

Abstract
Bromodomains have been pursued intensively over the past several years as emerging targets for the development of anti-cancer and anti-inflammatory agents. It has recently been shown that some kinase inhibitors are able to potently inhibit the bromodomains of BRD4. The clinical activities of PLK inhibitor BI-2536 and JAK2-FLT3 inhibitor TG101348 have been attributed to this unexpected poly-pharmacology, indicating that dual-kinase/bromodomain activity may be advantageous in a therapeutic context. However, for target validation and biological investigation, a more selective target profile is desired. Here we report that benzo[e]pyrimido-[5,4-b]diazepine-6(11H)-ones, versatile ATP-site directed kinase pharmacophores utilized in the development of inhibitors of multiple kinases including a number of previously reported kinase chemical probes, are also capable of exhibiting potent BRD4-dependent pharmacology. Using a dual kinase-bromodomain inhibitor of the kinase domains of ERK5 and LRRK2, and the bromodomain of BRD4 as a case study, we define the structure-activity relationships required to achieve dual kinase/BRD4 activity as well as how to direct selectivity towards inhibition of either ERK5 or BRD4. This effort resulted in identification of one of the first reported kinase-selective chemical probes for ERK5 (JWG-071), a BET selective inhibitor with 1 μM BRD4 IC50 (JWG-115), and additional inhibitors with rationally designed polypharmacology (JWG-047, JWG-069). Co-crystallography of seven representative inhibitors with the first bromodomain of BRD4 demonstrate that distinct atropisomeric conformers recognize the kinase ATP-site and the BRD4 acetyl lysine binding site, conformational preferences supported by rigid docking studies.
INTRODUCTION
BRD4 is a critical transcription co-activator protein that possesses two druggable acetyl lysine binding domains (bromodomains) targeting the protein to chromatin by binding acetylated histones.1 BRD4 is a master regulatory elongation factor, influencing function of CDK92, the kinase activity of which is essential to phosphorylate RNA Polymerase II to initiate transcription,3–5 and aberrant activity of BRD4 contributes to deregulated transcription, a hallmark of many cancers.6–8 BRD4 has been actively pursued as an oncology drug target with at least six compounds that target the acetyl lysine binding pocket currently under clinical investigation.9 These selective BET inhibitors have demonstrated phenotypic effects against a broad range of cancer cell lines and in models of inflammation, atherosclerosis and male fertility.
Synergy between BRD4 inhibition and a multitude of kinase inhibitors has also been identified in cancer (e.g. with FLT3TKI10, rapamycin11 and CX-494512), providing a rationale for development of therapeutics with potent dual BRD4-kinase activity.
Recently several groups have made the surprising discovery that many ATP-competitive kinase inhibitors can also bind potently to the acetyl-lysine binding domain of BRD4.13,14 Kinase inhibitors based on at least 10 different scaffolds have been shown to inhibit the BRD4 bromodomains. The clinical activities of the PLK inhibitor BI-2536 and the JAK2-FLT3 inhibitor TG101348 have been attributed to this unexpected poly-pharmacology, indicating that in a therapeutic context dual kinase-bromodomain activity may be desirable.
However, due to its ubiquitous expression and central importance to the regulation of transcription, uncharacterized offtarget BRD4 activity in chemical probes is undesirable, as it complicates the connection between the target and any phenotypes observed. Characterization of the BRD4 activity of classical kinase inhibitor scaffolds, and the trends governing the selectivity profile are important for unraveling the pharmacologically relevant targets for these compounds. To this end, extensive work has been invested in characterizing the factors governing the PLK and BRD4 activity of BI-2536 analogs based on a privileged ATP-site targeting pyrido[2,3-d] pyrimidine core.15,16
Here we characterize the SAR trends and structural basis for the kinase and bromodomain activity profile of the related benzo[e]pyrimido-[5,4-b] diazepine-6(11H)-ones and use these rules to generate selective chemical probes for ERK5/LRRK2 and BRD4, and to develop rationally designed dual kinase (ERK5/LRRK2)-BRD4 inhibitors. We also observed two distinct conformers exclusively recognize the kinase ATP-site and the BRD4 acetyl lysine-binding site and analyzed the interconversion rate for conformers with large substituents.
RESULTS AND DISCUSSION
ERK5 inhibitors XMD11–50 (LRRK2-IN-1) and XMD17–26 are dual kinase-bromodomain inhibitors.
Based on the privileged nature of the pyrido[2,3-d] pyrimidine core of BI2536 as an ATP-site targeting scaffold, we had built combinatorial libraries of analogs with a benzo[e]pyrimido-[5,4-b]diazepine-6(11H)-one scaffold for use in kinase inhibitor projects, resulting in the discovery of multiple kinase inhibitor tool compounds (Figure 1).17 In order to test if this series also inhibits BRD4, analogous to BI-2536, we performed a BRD4 AlphaScreen assay of our kinase-biased chemical library (data not shown).18 Many diazepinones were found to be active. Importantly some hits from this screen are published “selective” kinase chemical probes,19–24 and therefore this result provides valuable insight into their observed pharmacology25,26 (Figure 1). These molecules represent a new class of dual kinase-bromodomain inhibitors.
Figure 1.

BRD4 (BD1) inhibition of published kinase chemical probes. Kinase IC50s are as previously published in references. BRD4 IC50 values were measured by AlphaScreen™ binding assay and reported as the average of 2 replicates ± SE (conducted in Dana-Farber Cancer Institute, top data) and as the average of 4 replicates ± SD (conducted in University of Oxford, bottom data). (+)-JQ1 IC50s are 64 nM and 120 nM respectively.
We chose to investigate the structure activity relationship (SAR) of this scaffold against both ERK5/LRRK2 and BRD4, using BRD4 hit compounds XMD11–50 (LRRK2-IN-1) and XMD17–26 (published as an ERK5 inhibitor) as chemical starting points, aiming to develop a selective ERK5 inhibitor. This was motivated by the dual kinase-bromodomain inhibition profile of the compounds (Figure 1), the lack of availability of a selective ERK5 chemical probe, the poorly characterized biological function of ERK5, and the potential of ERK5 as a cancer drug target. Specifically, the MEK5/ERK5 pathway has been reported to be activated and/or overexpressed during tumor development, metastasis and tumor angiogenesis. 27–29
We began by confirming the BRD4 activity of the hit kinase inhibitors using isothermal titration calorimetry (ITC) and differential scanning fluorimetry (DSF) (Table 1, Supplemental Figure 1 and 2), using (+)-JQ1 as a BRD4 positive control. These data were in good agreement with IC50 values measured by AlphaScreen. Both compounds show comparable potency to other well characterized dual kinase/bromodomain inhibitors.13,14
Table 1.
Evaluation of initial BRD4 hits.
| Compound ID | IC50 (nM)a | IC50 (nM)b | Kd (nM)c | ΔTm (°C)d |
|---|---|---|---|---|
| XMD11–50 (LRRK2-IN-1) | 703 ± 17 | 1040 ± 180 | 192 ± 28 | 2.1 ± 0.37 |
| XMD17–26 | 760 ± 12 | 754 ± 110 | 156 ± 12 | 4.1 ± 0.24 |
| (+)-JQ1 | 64 | 120 | 49.0 ± 2.4* | 10.1 ± 0.11 |
IC50 values were measured by AlphaScreen™ binding assay and reported as the average of 2 replicates ± SE. Experiments were conducted in Dana-Farber Cancer Institute.
IC50 values were measured by AlphaScreen™ binding assay and reported as the average of 4 replicates ± SD. Experiments were conducted in University of Oxford.
Kd values were measured by ITC and reported as the average of 4 replicates ± SD.
ΔTm values were measured by DSF and reported as the average of 3 replicates ± SD.
The JQ1 Kd value is from a previous publication.
To understand how these compounds bind to BRD4-BD1, we determined X-ray co-crystal structures of BRD4-BD1 with both XMD11–50 and XMD17–26 to 1.2 Å and 1.6 Å resolution respectively. (Figure 2A, 2B and Supplemental Table 1). Both XMD11–50 and XMD17–26 were clearly defined in the electron density maps and bound in the acetyl lysine binding pocket (Supplemental Figure 3A, 3B and Figure 2A, 2B). The core of the benzo[e]pyrimido- [5,4-b] diazepine-6(11H)-one scaffold is buried deeply in the pocket and mediates most direct interactions with the bromodomain and associated structural waters. It forms two hydrogen bonds directly with the protein, one with the side chain of conserved Asn140, the other with the carbonyl oxygen of Pro82. The N-methyl amide group in the diazipinone ring fills a hydrophobic cavity in a manner analogous to N-acetyl group of an acetylated lysine residue.30 Structural waters are usually conserved and mediate a ligand-bromodomain interaction. In both structures, there are three key structural waters forming hydrogen bonds with the ligand directly, and bridging hydrogen-bond interactions with the side chains of Gln85 and Tyr97, the carbonyl oxygens of Pro86 and Gln85 (Figure 2A, 2B). In addition, the methoxyphenyl moiety of the aniline tail forms a hydrophobic interaction with the WPF (residue 81–83) shelf in the ZA loop (residue 78–105). The rest of the ligand tail consisting of the methyl-4-piperidine-4-yl piperazine moiety is solvent exposed, which suggests that it is not essential for ligand-protein recognition. The structure of XMD17–26 shares the same core interactions observed in the XMD11–50 structure (Figure 2B). In addition, the WPF shelf forms a hydrophobic interaction with the cyclopentyl R1 substituent of XMD17–26 and further stabilized by the side chain of Iso146 (Figure 2 C). The more bulky cyclopentyl substituent also causes a shift in the conformation of the tricyclic ring system. As a result, different angles between the planes of the scaffold are observed for the two compounds in the X-ray structures (Figure 2 C).
Figure 2.

(A) X-ray structure of XMD11–50 bound to BRD4-BD1. (B) X-ray structure of XMD17–26 bound to BRD4-BD1. (C) Overlay of the two compounds highlighting the different angular configuration of the diazipinone ring system. The hydrophobic pocked composed by the side chains of Try81, Pro82 and Iso146 is highlighted in cyan.
Modulating selectivity for BRD4, ERK5 and LRRK2.
We initiated our studies with a systematic structure-activity relationship analysis of the R1 substitution (Table 2). The R1 group influences the conformation of the 7-membered diazepine ring and mediates steric and hydrophobic interactions with BRD4. Thus, we anticipated that R1 substitutions would affect BRD4 activity and result in different selectivity profiles against BRD4, ERK5 and LRRK2. The newly synthesized compounds were tested for their ability to inhibit ERK5 and LRRK2 in vitro kinase activities, ERK5 cellular activity in human HeLa cells, BRD4-BD1 binding affinity and BRD4 cellular activity in the BRD4-dependant 797 NUT-midline carcinoma cells which harbors a BRD4-NUT fusion oncogene.31 Addition of progressively larger alkyl substituents at R1, from methyl (XMD11–50) to sec-butyl (JWG-071) resulted in gradually reduced binding affinity for BRD4 and LRRK2, but had almost no effect on ERK5 inhibition, which may be due to steric clashes with the αC-helix of BRD4, indicating this position is a crucial hotspot for dialing out bromodomain targets while maintaining ERK5 kinase activity. All attempts to synthesize 3-pentyl substituted analogs to explore the effect of larger substituents at R1 failed, possibly due to steric hindrance. Having identified a kinase class-selective compound, the kinome wide selectivity of JWG-071 was assessed using the KinomeScan methodology (DiscoverX) across a panel of 468 human kinases at a concentration of 1 μM. Only two kinases, ERK5 and DCAMKL2, were hits in this assay (Ambit scores of less than 10%, Supplemental Figure 4 and Supplemental data). This off-target kinase activity was confirmed in biochemical enzyme assays, where JWG-071 showed little selectivity for ERK5 relative to LRRK2 and DCAMKL2 in vitro (1.2 and 2.5 folds respectively). Compared to the previously published ERK5 tool compound XMD17–109 (ERK5-IN-1), JWG-071 has 2-fold improved ERK5 activity, and 10-fold improved BRD4 selectivity. Overall, these data show that JWG-071 will be a much-needed chemical probe for deconvoluting ERK5 and BRD4 pharmacology.
Table 2.
Activity profile of inhibitors with diazepinone scaffold
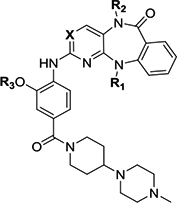 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound ID | R1 | R2 | R3 | X | Biochemical IC50 (BRD4, μM)a | Biochemical IC50 (BRD4, μM)b | Enzymatic IC50 (ERK5, μM)c | Enzymatic IC50 (LRRK2, μM)d | Viability IC50(BRD4, μM)e | Cellular EC50 (ERK5, μM)f | Inhibitor classificationg | Co-crystal (pdb number) |
| XMD11–50 (LRRK2-IN-1) | Me | Me | Me | N | 0.703 ± 0.017 | 1.04 ± 0.18 | 0.114 ±0.011 | 0.0034 ±0.0017 | 1.989 ± 0.299 | 0.16 ± 0.04 | BRD4/LRRK2 | Roco4(4YZM)32 BRD4 (5WA5) |
| JWG-048 | Et | Me | Me | N | 1.33 ± 0.05 | 2.41 ± 0.23 | 0.173 ±0.037 | 0.0044 ±0.0005 | 2.625 ± 0.540 | 0.022 ± 0.004 | LRRK2 | BRD4 (5W55) |
| JWG-046 | i-Pr | Me | Me | N | 4.59 ± 0.14 | 5.14 ± 0.59 | 0.125 ±0.026 | 0.054 ± 0.004 | ND | 0.014 ± 0.004 | ERK5/LRRK2 | BRD4 (6CD4) |
| JWG-071 | s-Bu | Me | Me | N | 5.42 ± 0.18 | 6.31 ± 1.93 | 0.088 ±0.005 | 0.109 ± 0.014 | >10 | 0.020 ± 0.003 | ERK5/LRRK2 | BRD4 (6CJ1) |
| JWG-069 | cyclobutyl | Me | Me | N | 0.201 ± 0.007 | 0.41 ± 0.01 | 0.075 ±0.009 | 0.021 ± 0.001 | 0.116 ± 0.001 | 0.019 ± 0.005 | BRD4/ERK5/LRRK2 | BRD4 (6CIY) |
| XMD17–26 | cyclopentyl | Me | Me | N | 0.760 ± 0.012 | 0.75 ± 0.11 | 0.082 ±0.009 | 0.095 ± 0.010 | 0.664 ± 0.014 | 0.08 ± 0.02 | BRD4/ERK5/LRRK2 | ERK5 (4B99)21 BRD4 (5CD5) |
| JWG-112 | s-Bu | H | Me | N | 25.7 ± 1.8 | >40 | >10 | 1.59 ± 0.37 | ND | ND | No activity | |
| JWG-049 | i-Pr | Me | Et | N | 2.50 ± 0.07 | 3.53 ± 0.82 | 0.069 ±0.007 | 0.114 ± 0.011 | ND | 0.025 ± 0.007 | ERK5/LRRK2 | |
| JWG-114 | i-Pr | Me | i-Pr | N | 0.862 ± 0.065 | 1.08 ± 0.13 | 0.421 ±0.007 | 0.235 ± 0.034 | ND | ND | BRD4/weak LRRK2 | |
| XMD17–109 (ERK5-IN-1) | cyclopentyl | Me | Et | N | 0.217 ± 0.008 | 0.68 ± 0.03 | 0.162 ±0.006 | 0.171 ± 0.030 | ND | 0.09 ± 0.03 | BRD4/ERK5/LRRK2 | |
| JWG-047 | cyclopentyl | Me | i-Pr | N | 0.135 ± 0.004 | 0.30 ± 0.05 | 0.160 ±0.008 | 0.296 ± 0.031 | 0.465 ± 0.012 | 0.032 ± 0.009 | BRD4/ERK5/LRRK2 | BRD4 (6CIS) |
| AX15836 | SO2Me | Me | Et | N | >100 | >40 | 0.012 ±0.002 | 0.942 ± 0.569 | ND | 0.036 ± 0.004 | ERK5 | |
| DFCI-2–208 | Me | Me | Me | C | 5.21 ± 0.14 | 4.64 ± 0.65 | >10 | >10 | ND | ND | No activity | |
| JWG-115 | cyclopentyl | Me | i-Pr | C | 1.08 ± 0.05 | 1.10 ± 0.05 | >10 | > 3.3 | ND | ND | BRD4 (weak) | |
| (+)-JQ-1 | 0.064 | 0.12 | >10 | >10 | 0.228 | ND | BRD4 | |||||
IC50 values were measured by AlphaScreen™ binding assay and reported as the average of 2 replicates ± SE. Experiments were conducted in Dana-Farber Cancer Institute.
IC50 values were measured by AlphaScreen™ binding assay and reported as the average of 4 replicates ± SD. Experiments were conducted in University of Oxford.
IC50 values were measured by in vitro assay and reported as the average of 2 replicates ± SD.
IC50 values were measured using Adapta assay format (ThermoFisher Scientific) and reported as the average of 2 replicates ± SD.
IC50 were determined in 797 NUT-midline carcinoma cells by cell numbers using Cell Titer Glo and reported as the average of 3 replicates ± SD.
EC50 values were measured by EGF-stimulated autophosphorylation of ERK5 in Hela cells and reported as the average of 2 replicates ± SD.
Considering that weak BET bromodomain inhibition has been shown to result in phenotypic responses, a BRD4 activity within 20 fold of JQ1 was defined as BET active. 0.3 μM was chosen as the threshold for kinase activity. A 10-fold activity difference among kinases was considered selective.
Surprisingly, the cyclobutyl and cyclopentyl R1 substituted compounds JWG-069 and XMD17–26 exhibited strong binding to BRD4. These cyclic substitutions were also well tolerated by ERK5 and LRRK2, indicating that these are optimal R1 groups for achieving BRD/Kinase polypharmacology. The activities of corresponding analogs with 2-methoxy-4(4-methylpiperazin-1-yl) aniline tails against ERK5, LRRK2 and BRD4 displayed a similar trend, although with slightly lower BRD4 binding, indicating that the activity difference is mainly due to the changes to interactions with the R1 substituent and the benzo[e]pyrimido- [5,4-b]diazepine-6(11H)-one core structure (Supplemental Table 2).
In order to gain a structural understanding of this piece of the SAR, we determined crystal structures of BRD4 with the R1 analogs (Table 2, entries 2–5, Supplemental Table 1, and Supplemental Figure 3C-3H), and compared them to the structures with XMD11–50 and XMD17–26. The binding mode and key interaction with Asn140 is conserved among all analogs. Alkyl substitutions at the R1 position introduce additional steric restrictions, changing the angular configuration of the “butterfly-shaped” tricyclic core as seen in the structure of XMD17–26 (Figure 3A).
Figure 3.

(A) Overlay of structures of XMD11–50 (green), JWG-048 (salmon), JWG-046 (orange), JWG-071(pink), JWG-069 (cyan) and XMD17–26 (yellow) highlighting the different curvature of the diazipinone ring system and deviation of the tail. (B) Overlay of Roco4/XMD11–50 (pdb code 4YZM, top), and ERK5/XMD17–26 (pdb code 4B99, bottom), crystal structure reveal differences in P-loop (red) conformation and explain observed R2 SAR. (C) Overlay of JWG-047 and XMD17–26 in complex with BRD4-BD1 reveal differences in local interactions and the presence of a new water molecule in the JWG-047. Composite-omit map of JWG-047 in 2Fo-Fc format with a σ-level at 2.0.
Since co-crystal structures of XMD17–26 with ERK5 kinase (PDB: 4B99)21 and XMD11–50 with Roco4 kinase (Dictyostelium disco homolog of LRRK2, PDB: 4YZM)32 have been solved previously, we were also able to examine differences between the BRD4/ligand complexes and kinase/ligand complexes for these molecules. The crystal structure of JWG-069 (R1 = cyclobutyl) in complex with BRD4-BD1 shows that additional hydrophobic interactions are formed by the R1-group and the side chains of Trp81 and Pro82. Such interactions are further stabilized by Iso146 as well as the aniline tail of the compound, as was observed in the XMD17–26 structure (R1 = cyclopentyl, Figure 2C). However, such hydrophobic stacking interactions of the R1 group are not present in the ERK5/ XMD17–26 structure, leaving the ability to inhibit the kinases effectively unaltered when compared with simple acyclic alkyl R1-groups. Consistent with previously published results, a gradual decrease of inhibitory activity against LRRK2 was observed upon increasing the size of the R1-group.19,20 Structural analysis of XMD11–50 with Roco4 kinase shows that the methyl group on R1 is accommodated by a small hydrophobic pocket formed by Leu99, Val107, and is very close to the P-loop. Larger substitutions do not fit into this space, due to a steric clash with the P-loop. However, in ERK5, the P-loop swings upward and opens this space, enabling accommodation of larger substituents (Figure 3B). These structures provide insight into why LRRK2 activity is changed in response to substitution at the R1 position, whilst there is no change to ERK5 activity.
The observations i) that the N-methyl amide group of the diazipinone ring fills the hydrophobic cavity of BRD4-BD1 and ii) that the ACK probe XMD8–87 which lacks a methyl group at this position shows much weaker inhibition of BRD4-BD1 encouraged us to explore the effect of N-methylation at the R3 position. Four pairs of compounds (R2=Me/H) with different R1 substitution were first compared (Supplemental Table 2). Compounds with R2=H show lower affinities for both BRD4 and ERK5 with the exception of ACK. The same phenomenon was observed for XMD11–50 (LRRK2 probe), XMD12–1 (Aurora A probe) and their pairs. These activity patterns indicate that the N-methyl amide pharmacophore is essential for both BRD4 and ERK5 binding. JWG-112, the hydrogen analog of the selective ERK5 inhibitor JWG-071, was also synthesized and tested. As expected, inhibitory activity toward both BRD4 and ERK5 decreased dramatically.
The ligand bound BRD4 structures described above show that the methoxyphenyl moiety (R3) participates in bromodomain binding, and the kinase structures Roco4/XMD11–50 and ERK5/XMD17–26 also show direct interaction between the kinase and this moiety. Therefore, we next investigated how substitution at the R3 position modulates protein selectivity (Table 2). Compounds with acyclic alkyl (JWG-046, JWG049, and JWG-114) or cycloalkyl substituents (XMD17–26, XMD17–109, and JWG-047) at the R1 position have been tested. Increasing the size of R3 slightly strengthened binding to BRD4, while decreasing inhibition of ERK5 and LRRK2. The structure of JWG-047 with BRD4-BD1 revealed a small shift at the isopropyloxyphenyl moiety compared to the methoxyphenyl group of XMD17–26, resulting in an additional structural water participating in the ligand-protein interaction and increased van de Waals interactions between the R3 isopropyl group and Gln85 (Figure 3C). We did not explore R3 substitution further because of its modest effect on binding. Ultimately, the variation of substituents at the three tested positions resulted in a dual kinase-bromodomain inhibitor JWG-047 with BRD4 activity comparable to JQ1 (2.2 – 2.5 fold less active than JQ1).
Finally, we used the structural information to design BRD4 selective inhibitors by modifying the scaffold. Since the nitrogen atom at the 3-position of the pyrimidine is known to be critical for kinase activity by forming a key hydrogen bond with the kinase hinge region, substituting the pyrimidine for a pyridine ring should result in a compound with much lower kinase activity and this is likely to hold true across the kinome, rather than as a LRRK2/ERK5 specific effect.21,32 As expected, both DFCI-2–208 and JWG-115 lost activity against the kinase targets. However, their activity against BRD4 also decreased 4–8 fold compared to the pyrimidine analogs XMD11–50 and JWG-047, demonstrating pyrimidine core is favorable even for BRD binding (Table 2). To assess the overall bromodomain selectivity, two sample compounds, the BRD selective compound JWG-115 and triple-targets inhibitor JWG-047 were first screened against a panel of 32 bromodomains that comprised representative members of all subfamilies at a concentration of 10 μM (Bromoscan from DiscoverX) (Supplemental Figure 5). Both compounds showed strong binding among BET family and no binding outside BET family. The seven BRD4 active analogs in Table 2 were also tested by AlphaScreen assay against BRD2(1), BRD3(1), BRDT(1), TRIM24/TIF1A (group V), TRIM33 (group V), and SMARCA4 (group VIII) (Supplemental Table 3). Most compounds show similar activity against BRD4 (1) and BRD3 (1), lower activity against BRD2 (1) and BRDT (1), and no activity on other bromodomain group members, in agree with BROMOscan screening result. JWG-115 therefore is a modestly potent (1 μM of IC50) BET-selective probe against the tested kinases ERK5/LRRK2.
Different atropisomeric conformers bound to BRD4 and kinases.
Strikingly, when comparing kinase and BRD4 co-crystal structures, we observed that both XMD11–50 and XMD17–26 exhibited different conformations when bound to BRD4 than when bound to kinase targets (Figure 4A, 4B, PDB: 4YZM and 4B99). These conformers arise due to the axial chirality about the C-N bond in the rotationally hindered 7-membered ring (Figure 4C). In the kinase structures the (Ra) conformer is observed, whereas in the BRD4 structures solved here the (Sa) conformer is observed.
Figure 4.

View showing different conformers bound to BRD4 and Kinase; (A) XMD11–50 bound to BRD4-BD1 (upper panel) and Roco4, pdb code 4YZM (lower panel). (B) XMD17–26 bound to BRD4-BD1 (upper panel) and ERK5, pdb code 4B99 (lower panel). (C) Axis of chirality can rotate to generate two different conformations of the tricyclic ring system.
As we did not observe separation of entries 1 – 6, Table 2, by chiral HPLC separation, we determined t1/2 rac using NMR spectroscopy, in order to characterize whether these molecules exist as conformers or separable atropisomers. When the half-life (t1/2) for interconversion of the two conformations is greater than 1000 seconds at room temperature, which corresponds to an interconversion energy barrier greater than 20 kcal/mol, the molecules exist as a mixture of two distinct atropisomers and each atropisomer can, in theory, be isolated. 33,34 In this case, each atropisomer should be evaluated separately for its ability to bind to BRD4, ERK5 and LRRK2. We anticipated the t1/2 in this series to be influenced by the steric bulk of R1. In order to generate diasteromers that exhibit distinct 1H NMR resonances which would enable characterization of the t1/2 of racemization using variable temperature 1H NMR experiments we synthesized the corresponding (−)-menthol derivatives of R1 variants. Considering menthol and methyl-4-(piperidine-4-yl) piperazine moieties at the tail position are likely to have little effect on the 7-membered ring conformation, we hypothesized that the parent molecules have similar interconversion temperatures as (−)-menthol derivatives. JWG-071 (R1 = sec-butyl) contains a stereocenter and the potential for four diastereomers. In this molecule we could directly observe two sets of NMR signals, therefore we could measure the coalescence temperature directly (60 oC) without needing to resort to the preparation of chiral menthol esters.
At room temperature, JWG-088 and JWG-091 exhibited only a single set of NMR resonances indicating that they are achiral compounds whose conformers interconvert rapidly. Once R1 was increased, the compounds exhibited two sets of resonances with equal abundance (JWG-082, JWG-089, and JWG-090). Hence, we performed variable temperature 1H NMR experiments from 25 °C to 120 °C in DMSO-d6 (Supplemental Spectra) and observed that the coalescence temperatures of isopropyl, cyclobutyl, and cyclopentyl analogs were 47 °C, 100 °C and 68 °C respectively (Table 3 and Supplemental Spectra). During the course of this work, another group reported modification of XMD17–109 to generate AX15836 that possess excellent ERK5 activity and selectivity against BRD4.26 We independently synthesized and confirmed the activity and selectivity of this inhibitor (Table 2). We also synthesized the corresponding menthol ester JWG-098 (R1 = methylsulfonyl) and observed its coalescence temperature to be 120 °C (Table 3).
Table 3.
Relationship between R1 and observed coalescence temperature, calculated half-life, and energy barrier of interconversion.
| Compound ID | Corresponding parent molecule | Structure | R1 | Tco (°C) | Calc t1/2 rac(s) | Calc ΔErac (kcal/mol) |
|---|---|---|---|---|---|---|
| JWG-088 | XMD11–50 (LRRK2-IN-1) | 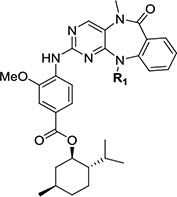 |
Me | <25 | - | - |
| JWG-091 | JWG-048 | Et | <25 | - | - | |
| JWG-082 | JWG-046 | i-Pr | 47 | 0.07 | 16.3 | |
| JWG-089 | JWG-069 | cyclobutyl | 100 | 2.66 | 18.4 | |
| JWG-090 | XMD17–26 | cyclopentyl | 68 | 0.35 | 17.2 | |
| JWG-098 | AX15836 | −SO2Me | 120 | 31.0 | 19.9 | |
| JWG-071 | As in table 2 | s-Bu | 60 | 0.04 | 15.9 |
We next calculated the half-lives and energy barriers for interconversion of the compounds. As we expected, the R1 substituent affected the energy barrier and rate of interconversion between conformers, and this conclusion was supported by quantum mechanical analysis of the ground and transition states of the conformers of XMD11–50 and XMD17–26 (Table 3, Supplemental Figure 6). Nevertheless, all analogs had energy barriers lower than 20 kcal/mol, indicating that these analogs interconverted with half-lives measured in seconds and can therefore not be separated at room temperature.
In the absence of separable atropisomers, we used docking studies to explore the observed isomer preferences of kinases and bromodomains. Using the dual kinase-bromodomain targeting analogs XMD11–50 and XMD17–26, the Ra and Sa conformations of each compound were frozen and docked into apo BRD4 bromodomain and ERK5 kinase domain structures. For both compounds, low energy solutions were found for the Sa confomers docked into BRD4, but no solutions were found for the Ra conformer. Similarly, low energy solutions were found for the Ra conformer docked into ERK5, but no solutions were found for the Sa confomer, indicating that the observed preferences in crystallography are due to differing binding affinities of the conformers for each target (Supplemental Figure 7).We anticipate that compounds that introduce further steric constraints to the diazepine core might possess atropisomers that could be separated. In contract to JWG-071, JWG-112 displays only one set of NMR resonances at room temperature, suggesting substitution at R2 position also increases steric hindrance and can be a further modification spot. Our computational analysis indicates that separable atropisomers may display kinase selectivity (Ra) or BRD selectivity (Sa) dependent on their chirality.
Conclusions.
In this paper, we identify a new class of dual kinase-bromodomain inhibitors with a shared hetero-tricyclic core. Through SAR studies and complementary structural studies, we defined a blueprint for tuning in or out kinase or bromodomain activity (Figure 5). This resulted in identification of an ERK5 selective chemical probe (JWG-071) and a BRD4 selective probe with 1 μM BRD4 IC50 (JWG-115) as well as inhibitors with rationally designed polypharmacology (JWG-047, JWG-069). We also observed different conformational isomers in bromodomain vs kinase X-ray structures, leading to investigation of the atropisomeric properties of this scaffold. Atropisomerism has previously been exploited to improve the subfamily selectivity of kinase inhibitors.35 In this work we report the first example of different protein target classes distinguishing between atropisomeric conformers. Since the 7-membered diazepine ring is widely used in drug development and exists in natural products36, this newly discovered contribution of atropisomers to protein target selectivity is a highly relevant topic warranting further investigation. Future work will focus on synthesis of enantiomerically pure analogs with slower interconversion rates to evaluate the binding ability to BRD4 and kinases of each isomer.
Figure 5.

SAR rules for benzo[e]pyri(mi)do-[5,4-b]diazepine-6(11H)-one activity against BRD4 and ERK5 targets.
METHODS
Protein Preparation.
The first bromodomain of Brd4 (BRD4-BD1, residues 42–168) was subcloned into a modified pET-15b(+) vector with an N-terminal His6 tag followed by a Tobacco Etch Virus cleavage site. The encoded protein was expressed in the E. coli BL21(DE3) strain. E. coli cells transformed with the vector were grown 4 hours at 37 °C to an OD600 of 1.0, in 1 liter LB media containing ampicillin (0.1 mg/ml). After 0.1 mM IPTG induction at 20 °C, cells were cultured overnight, and collected by centrifugation at 4,000 g. The pellet was suspended in 40 ml lysis buffer containing 50 mM HEPES, pH 7.4, 500 mM NaCl, 5 mM β-mercaptoethanol, and 1 mM PMSF. Cells were lysed on ice by sonication and cell debris was precipitated by centrifugation at 15,000 g for 30 minutes. BRD4-BD1 was purified by affinity chromatography on an Ni-NTA agarose column (Qiagen), using an elution buffer of 50 mM HEPES, pH 7.4, 500 mM NaCl, 5 mM β mercaptoethanol, and 250 mM of imidazole. After overnight TEV cleavage of the His6-tag, the cleaved tag was captured on affinity resin, and BRD4-BD1 was then purified to homogeneity by gel filtration before concentration to roughly 15 mg/ml in storage buffer (25mM HEPES, pH 7.4, and 150 mM NaCl).
Bromodomain BRD4–1 AlphaScreen binding assay in Dana-Farber Cancer Institute.
Assays were performed with minor modifications from the manufacturer’s protocol (Perkin Elmer, USA). All reagents were diluted in AlphaScreen™ buffer (50 mM HEPES, 150 mM NaCl, 0.01% v/v Tween-20, 0.1% w/v BSA, pH = 7.4). After addition of Alpha beads to master solutions, all subsequent steps were performed under low light conditions. A 2x solution of components with final concentrations of His-Brd4(1) at 0.020 μM, Ni-coated Acceptor Bead at 10 μg/ml, and biotinylated-(+)-JQ1 at 0.010 μM were added in 10 μL to 384-well plates (AlphaPlate-384, PerkinElmer) using an EL406 liquid handler (Biotek, USA). Plates were spun down at 1000 rpm. A 10-point 1: 3 serial dilution of compounds in DMSO was prepared at 200x the final concentration. 100 nL of compound from these stock plates were added by pin transfer using a Janus Workstation (PerkinElmer). A 2x solution of streptavidin-coated donor beads with a final concentration of 10 μg/ml was added in a 10 μL volume. The plates were spun down again at 1000 rpm and sealed with foil to prevent light exposure and evaporation. The plates were then incubated at room temperature for 1 h and read on an Envision 2104 (PerkinElmer) using the manufacturer’s protocol. IC50 values were calculated using a 4-parameter logistic curve in Prism 6 (GraphPad Software, USA) after normalization to DMSOtreated negative control wells (0.2% DMSO, v/v).
Bromodomain BRD4–1 AlphaScreen binding assay in University of Oxford.
Assays were performed as described previously with minor modifications from the manufacturer’s protocol (PerkinElmer, USA). All reagents were diluted in 25 mM HEPES, 100 mM NaCl, 0.1 % BSA, pH 7.4 and 0.05 % CHAPS and allowed to equilibrate to room temperature prior to addition to plates. A 11-point 1:2.0 serial dilution of the ligands was prepared on low-volume 384-well plates (ProxiPlateTM384 Plus, PerkinElmer, USA) using LabCyte Eco liquid handler. Plates filled with 5 uL of the assay buffer followed by 7 uL of biotinylated peptide [H-YSGRGKacGGKacGLGKac-GGAKacRHRK(Biotin)-OH and His-tagged protein to achieve final assay concentrations of 25 nM. Plates were sealed and incubated for a further 60 minutes, before the addition of 8 μl of the mixture of streptavidin-coated donor beads (12.5 μg/ml) and nickel chelate acceptor beads (12.5 μg/ml) under low light conditions. Plates were foil-sealed to protect from light, incubated at room temperature for 60 minutes and read on a PHERAstar FS plate reader (BMG Labtech, Germany) using an AlphaScreen 680 excitation/570 emission filter set. IC50 values were calculated in Prism 6 (GraphPad Software, USA) after normalization against corresponding DMSO controls and are given as the final concentration of compound in the 20 μl reaction volume.
Isothermal Titration Calorimetry.
Experiments were performed using a MicroCal ITC200 calorimeter (GE Healthcare). All experiments were carried out at 25 °C with1000 RPM stirring speed, in a buffer containing 25mM HEPES pH7.5 and 100 mM NaCl. The injection syringe was loaded with a solution of the protein sample at 200 uM concentration and the cell was filled with compound at 20uM concentration. Single binding site model in Origin software (OriginLab, Northampton MA USA) was employed to calculate dissociation constants and thermodynamic parameters.
Protein Stability Assay.
Differential scanning fluorimetry (DSF) was used to measure stabilization of BRD4-BD1 at 2 μM concentration in the presence of 10 μM compound in a volume of 20 μl. SYPRO Orange (Molecular Probes) was included (diluted 1:1000) as the fluorescence probe, with excitation and emission wavelength set to 465nm and 590nm, respectively. Fluorescence measurements were taken regularly over a thermal gradient from 25 °C to 95 °C using a ramp rate of 3 °C per minute. The data were plotted using non-linear least squares fitting, and temperature shifts were calculated as the difference between the transition midpoints of the protein with and without compounds.
ERK5 kinase activity in vitro assay.
A 40 μL reaction mixture was prepared containing 200 ng of pure active ERK5 and the indicated amount of inhibitor in 50 mM Tris-HCl, pH 7.5, 0.1 mM EGTA, 1 mM 2-mercaptoethanol. The reaction was initiated by adding 10 mM magnesium acetate, and 50 μM [γ32P]-ATP (500 cpm/pmol) and 200 μM PIMtide (ARKKRRHPSGPPTA) as substrates. Assays were carried out for 20 min at 30 oC, terminated by applying the reaction mixture onto p81 paper and the incorporated radioactivity measured as described previously. The expression and purification of ERK5 was carried out as described previously. 37
LRRK2 kinase activity in vitro assay.
An in vitro assay of LRRK2 kinase activity was performed in Adapta kinase assay format at Thermo Fisher Scientific (Madison, WI) using the SelectScreen Kinase Profiling Service. All protocols are available from the Thermo Fisher Scientific website.
797 NMC Cell Viability Assay.
Cells were plated in a 96-well plate at a density of 5,000 cells/well and incubated for 3 days in the presence of various concentration of compounds or DMSO. Relative cell numbers were determined in 6 replicates using Cell Titer Glo (Promega, Madison, WI) according to the manufacturer’s instructions.
Cellular assay of ERK5.
Human HeLa cells were serum starved overnight followed by treatment with the indicated inhibitor for one hour. Cells were then stimulated with EGF (50 ng/mL) for 20 min and harvested in RIPA buffer (1X PBS, 1% NP40, 0.5% sodium deoxycholate, 0.1% SDS, 0.1 mg/ml PMSF and 1mM sodium orthovanadate). Proteins from total cell lysates were resolved by 6% sodium dodecyl sulfate (SDS)polyacrylamide gel electrophoresis (PAGE), transferred to nitrocellulose membrane, blocked in 5% nonfat milk, and blotted with anti-ERK5 antibody (Cell Signaling Technology, Boston, MA). Active ERK5 activity was calculated by monitoring the amount of retarded, autophosphorylated band in the immunoblots, as described elsewhere.
Crystallization and Data Collection.
BRD4-BD1 complexes were crystallized using the hanging drop vapor diffusion method at room temperature, using either co-crystallization or soaking methods. For soaking experiments, inhibitor-free crystals were first obtained in a drop with equal volumes of BD1 at 4 mg/ml and a precipitant solution containing 100mM sodium nitrate, 5% ethylene glycol, and 18% (w/v) PEG3350, as precipitant. Rod-like crystals obtained in 10 days were transferred and soaked in the same crystallization buffer pulsing compounds at 0.5 mM concentration for 5–7 days. For co-crystallization experiments, protein was first incubated with compounds at molar ratio of 1:3 and then mixed with the same precipitant solution as in soaking experiments. For data collection, a single crystal was picked and flash-frozen with a precipitant solution containing 20% (v/v) glycerol. Diffraction data for each complex were collected from a flash-cooled crystal at 100 °K using the NE-CAT 24ID-C or 24ID-E beam lines at Argonne National Laboratory. Data were processed, integrated, and scaled using HKL2000 (Otwinowski, Z., and Minor, W. (1997)).
Structure determination and refinement.
Structures of BRD4-BD1 complexes were solved by molecular replacement (MR) in Phenix38 using the BD1-UMB32 structure (PDB 4WIV) as a search model. Structure refinement was carried out using conjugate-gradient energy minimization, torsion-constrained molecular dynamics simulated annealing, group B factor refinement, and individual B factor refinement protocols in Phenix with 5% of the reflections omitted for free R factor calculation. Electron density peaks above 3σ in difference Fourier maps were assigned to water molecules in later refinement stages if they had reasonable geometry in relation to hydrogen bond donors or acceptors and their B-factors did not rise above 50 Å2 during subsequent refinement. Model building was performed in Coot guided by σA-weighted 2Fo-Fc and Fo-Fc maps and composite omit maps.
Kinome Profiling.
Kinome profiling was performed using KinomeScan ScanMAX at compound concentration of 1μM. Data was reported in Supplemental data. Protocols are available from DiscoverX.
Bromodomain Profiling.
Bromodomain profiling was performed using BROMOscan at compound concentration of 10μM. Data was reported in Supplemental data. Protocols are available from DiscoverX.
Supplementary Material
ACKNOWLEDGMENTS
We thank P. Cohen for helpful discussion. We also thank E. Megias for technical support.
Funding Sources
This work was supported by the NIH LINCS Program grant U54HL127365 (to N. S. Gray and J. Wang), NIH P50 GM107618 (to X. Xu and S. C. Blacklow), NIH U54 HD093540 and P01 CA066996 (to J. Qi), Medical Research Council MC_UU_12016/2 (to D. R. Alessi), the Spanish Ministerio de Economia y Competitividad (MINECO) grant SAF2015-60268R (to J.M. Lizcano) and co-funded by Fondo Europeo de Desarrollo Regional (FEDER) funds. D.L. Buckley was supported as a Merck Fellow of Damon Runyon Cancer Research Foundation (DRG-2196-14).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures, supporting figures, supporting tables; kinase selectivity profile of JWG-071; and variable temperature 1H NMR spectra (PDF)
REFERENCES
- (1).Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert J-P, Barsyte-Lovejoy D, Felletar I, Volkmer R, Müller S, Pawson T, Gingras A-C, Arrowsmith Cheryl H., and Knapp S (2012) Histone Recognition and Large-Scale Structural Analysis of the Human Bromodomain Family, Cell 149, 214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Winter GE, Mayer A, Buckley DL, Erb MA, Roderick JE, Vittori S, Reyes JM, di Iulio J, Souza A, Ott CJ, Roberts JM, Zeid R, Scott TG, Paulk J, Lachance K, Olson CM, Dastjerdi S, Bauer S, Lin CY, Gray NS, Kelliher MA, Churchman LS, and Bradner JE (2017) BET Bromodomain Proteins Function as Master Transcription Elongation Factors Independent of CDK9 Recruitment, Mol Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Yang Z, Yik JHN, Chen R, He N, Jang MK, Ozato K, and Zhou Q (2005) Recruitment of P-TEFb for Stimulation of Transcriptional Elongation by the Bromodomain Protein Brd4, Molecular Cell 19, 535–545. [DOI] [PubMed] [Google Scholar]
- (4).Itzen F, Greifenberg AK, Bosken CA, and Geyer M (2014) Brd4 activates P-TEFb for RNA polymerase II CTD phosphorylation, Nucleic Acids Res. 42, 7577–7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Jonkers I, and Lis JT (2015) Getting up to speed with transcription elongation by RNA polymerase II, Nat Rev Mol Cell Biol 16, 167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MG, Qi J, Rahl PB, Sun HH, Yeda KT, Doench JG, Reichert E, Kung AL, Rodig SJ, Young RA, Shipp MA, and Bradner JE (2013) Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma, Cancer Cell 24, 777–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Ott CJ, Kopp N, Bird L, Paranal RM, Qi J, Bowman T, Rodig SJ, Kung AL, Bradner JE, and Weinstock DM (2012) BET bromodomain inhibition targets both c-Myc and IL7R in high-risk acute lymphoblastic leukemia, Blood 120, 2843–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, and Young RA (2013) Selective inhibition of tumor oncogenes by disruption of super-enhancers, Cell 153, 320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Tanaka M, Roberts JM, Qi J, and Bradner JE (2015) Inhibitors of emerging epigenetic targets for cancer therapy: a patent review (2010–2014), Pharm Pat Anal 4, 261–284. [DOI] [PubMed] [Google Scholar]
- (10).Fiskus W, Sharma S, Qi J, Shah B, Devaraj SG, Leveque C, Portier BP, Iyer S, Bradner JE, and Bhalla KN (2014) BET protein antagonist JQ1 is synergistically lethal with FLT3 tyrosine kinase inhibitor (TKI) and overcomes resistance to FLT3-TKI in AML cells expressing FLT-ITD, Mol Cancer Ther 13, 2315–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Lee DH, Qi J, Bradner JE, Said JW, Doan NB, Forscher C, Yang H, and Koeffler HP (2015) Synergistic effect of JQ1 and rapamycin for treatment of human osteosarcoma, Int J Cancer 136, 2055–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Shu S, Lin CY, He HH, Witwicki RM, Tabassum DP, Roberts JM, Janiszewska M, Huh SJ, Liang Y, Ryan J, Doherty E, Mohammed H, Guo H, Stover DG, Ekram MB, Peluffo G, Brown J, D’Santos C, Krop IE, Dillon D, McKeown M, Ott C, Qi J, Ni M, Rao PK, Duarte M, Wu SY, Chiang CM, Anders L, Young RA, Winer EP, Letai A, Barry WT, Carroll JS, Long HW, Brown M, Liu XS, Meyer CA, Bradner JE, and Polyak K (2016) Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer, Nature 529, 413–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Ember SW, Zhu JY, Olesen SH, Martin MP, Becker A, Berndt N, Georg GI, and Schonbrunn E (2014) Acetyl-lysine binding site of bromodomain-containing protein 4 (BRD4) interacts with diverse kinase inhibitors, ACS Chem Biol 9, 1160–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ciceri P, Muller S, O’Mahony A, Fedorov O, Filippakopoulos P, Hunt JP, Lasater EA, Pallares G, Picaud S, Wells C, Martin S, Wodicka LM, Shah NP, Treiber DK, and Knapp S (2014) Dual kinase-bromodomain inhibitors for rationally designed polypharmacology, Nat Chem Biol 10, 305–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Chen L, Yap JL, Yoshioka M, Lanning ME, Fountain RN, Raje M, Scheenstra JA, Strovel JW, and Fletcher S (2015) BRD4 Structure-Activity Relationships of Dual PLK1 Kinase/BRD4 Bromodomain Inhibitor BI-2536, ACS Med Chem Lett 6, 764–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Koblan LW, Buckley DL, Ott CJ, Fitzgerald ME, Ember SW, Zhu JY, Liu S, Roberts JM, Remillard D, Vittori S, Zhang W, Schonbrunn E, and Bradner JE (2016) Assessment of Bromodomain Target Engagement by a Series of BI2536 Analogues with Miniaturized BET-BRET, ChemMedChem 11, 2575–2581. [DOI] [PubMed] [Google Scholar]
- (17).Miduturu CV, Deng X, Kwiatkowski N, Yang W, Brault L, Filippakopoulos P, Chung E, Yang Q, Schwaller J, Knapp S, King RW, Lee JD, Herrgard S, Zarrinkar P, and Gray NS (2011) High-throughput kinase profiling: a more efficient approach toward the discovery of new kinase inhibitors, Chem Biol 18, 868–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Roberts JM, and Bradner JE (2015) A BeadBased Proximity Assay for BRD4 Ligand Discovery, Curr Protoc Chem Biol 7, 263–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Deng X, Dzamko N, Prescott A, Davies P, Liu Q, Yang Q, Lee JD, Patricelli MP, Nomanbhoy TK, Alessi DR, and Gray NS (2011) Characterization of a selective inhibitor of the Parkinson’s disease kinase LRRK2, Nat Chem Biol 7, 203–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Deng X, Elkins JM, Zhang J, Yang Q, Erazo T, Gomez N, Choi HG, Wang J, Dzamko N, Lee JD, Sim T, Kim N, Alessi DR, Lizcano JM, Knapp S, and Gray NS (2013) Structural determinants for ERK5 (MAPK7) and leucine rich repeat kinase 2 activities of benzo[e]pyrimido[5,4-b]diazepine-6(11H)-ones, Eur. J. Med. Chem 70, 758–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Elkins JM, Wang J, Deng X, Pattison MJ, Arthur JS, Erazo T, Gomez N, Lizcano JM, Gray NS, and Knapp S (2013) X-ray crystal structure of ERK5 (MAPK7) in complex with a specific inhibitor, J. Med. Chem 56, 4413–4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Yang Q, Deng X, Lu B, Cameron M, Fearns C, Patricelli MP, Yates JR Iii, Gray NS, and Lee J-D (2010) Pharmacological Inhibition of BMK1 Suppresses Tumor Growth through Promyelocytic Leukemia Protein, Cancer Cell 18, 258–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Kwiatkowski N, Deng X, Wang J, Tan L, Villa F, Santaguida S, Huang HC, Mitchison T, Musacchio A, and Gray N (2012) Selective aurora kinase inhibitors identified using a taxol-induced checkpoint sensitivity screen, ACS Chem Biol 7, 185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Kwiatkowski N, Jelluma N, Filippakopoulos P, Soundararajan M, Manak MS, Kwon M, Choi HG, Sim T, Deveraux QL, Rottmann S, Pellman D, Shah JV, Kops GJ, Knapp S, and Gray NS (2010) Small-molecule kinase inhibitors provide insight into Mps1 cell cycle function, Nat Chem Biol 6, 359–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Sureban SM, May R, Weygant N, Qu D, Chandrakesan P, Bannerman-Menson E, Ali N, Pantazis P, Westphalen CB, Wang TC, and Houchen CW (2014) XMD8–92 inhibits pancreatic tumor xenograft growth via a DCLK1-dependent mechanism, Cancer Lett 351, 151–161. [DOI] [PubMed] [Google Scholar]
- (26).Lin EC, Amantea CM, Nomanbhoy TK, Weissig H, Ishiyama J, Hu Y, Sidique S, Li B, Kozarich JW, and Rosenblum JS (2016) ERK5 kinase activity is dispensable for cellular immune response and proliferation, Proc Natl Acad Sci U S A 113, 11865–11870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Montero JC, Ocana A, Abad M, Ortiz-Ruiz MJ, Pandiella A, and Esparis-Ogando A (2009) Expression of Erk5 in early stage breast cancer and association with disease free survival identifies this kinase as a potential therapeutic target, PLoS One 4, e5565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Sticht C, Freier K, Knopfle K, Flechtenmacher C, Pungs S, Hofele C, Hahn M, Joos S, and Lichter P (2008) Activation of MAP kinase signaling through ERK5 but not ERK1 expression is associated with lymph node metastases in oral squamous cell carcinoma (OSCC), Neoplasia 10, 462470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Hayashi M, Fearns C, Eliceiri B, Yang Y, and Lee JD (2005) Big mitogen-activated protein kinase 1/extracellular signal-regulated kinase 5 signaling pathway is essential for tumor-associated angiogenesis, Cancer Res 65, 7699–7706. [DOI] [PubMed] [Google Scholar]
- (30).Vidler LR, Filippakopoulos P, Fedorov O, Picaud S, Martin S, Tomsett M, Woodward H, Brown N, Knapp S, and Hoelder S (2013) Discovery of novel smallmolecule inhibitors of BRD4 using structure-based virtual screening, J. Med. Chem 56, 8073–8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).French CA, Miyoshi I, Kubonishi I, Grier HE, Perez-Atayde AR, and Fletcher JA (2003) BRD4-NUT fusion oncogene: a novel mechanism in aggressive carcinoma, Cancer Res 63, 304–307. [PubMed] [Google Scholar]
- (32).Gilsbach BK, Messias AC, Ito G, Sattler M, Alessi DR, Wittinghofer A, and Kortholt A (2015) Structural Characterization of LRRK2 Inhibitors, J. Med. Chem 58, 3751–3756. [DOI] [PubMed] [Google Scholar]
- (33).Laplante SR, L, Fandrick KR, Fandrick DR, Hucke O, Kemper R, Miller SP, and Edwards PJ (2011) Assessing atropisomer axial chirality in drug discovery and development, J. Med. Chem 54, 7005–7022. [DOI] [PubMed] [Google Scholar]
- (34).LaPlante SR, Edwards PJ, Fader LD, Jakalian A, and Hucke O (2011) Revealing atropisomer axial chirality in drug discovery, ChemMedChem 6, 505–513. [DOI] [PubMed] [Google Scholar]
- (35).Smith DE, Marquez I, Lokensgard ME, Rheingold AL, Hecht DA, and Gustafson JL (2015) Exploiting Atropisomerism to Increase the Target Selectivity of Kinase Inhibitors, Angew. Chem. Int. Ed. Engl 54, 11754–11759. [DOI] [PubMed] [Google Scholar]
- (36).Smyth JE, Butler NM, and Keller PA (2015) A twist of nature - the significance of atropisomers in biological systems, Nat. Prod. Rep 32, 1562–1583. [DOI] [PubMed] [Google Scholar]
- (37).Erazo T, Moreno A, Ruiz-Babot G, RodriguezAsiain A, Morrice NA, Espadamala J, Bayascas JR, Gomez N, and Lizcano JM (2013) Canonical and kinase activity-independent mechanisms for extracellular signal-regulated kinase 5 (ERK5) nuclear translocation require dissociation of Hsp90 from the ERK5-Cdc37 complex, Mol Cell Biol 33, 1671–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, and Zwart PH (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution, Acta Crystallogr D Biol Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.