatal testing: population-based study from a country with existing first-trimester screening. Ultrasound Obstet Gynecol. 2014;43:265–271. doi: 10.1002/uog.13270. [DOI] [PubMed] [Google Scholar]
- 36.Tuke MA, Ruth KS, Wood AR, Beaumont RN, Tyrrell J, Jones SE, et al. Mosaic turner syndrome shows reduced penetrance in an adult population study. Genet Med. 2018 [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- 37.El-Mansoury M, Barrenas ML, Bryman I, Hanson C, Larsson C, Wilhelmsen L, et al. Chromosomal mosaicism mitigates stigmata and cardiovascular risk factors in turner syndrome. Clin Endocrinol. 2007;66:744–751. doi: 10.1111/j.1365-2265.2007.02807.x. [DOI] [PubMed] [Google Scholar]
- 38.Aksglaede L, Garn ID, Hollegaard MV, Hougaard DM, Rajpert-De Meyts E, Juul A. Detection of increased gene copy number in DNA from dried blood spot samples allows efficient screening for Klinefelter syndrome. Acta Paediatr. 2012;101:e561–e563. doi: 10.1111/apa.12008. [DOI] [PubMed] [Google Scholar]
- 39.Bojesen A, Juul S, Birkebaek NH, Gravholt CH. Morbidity in Klinefelter syndrome: a Danish register study based on hospital discharge diagnoses. J Clin Endocrinol Metab. 2006;91:1254–1260. doi: 10.1210/jc.2005-0697. [DOI] [PubMed] [Google Scholar]
- 40.Bardsley MZ, Kowal K, Levy C, Gosek A, Ayari N, Tartaglia N, et al. 47,XYY syndrome: clinical phenotype and timing of ascertainment. J Pediatr. 2013;163:1085–1094. doi: 10.1016/j.jpeds.2013.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Otter M, Schrander-Stumpel CTRM, Curfs LMG. Triple X syndrome: a review of the literature. Eur J Hum Genet. 2010;18:265–271. doi: 10.1038/ejhg.2009.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Skakkebaek A, Gravholt CH, Rasmussen PM, Bojesen A, Jensen JS, Fedder J, et al. Neuroanatomical correlates of Klinefelter syndrome studied in relation to the neuropsychological profile. Neuroimage Clin. 2014;4:1–9. doi: 10.1016/j.nicl.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tartaglia NR, Howell S, Sutherland A, Wilson R, Wilson L. A review of trisomy X (47,XXX) Orphanet J Rare Dis. 2010;5:8. doi: 10.1186/1750-1172-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Collaer ML, Geffner ME, Kaufman FR, Buckingham B, Hines M. Cognitive and behavioral characteristics of turner syndrome: exploring a role for ovarian hormones in female sexual differentiation. Horm Behav. 2002;41:139–155. doi: 10.1006/hbeh.2001.1751. [DOI] [PubMed] [Google Scholar]
- 45.Bishop DV, Jacobs PA, Lachlan K, Wellesley D, Barnicoat A, Boyd PA, et al. Autism, language and communication in children with sex chromosome trisomies. Arch Dis Child. 2011;96:954–959. doi: 10.1136/adc.2009.179747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grosse SD, Rogowski WH, Ross LF, Cornel MC, Dondorp WJ, Khoury MJ. Population screening for genetic disorders in the 21st century: evidence, economics, and ethics. Public Health Genomics. 2010;13:106–115. doi: 10.1159/000226594. [DOI] [PubMed] [Google Scholar]
- 47.Barr ML, Sergovich FR, Carr DH, Saver EL. The triplo-X female: an appraisal based on a study of 12 cases and a review of the literature. Can Med Assoc J. 1969;101:247–258. [PMC free article] [PubMed] [Google Scholar]
- 48.Davenport ML. Approach to the patient with turner syndrome. J Clin Endocrinol Metab. 2010;95:1487–1495. doi: 10.1210/jc.2009-0926. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the DCCR and Statistics Denmark but restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available. Data are however available from the DCCR and Statistics Denmark upon request if permission from relevant authorities in Denmark has been obtained.