

Abstract
Key points
Exercise/exercise training can enhance insulin sensitivity through adaptations in skeletal muscle, the primary site of insulin‐mediated glucose disposal; however, in humans the range of improvement can vary substantially.
The purpose of this study was to determine if obesity influences the magnitude of the exercise response in relation to improving insulin sensitivity in human skeletal muscle.
Electrical pulse stimulation (EPS; 24 h) of primary human skeletal muscle myotubes improved insulin action in tissue from both lean and severely obese individuals, but responses to EPS were blunted with obesity.
EPS improved insulin signal transduction in myotubes from lean but not severely obese subjects and increased AMP accumulation and AMPK Thr172 phosphorylation, but to a lesser degree in myotubes from the severely obese.
These data reveal that myotubes of severely obese individuals enhance insulin action and stimulate exercise‐responsive molecules with contraction, but in a manner and magnitude that differs from lean subjects.
Abstract
Exercise/muscle contraction can enhance whole‐body insulin sensitivity; however, in humans the range of improvements can vary substantially. In order, to determine if obesity influences the magnitude of the exercise response, this study compared the effects of electrical pulse stimulation (EPS)‐induced contractile activity upon primary myotubes derived from lean and severely obese (BMI ≥ 40 kg/m2) women. Prior to muscle contraction, insulin action was compromised in myotubes from the severely obese as was evident from reduced insulin‐stimulated glycogen synthesis, glucose oxidation, glucose uptake, insulin signal transduction (IRS1, Akt, TBC1D4), and insulin‐stimulated GLUT4 translocation. EPS (24 h) increased AMP, IMP, AMPK Thr172 phosphorylation, PGC1α content, and insulin action in myotubes of both the lean and severely obese subjects. However, despite normalizing indices of insulin action to levels seen in the lean control (non‐EPS) condition, responses to EPS were blunted with obesity. EPS improved insulin signal transduction in myotubes from lean but not severely obese subjects and EPS increased AMP accumulation and AMPK Thr172 phosphorylation, but to a lesser degree in myotubes from the severely obese. These data reveal that myotubes of severely obese individuals enhance insulin action and stimulate exercise‐responsive molecules with contraction, but in a manner and magnitude that differs from lean subjects.
Keywords: Contractile Activity, Exercise, Insulin Signaling, Skeletal Muscle
Key points
Exercise/exercise training can enhance insulin sensitivity through adaptations in skeletal muscle, the primary site of insulin‐mediated glucose disposal; however, in humans the range of improvement can vary substantially.
The purpose of this study was to determine if obesity influences the magnitude of the exercise response in relation to improving insulin sensitivity in human skeletal muscle.
Electrical pulse stimulation (EPS; 24 h) of primary human skeletal muscle myotubes improved insulin action in tissue from both lean and severely obese individuals, but responses to EPS were blunted with obesity.
EPS improved insulin signal transduction in myotubes from lean but not severely obese subjects and increased AMP accumulation and AMPK Thr172 phosphorylation, but to a lesser degree in myotubes from the severely obese.
These data reveal that myotubes of severely obese individuals enhance insulin action and stimulate exercise‐responsive molecules with contraction, but in a manner and magnitude that differs from lean subjects.
Introduction
Obesity is associated with impaired insulin action in skeletal muscle, the primary site of insulin‐mediated glucose disposal (DeFronzo et al. 1985; Bouzakri et al. 2005; Ehrenborg & Krook, 2009). Exercise/exercise training improves insulin action at the level of both the whole‐body and skeletal muscle in healthy and insulin‐resistant populations (Christ‐Roberts et al. 2003; Yates et al. 2007; Kjobsted et al. 2016). However, there is considerable variation in the magnitude of this response (Teran‐Garcia et al. 2005); for example, review papers (Stephens & Sparks, 2015; Malin et al. 2016) have reported that 15–20% of individuals with type 2 diabetes fail to improve indices of metabolic health, including insulin sensitivity, with exercise training. Similarly, in insulin‐resistant individuals, AMPK phosphorylation and PGC1α mRNA content, both of which can be linked with insulin action, were depressed compared to lean controls following a single bout of exercise (Sriwijitkamol et al. 2007; De Filippis et al. 2008). Such data suggest that metabolic impairments such as obesity and insulin resistance may influence the magnitude of the response to the exercise stimulus (Stephens & Sparks, 2015; Malin et al. 2016).
Individuals with severe obesity (BMI ≥ 40 kg/m2) exhibit insulin resistance in skeletal muscle along with a compromised capacity for fat oxidation (Kim et al. 2000; Hulver et al. 2005; Bikman et al. 2010). These deficiencies are retained in primary human skeletal muscle cell cultures differentiated into myotubes (Hulver et al. 2005; Jorgensen et al. 2005; Ehrenborg & Krook, 2009). In primary myotubes, electrical pulse stimulation (EPS) can produce biological adaptations similar to endurance‐oriented exercise/exercise training (Nedachi et al. 2008; Lambernd et al. 2012; Nikolic et al. 2012). An advantage of the primary skeletal muscle culture/EPS system is that responses to an identical absolute contractile stimulus can be studied without the confounding factors inherent with the compromised cardiovascular capacity evident with severe obesity (i.e. reduced cardiorespiratory fitness and exercise tolerance; Guesbeck et al. 2001). In addition, contractile activity can be studied devoid of in vivo influences such as hormone concentrations, which can differ with obesity.
The intent of the current study was to use the primary culture system to compare the responses to 24 h of EPS (contractile activity) in primary myotubes from severely obese, insulin‐resistant individuals and lean controls. We tested the hypothesis that EPS would improve insulin action (i.e. insulin‐stimulated glycogen synthesis, glucose oxidation, glucose transport, insulin signal transduction, GLUT4 translocation) regardless of the initial degree of insulin resistance and obesity. AMPK signalling plays an important role in regulating fuel and insulin action during and after contractile activity (Jorgensen et al. 2005; Kjobsted et al. 2015, 2016). While some studies have shown an impairment in AMPK signalling with exercise in insulin‐resistant obese individuals (Sriwijitkamol et al. 2007; De Filippis et al. 2008), others have not (Musi et al. 2001; Kjobsted et al. 2016). Thus, we also experimentally tested if the effects of EPS on AMPK activation differed with obesity.
Methods
Ethical approval
Lean (BMI < 25 kg/m2) and severely obese (BMI ≥ 40 kg/m2) Caucasian women were recruited; all participants were sedentary (no structured exercise for the previous 6 months), and not taking medications affecting glucose metabolism. We focused upon a single gender, as there are sex differences in glucose metabolism (Chella Krishnan et al. 2018). In addition, the severely obese subjects were recruited from bariatric surgery clinics; ∼80% of patients undergoing bariatric surgery are women and the incidence of severe obesity is substantially higher in women than men (Hales et al. 2018). We focused upon Caucasians, as we have previously reported (Cortright et al. 2006) ethnicity differences in muscle metabolism which may compromise the exercise response. Written informed consent was obtained prior to any experimental procedures. All procedures were approved by East Carolina University Policy and Review Committee on Human Research (approval reference: CR00004965) and conformed to the Declaration of Helsinki except for registration in a data base.
Study design
The intent of this study was to determine the responses of primary human skeletal muscle cells (myotubes) derived from lean and severely obese donors to 24 h of EPS. The primary functional outcomes were indices of insulin action (insulin‐stimulated glycogen synthesis, glucose oxidation, glucose uptake). Components of insulin action (insulin signal transduction, GLUT4 translocation), indices of contraction/energy demand (nucleotide concentration, AMPK signalling, glycogen content), and exercise‐responsive protein expression (PGC1α, citrate synthase, GLUT4) were examined as a possible means to explain differences observed.
Subject characteristics
Subjects reported to the laboratory in the morning (07.00–08.30 h) in the overnight (8–14 h) fasted condition. A venous blood sample was obtained, frozen and subsequently analysed for glucose, insulin and HbA1C (Access Immunoassay System, Beckman Coulter; Fullerton, CA, USA) and a HOMA‐IR calculated (Bonora et al. 1998). Both stature and body mass were obtained with shoes off while fully clothed.
Skeletal muscle cell cultures
As described previously (Berggren et al. 2007; Aas et al. 2013), satellite cells were isolated from muscle biopsies obtained from the vastus lateralis and amplified on type‐I collagen‐coated plates until reaching approximately 80–90% confluence in growth media (DMEM low glucose medium supplemented with 10% FBS, 0.5 mg/ml BSA, 0.05% fetuin, 20 ng/ml human epidermal growth factor, 0.39 μg/ml dexamethasone, and 100 μg/ml penicillin/streptomycin) in a 5% CO2 and 37°C humidified atmosphere. Upon reaching 80–90% confluence, myoblasts were differentiated to myotubes by switching to differentiation media (DMEM low glucose medium supplemented with 2% horse serum, 0.3% BSA, 0.05% fetuin, and 100 μg/ml penicillin/streptomycin). Experiments/EPS was conducted on days 5–6 of differentiation.
EPS
Mature myotubes were electrically stimulated for 24 h in 2 ml of media. The pulse generator (C‐Dish, IonOptix LLC, Milton, MA, USA) provided bipolar stimuli at 11.5 V, 1 Hz and 2 ms, similar to other protocols (Nedachi et al. 2008; Lambernd et al. 2012; Nikolic et al. 2012). Cell lysates were collected immediately after the 24 h of EPS to measure nucleotides, AMPK and ACC phosphorylation, and glycogen content. Following 3 h of serum starvation, myotubes were incubated with 100 nM insulin for 10 min to examine indices of insulin action, AMPK phosphorylation and protein abundance. As in other studies (Treebak et al. 2009; Feng et al. 2015), we utilized a ∼3 h period after contractile activity to minimize the insulin‐independent effects on glucose transport seen during and immediately after muscle contractions and to also permit the myotubes to return to a basal state with the removal of serum‐related factors (i.e. residual insulin etc.).
Glycogen synthesis
Following 3 h of serum starvation, myotubes were incubated with media containing d‐[U‐14C] glucose (Perkin‐Elmer, MA, USA; 1 μCi/ml, 5.0 mM glucose) in the presence or absence of insulin for 2 h at 37° C (Al‐Khalili et al. 2003). Myotubes were then washed twice with ice‐cold DPBS and solubilized with 0.05% SDS. Lysates were combined with carrier glycogen (2 mg) and hydrolysed at 100°C for 1 h, followed by cooling on ice for 30 min. The remaining lysate was used to measure protein concentration using the bicinchoninic acid (BCA) assay (Pierce Biotechnology, Rockford, IL, USA). Ice‐cold 100% ethanol was added to the hydrolysed lysates, and samples rotated overnight at 4°C for the precipitation of glycogen. Glycogen pellets were centrifuged at 11,100 g for 15 min at 4°C and washed with 70% ethanol followed by centrifugation. The glycogen pellets were re‐suspended with dH2O and incorporation of radioactive glucose into glycogen was determined with liquid scintillation.
Glucose oxidation
Following 3 h of serum starvation, myotubes were incubated in a sealed plate with media containing d‐[U‐14C] glucose (Perkin‐Elmer; 1 μCi/ml, 5.0 mM glucose) in the presence or absence of 100 nM insulin for 2 h at 37°C. Immediately after incubation, radioactive media was transferred into a customized 48‐well trapping plate with fabricated grooves between two adjoining wells to allow for acid‐driven 14CO2 to be trapped by fresh 1 M NaOH (Kim et al. 2000). Incorporation of radioactive glucose into CO2 was determined with liquid scintillation. Myotubes were washed with ice‐cold PBS and solubilized in 0.05% SDS to measure protein concentration and the BCA assay for normalization
Glucose uptake
Following 3 h of serum starvation, myotubes underwent a 1 h pre‐incubation incubation at 37°C with Krebs‐Henselet buffer (KHB; 118 mM sodium chloride, 4.7 mM potassium chloride, 1.18 mM magnesium sulfate, 2.5 mM calcium chloride, 1.17 mM potassium phosphate, and 25 mM sodium bicarbonate) with 1% BSA and 1 mM pyruvate. Immediately after the last 15 min of insulin stimulation, myotubes were incubated in KHB with both cold 2‐deoxyglucose (DOG; 10 mM/well final concentration) and radioactive 2‐[1,2‐3H(N)] DOG (Perkin‐Elmer; 1 μCi/mL, 5.0 mM of 2‐DOG final concentration) in the presence or absence of 100 nM insulin and d‐[1‐14C] mannitol (0.1 μCi/mL, 20 mM of mannitol final concentration) to account for non‐facilitated diffusion. After the 1 h incubation, 50 μl of 20% dextrose (10 mM/well final concentration) was added and radioactive media was transferred to scintillation vials to quantify 2‐DOG uptake. (Nikolic et al. 2012). Remaining lysates were used to measure protein concentration by BCA assay and data normalized to total protein content.
GLUT4 translocation
GLUT4 translocation was determined with a modified plasma membrane sheet assay (Knight & Olson, 2003). During the last 10 min of insulin stimulation, myotubes were rinsed twice with ice‐cold DPBS and incubated three times for 30 s with 5 ml of swelling buffer (KHMgE buffer; 70 mM potassium chloride, 30 mM HEPES, 5 mM magnesium chloride, 3 mM EGTA, pH 7.5). To remove the cytosolic portion, myotubes underwent a sonic disruption at 1.3 arbitrary intensity of a sonicator (Branson Sonifier, Fisher Scientific, Waltham, MA, USA) using a microtip in 15 ml of sonication solution (1× KHMgE buffer containing 1 mM dithiothreitol, 1 mM polymethylsulfonyl fluoride). To remove cell debris, myotubes were rinsed three times with sonication solution. Immediately after the rinsing, 100 μl of ice‐cold lysis buffer was added and Western blot analyses were used to detect plasma membrane GLUT4.
Non‐oxidized glycolytic metabolites
This assay measured the non‐oxidized ionized intermediates from glycolysis (lactate, pyruvate and alanine). These non‐oxidized glycolytic (NOG) metabolites were measured using Whatman, Grade DE81 ion‐exchange cellulose paper (GE Healthcare Life Sciences, PA, USA) as described previously (Zou et al. 2018). Briefly, 100 μl of incubation media was collected immediately 2 h after incubating with media containing d‐[U‐14C] glucose following EPS and centrifuged for 5 min at 9500 g. An aliquot of the supernatant was added to the ion‐exchange cellulose paper, followed by 30 min of drying and 4 washes of 10 min each with dH2O. After washing, the paper was collected in 4 ml liquid scintillation fluid and 14C‐labelled glucose incorporation into NOG was determined by liquid scintillation counting. The non‐ionized glucose tracer in the media was not retained in this fraction, which was validated by applying 14C‐labelled glucose to the filter paper and scintillation counting. Water after each wash was collected and tested to verify there was no non‐ionized product left on the paper following the 4 washes. Data were normalized to total protein content (BCA assay).
Immunoblot analyses
Homogenized cell lysates were utilized for immunoblot analysis as previously described (Bikman et al. 2010). Primary antibodies were: acetyl CoA carboxylase (ACC) (Ser79) (3661; Cell Signaling, Beverly, MA, USA); ACC protein (3662; Cell Signaling); IRS1 Tyr632 (09‐433; Millipore, Billerica, MA, USA); IRS1 protein (sc‐559; Santa Cruz Biotechnology, Dallas, TX, USA); Akt (Ser473, Thr308) (9271, 9275; Cell Signaling); Akt protein (9272; Cell Signaling); AMP‐activated protein kinase (AMPK) (Thr172) (2531; Cell Signaling); AMPK protein (2532; Cell Signaling); Akt‐substrate at 160 kDa (TBC1D4) (Thr642) (ab59173; Abcam, Cambridge, MA, USA); TBC1D4 (Ser588, Ser318, Ser341, Ser704) (customized by Capra Science, Sweden); TBC1D4 protein (07‐741; Millipore, Billerica, MA, USA); β actin (926‐42210; LI‐COR Biosciences, Lincoln, NE, USA); GLUT4 (sc‐53566; Santa Cruz Biotechnology); glycogen synthase kinase 3α/β (GSK‐3α/β) (Ser21/9) (8566; Cell Signaling); GSK‐3β (27C10) protein (9315; Cell Signaling); peroxisome proliferator‐activated receptor γ coactivator α (PGC1α) (ab106814; Abcam); and citrate synthase (ab96600; Abcam). Following overnight incubation with primary antibodies, membranes were probed with IRDye secondary antibodies (LI‐COR Biosciences) and scanned (Odyssey 9120, LI‐COR Biosciences).
Glycogen content
As previously described (Manabe et al. 2012), immediately after EPS myotubes were harvested, sonicated and supernatant applied to an Ultrafree‐MC10 (Millipore, Bedford, MA, USA) for filtrating residue protein. The flow‐through samples were neutralized and glucose content measured using a hexokinase kit (Thermo Fisher Scientific, Waltham, MA, USA).
Nucleotides
As previously described (Brault et al. 2013), myotubes were harvested with ice‐cold 0.5 N perchloric acid (PCA), sonicated for 10 s and centrifuged at 13,000 g for 10 min at 4°C. Extracts were neutralized with 1 N KOH, centrifuged at a maximal speed for 10 min at 4°C to remove the perchlorate salt and stored at −80°C until analysis. Samples were separated by ultra‐performance liquid chromatography (Waters Acquity H‐Class system) to determine the concentration of adenine nucleotides (adenosine triphosphate (ATP), adenosine diphosphate (ADP), adenosine monophosphate (AMP), and a degradation product (inosine monophosphate (IMP)). Data were expressed relative to the number of initially plated cells.
Calcium fluorescence
Calcium assay buffer and fluo‐8 dye loading solution (AAT Bioquest Inc., Sunnyvale, CA, USA) were added and incubated for the last 30 min of the 24 h of EPS. Calcium transient was observed under an Evos fluorescent microscope (Life Technologies EVOS FL Auto Fluorescence) at excitation and emission wavelengths of 490 and 525 nm, respectively.
Contractile activity
Mechanical contraction was determined by the changes in distance between two selected points on a myotube using a tracker video analyser (Open Source Physics, USA). An active manner of mechanical contraction was observed after a few hours of EPS as previously described (Nedachi et al. 2008; Lambernd et al. 2012).
Cell viability
Cell viability was determined using MTT (3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide as described previously (Lambernd et al. 2012). Following EPS myotubes were incubated for 2 h at 37°C in 1.2 mM MTT solution in DMEM low glucose phenol‐free medium. Myotubes were rinsed with PBS, mixed with 500 μl DMSO, and incubated for 10 min at 37°C. Absorbance at 540 nm was determined using a plate reader (Synergy H1 Hybrid Reader, Winooski, VT, USA).
Proliferation and differentiation
Myoblast proliferation was determined by lifting the cells (0.05% Trypsin/EDTA) at 24, 48, and 72 h after plating. Total cell count and cell viability were assessed using an automated cell counter (Beckman Coulter Vi‐Cell, Brea, CA, USA). Cells were also counted using the MTT cell proliferation assay (Molecular Probes Inc., Eugene, OR, USA).
On day 7 of differentiation, myotubes were labelled with myosin heavy chain antibody (MHC; MF20, Developmental Studies Hybridoma Bank, Iowa City, IA, USA) overnight at 4°C. Nuclei were incubated with IgG2b Alexa Fluor 488 secondary antibody for 1 h, washed twice in PBS, and stained with 4′,6‐diamidino‐2‐phenylindole ((DAPI); Sigma‐Aldrich, St Louis, MO, USA) and using ImageJ64 (NIH, Bethesda, MD, USA) a fusion index and myotube area were obtained. Myotube area was quantified by analysing the amount of MHC covering the culture area using the method of Bollinger et al. (2015). In addition, cell lysates were assayed for markers of differentiation (MyoD (M‐318); Santa Cruz Biotechology) and myosin heavy chain content (MHC (MF20); Developmental Studies Hybridoma Bank, Iowa City, IA, USA).
Statistical analysis
Student's unpaired or paired two‐tailed t tests or two‐ or three‐way ANOVA were utilized to determine statistical significance. Factors were group (lean vs. severely obese), presence or absence of insulin, and condition (control or EPS) or solely group (differentiation and proliferation characteristics). Statistical significance was set as P ≤ 0.05. When significance was detected in either main effects or interactions, Student's two‐tailed t tests were performed for distinguishing where differences existed. Comparisons of interest were selected a priori to minimize type 1 error. All data are expressed as means ± SEM.
Results
Subject characteristics
Lean and severely obese Caucasian women were matched by age (n = 8 per group). As presented in Table 1, the severely obese group exhibited an elevated BMI, fasting insulin and HOMA‐IR compared to the lean participants.
Table 1.
Characteristics of lean and severely obese women
| Lean | Severely obese | P value | |
|---|---|---|---|
| n | 8 | 8 | |
| Sex (male/female) | 0/8 | 0/8 | |
| Ethnicity | 8 C | 8 C | |
| Age (years) | 29.5 ± 2.6 | 30.8 ± 1.6 | 0.82 |
| BMI (kg/m2) | 23.1 ± 0.7 | 44.9 ± 2.5 | P < 0.01 |
| Fasting glucose (mg/dl) | 87.0 ± 2.6 | 86.8 ± 2.6 | 0.62 |
| HbA1c (%) | 4.5 ± 0.1 | 4.5 ± 0.1 | 0.62 |
| Fasting insulin (μU/ml) | 7.9 ± 0.8 | 17.4 ± 2.3 | P < 0.01 |
| HOMA‐IR | 1.7 ± 0.2 | 3.7 ± 0.4 | P < 0.01 |
C = Caucasian. Data are means ± SEM.
Proliferation and differentiation
The MTT assay and cell counts revealed that the number of myoblasts in both groups significantly increased at 48 (P < 0.001) and 72 h (P < 0.001) compared to 24 h of proliferation, with no differences between the lean (n = 4) and severely obese (n = 5) groups (Fig. 1 A). Cell viability increased in both the lean and severely obese groups at 48 (P = 0.01) and 72 h (P = 0.02) with no differences between the groups (data not shown).
Figure 1. Indices of myoblast proliferation, myotube differentiation and cell viability.
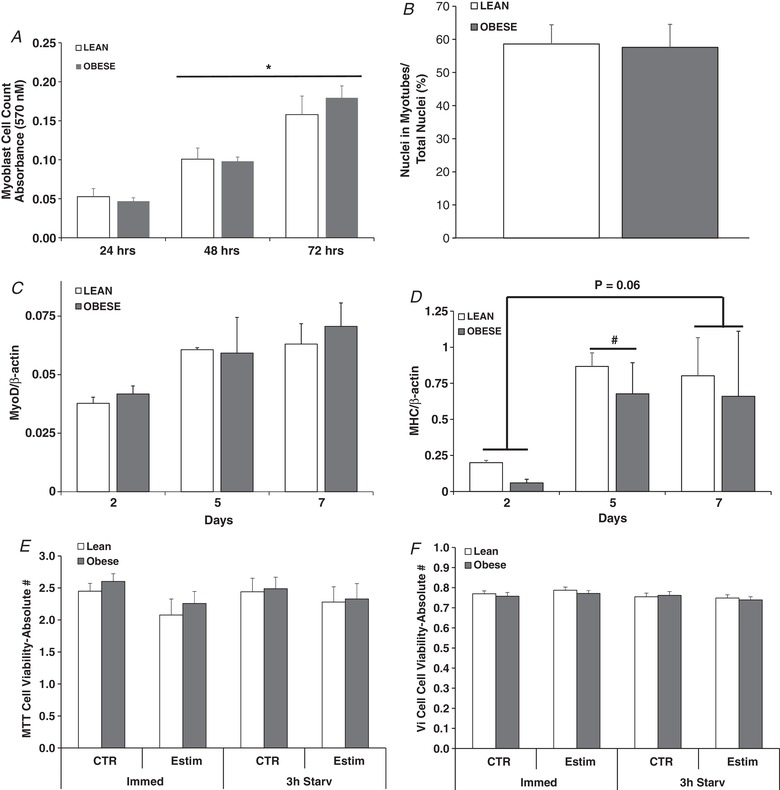
A, myoblast cell count (MTT assay) at 24, 48 and 72 h of proliferation. B, fusion index in myotubes at day 7 of differentiation. C, MyoD expression in myotubes at days 2, 5 and 7 of differentiation. D, myosin heavy chain expression in myotubes at days 2, 5 and 7 of differentiation. E, cell viability in myotubes determined by MTT under control and after 24 h of EPS (Estim) either immediately after EPS (Immed) or after EPS + 3 h of serum starvation (Starv). F, cell viability in myotubes determined by Vi‐Cell visualization under control and after 24 h of EPS (Estim) either immediately after EPS (Immed) or after EPS + 3 h of serum starvation (Starv). Values are means ± SEM. n = 4 lean and 5 obese. * P < 0.05 for 48 and 72 versus 24 h. # P < 0.05 for 5 versus 2 days.
At day 7 of differentiation, both the fusion index (Fig. 1 B) and myotube area (data not shown) did not differ between the lean and severely obese groups. At days 2, 5 and 7 of differentiation, neither MyoD nor total myosin heavy chain protein content differed between subject groups (Fig. 1 C and D). Total myosin protein content increased in both groups at day 5 of differentiation (P = 0.01) in comparison to day 2 (Fig. 1 D). Total myosin protein content at day 7 tended to be higher compared to day 2 (P = 0.06).
Cell viability with EPS
Cell viability determined immediately after 24 h of EPS did not differ between groups (Fig. 1 E and F) nor changed with EPS. Similar results were obtained when viability was determined after 24 h of stimulation + 3 h of serum starvation (Fig. 1 and F).
Glycogen and nucleotide content
Glycogen content was reduced in myotubes from both groups of subjects immediately after 24 h of EPS (P < 0.05), with no differences between groups (Fig. 2 A). As presented in Fig. 2 B, ATP and ADP content immediately after EPS did not differ between groups. AMP concentration was elevated with EPS (P < 0.05) and was significantly higher in myotubes from lean (P < 0.05) compared to severely obese subjects. IMP content increased after 24 h of EPS independent of donor type.
Figure 2. Glycogen and adenine nucleotide content after 24 h of EPS (Estim) in myotubes from lean and severely obese subjects.
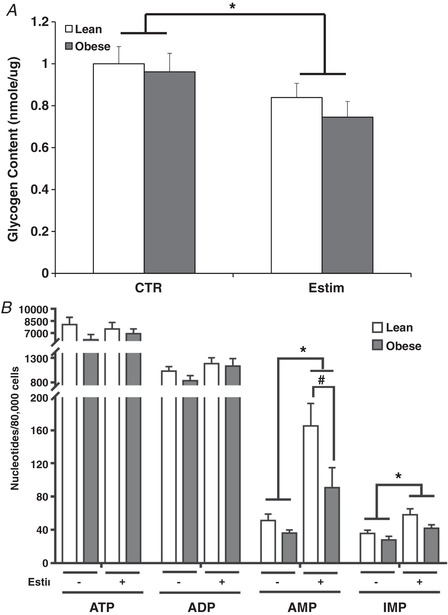
A, glycogen content under control (CTR) and immediately after 24 h of EPS. B, adenine nucleotides (ATP, ADP, AMP) and IMP content immediately after 24 h of EPS. Values are means ± SEM. n = 8 per group. * P < 0.05 for difference with EPS versus control condition. # P < 0.05 for difference between lean and obese.
AMPK and ACC
AMPK (Thr172) phosphorylation (Fig. 3 A) increased when determined immediately after 24 h of EPS (P < 0.05) in both groups, but to a lesser extent in myotubes from the obese subjects (P < 0.05). ACC (Ser79) phosphorylation increased with EPS (P < 0.05) in both groups (Fig. 3 B) and demonstrated a similar pattern of change to AMPK (Fig. 3 A and B).
Figure 3. AMPK activity under control and immediately after 24 h of EPS (Estim) in myotubes from lean and severely obese subjects.
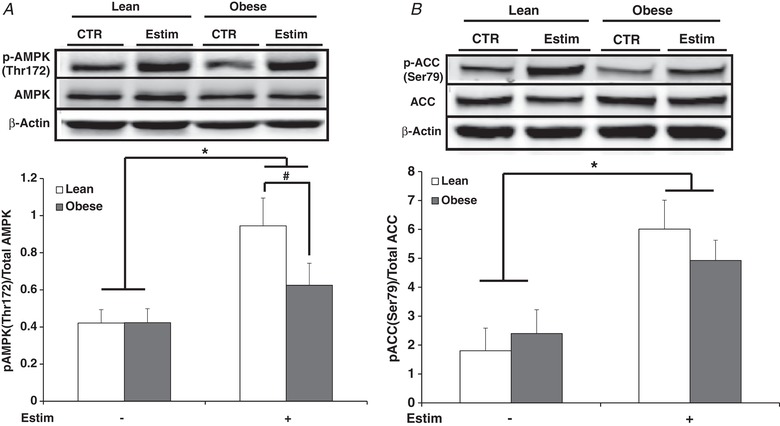
A, phosphorylation of AMPK Thr172. B, phosphorylation of ACC Ser79. Values are means ± SEM. n = 8 per group. * P < 0.05 for increase with EPS versus control condition. # P < 0.05 for difference between lean and obese.
PGC1α, citrate synthase, GLUT4 and AMPK content following (3 h) EPS
PGC1α protein content increased with EPS + 3 h of serum starvation (P < 0.05; Fig. 4 A). Citrate synthase content exhibited no change with EPS + serum starvation; however, protein expression was elevated in myotubes from the lean compared to the obese subjects after EPS (P < 0.05; Fig. 4 B). Total GLUT4 protein content did not differ between groups nor was it affected by EPS (Fig. 4 C). After EPS + 3 h serum starvation, AMPK Thr172 phosphorylation (Fig. 4 D) as well as ACC phosphorylation (data not shown) did not differ. GSK3α (Ser21/9) phosphorylation was not altered with EPS (data not shown).
Figure 4. Protein content in myotubes from lean and severely obese subjects after 24 h of EPS + 3 h of serum starvation (Estim).
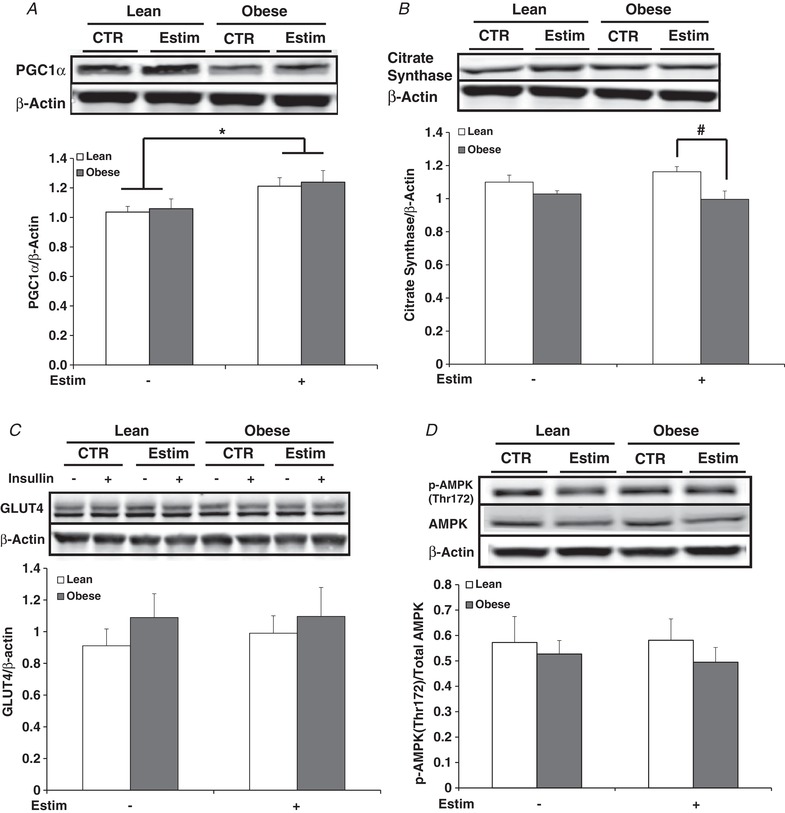
A, PGC1α. B, citrate synthase. C, GLUT4. D, p‐AMPK Thr172. Values are means ± SEM. n = 8 per group. * P < 0.05 for increase with EPS versus control condition. # P < 0.05 for difference between lean and obese.
Indices of insulin action
Insulin‐stimulated glycogen synthesis was depressed in myotubes from the severely obese subjects (P < 0.05; Fig. 5 A). EPS increased insulin‐stimulated glycogen synthesis in both groups of subjects (Fig. 5 A and B). In myotubes from the severely obese subjects, insulin‐stimulated glycogen synthesis increased with EPS to the extent that it exceeded that in the lean subjects under the control, non‐EPS condition (P < 0.05; Fig. 5 A). However, insulin‐stimulated glycogen synthesis with EPS remained depressed in myotubes from the severely obese compared to lean donors (Fig. 5 A) and the relative increase in insulin‐stimulated glycogen synthesis with EPS was suppressed in myotubes from obese compared to lean subjects (P < 0.05; Fig. 5 B). There were significant (P < 0.05) main effects for insulin (basal vs. insulin stimulated) and subject group (lean vs. obese).
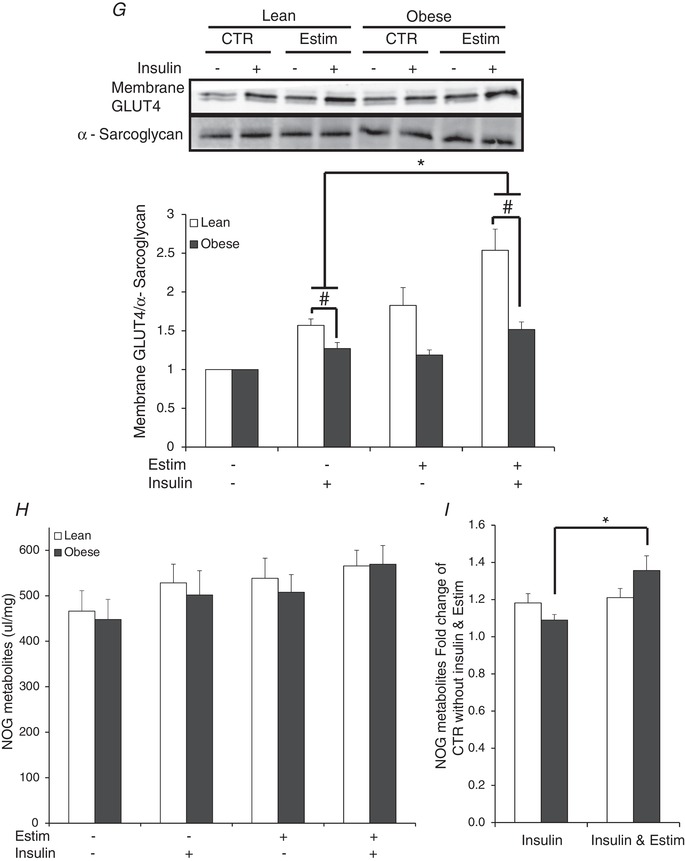
Figure 5. Indices of insulin action in the absence or presence of 100 nM insulin in myotubes from lean and severely obese subjects after 24 h of EPS + 3 h serum starvation (Estim).
A, absolute values of glycogen synthesis. B, fold change in glycogen synthesis over non‐insulin, non‐Estim condition. C, absolute values of glucose oxidation. D, fold change in glucose oxidation over non‐insulin, non‐Estim condition. E, absolute values of glucose uptake. F, fold change in glucose uptake over non‐insulin, non‐Estim condition. G, membrane GLUT4. H, absolute values of non‐oxidized glycolytic products (NOG) in the media. I, fold change in media NOG over non‐insulin, non‐Estim condition. Values are means ± SEM. n = 8 per group. * P < 0.05 for difference with EPS vs. control condition. # P < 0.05 for difference between lean and obese. Data were also compared between the insulin‐stimulated values in the lean subjects versus the insulin‐stimulated + EPS values in the obese subjects to determine if EPS normalized insulin‐stimulated metabolism despite the presence of obesity. In panels A, C and E, there were significant (P < 0.05) main effects for insulin (basal vs. insulin stimulated) and subject group (lean vs. obese).
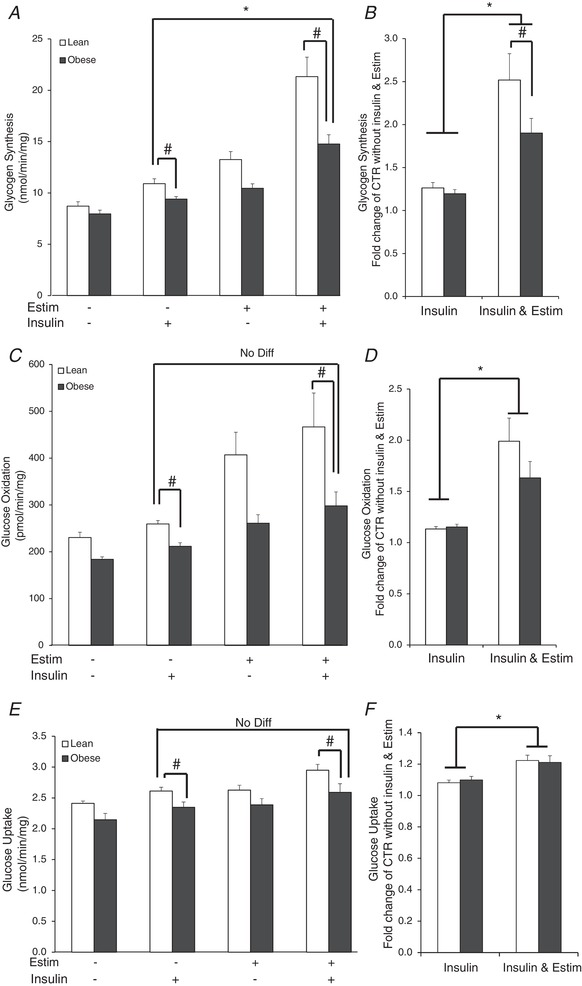
Insulin‐stimulated glucose oxidation was reduced in myotubes from obese donors under control conditions (P < 0.05; Fig. 5 C). Twenty‐four hours of EPS increased insulin‐stimulated glucose oxidation in both groups (Fig. 5 C and D); in the severely obese subjects, insulin‐stimulated glucose oxidation increased with EPS to the extent that it approximated that in the lean subjects under insulin‐stimulated non‐EPS conditions (Fig. 5 C). However, after EPS, values remained depressed in myotubes of the obese compared to lean subjects (P < 0.05; Fig. 5 C). There were significant (P < 0.05) main effects for insulin and subject group.
Insulin‐stimulated glucose uptake was reduced in myotubes from obese donors under control conditions (P < 0.05; Fig. 5 E). EPS increased insulin‐stimulated glucose uptake in both groups (Fig. 5 F). In myotubes from severely obese subjects, EPS increased insulin‐stimulated glucose uptake to the extent that it did not differ from the lean subjects under control, non‐EPS conditions (Fig. 5 E). However, insulin‐stimulated glucose uptake remained depressed after EPS in the myotubes from obese compared to lean subjects (P < 0.05; Fig. 5 E). There were significant (P < 0.05) main effects for insulin and subject group.
Under non‐EPS conditions, insulin‐stimulated membrane GLUT4 content was depressed in myotubes from the severely obese compared to the lean subjects (P < 0.05; Fig. 5 G). EPS increased insulin‐stimulated membrane GLUT4 content (P < 0.05; Fig. 5 G). However, insulin‐stimulated membrane GLUT4 content remained suppressed after EPS in the severely obese compared to lean subjects (P < 0.05). The relative increase in NOG concentration with insulin + EPS was higher than insulin alone in myotubes from the severely obese subjects (P < 0.05; Fig. 5 I), which suggests that glucose was preferentially partitioned to NOG rather than oxidation or storage in response to EPS.
Insulin signalling
Insulin signalling data are presented as fold‐changes, as there were no differences in basal protein content. EPS increased insulin‐stimulated IRS1 Tyr632 phosphorylation in only the lean subjects (P < 0.05; Fig. 6 A). For Akt Ser473, EPS increased insulin‐stimulated phosphorylation in only the lean subjects (P < 0.05); insulin‐stimulated phosphorylation was depressed in myotubes from the obese compared to the lean subjects. Insulin‐stimulated Akt Thr308 was depressed in myotubes from the severely obese compared to the lean subjects and not altered with EPS.
Figure 6. Insulin signal transduction in the absence or presence of 100 nM insulin in myotubes from lean and severely obese subjects after 24 h of EPS + 3 h serum starvation (Estim).
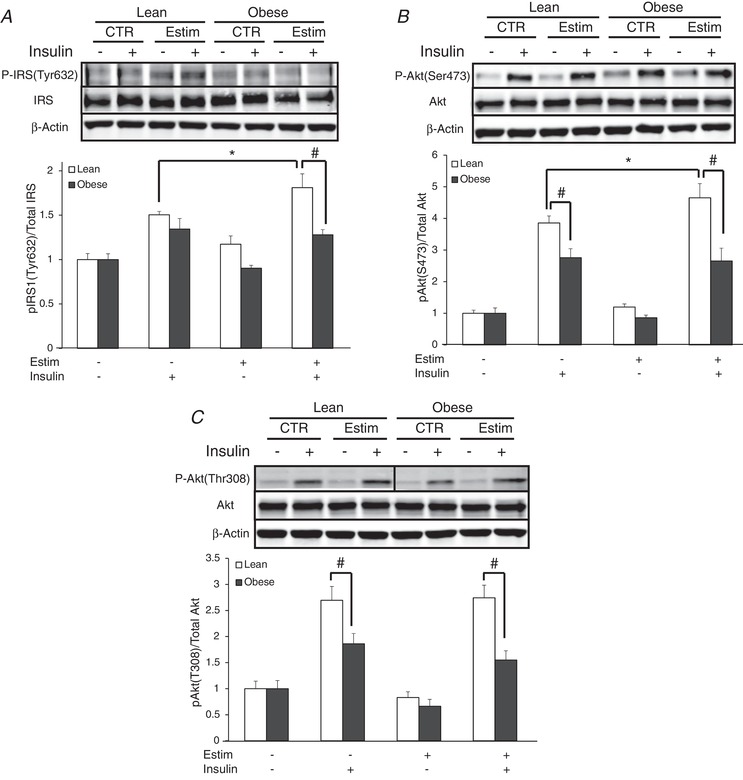
A, IRS1 Tyr632 phosphorylation. B, Akt Ser473 phosphorylation. C, Akt Thre308 phosphorylation. Values are means ± SEM. n = 8 per group. * P < 0.05 for difference with EPS versus control condition. # P < 0.05 for difference between lean and obese.
EPS increased insulin‐stimulated TBC1D4 Thr642, Ser588 and Ser341 phosphorylation in only the lean subjects (P < 0.05; Fig. 7 A–C). Insulin‐stimulated TBC1D4 Ser341 and Ser704 phosphorylation was elevated after EPS in the lean versus obese group (P < 0.05; Fig. 7 C and D). Insulin‐stimulated TBC1D4 Ser318 phosphorylation did not differ between groups nor change with EPS (Fig. 7 E).
Figure 7. Insulin signal transduction at the level of TBC1D4 in the absence or presence of 100 nM insulin in myotubes from lean and severely obese subjects after 24 h of EPS + 3 h serum starvation (Estim).
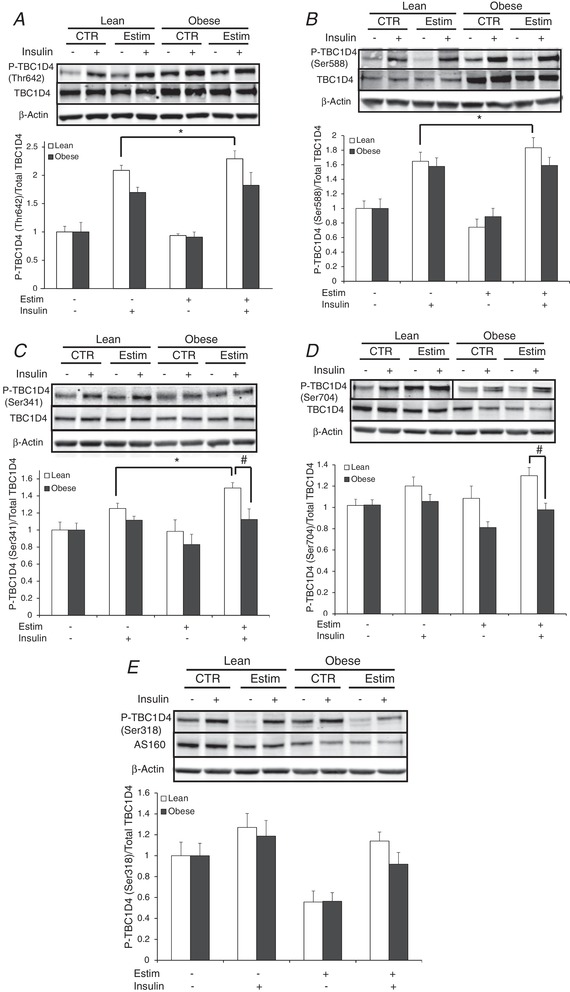
A, TBC1D4 Thr642 phosphorylation. B, TBC1D4 Ser588 phosphorylation. C, TBC1D4 Ser341 phosphorylation. D, TBC1D4 Ser704 phosphorylation. E, TBC1D4 Ser318 phosphorylation. Values are means ± SEM. n = 8 per group. * P < 0.05 for difference with EPS versus control condition. # P < 0.05 for difference between lean and obese.
Discussion
The main finding of the current study was that despite being initially depressed, 24 h of EPS improved functional indices of insulin action (insulin‐stimulated glycogen synthesis, glucose oxidation and glucose uptake) in primary myotubes from severely obese, insulin‐resistant donors to an extent that values exceeded or were equivalent to myotubes from lean individuals under control (non‐EPS) conditions (Fig. 5). An advantage of the EPS model is that the myotubes from the lean and severely obese individuals received the same absolute contractile stimulus. This is an important consideration due to the compromised cardiorespiratory fitness of obese/severely obese individuals, which can require in vivo exercise to be performed at either a lower absolute workload (i.e. watts) when matching relative exercise intensity (% ) or at a higher relative intensity of when absolute workload is matched to a lean control group. In addition, EPS permits skeletal muscle metabolism to be studied in the absence of in vivo systemic influences (i.e. hormones, neural input, blood flow, etc.), which may differ with obesity. Our findings suggest that although the skeletal muscle of severely obese individuals can be metabolically remodelled, the tissue may not be as intrinsically responsive as in lean individuals when faced with contractile stimuli. The observation that functional indices of insulin action improved with contractile activity in myotubes from severely obese subjects, but remained depressed compared to lean subjects (Fig. 5) suggests that properties specific to skeletal muscle may be linked with the dampened in vivo responses after exercise noted by others in obese and/or insulin resistant individuals (Sriwijitkamol et al. 2007; De Filippis et al. 2008; Stephens & Sparks, 2015; Malin et al. 2016). These data also, however, reinforce the efficacy of interventions utilizing contractile activity in alleviating insulin resistance and/or improving insulin sensitivity in human skeletal muscle regardless of being lean or obese (Christ‐Roberts et al. 2003; Frosig et al. 2007; Hawley & Lessard, 2008; Vind et al. 2011).
A possible mechanism linked with the dampened ability to increase insulin action in severe obesity may involve the inability of contractile activity to potentiate insulin signal transduction. In other studies, insulin‐stimulated Akt Ser473 or Thr308 phosphorylation was potentiated after a bout of cycle or single‐leg exercise in healthy, non‐obese subjects (Christ‐Roberts et al. 2003; Howlett et al. 2006; Treebak et al. 2014) but not in obese insulin‐resistant subjects (Christ‐Roberts et al. 2003). The present data agree in relation to Akt Ser473 (Fig. 6 B) but not Akt Thr308 (Fig. 6 C). However, other human and rodent studies indicate an absence of enhanced insulin signalling after contraction at the level of Akt Thr308 and Ser473 (Wojtaszewski et al. 1997, 2000; Hamada et al. 2006). The present data (Fig. 6 A) also indicate a minimal involvement of insulin signal transduction at the level of IRS1 for the improvement in insulin action seen in muscle from obese, insulin‐resistant individuals in response to contractile activity.
TBC1D4 appears to be a point of convergence of signalling events initiated with insulin‐stimulation and muscle contractile activity (Treebak et al. 2009, 2014; Cartee, 2015 b). While the phosphorylation of the nine confirmed sites of TBC1D4 can be altered when determined immediately after exercise (Treebak et al. 2014; Cartee, 2015 b), the current experiment was designed to focus upon the enhancement in insulin action evident in the hours (3 h in the present study) after contractile activity. Herein, we report that under control (no EPS) conditions Thr642, Ser318, Ser341, Ser588 and Ser704 were all stimulated by insulin (Fig. 7). In agreement, Thr642, Ser318, Ser341, Ser588 and Ser704 are insulin responsive, and all but Ser588 exhibit consistent insulin‐stimulated phosphorylation in populations ranging from lean to obese or obese individuals with type 2 diabetes (Treebak et al. 2009; Pehmoller et al. 2012; Albers et al. 2015; Cartee, 2015 b). In lean subjects, acute in vivo exercise followed by insulin stimulation potentiated TBC1D4 phosphorylation at Ser318, Ser341, Ser588, Ser704, Ser751 and Thr642 in skeletal muscle (Treebak et al. 2009, 2014; Vind et al. 2011; Pehmoller et al. 2012; Cartee, 2015 b). In the present study, in primary myotubes from lean subjects EPS increased the insulin‐mediated phosphorylation of Thr642, Ser341 and Ser588 but did not potentiate the phosphorylation of other residues (Ser318, Ser704; Fig. 7) in contrast to other data (Treebak et al. 2009, 2014; Vind et al. 2011; Pehmoller et al. 2012; Cartee, 2015 b). However, it is difficult to definitively conclude which TBC1D4 sites are phosphorylated with exercise + insulin stimulation; for example, virtually identical leg extensor exercise for 1 h induced different phosphorylation signatures in young, healthy individuals (Treebak et al. 2009, 2014). The current data indicate that in general, insulin‐stimulated phosphorylation patterns of TBC1D4 after in vitro contractile activity (EPS) are similar to those reported with in vivo exercise. However, the novel and important information provided from the current data is that the ability of contractile activity to potentiate insulin‐stimulated TBC1D4 phosphorylation was limited to skeletal muscle from lean individuals and was not present in severely obese individuals.
The fuel‐sensing AMPK signalling network can be activated with contractile activity, which in turn may increase insulin sensitivity via activation of TBC1D4 (Cartee, 2015 a; Kjobsted et al. 2015, 2016, 2017). For example, activation of AMPK via AICAR (an AMP analogue) led to an increase in insulin‐stimulated glucose uptake in skeletal muscle, suggesting that AMPK activation may enhance post‐exercise insulin sensitivity (Fisher et al. 2002; Kjobsted et al. 2015). In the present study despite the same contractile stimulus and degree of energy demand (as indicated by glycogen depletion; Fig. 2), myotubes from the severely obese had suppressed AMPK activation when determined immediately after the 24 h of EPS compared to the lean subjects; ACC, a downstream substrate of AMPK demonstrated a similar pattern (Fig. 3). These data agree with the observations that insulin‐resistant obese adults (Sriwijitkamol et al. 2007; De Filippis et al. 2008) and obese individuals with type 2 diabetes (Sriwijitkamol et al. 2007) exhibited a depressed ability to phosphorylate AMPK in response to in vivo exercise. An intriguing aspect of the current study is that this suppressed AMPK activation with obesity could be due to a lower concentration of AMP with contractile activity (Fig. 2 B) rather than factors such as differences in AMPK abundance (Figs 3 A and 4 D). This lower concentration of AMP could not be attributed to increased production of IMP (a degradation product of AMP) via AMP deaminase with severe obesity (Fig. 2 B); thus, it is not evident why AMP accumulation was dampened with severe obesity. It should be considered that other factors such as liver kinase B (LKB) and calcium flux can influence AMPK activity during muscle contraction and could be involved with the reduced phosphorylation we observed with obesity (O'Neill, 2013). Regardless, the present study provides the novel data that human skeletal muscle myotubes from severely obese individuals, in comparison to lean subjects, exhibit a lower abundance of AMP coupled with suppressed AMPK phosphorylation in response to an identical contractile stimulus. It is important to note that the increase in AMPK phosphorylation returned to resting levels at 3 h after contraction ceased (Fig. 4 D), which suggests that the transient activation of AMPK with contractile activity initiates subsequent processes which may in turn contribute to enhancing insulin action. It is also acknowledged that in human skeletal muscle there are several heterotrimeric combinations of AMPK, with some isoforms more contraction responsive than others (Kjobsted et al. 2017); differences between key signalling components of AMPK may have thus been obscured by only determining overall AMPK phosphorylation in the current study (Fig. 3).
An AMPK‐TBC1D4 signalling axis has been proposed to be a mechanism by which prior exercise potentiates insulin sensitivity (Treebak et al. 2009, 2014; Vind et al. 2011; Cartee, 2015 a,b; Kjobsted et al. 2017). The present data support this hypothesis but only in the skeletal muscle of lean, insulin‐sensitive individuals. Our unanticipated finding was that the improvement in insulin action with contractile activity in muscle from severely obese, insulin‐resistant individuals appeared to involve a different mechanism, as despite increased AMPK phosphorylation (Fig. 3) and insulin action (Fig. 5), there was no overt potentiation of insulin signal transduction at the level of TBC1D4 or upstream with EPS (Figs 6 and 7). In contrast, an intact AMPK signalling network has been reported in the skeletal muscle of insulin‐resistant individuals (Kjobsted et al. 2016); however, it is important to note that Kjøbsted et al. examined middle‐aged (mean of 55 years) overweight/obese subjects with and without type 2 diabetes, with no comparison to lean, insulin‐sensitive individuals. As muscle glycogen content can influence insulin action (Richter et al. 2001), the decline in glycogen concentration after EPS (Fig. 2) may have, at least in part, have contributed to the improvement in insulin action in the severely obese donors. An intriguing finding was that the pattern of changes in membrane‐bound GLUT 4 protein content after EPS matched functional indices of insulin action (glycogen synthesis, glucose oxidation) more closely than indices of insulin signal transduction (Figs 5, 6, 7). This suggests that contractile activity may produce an intracellular environment more conducive to GLUT4 translocation with insulin, independent of insulin signalling. This premise agrees with other findings indicating that prior contractile activity improved insulin action independent of insulin signal transduction (Geiger et al. 2005) and a reorientation of insulin‐responsive GLUT4 in skeletal muscle following exercise (Jensen & Richter, 2012).
The current data demonstrate the general utility of the EPS model for inducing contraction‐related responses and adaptations. Glycogen concentration decreased, PGC1α content increased, and nucleotide content was altered in a manner indicative of increased energy demand and muscle contraction. In addition, AMPK and ACC phosphorylation were elevated when determined immediately after EPS. Although non‐quantitative, during EPS, we could observe physical movement and calcium flux. These findings suggest that an in vitro contraction system may be an effective model for studying the effects of contractile activity in human skeletal muscle. A 24 h period of EPS was selected to optimize the contractile response based upon other EPS/myotube studies (Nedachi et al. 2008; Lambernd et al. 2012; Manabe et al. 2012; Nikolic et al. 2012; Feng et al. 2015) and our own preliminary experiments. Muscle glycogen content decreased by ∼20% (Fig. 2), which would represent a fairly modest exercise stimulus when compared to in vivo human findings (Vollestad & Blom, 1985) and that the EPS protocol approximated mild/moderate exercise. In relation to responses to insulin action, the present data are in contradiction with those of Feng et al. who reported that insulin‐stimulated glycogen synthesis did not change with EPS in primary myotubes from lean subjects and only improved in muscle from severely obese individuals (Feng et al. 2015). Explanations for this discrepancy may involve differences in the duration of the contraction (48 vs. the current 24 h), strength of stimulation (30 vs. current 11.5 V), or age of the lean subjects (49 vs. the current 30 years) (Feng et al. 2015). In the current study, differences between the lean and obese groups could not be attributed to differences in myoblast proliferation, myotube differentiation or cell viability (Fig. 1).
In conclusion, electrical pulsed stimulation of primary myotubes from lean and severely obese subjects induced improvements in insulin action, but to a lesser extent with severe obesity. However, contractile activity in severely obese subjects normalized insulin action to the levels of their lean counterparts under control conditions (non‐EPS). Insulin signal transduction was enhanced after EPS in myotubes from lean but not severely obese subjects, implying that different mechanisms were involved with the improvements in insulin‐mediated metabolism with obesity. The current findings suggest that the suppressed exercise adaptation with severe obesity may be linked to a reduced activation of AMPK in response to contractile stimuli. It is not evident if the skeletal muscle of women with lesser degrees of obesity demonstrate the same impairments, nor if the dampened response to exercise is linked to obesity, insulin resistance, or a combination of both. However, despite these limitations, the current data reveal that myotubes of severely obese individuals enhance insulin action with contraction, but in a manner and magnitude that differs from lean subjects.
Additional information
Competing interests
The authors have no conflicts of interest, financial or otherwise, to declare.
Author contributions
J.A.H. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. S.P. and J.A.H. did the conception and design of the research; S.P., K.T., D.Z., J.J.B., K.Z., C.J.T and J.A.H. performed the experiments; S.P., K.T., D.Z., and J.B.B. analysed the data; S.P., K.T., D.Z., J.J.B., K.Z., T.S.N., A.C., J.T.T. and J.A.H. interpreted the results of the experiments; S.P. prepared the figures; S.P., A.C., J.T.T., and J.A.H. drafted the manuscript; S.P., A.C., J.T.T. and J.A.H. edited and revised the manuscript; all authors approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
Funding for this work was provided by grants from the National Institute of Diabetes and Digestive and Kidney Diseases (DK‐056112, J. A. Houmard), National Institute of Arthritis and Musculoskeletal and Skin Diseases (AR‐070200, J. J. Brault), and a student research grant from the American College of Sports Medicine (S. Park).
Acknowledgements
We thank Angela Clark and Gabriel Dubis for assisting with specimen collection and subject recruitment. We thank the subjects for their participation and the ECU Department of Surgery for permitting access to their patients.
Biography
Sanghee Park is a post‐doctoral researcher at the University of Arkansas for Medical Sciences where he works on projects examining in vivo anabolic responses with a variety of protein meals in humans using stable isotope tracers. For his doctoral works at East Carolina University, he worked under Dr Houmard to investigate if contractile activity induced by electrical pulse stimulation can provide the same beneficial effects as exercise in myotubes obtained from severely obese and lean individuals.

Edited by: Michael Hogan & Bettina Mittendorfer
References
- Aas V, Bakke SS, Feng YZ, Kase ET, Jensen J, Bajpeyi S, Thoresen GH & Rustan AC (2013). Are cultured human myotubes far from home? Cell Tissue Res 354, 671–682. [DOI] [PubMed] [Google Scholar]
- Albers PH, Pedersen AJ, Birk JB, Kristensen DE, Vind BF, Baba O, Nohr J, Hojlund K & Wojtaszewski JF (2015). Human muscle fiber type‐specific insulin signaling: impact of obesity and type 2 diabetes. Diabetes 64, 485–497. [DOI] [PubMed] [Google Scholar]
- Al‐Khalili L, Chibalin AV, Kannisto K, Zhang BB, Permert J, Holman GD, Ehrenborg E, Ding VDH, Zierath JR & Krook A (2003). Insulin action in cultured human skeletal muscle cells during differentiation: assessment of cell surface GLUT4 and GLUT1 content. Cell Mol Life Sci 60, 991–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berggren JR, Tanner CJ & Houmard JA (2007). Primary cell cultures in the study of human muscle metabolism. Exerc Sport Sci Rev 35, 56–61. [DOI] [PubMed] [Google Scholar]
- Bikman BT, Zheng D, Reed MA, Hickner RC, Houmard JA & Dohm GL (2010). Lipid‐induced insulin resistance is prevented in lean and obese myotubes by AICAR treatment. Am J Physiol Regul Integr Comp Physiol 298, R1692–R1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollinger LM, Powell JJ, Houmard JA, Witczak CA & Brault JJ (2015). Skeletal muscle myotubes in severe obesity exhibit altered ubiquitin‐proteasome and autophagic/lysosomal proteolytic flux. Obesity (Silver Spring) 23, 1185–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonora E, Kiechl S, Willeit J, Oberhollenzer F, Egger G, Targher G, Alberiche M, Bonadonna RC, Muggeo M (1998). Prevalence of insulin resistance in metabolic disorders: the Bruneck Study. Diabetes 47, 1643–1649. [DOI] [PubMed] [Google Scholar]
- Bouzakri K, Koistinen HA & Zierath JR (2005). Molecular mechanisms of skeletal muscle insulin resistance in type 2 diabetes. Curr Diabetes Rev 1, 167–174. [DOI] [PubMed] [Google Scholar]
- Brault JJ, Pizzimenti NM, Dentel JN & Wiseman RW (2013). Selective inhibition of ATPase activity during contraction alters the activation of p38 MAP kinase isoforms in skeletal muscle. J Cell Biochem 114, 1445–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartee GD (2015. a). AMPK‐TBC1D4‐dependent mechanism for increasing insulin sensitivity of skeletal muscle. Diabetes 64, 1901–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartee GD (2015. b). Roles of TBC1D1 and TBC1D4 in insulin‐ and exercise‐stimulated glucose transport of skeletal muscle. Diabetologia 58, 19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chella Krishnan K, Mehrabian M, Lusis AJ (2018). Sex differences in metabolism and cardiometabolic disorders. Current Opin Lipidol 29, 404–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christ‐Roberts CY, Pratipanawatr T, Pratipanawatr W, Berria R, Belfort R & Mandarino LJ (2003). Increased insulin receptor signaling and glycogen synthase activity contribute to the synergistic effect of exercise on insulin action. J Appl Physiol 95, 2519–2529. [DOI] [PubMed] [Google Scholar]
- Cortright RN, Sandhoff KM, Basilio JL, Berggren JR, Hickner RC, Hulver MW, Dohm GL & Houmard JA (2006). Skeletal muscle fat oxidation is increased in African‐American and White women after 10 days of endurance exercise training, Obesity 14, 1201–1210. [DOI] [PubMed] [Google Scholar]
- De Filippis E, Alvarez G, Berria R, Cusi K, Everman S, Meyer C & Mandarino LJ (2008). Insulin‐resistant muscle is exercise resistant: evidence for reduced response of nuclear‐encoded mitochondrial genes to exercise. Am J Physiol Endocrinol Metab 294, E607–E614. [DOI] [PubMed] [Google Scholar]
- DeFronzo RA, Gunnarsson R, Bjorkman O, Olsson M & Wahren J (1985). Effects of insulin on peripheral and splanchnic glucose metabolism in noninsulin‐dependent (type II) diabetes mellitus. J Clin Invest 76, 149–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenborg E & Krook A (2009). Regulation of skeletal muscle physiology and metabolism by peroxisome proliferator‐activated receptor delta. Pharmacol Rev 61, 373–393. [DOI] [PubMed] [Google Scholar]
- Feng YZ, Nikolić N, Bakke SS, Kase ET, Guderud K, Hjelmesæth J, Aas V, Rustan AC & Thoresen GH (2015). Myotubes from lean and severely obese subjects with and without type 2 diabetes respond differently to an in vitro model of exercise. Am J Physiol Cell Physiol 308, C548–C556. [DOI] [PubMed] [Google Scholar]
- Fisher JS, Gao J, Han DH, Holloszy JO & Nolte LA (2002). Activation of AMP kinase enhances sensitivity of muscle glucose transport to insulin. Am J Physiol Endocrinol Metab 282, E18–E23. [DOI] [PubMed] [Google Scholar]
- Frosig C, Rose AJ, Treebak JT, Kiens B, Richter EA & Wojtaszewski JF (2007). Effects of endurance exercise training on insulin signaling in human skeletal muscle: interactions at the level of phosphatidylinositol 3‐kinase, Akt, and AS160. Diabetes 56, 2093–2102. [DOI] [PubMed] [Google Scholar]
- Geiger PC, Wright DC, Han DH & Holloszy JO (2005). Activation of p38 MAP kinase enhances sensitivity of muscle glucose transport to insulin. Am J Physiol Endocrinol Metab 288, E782–E788. [DOI] [PubMed] [Google Scholar]
- Guesbeck NR, Hickey MS, MacDonald KG, Pories WJ, Harper I, Ravussin E, Dohm GL & Houmard JA (2001). Substrate utilization during exercise in formerly morbidly obese women. J Appl Physiol (1985) 90, 1007–1012. [DOI] [PubMed] [Google Scholar]
- Hales CM, Fryar CD & Carroll MD (2018). Trends in obesity and severe obesity prevelance in US youths and adults by sex and age, 2007–2008 to 2015—2016. JAMA 319:1723–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada T, Arias EB & Cartee GD (2006). Increased submaximal insulin‐stimulated glucose uptake in mouse skeletal muscle after treadmill exercise. J Appl Physiol 101, 1368–1376. [DOI] [PubMed] [Google Scholar]
- Hawley JA & Lessard SJ (2008). Exercise training‐induced improvements in insulin action. Acta Physiol (Oxf) 192, 127–135. [DOI] [PubMed] [Google Scholar]
- Howlett KF, Sakamoto K, Yu H, Goodyear LJ & Hargreaves M (2006). Insulin‐stimulated insulin receptor substrate‐2‐associated phosphatidylinositol 3‐kinase activity is enhanced in human skeletal muscle after exercise. Metabolism 55, 1046–1052. [DOI] [PubMed] [Google Scholar]
- Hulver MW, Berggren JR, Carper MJ, Miyazaki M, Ntambi JM, Hoffman EP, Thyfault JP, Stevens R, Dohm Gis, Houmard JA & Muoio DM (2005). Elevated stearoyl‐CoA desaturase‐1 expression in skeletal muscle contributes to abnormal fatty acid partitioning in obese humans. Cell Metab 2, 251–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen TE & Richter EA (2012). Regulation of glucose and glycogen metabolism during and after exercise. J Physiol 590, 1069–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen SB, Wojtaszewski JF, Viollet B, Andreelli F, Birk JB, Hellsten Y, Schjerling P, Vaulont S, Neufer PD, Richter EA & Pilegaard H (2005). Effects of alpha‐AMPK knockout on exercise‐induced gene activation in mouse skeletal muscle. FASEB J 19, 1146–1148. [DOI] [PubMed] [Google Scholar]
- Kim JY, Hickner RC, Cortright RL, Dohm GL & Houmard JA (2000). Lipid oxidation is reduced in obese human skeletal muscle. Am J Physiol Endocrinol Metab 279, E1039–E1044. [DOI] [PubMed] [Google Scholar]
- Kjobsted R, Munk‐Hansen N, Birk JB, Foretz M, Viollet B, Bjornholm M, Zierath JR, Treebak JT & Wojtaszewski JF (2017). Enhanced muscle insulin sensitivity after contraction/exercise is mediated by AMPK. Diabetes 66, 598–612. [DOI] [PubMed] [Google Scholar]
- Kjobsted R, Pedersen AJ, Hingst JR, Sabaratnam R, Birk JB, Kristensen JM, Hojlund K & Wojtaszewski JF (2016). Intact regulation of the AMPK signaling network in response to exercise and insulin in skeletal muscle of male patients with type 2 diabetes: illumination of AMPK activation in recovery from exercise. Diabetes 65, 1219–1230. [DOI] [PubMed] [Google Scholar]
- Kjobsted R, Treebak JT, Fentz J, Lantier L, Viollet B, Birk JB, Schjerling P, Bjornholm M, Zierath JR & Wojtaszewski JF (2015). Prior AICAR stimulation increases insulin sensitivity in mouse skeletal muscle in an AMPK‐dependent manner. Diabetes 64, 2042–2055. [DOI] [PubMed] [Google Scholar]
- Knight JB & Olson AL (2003). Visualization and quantitation of integral membrane proteins using a plasma membrane sheet assay. Methods Mol Med 83, 113–118. [DOI] [PubMed] [Google Scholar]
- Lambernd S, Taube A, Schober A, Platzbecker B, Gorgens SW, Schlich R, Jeruschke K, Weiss J, Eckardt K & Eckel J (2012). Contractile activity of human skeletal muscle cells prevents insulin resistance by inhibiting pro‐inflammatory signalling pathways. Diabetologia 55, 1128–1139. [DOI] [PubMed] [Google Scholar]
- Malin SK, Liu Z, Barrett EJ & Weltman A (2016). Exercise resistance across the prediabetes phenotypes: Impact on insulin sensitivity and substrate metabolism. Rev Endocr Metab Disord 17, 81–90. [DOI] [PubMed] [Google Scholar]
- Manabe Y, Miyatake S, Takagi M, Nakamura M, Okeda A, Nakano T, Hirshman MF, Goodyear LJ & Fujii NL (2012). Characterization of an acute muscle contraction model using cultured C2C12 myotubes. PLoS One 7, e52592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musi N, Fujii N, Hirshman MF, Ekberg I, Froberg S, Ljungqvist O, Thorell A & Goodyear LJ (2001). AMP‐activated protein kinase (AMPK) is activated in muscle of subjects with type 2 diabetes during exercise. Diabetes 50, 921–927. [DOI] [PubMed] [Google Scholar]
- Nedachi T, Fujita H & Kanzaki M (2008). Contractile C2C12 myotube model for studying exercise‐inducible responses in skeletal muscle. Am J Physiol Endocrinol Metab 295, E1191–E1204. [DOI] [PubMed] [Google Scholar]
- Nikolic N, Bakke SS, Kase ET, Rudberg I, Flo Halle I, Rustan AC, Thoresen GH & Aas V (2012). Electrical pulse stimulation of cultured human skeletal muscle cells as an in vitro model of exercise. PLoS One 7, e33203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill HM (2013). AMPK and exercise: glucose uptake and insulin sensitivity. Diabetes Metab J 37, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pehmoller C, Brandt N, Birk JB, Hoeg LD, Sjoberg KA, Goodyear LJ, Kiens B, Richter EA & Wojtaszewski JF (2012). Exercise alleviates lipid‐induced insulin resistance in human skeletal muscle‐signaling interaction at the level of TBC1 domain family member 4. Diabetes 61, 2743–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter EA, Derave W & Wojtaszewski JF (2001). Glucose, exercise and insulin: emerging concepts. J Physiol 535, 313–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriwijitkamol A, Coletta DK, Wajcberg E, Balbontin GB, Reyna SM, Barrientes J, Eagan PA, Jenkinson CP, Cersosimo E, DeFronzo RA, Sakamoto K & Musi N (2007). Effect of acute exercise on AMPK signaling in skeletal muscle of subjects with type 2 diabetes: a time‐course and dose‐response study. Diabetes 56, 836–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens NA & Sparks LM (2015). Resistance to the beneficial effects of exercise in type 2 diabetes: are some individuals programmed to fail? J Clin Endocrinol Metab 100, 43–52. [DOI] [PubMed] [Google Scholar]
- Teran‐Garcia M, Rankinen T, Koza RA, Rao DC & Bouchard C (2005). Endurance training‐induced changes in insulin sensitivity and gene expression. Am J Physiol Endocrinol Metab 288, E1168–E1178. [DOI] [PubMed] [Google Scholar]
- Treebak JT, Frøsig C, Pehmøller C, Chen S, Maarbjerg SJ, Brandt N, MacKintosh C, Zierath JR, Hardie DG, Kiens B, Richter EA, Pilegaard H & Wojtaszewski JFP (2009). Potential role of TBC1D4 in enhanced post‐exercise insulin action in human skeletal muscle. Diabetologia 52, 891–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treebak JT, Pehmoller C, Kristensen JM, Kjobsted R, Birk JB, Schjerling P, Richter EA, Goodyear LJ & Wojtaszewski JF (2014). Acute exercise and physiological insulin induce distinct phosphorylation signatures on TBC1D1 and TBC1D4 proteins in human skeletal muscle. J Physiol 592, 351–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vind BF, Pehmoller C, Treebak JT, Birk JB, Hey‐Mogensen M, Beck‐Nielsen H, Zierath JR, Wojtaszewski JF & Hojlund K (2011). Impaired insulin‐induced site‐specific phosphorylation of TBC1 domain family, member 4 (TBC1D4) in skeletal muscle of type 2 diabetes patients is restored by endurance exercise‐training. Diabetologia 54, 157–167. [DOI] [PubMed] [Google Scholar]
- Vollestad NK & Blom PCS (1985). Effect of varying exercise intensity on glycogen depletion in human muscle fibers. Acta Physiol Scand 125, 395–405. [DOI] [PubMed] [Google Scholar]
- Wojtaszewski JF, Hansen BF, Gade, Markuns JF, Goodyear LJ & Richter EA (2000). Insulin signaling and insulin sensitivity after exercise in human skeletal muscle. Diabetes 49, 325–331. [DOI] [PubMed] [Google Scholar]
- Wojtaszewski JF, Hansen BF, Kiens B & Richter EA (1997). Insulin signaling in human skeletal muscle: time course and effect of exercise. Diabetes 46, 1775–1781. [DOI] [PubMed] [Google Scholar]
- Yates T, Khunti K, Bull F, Gorely T & Davies MJ (2007). The role of physical activity in the management of impaired glucose tolerance: a systematic review. Diabetologia 50, 1116–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou K, Hinkley JM, Park S, Zheng D, Jones TE, Pories WJ, Hornby PJ, Lenhard J, Dohm GL & Houmard JA (2018). Altered tricarboxylic acid cycle flux in primary myotubes from severely obese humans. Int J Obes (Lond); 10.1038/s41366-018-0137-7. [DOI] [PubMed] [Google Scholar]