

Key Clinical Message
Paroxysmal nocturnal hemoglobinuria (PNH), a rare benign hematological disorder, presents with a wide variety of clinical symptoms. A direct Coombs‐negative hemolytic anemia combined with an increased LDH = Lactate dehydrogenase level are signs to test for PNH. Follow‐up does not need any microscopic review's only flow cytometric PNH clone size.
Keywords: abdominal pain, Coombs‐negative hemolysis, flow cytometry, hemolytic anemia, Paroxysmal nocturnal hemoglobinuria
1. INTRODUCTION
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare, acquired, and benign hematologic genetic disorder of hematopoietic stem cells (1‐2 cases per million per year). PNH is defined by a cluster of blood cells (red and/or white) lacking expression of the GPI‐anchor protein and its binding to CD55 and/or CD59 on the cell membrane, referred to as a PNH clone. The main clinical presentation of PNH patients is in the third decade of life, with a median survival time of 22 years.1 At present, 125 patients who are diagnosed with PNH are currently registered in the Netherlands, but this is considered an underestimation.2
A PNH clone develops if hematopoietic stem cells acquire a somatic mutation in the X‐linked phosphatidylinositol glycan protein A (PIG‐A) gene. Mutations of the PIG‐A gene interfere with the formation of glycosylphosphatidylinositol (GPI)‐anchors, and therefore, the binding of the CD55 and CD59 proteins to GPI‐anchors on the cell membrane are inhibited.3 Both GPI‐linked proteins (CD55 and CD59) are involved in the inactivation of the innate immune system, the alternative pathway of the complement cascade, inhibiting hemolysis.4 CD55 inhibits C3 convertase, preventing extravascular hemolysis, while CD59 is involved in the inhibition of the complement activated membrane (CAM), preventing intravascular hemolysis.3, 5 Therefore, the absence of GPI‐linked proteins, CD55 and CD59, activate the immune system and induce hemolysis in PNH patients.
Paroxysmal nocturnal hemoglobinuria patients are at increased risk for thrombosis. A multifactor mechanism for thrombosis exists in these patients mainly triggered by the complement system. Approximately half of patients develop thrombosis, both venous and arterial, which is the main cause of morbidity and death.6 PNH clone size correlates with the risk of developing thrombo‐embolic complications as does an increased LDH level (>1.5 x Upper Limit of Normal (ULN); 250 IU/L). Interestingly, a combination of increased LDH level, which is a parameter of hemolysis, and clinical symptoms such as abdominal pain, chest pain, dyspnea, or hemoglobinuria are associated with an increased risk of thrombosis.7 However, thrombosis occurs in PNH patients with a small clone size or a normal LDH also.
In 2007, the drug eculizumab (Soliris), a humanized monoclonal anti‐C5 antibody, was approved for symptomatic PNH patients by the European Medicines Agency (EMA) and the US Food and Drug Administration (FDA). Significant improvements in hemoglobin levels, lower transfusion demand, quality of life, and overall survival are observed in PNH patients after treatment with eculizumab, along with a decrease in hemolysis and thrombosis.8
The diagnosis PNH can be challenging and cannot be confirmed by general clinical chemistry parameters only. According to the international PNH guideline, flow cytometry using fluorescent labeled aerolysin (FLAER) as main antibody on a fresh peripheral blood sample is the primary diagnostic tool for confirmation of PNH.9 The FLAER antibody binds directly to the GPI anchor on the membrane of blood cells, detecting a PNH clone on a specific and sensitive (threshold cell clone size is 0.5%) manner.10, 11
After diagnosis of PNH, patients should be monitored regularly using microscopic and flow cytometric diagnostics for follow‐up of the evolution of the PNH clone.12 Annual monitoring is sufficient if the disease is stable. Change in the clinical or hematological parameters requires more frequent monitoring; however, the exact frequency is not specified by the international guideline. 9, 11
In this case report, two cases are described to highlight the fact that a combination of clinical symptoms and clinical chemistry parameters should alert the physicians to test for PNH. We emphasize the importance of optimal and multidisciplinary collaboration of clinical and laboratory physicians in order to shorten the time between presentation and diagnosis.
2. CASE I: FEMALE, 23 YEARS
A 23‐year‐old female without prior medical history was admitted to the emergency department due to bleedings of the jawbone after inflammation. During physical examination, she reported significant abdominal pain. Laboratory tests (Table 1) revealed decreased hemoglobin, increased reticulocytes and decreased haptoglobin levels, confirming the presence of hemolytic anemia. The direct Coombs test was negative, excluding an autoimmune hemolytic anemia. LDH was strongly elevated, and levels of bilirubin were 3x >ULN, while thrombocytes, protrombine time (PT), and activated partial trombine time (aPTT) were within normal ranges. A urine sample tested positive for hemoglobinuria (≥10 red blood cells per high‐power field). Initially, a thrombotic thrombocytopenic purpura (TTP) or hemolytic‐uremic syndrome (HUS) were suspected, but no significant number of schistocytes were detected in the blood smear, thus making TTP or HUS unlikely. Within a few hours, the patient observed a darkening of her urine indicating that the hematuria was ongoing. Consequently, the decision was made to transfer the patient to the department of internal medicine for further diagnostics after contact with the laboratory specialist and treatment. A high dose of prednisone was administered, but hemoglobin levels did not improve. Flow cytometry was ordered, which showed a PNH clone of 74% in neutrophils (Figure 1), 60% in monocytes as determined using the reagent: FLAER, CD24, and CD14 (Figure 2). Hereafter, the diagnosis of PNH was made.
Table 1.
Biochemical Results
| Case I | Case II | Reference interval | ||
|---|---|---|---|---|
| After 2 mo | ||||
| Hemoglobin | 6.4 (10.3) | 6.2 (10.0) | 6.5 (10.5) | 7.5‐10.0 mmol/L (12‐18 g/dL) |
| Reticulocytes | 3.6 | n.d. | 3.9 | 0.0‐2.5% |
| Thrombocytes | 222 × 109 | 248 × 109 | 267 × 109 | 150‐400 × 109/L |
| PT | 10 | n.d. | n.d. | 8‐11 s |
| aPTT | 22 | n.d. | n.d. | <32 s |
| Haptoglobin | < 0.1 | n.d. | < 0.1 | 0.3‐2.0 g/L |
| LDH | 2399 | 1214 | 1183 | <250 IU/L |
| Bilirubin | 3.1 | 0.7 | 0.5 | <1 mg/dL |
| Ferritin | 104 | n.d. | 13 | 20‐150 μg/L |
| Amylase | 67 | 51 | n.d. | <115 IU/L |
| Direct Coombs | Negative | n.d. | Negative | Negative |
n.d., not determined.
Figure 1.
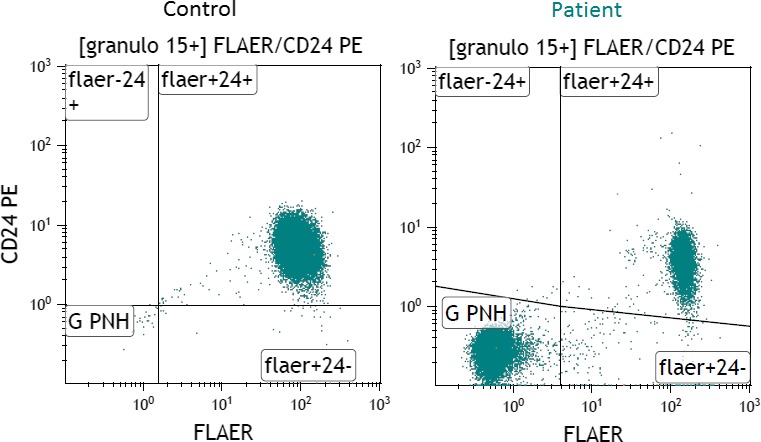
Results of flow cytometry on a control sample (left) showing CD24 binding on the granulocytes, and the patient described in Case I (right) with the detected PNH clone displaying negative CD24 binding on the granulocytes. As a control, a random sample of a Caucasian patient with a high level of leukocytes was used
Figure 2.
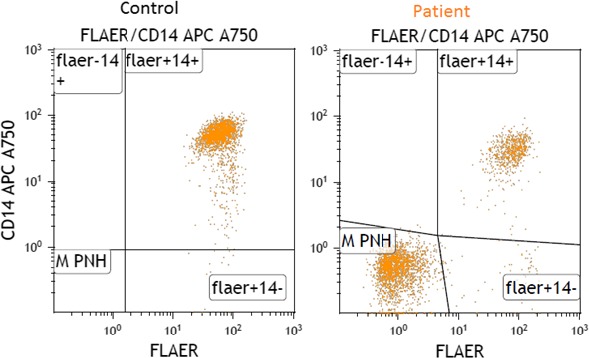
Results of flowcytometry on a control sample (left) showing CD14 binding on the monocytes, and the patient described in case I (right) with the detected PNH clone displaying negative CD14 binding on the monocytes. As a control, a random sample of a Caucasian patient with a high level of leukocytes was used
The patient underwent a full body scan to detect any possible thrombo‐embolic events, but none were observed. The patient's condition improved, and she was discharged to the outpatient clinic. Despite the presence of the risk factors of a strongly elevated LDH level, paroxysmal abdominal pain and a large PNH clone, no thrombo‐embolic events have been observed in this patient up to present.7, 10 The patient was consequently referred to the Dutch center of expertise for PNH (Radboud University Hospital, Nijmegen).
3. CASE II: FEMALE, 48 YEARS
The second case describes a 48‐year‐old female patient, without prior medical history, who presented at the gastroenterological department with complaints of severe abdominal pain for the past 4 months. Laboratory tests revealed anemia and elevated levels of LDH. An abdominal ultrasound revealed no abnormalities, while a colonoscopy showed mild inflammation. However, no explanation was found for the paroxysmal abdominal pain which led to a working diagnosis of constipation combined with inflammation of unknown origin.
Two months later, the patient presented to the emergency department of our hospital, complaining of severe abdominal pain again. This time, an acute pancreatitis was suspected but a normal amylase level made this diagnosis unlikely. Laboratory results showed a Coombs‐negative hemolytic anemia in combination with elevated LDH levels. After gastroscopy and CT‐abdomen, no explanation was found for the patient's clinical symptoms. Only an iron deficiency anemia was diagnosed based on ferritin levels, but supplementation did not resolve the iron deficiency and the anemia remained.
Due to the persistent anemia, the patient was referred to the department of internal medicine and laboratory specialists were consulted. Blood smear analysis showed the absence of schistocytes, excluding TTP or HUS. Based on the combination of laboratory results and symptoms, the hematologist suspected the presence of PNH. Immunophenotyping consequently showed a PNH clone of 60% in neutrophils and a clone of 50% in monocytes using FLAER, CD24, and CD14, confirming the diagnosis of PNH. As with case I, the patient was referred to the Dutch center of expertise for PNH (Radboud University Hospital, Nijmegen).
4. DISCUSSION
As illustrated by the two described cases here, the diagnosis of PNH is not always easily determined. Probably due to the broad spectrum of nonspecific clinical symptoms, a PNH patient often consults several different specialists before the diagnosis of PNH is made. The patients described in this case report both suffered from extreme abdominal pain, which is a symptom reported in 44% of PNH patients.13 A combination of unexplained abdominal pain and unexplained Coombs‐negative hemolytic anemia should alert physicians to the possibility of PNH. Optimal collaboration with clinical laboratory specialists can speed up early diagnosis of PNH.
The international guideline for PNH recommends several clinical indications which should elicit testing for the disease, including presentation of an unexplained Coombs‐negative hemolytic anemia, and/or hemoglobinuria, see Table 2.9, 11 Both cases described here demonstrated at least one of these signals, although testing for PNH was not performed immediately after first presentation.
Table 2.
Clinical indications for PNH testing
| Intravascular hemolysis as evidence by hemoglobinuria or elevated plasma hemoglobin. |
| Evidence of unexplained hemolysis with accompanying: |
| Iron‐deficiency, OR |
| Abdominal pain or esophageal spasm, OR |
| Thrombosis, OR |
| Granulocytopenia and/or thrombocytopenia |
| Other acquired Coombs‐negative, nonschistocytic, noninfectious hemolytic anemia |
| Thrombosis with unusual features: |
| Unusual sites |
| Hepatic veins (Budd‐Chiari Syndrome) |
| Other intra‐abdominal veins (portal, splenic, splanhnic) |
| Cerebral sinuses |
| Dermal veins |
| With signs of accompanying hemolytic anemia |
| With unexplained cytopenia |
| Evidence of bone marrow failure: |
| Suspected or proven aplastic or hypoplastic anemia |
| Refractory cytopenia with unilineage dysplasia |
| Other cytopenias of unknown etiology after adequate workup |
Borowitz et al.9
In conclusion, the aim of this report is to raise awareness on the combination of unexplained Coombs‐negative hemolytic anemia with elevated LDH levels, and unexplained clinical symptoms of abdominal pain to suspect a diagnosis of PNH. As flow cytometry is available in most clinical laboratories and the diagnostic strategy with FLAER antibodies may be standard practice, the clinical laboratory is able to diagnose PNH in a sensitive, specific, and rapid manner. Finally, we advise the development of an automated algorithm in the laboratory system based on clinical and chemistry parameters such as hemoglobin, direct Coombs, haptoglobin, and LDH. The idea is that this algorithm automatically flags in the computer system of the laboratory system and the clinical laboratory could advice the doctor to test for PNH. Such an algorithm is a useful tool for early diagnosis of PNH. To our knowledge, such algorithm or decision scheme has not been developed yet, but may help to prevent potentially lethal complications of PNH.
CONFLICT OF INTEREST
None declared.
AUTHOR CONTRIBUTIONS
NE: assistant Clinical Chemist, the corresponding author, acquired the diagnose of the PNH disease and clinical data. JR and HJV: Clinical chemist conducted the flow cytometric analyses for the diagnosis of PNH, supervised the study. KS: supported author. MDL and ME: the hematological provided the clinical data. FHJW: MDL doctor of the patient in case II.
ACKNOWLEDGMENTS
The support of Dr. S. Mussche, member of Alexion.
Elias N, Riedl J, Stouten K, Levin M‐D, Eefting M, Vermeer HJ. Abdominal pain in combination with an unexplained hemolytic anemia are crucial signs to test for paroxysmal nocturnal hemoglobinuria: A case report. Clin Case Rep. 2019;7:175‐179. 10.1002/ccr3.1771
REFERENCES
- 1. de Latour RP, Mary JY, Salanoubat C, et al. Paroxysmal nocturnal hemoglobinuria: natural history of disease subcategories. Blood. 2008;112(8):3099‐3106. [DOI] [PubMed] [Google Scholar]
- 2. Muus P, Langemeijer S, Halkes S, et al. Richtlijn Paroxysmale Nachtelijke Hemoglobinurie (23 November 2016).
- 3. Risitano AM. Paroxysmal nocturnal hemoglobinuria and the complement system: recent insights and novel anticomplement strategies. Adv Exp Med Biol. 2013;735:155‐172. [DOI] [PubMed] [Google Scholar]
- 4. Parker CJ. Management of paroxysmal nocturnal hemoglobinuria in the era of complement inhibitory therapy. Hematology Am Soc Hematol Educ Program. 2011;2011:21‐29. [DOI] [PubMed] [Google Scholar]
- 5. Parker CJ. Update on the diagnosis and management of paroxysmal nocturnal hemoglobinuria. Hematology Am Soc Hematol Educ Program. 2016;2016(1):208‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hill A, Kelly RJ, Hillmen P. Thrombosis in paroxysmal nocturnal hemoglobinuria. Blood. 2013;121(25):4985‐4996; quiz 5105. [DOI] [PubMed] [Google Scholar]
- 7. Lee JW, Jang JH, Kim JS, et al. Clinical signs and symptoms associated with increased risk for thrombosis in patients with paroxysmal nocturnal hemoglobinuria from a Korean Registry. Int J Hematol. 2013;97(6):749‐757. [DOI] [PubMed] [Google Scholar]
- 8. Hillmen P, Muus P, Roth A, et al. Long‐term safety and efficacy of sustained eculizumab treatment in patients with paroxysmal nocturnal haemoglobinuria. Br J Haematol. 2013;162(1):62‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Borowitz MJ, Craig FE, Digiuseppe JA, et al. Guidelines for the diagnosis and monitoring of paroxysmal nocturnal hemoglobinuria and related disorders by flow cytometry. Cytometry B Clin Cytom. 2010;78(4):211‐230. [DOI] [PubMed] [Google Scholar]
- 10. Pu JJ, Brodsky RA. Paroxysmal nocturnal hemoglobinuria from bench to bedside. Clin Transl Sci. 2011;4(3):219‐224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sutherland DR, Illingworth A, Keeney M, Richards SJ. High‐sensitivity detection of pnh red blood cells, red cell precursors, and white blood cells. Curr Protoc Cytom. 2015;72:6.37.1‐30. [DOI] [PubMed] [Google Scholar]
- 12. Maciejewski JP, Rivera C, Kook H, Dunn D, Young NS. Relationship between bone marrow failure syndromes and the presence of glycophosphatidyl inositol‐anchored protein‐deficient clones. Br J Haematol. 2001;115(4):1015‐1022. [DOI] [PubMed] [Google Scholar]
- 13. Schrezenmeier H, Muus P, Socie G, et al. Baseline characteristics and disease burden in patients in the International Paroxysmal Nocturnal Hemoglobinuria Registry. Haematologica. 2014;99(5):922‐929. [DOI] [PMC free article] [PubMed] [Google Scholar]