

Abstract
As the closest living relatives of animals, choanoflagellates offer unique insights into animal origins and core mechanisms underlying animal cell biology. However, unlike traditional model organisms, such as yeast, flies, and worms, choanoflagellates have been refractory to DNA delivery methods for expressing foreign genes. Here we report a robust method for expressing transgenes in the choanoflagellate Salpingoeca rosetta, overcoming barriers that have previously hampered DNA delivery and expression. To demonstrate how this method accelerates the study of S. rosetta cell biology, we engineered a panel of fluorescent protein markers that illuminate key features of choanoflagellate cells. We then investigated the localization of choanoflagellate septins, a family of GTP-binding cytoskeletal proteins that are hypothesized to regulate multicellular rosette development in S. rosetta. Fluorescently tagged septins localized to the basal poles of S. rosetta single cells and rosettes in a pattern resembling septin localization in animal epithelia. The establishment of transfection in S. rosetta and its application to the study of septins represent critical advances in the use of S. rosetta as an experimental model for investigating choanoflagellate cell biology, core mechanisms underlying animal cell biology, and the origin of animals.
INTRODUCTION
First described in the mid-nineteenth century, choanoflagellates inspired great debate regarding animal taxonomy (James-Clark, 1868; Kent, 1871; Leadbeater, 2015). The most diagnostic morphological feature of choanoflagellates, a “collar complex” composed of a single apical flagellum surrounded by a collar of actin-filled microvilli (Figure 1), was interpreted as evidence of a special relationship between choanoflagellates and sponges, whose choanocytes (or “collar cells”) each bear a collar complex. Subsequent phylogenetic analyses and the discovery of cells with a collar complex in nearly all animal phyla have revealed that sponges and all other animals are monophyletic, with choanoflagellates as their closest living relatives (Figure 1; Lang et al., 2002; King et al., 2008; Ruiz-Trillo et al., 2008; Burger et al., 2003; Fairclough et al., 2013; Brunet and King, 2017). Moreover, comparative genomic analyses have revealed that choanoflagellates, animals, and other holozoans express genes required for animal multicellularity and embryogenesis (Sebe-Pedros et al., 2017; Richter et al., 2018), including cadherins (Abedin and King, 2008), tyrosine kinases (Manning et al., 2008; Suga et al., 2012), and Myc (Young et al., 2011). Thus, comparisons between animals and choanoflagellates have the potential to provide unique insights into animal origins and core features of animal cell biology that are not conserved in other experimental models, such as yeast (King, 2004).
FIGURE 1:
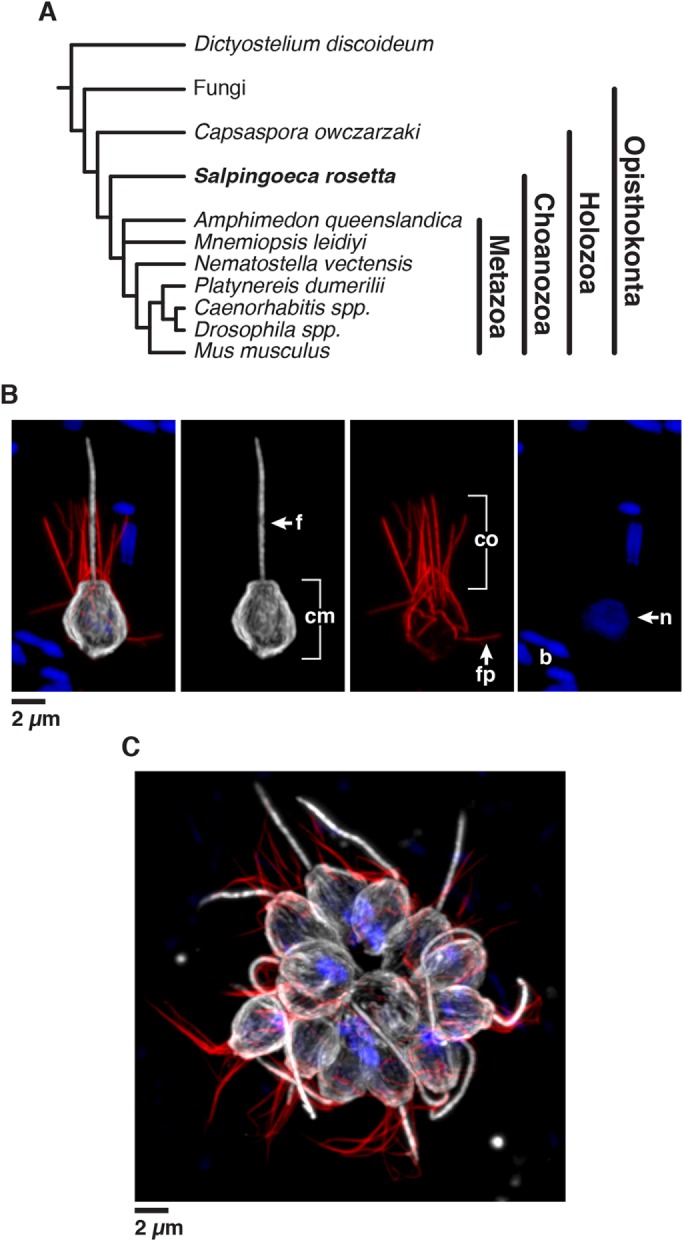
Introduction to Salpingoeca rosetta, an experimentally tractable model choanoflagellate. (A) S. rosetta and other choanoflagellates are the closest living relatives of animals (Metazoa), which together with animals comprise the clade Choanozoa. (B, C) S. rosetta has a complex life history that includes single cells (B) and multicellular rosettes (C). Immunofluorescence in fixed, permeabilized single cells (B) highlights the diagnostic cellular architecture of choanoflagellates, including a single apical flagellum (f) made of microtubules (white) surrounded by a collar (co) filled with F-actin (red) of microvilli. Staining for tubulin also illuminates cortical microtubules (cm) that run in parallel tracks along the cell periphery from the apical to the basal poles of each cell. DNA staining (blue) highlights the choanoflagellate nucleus (n) and the nucleoids of bacterial prey (b) present in choanoflagellate cultures. In multicellular rosettes (C, stained as in B), the basal poles of cells are oriented toward the interior of the rosette and the apical flagella point outward.
The choanoflagellate Salpingoeca rosetta—previously named Proterospongia sp. (King et al., 2003)—has recently emerged as an experimentally tractable model. S. rosetta develops from a single founding cell into a spherical, multicellular “rosette” (Figure 1C) through serial rounds of cell division in a process that evokes the earliest stages of animal embryogenesis (Fairclough et al., 2010). Since the establishment of the first S. rosetta cultures almost 20 years ago, S. rosetta has become increasingly amenable to cell and molecular biological approaches due to the sequencing of its genome (Fairclough et al., 2013), the establishment of forward genetic screens (Levin and King, 2013; Levin et al., 2014), the ability to experimentally control key events in its life history (Dayel et al., 2011), and the discovery that environmental bacteria induce multicellular rosette development and mating (Dayel et al., 2011; Alegado et al., 2012; Woznica et al., 2016, 2017).
An important remaining barrier to the study of molecular and cellular mechanisms in S. rosetta has been the inability to perform transfection and transgene expression. Furthermore, the absence of the RNA interference pathway in S. rosetta has precluded gene knockdowns (Fairclough et al., 2013; Richter et al., 2018). Here we report the establishment of a robust nucleofection-based method of transfecting and expressing transgenes in S. rosetta. By engineering plasmids with S. rosetta regulatory sequences driving the expression of fluorescently tagged S. rosetta proteins, we have developed a broad panel of markers for the study of choanoflagellate cell biology in vivo. As a first application, we used transgene expression to characterize septins, genes with conserved roles in fungal (Helfer and Gladfelter, 2006; Berepiki and Read, 2013) and animal development (Neufeld and Rubin, 1994; Adam et al., 2000; Kim et al., 2010; Fairclough et al., 2013; O’Neill and Clark, 2013) that have been hypothesized to regulate rosette development (Fairclough et al., 2013). By imaging fluorescently tagged septins in live cells, we show that their localization in S. rosetta resembles that in animal epithelia, providing a potential evolutionary link between the mechanisms underlying animal and choanoflagellate multicellularity.
RESULTS
A robust method for transfecting S. rosetta
To detect successful transfection, we started by engineering four different DNA plasmid constructs, each with different S. rosetta regulatory sequences fused to a gene, nanoluc (Hall et al., 2012), encoding a highly sensitive luciferase (Supplemental Figure S1B). Because no choanoflagellate promoters had previously been mapped, we increased the likelihood of cloning sequences that would drive robust gene expression by fusing nanoluc to noncoding sequences flanking a set of genes—elongation factor L (efl), α-tubulin (tub), non-muscle actin (act), and histone H3 (H3)—that each exhibit high expression, lack introns in their open reading frames, and have well-annotated 5′- and 3′-untranslated regions (Supplemental Figure S1A; Fairclough et al., 2013).
Next, we set out to deliver these DNA plasmid constructs into S. rosetta cells using nucleofection, an electroporation-based technique that has proven particularly effective for transfection of diverse eukaryotes (Janse et al., 2006b; Caro et al., 2012; Vinayak et al., 2015), including mammalian primary cells that are resistant to transfection (Gresch et al., 2004; Hamm et al., 2002). To quantify transfection efficiency, we performed luciferase assays on cell lysates. Through many trials, we eventually achieved a low level of transfection with nucleofection by improving conditions for culturing S. rosetta cells (Supplemental Figure S2), modifying approaches for handling cells throughout the nucleofection procedure (Supplemental Information), and screening 30 unique combinations of electrical pulses and buffers (Supplemental Figure S3).
Optimization around these initial conditions culminated in a procedure that provided robust and reproducible transfection of S. rosetta (Figure 2A; Materials and Methods and www.protocols.io/groups/king-lab). When used in the optimized transfection procedure, all four transfection reporters drove strong expression of nanoluc protein, producing luminescence signals that were more than three orders of magnitude above the detection limit (Figure 2B).
FIGURE 2:

Robust procedure for transfecting S. rosetta. (A) Summary of the stepwise procedure to transfect S. rosetta with DNA plasmids. To prepare S. rosetta for transfection, cells were harvested at mid–log phase and then washed to remove bacteria (depicted as gray ovals). S. rosetta cells (depicted with an apical collar, flagellum, and nucleus; n) were primed for nucleofection (step 1) through incubation in a buffer that degrades extracellular material. A DNA plasmid encoding a highly sensitive luciferase, nanoluc, or a fluorescent protein was then transfected into the nucleus with a nucleofector (step 2). Immediately after transfection, the cells rested in a buffer that promotes membrane closure (step 3). Finally, the cells were transferred into 1× High Nutrient Medium prepared with AK seawater for 2 d (step 4) before we assayed the expression of nanoluc or fluorescent proteins from the transfected DNA. (B) Plasmids with noncoding DNA sequences flanking the coding sequences for S. rosetta elongation factor L (pEFL), α-tubulin (pTub), β-actin (pAct), and histone H3 (pH3) genes drive the expression of a codon-optimized nanoluc reporter gene. pEFL-nanoluc, pTub-nanoluc, pAct-nanoluc, and pH3-nanoluc reporter plasmids (2.5 µg) were each transfected into S. rosetta, and the cells were subsequently assayed for luciferase expression. Each reporter produced a luminescence signal that was at least three orders of magnitude greater than the detection limit (dotted line) and significantly greater (one-way analysis of variance, p < 0.001) than the background from a negative control, in which cells were transfected with an empty pUC19 vector (None). See Materials and Methods for details on replicates and statistical tests. (C) Systematically omitting each step of the transfection procedure revealed critical steps for the delivery and expression of plasmid DNA in S. rosetta cells. As a baseline for comparison, cells with 2.5 µg of pH3-nanoluc reporter (row b) produced a luciferase signal that was three orders of magnitude greater than the background detected from cells transfected without the reporter plasmid (row a). Omitting the priming step by incubating cells in artificial seawater instead of priming buffer (row c) decreased luciferase signal by over two orders of magnitude. Nucleofection without carrier DNA (row d) or the application of the CM156 electrical pulse (row e) resulted in a complete loss of luciferase signal, indicating that both were essential for successful transfection. Directly transferring cells to sea water after nucleofection instead of to a buffer that promotes membrane resealing during the rest step (row f) decreased the luciferase signal almost 10-fold. Finally, despite the fact that most prey bacteria were washed out prior to nucleofection, addition of fresh prey bacteria did not appear to be necessary. Supplementing transfected cells with fresh prey bacteria at the start of the recovery step had seemingly little effect on transfection success (row g), probably due to the persistence of a small number of live bacteria throughout the nucleofection procedure. (D, E) Fluorescent reporters mark transfected cells. Live cells transfected with a pAct-mWasabi reporter construct could be observed by fluorescence microscopy (D) and quantified by flow cytometry (E). Untransfected cells were used to draw a gate that includes 99.99% of cells, or four SDs above the mean fluorescence value (left). The same gate was applied to a population of transfected cells (right) to categorize the mWasabi- population. Cells with higher values of green fluorescence that lay outside of the mWasabi- gate are categorized as mWasabi+. The transfection efficiency, as quantified by three independent flow cytometry experiments, was ∼1% in a population of 1 million cells.
Because this was the first example, to our knowledge, of successful transgene expression in any choanoflagellate, we sought to identify which steps in the optimized protocol were most essential. Using the pH3-nanoluc transfection reporter, we quantified how the omission of each step impacted transfection efficiency (Figure 2C). In addition to the use of an optimal electrical pulse during nucleofection, the two most important steps were priming the cells by degrading the extracellular matrix prior to nucleofection (Figure 2A, step 1; Supplemental Figure S4) and the inclusion of carrier DNA during nucleofection (Figure 2A, step 2; Supplemental Figure S5); eliminating either of these steps resulted in a nearly complete loss of signal.
Priming the cells for nucleofection was a novel step motivated by our observation that S. rosetta cells are surrounded by a potentially protective extracellular coat that might inhibit DNA transfection. Evidence for the extracellular coat came from the presence of genes in choanoflagellate genomes that appear homologous to genes that encode animal extracellular matrix proteins (King et al., 2008; Fairclough et al., 2013), the visualization of an extracellular coat with the lectin binding protein wheat germ agglutinin (Dayel et al., 2011), the basal secretion of a C-type lectin called Rosetteless into the interiors of rosettes as part of an extracellular matrix that may mediate intercellular adhesion (Levin et al., 2014), and historical descriptions of a jelly or hyaline coating covering choanoflagellate cells (Leadbeater, 2015). The buffer that primes cells for transfection uses a chaotrope, chelator, reducing agent, and protease to break down the extracellular coat before transfection (Supplemental Text, Supplemental Figure S4, and Supplemental Figure S10). The inclusion of carrier DNA (pUC19) in nucleofection reactions greatly reduced the amount of reporter plasmid required for detecting successful transfection (Supplemental Figure S5). Another improvement was the development of a recovery buffer that enhanced transfection 10-fold (Figure 2A, step 3), presumably by promoting membrane resealing after nucleofection (Rols and Teissie, 1989, 1990).
Luciferase assays performed on cell lysates gave a sensitive read-out of populationwide nanoluc expression but did not allow the examination of live, transfected cells nor reveal the proportion of cells that were successfully transfected. Therefore, we next engineered eight reporters with different fluorescent proteins placed under the control of regulatory sequences from an S. rosetta actin homologue. Fluorescence was readily detected from cells transfected with reporters encoding mTFP1 (Ai et al., 2006), mWasabi (Ai et al., 2008), sfGFP (Pedelacq et al., 2006), mNeonGreen (Shaner et al., 2013), mPapaya (Hoi et al., 2013), TagRFP-T (Shaner et al., 2008), mCherry (Shaner et al., 2004), and tdTomato (Shaner et al., 2004). In contrast, an eGFP (Yang et al., 1996) reporter failed to yield fluorescent cells, likely due to protein misfolding, as cells transfected with the “super-folder” variant of GFP (sfGFP) did fluoresce properly. In transfected cells, the fluorescent signal was distributed throughout the nucleus and cytosol, yet excluded from membrane-bound compartments (Figure 3A).
FIGURE 3:

Fluorescent markers illuminate the cell biology of S. rosetta in live cells. Fluorescent subcellular markers expressed from reporter plasmids in live S. rosetta cells were constructed by fusing mCherry in frame to genes encoding localization peptides and proteins (Supplemental Datasets S1 and S3). Twenty-four hours after cotransfection of cells with 5 µg of a plasmid encoding a subcellular marker fused to the mCherry protein and 5 µg of a plasmid encoding untagged mTFP1 that served as a whole cell marker, live cells were visualized by superresolution microscopy with a Zeiss LSM 880 Airyscan. The variation in localization of the whole cell mTFP1 marker stems from cell-to-cell differences in the number and localization of vacuoles, which exclude mTFP1. In panels A–I, the cells are oriented with the apical flagellum at the top and the nucleus, when included in the plane of focus (A″–F″), is indicated with a dotted white line. (A) Without localization signals (None), fluorescent proteins (mCherry, A′, and mTFP1, A″) were distributed throughout the cell with a slight enrichment in the nucleus and complete exclusion from other membrane-bound compartments. (B, C) A fusion of mCherry to the carboxy terminus of Histone H3, B′, or the amino terminus of a simian virus 40 nuclear localization signal (NLS), C′, was confined to the nucleus, whereas mCherry fused to the carboxy terminus of elongation factor L (EFL; D) was excluded from the nucleus and restricted to the cytosol. (E) The endoplasmic reticulum (ER) was highlighted by fusing the signal sequence from Rosetteless (PTSG_03555) and an ER retention sequence (HDEL from PTSG_07223) to the amino and carboxy termini of mCherry, respectively. (F) The mitochondrial network was highlighted by fusing a targeting sequence from S. cerevisiae CoxIV to the amino terminus of mCherry. (G) A Lifeact peptide fused to the amino terminus of mCherry marked filamentous actin (F-actin) that forms filipodia (arrowhead) and actin filaments in the cell body that coalesce to form the collar (arrow). (H) Fusing mCherry to the amino terminus of α-tubulin highlighted parallel tracks of microtubules (arrowhead) that extended subcortically from the apical poles to the basal poles of cells and microtubules that emerged from the apical poles of the cell bodies to form the flagella. Flagella undulate rapidly in live cells and can be difficult to image in total; in this cell the most distal tip of the flagellum is captured in the plane of focus (arrow). (I) A plasma membrane marker constructed by fusing a geranyl-geranylation sequence (PTSG_00306) to the carboxy terminus of mCherry outlined the entire cell shape, including the collar, flagellum, and cell body. The membrane marker also weakly highlighted the Golgi (arrowhead). The food vacuole (asterisk) was often visualized due to autofluorescence from ingested bacteria or through accumulation of the fluorescent markers in the food vacuole, perhaps through autophagy. (J–L) Orthogonal views along the xy and xz axes from confocal micrographs showed fine details of cell architecture that were highlighted by transfecting cells with F-actin, microtubule, and plasma membrane markers fused to mCherry (magenta). In xz views, each cell is oriented with the flagellum facing toward the top of the micrograph; flagella appeared shorter and blurred because of the sigmoidal shape of the flagellar beat. Lifeact (J) and the plasma membrane (L) markers fused to mCherry showed the microvilli (arrowheads). (K) The α-tubulin-mCherry marker showed the subcortical tracks of microtubules at the cell periphery (arrowhead) and the microtubule organizing center (arrow).
We observed that transfected cells resembled untransfected cells in their shape, motility, and ability to propagate, indicating that transfection did not irreparably harm S. rosetta. Fluorescence persisted through multiple cell divisions, yet the diminishing signal in daughter cells indicated that transfection was transient (Supplemental Figure S6A). Importantly, using flow cytometry 1–2 d after transfection, we found that ∼1% of the population was reproducibly transfected, and fluorescence-activated cell sorting enriched this transfected cell population (Supplemental Figure S6B). This transfection frequency is comparable to high-frequency episomal transformation of the model yeast Saccharomyces cerevisiae, which ranges from 1 to 10% (Schiestl and Gietz, 1989; Kawai et al., 2010), and similar transfection frequencies are achieved in model apicomplexans (Janse et al., 2006a; Caro et al., 2012).
Fluorescent markers illuminate the cell architecture of S. rosetta
To demonstrate the versatility of the new method for transfection and simultaneously explore the cell biology of S. rosetta in vivo, we designed a set of fluorescent reporters to mark key features of S. rosetta cells: the nucleus, cytoplasm, collar, filopodia, flagellum, membrane, mitochondria, and endoplasmic reticulum (ER). For each fluorescent reporter, the mCherry gene was fused in-frame to S. rosetta DNA sequences encoding conserved proteins or peptides that localize to specific organelles or subcellular regions in yeast and mammalian cells. To benchmark each fluorescent marker, we compared its localization in transfected cells to cellular landmarks known from electron and immunofluorescence micrographs (Supplemental Figure S7; Abedin and King, 2008; King et al., 2008, 2009; Sebe-Pedros et al., 2013; Leadbeater, 2015).
Electron micrographs have revealed two distinct regions in the nucleus: the darkly stained nucleolus positioned in the center and the surrounding, more lightly stained nucleoplasm (Burkhardt et al., 2014; Leadbeater, 2015). As predicted, mCherry fused to either the carboxy terminus of H3 or the amino terminus of the simian virus 40 nuclear localization signal localized primarily to the S. rosetta nucleoplasm and was excluded from the cytoplasm (Figure 3, B and C; Kalderon et al., 1984; Kanda et al., 1998). In contrast, the cytoplasmic marker EFL-mCherry (Huh et al., 2003) localized to the cytosol and was excluded from the nucleus (Figure 3D).
Two of the most diagnostic features of the choanoflagellate cell are the actin-filled collar and the flagellum, which is composed of microtubules (Karpov and Leadbeater, 1998). A fusion of mCherry to the filamentous actin-binding peptide Lifeact (Riedl et al., 2008) highlighted the parallel arrangement of straight microvilli in the collar (Figure 3, G and J), as well as filopodia extending from the basal pole of the cell (Figure 3G, bottom arrow; Karpov and Leadbeater, 1998; Sebe-Pedros et al., 2013). In live cells, Lifeact-mCherry revealed the native structure of the collar, which can be distorted in cells fixed for staining with fluorescent phalloidin or actin antibodies (Sebe-Pedros et al., 2013). Lifeact-mCherry also showed details of actin filament organization that have not previously been evident, such as the existence of actin filaments that originate in the cell body and coalesce at the base of the collar to form each microvillus (Figure 3G’, top arrow; improved immunofluorescence techniques also preserve these cortical actin filaments, Figure 1B). A fusion of α-tubulin to mCherry (Straight et al., 1997) illuminated individual cortical microtubules emanating from the base of the flagellum to the basal pole of the cell (Figure 3K’, arrow) and allowed visualization of the rapidly beating flagellum in live cells (Figure 3, H and K).
A cell membrane marker, with a geranyl-geranylation sequence fused to mCherry (Reid et al., 2004; Wang and Casey, 2016), outlined the entire cell, including the flagellum, collar, and cell body (Figure 3, I and L), and faintly marked the Golgi apparatus (Figure 3I’, arrow). In live cells, the cell membrane marker captured the formation of a phagocytic cup engulfing bacterial prey (Supplemental Figure S8; Dayel and King, 2014). The ER marker (Friedman et al., 2011), which included the amino terminal signal sequence from the secreted protein Rosetteless (Levin et al., 2014) and a carboxy terminal ER retention sequence from the ER resident chaperone BiP (PTSG_07223), highlighted the continuity of the ER with the nuclear envelope and the distribution of ER throughout the cell, including around vacuoles (Figure 3E). A mitochondrial marker (Friedman et al., 2011) with an amino terminal targeting sequence from S. cerevisiae Cytochrome C Oxidase, Subunit IV revealed a network of mitochondria (Nunnari et al., 1997) that is enriched around the nucleus and extends throughout the cell (Figure 3F). Taken together, these fluorescent markers demonstrate new experimental capabilities to rapidly tag proteins and to monitor their localization in distinct cellular compartments and locales.
Transgenesis reveals septin localization in live S. rosetta single cells and rosettes
A major motivation for establishing transgenics in S. rosetta was to rapidly characterize candidate genes for multicellularity. Therefore, we investigated the localization of septins, a family of paralogous genes hypothesized to contribute to multicellular development in S. rosetta (Fairclough et al., 2013). Multiple sequence alignment, structural modeling, and phylogenetic analyses previously demonstrated the homology of three predicted S. rosetta septins—PTSG_04106, PTSG_06009, and PTSG_07215—with animal and fungal septins in Groups 1A (mammalian Septin 3 family), 1B (mammalian Septin 6 family), and 4 (S. cerevisiae Cdc12 family), respectively (Pan et al., 2007; Fairclough et al., 2013). In fungi and animals, the assembly of septin monomers into higher order structures is important for septin localization (McMurray et al., 2011) and for the conserved roles of septins in regulating cytokinesis and cell polarity. In animals, septins also function in phagocytosis (Huang et al., 2008), ciliogenesis (Hu et al., 2010), and planar cell polarity (Kim et al., 2010). Septin proteins have a characteristic domain architecture with a diagnostic amino terminal guanosine triphosphate–binding domain (G-domain), and most septins also have a carboxy terminal coiled-coil domain (Figure 4A; Pan et al., 2007; Nishihama et al., 2011). Septin paralogues interact directly through their G-domains to form heteromeric filaments, and these heteromeric filaments interact with each other through the septin coiled-coil domains to form higher-order assemblies (Sirajuddin et al., 2007; Bertin et al., 2008; Garcia et al., 2011).
FIGURE 4:

Septins assemble at the basal poles of S. rosetta cells. (A) SrSeptin2 has a prototypical protein domain architecture of septins, with an amino-terminal Septin G-domain that mediates filament formation and a carboxy-terminal coiled-coil domain that mediates higher-order assembly of septin filaments. To investigate the localization of SrSeptin2, we engineered fusions with mTFP1 at the amino terminus and created a truncation of the coiled-coil domain (∆CC). (B) A mTFP1-SrSeptin2 fusion protein localized to the basal pole of unicellular cells (B′, arrowhead). Cotransfecting cells with mTFP1-SrSeptin2 and a plasma membrane marker revealed SrSeptin2 distributed throughout the cytosol and enriched at the basal pole in confocal slices through the center of the cell. (C) mTFP1-SrSeptin6 mirrored the enrichment of mTFP1-SrSeptin2 at the basal pole (C′, arrowhead). The overlapping localization of SrSeptin2 and SrSeptin6 was compatible with these proteins forming heteromeric filaments with each other and other septin paralogues. (D) Consistent with the coiled-coil domain mediating the localization of septins through the formation of higher-order structures, SrSeptin2∆CC localized throughout the cytoplasm, with no visible enrichment at the basal pole. Surprisingly, the deletion also caused ectopic filaments (D′, arrowheads) to form around membrane-bound vesicles that were, based on their size and position in the cell, presumably food vacuoles. (E) In rosettes, mTFP1-SrSeptin2 localized to points of cell–cell contact corresponding to the basal poles of cells (E′; arrowhead). (F) As in single cells, mTFP1-SrSeptin2∆CC in rosettes was distributed throughout the cytosol and formed ectopic filaments (F′; arrowheads) around vacuoles. In panels E and F, S. rosetta single cells were transfected as in panels B and C, immediately induced to develop into rosettes (Woznica et al., 2016), and imaged the next day. (G) SrSeptin2 intercalated between microtubules at the basal pole of the cell. Cotransfecting cells with mTFP1-SrSeptin2 and the α-tubulin marker showed SrSeptin2 filaments intercalated between microtubules at the basal pole in confocal slices that capture the cell cortex to easily visualize microtubule tracks (G′, G″, G′″; box). G″″ shows a 4× magnification of the basal pole of a representative cell (boxed region from G′, G″, G′″). In panels B–F, autofluorescence from ingested bacteria or through accumulation of the fluorescent markers highlights the food vacuole (asterisk).
We started by examining the localization of the S. rosetta septin protein SrSeptin2 (PTSG_07215), a septin with the diagnostic G-domain and coiled-coil domain that typify human and fungal septins (Fairclough et al., 2013) and that are necessary for septin filament formation (Sirajuddin et al., 2007; Bertin et al., 2008). Strikingly, mTFP1-SrSeptin2 was enriched at the basal poles of single and rosettes cells (Figure 4, B and E) and at points of contact between adjacent cells in rosettes (Figure 4E). This mTFP1-SrSeptin2 fusion likely revealed the native localization of SrSeptin2 in S. rosetta because septins visualized by immunofluorescence microscopy in yeast (Haarer and Pringle, 1987; Ford and Pringle, 1991; Kim et al., 1991; Cid et al., 1998), Drosophila (Neufeld and Rubin, 1994; Adam et al., 2000; Silverman-Gavrila et al., 2008), and mammalian cells (Spiliotis et al., 2008) display the same localization as septins tagged with fluorescent proteins.
To investigate whether SrSeptin2 localized to the basal pole of cells might be part of heteromeric septin filaments, we examined the localization of another septin paralogue, SrSeptin6 (PTSG_06009), and found that SrSeptin6 displays the same basal localization as SrSeptin2 (Figure 4C). Such colocalization (Supplemental Figure S9) and the sequence homology with septins that have previously been shown to form heteromeric filaments (Sirajuddin et al., 2007) strongly suggest that SrSeptin2 and SrSeptin6 assemble together at the basal pole. We further found that the basal localization of SrSeptin2 requires the coiled-coil domain, as a complete deletion of the coiled-coil domain (SrSeptin2∆CC; Figure 4, A, D, and F) eliminated SrSeptin2 enrichment at the basal pole when expressed in wild-type cells. Unexpectedly, mTFP1-SrSeptin2∆CC formed ectopic rings around vesicles in the cytosol in wild-type cells (Figure 4, D and F). The localization of mTFP1-SrSeptin2∆CC resembled the formation of ectopic septin filaments at convex membranes and the depletion of septins at the concave membranes in the filamentous fungi Ashbya gosypii (Meseroll et al., 2012) upon the deletion of the coiled-coil domain of the fungal septin Shs1p. Similarly, vesicles to which mTFP1-SrSeptin2∆CC localized have convex membranes, as opposed to the concave membrane at the basal end of S. rosetta where wild-type SrSeptin2 localized.
The basal and lateral localization of SrSeptin2 and SrSeptin6 in rosettes is reminiscent of septin localization in polarized epithelial cells (Fares et al., 1995; Spiliotis et al., 2008), in which septins interact with the positive ends of microtubules that are growing toward the basal pole (Bowen et al., 2011). In choanoflagellates, microtubules radiate down from the apical microtubule organizing centers (Karpov and Leadbeater, 1998), with the plus ends meeting at the basal pole of each cell, similar to the orientation of microtubule plus ends toward the basal pole in animal epithelia (Meads and Schroer, 1995). To examine whether septins also interact with the plus ends of microtubules in S. rosetta, we cotransfected cells with mTFP1-SrSeptin2 and the tubulin marker α-tubulin-mCherry (Figure 4G). Fluorescence microscopy showed that septin filaments intercalate between cortical microtubules at the basal pole of the cell (Figure 4G). These data are consistent with conserved interactions between septins and microtubules from yeast to animals (Kusch et al., 2002; Kremer et al., 2005; Sellin et al., 2011; Spiliotis et al., 2008), including at the plus-ends of microtubules in choanoflagellates and animal epithelia (Bowen et al., 2011).
DISCUSSION
By synthesizing our growing knowledge of S. rosetta biology with a rigorous characterization and optimization of each step in the transfection procedure, we have developed a robust method for transgenesis in S. rosetta that can easily be implemented by other laboratories. This method overcomes numerous barriers that prevented efficient DNA delivery in our prior attempts using diverse methods, including standard electroporation, lipofection, bombardment, and cell-penetrating peptides. A key breakthrough for this study was the discovery that the extracellular coat surrounding S. rosetta might present a barrier to transfection, which motivated the development of a method for gently removing the extracellular material, thereby sensitizing cells for transfection. Additional improvements to the transfection procedure, such as a step promoting the closure of the plasma membrane after electrical pulsation, were designed to address the unique challenges that arise from culturing S. rosetta in sea water. Just as our method was informed by approaches developed in model microeukaryotes (Chlamydomonas and yeast), the methods we have established in S. rosetta will likely aid gene delivery in diverse nonmodel marine microeukaryotes. Overall, the gestalt of continually improving choanoflagellate husbandry (Levin and King, 2013), developing protocols for priming and recovering cells during nucleofection, and extensively optimizing transfection based on a quantitative assay produced a robust method of gene delivery in S. rosetta.
This work also provides a foundational set of vectors for expressing transgenes in S. rosetta (Supplemental Dataset S1). In these vectors, the expression of luciferase or fluorescent proteins was placed under the control of native regulatory elements. From these vectors, we constructed a panel of fluorescently tagged subcellular markers that serve as references for monitoring the localization of other proteins in S. rosetta. For example, through our pilot study of SrSeptin2 and SrSeptin6, the use of these new transgenic tools revealed that septins localize to the basal pole of choanoflagellates, mirroring their localization in animal epithelial cells (Fares et al., 1995; Spiliotis et al., 2008).
Observing septin localization in S. rosetta contributes to our understanding of how septin functions evolved prior to the evolution of an epithelium in stem animals. The tightly connected cells that form animal epithelia each have an apical and basal pole comprising distinct lipids, proteins, and cytoskeletal structures (Rodriguez-Boulan and Macara, 2014). Septins help shape epithelia and other types of cells by serving as buttresses (Tanaka-Takiguchi et al., 2009; Tooley et al., 2009) and diffusion barriers (Barral et al., 2000; Takizawa et al., 2000; Hu et al., 2010) for membranes that have distinct geometries (Bridges et al., 2016; Cannon et al., 2018) and lipid compositions (Casamayor and Snyder, 2003; Tanaka-Takiguchi et al., 2009; Bertin et al., 2010; Bridges et al., 2014) and by interacting with microtubules (Spiliotis, 2010) and filamentous actin (Mavrakis et al., 2014). While comparisons among animals and fungi have revealed conserved functions of septins in cell organization (Spiliotis and Gladfelter, 2012), the divergence of the lineages that gave rise to fungi and animals over one billion years ago (Parfrey et al., 2011) resulted in important differences in fungal and animal cell biology (Stajich et al., 2009). In fungi, septins facilitate polarized cell growth toward the new daughter cell (Kim et al., 1991; Berepiki and Read, 2013), compartmentalize connected daughter cells (Barral et al., 2000; Takizawa et al., 2000; Helfer and Gladfelter, 2006), and mediate cytokinesis (Hartwell, 1971). In addition to their conserved roles in cell division (Neufeld and Rubin, 1994), animal septins have specific functions in epithelia that maintain apical-basal polarity (Spiliotis et al., 2008), planar cell polarity (Kim et al., 2010), intercellular adhesion (Park et al., 2015; Kim and Cooper, 2018), and ciliogenesis (Hu et al., 2010; Kim et al., 2010). The basal localization of septins in S. rosetta suggests that septin filaments organize a distinct region at the basal end of the cell, perhaps supporting intercellular contacts at the basal ends of cells in rosettes (Figure 4E). Consistent with this hypothesis is the observation that the Rosetteless protein, which is necessary for rosette development, localizes to the basal end of cells before secretion into the interiors of rosettes, where the basal ends of cells meet (Levin et al., 2014). Continued study of septin function in S. rosetta will establish the mechanisms by which septins facilitate multicellular development, and further comparisons with nonmetazoan holozoans such as Capsaspora owczarzaki (Parra-Acero et al., 2018) and Creolimax fragrantissima (Suga and Ruiz-Trillo, 2013) by recently established transgenic methods will provide further insights into the ancestral functions of septins.
Previous analyses of gene function in choanoflagellates relied on custom antibodies (Abedin and King, 2008; Young et al., 2011; Burkhardt et al., 2014; Levin et al., 2014), laborious forward genetic screens (Levin et al., 2014), and in vitro biochemistry (Burkhardt et al., 2014). The ability to express transgenes in S. rosetta will accelerate studies of the ancestral functions of animal genes that are conserved in choanoflagellates. We anticipate that future work will build on this approach, eventually leading to the development of methods for stable transgenesis and genome editing in S. rosetta. Combining an expanded repertoire of approaches for investigating gene function in depth in S. rosetta by comparisons to other experimentally tractable choanoflagellates (Li et al., 2018; Richter et al., 2018) and nonchoanozoans (Suga and Ruiz-Trillo, 2013; Parra-Acero et al., 2018) promises to yield increasingly mechanistic insights into the ancestry of animal cell biology.
MATERIALS AND METHODS
Cell culture and media preparation
S. rosetta was cultured with a single bacterial species, Echinicola pacifica (Nedashkovskaya et al., 2006), that serves as a food source (Levin and King, 2013; American Type Culture Collection, Manassas, VA; Cat. No. PRA-390). Media recipes are provided in Supplemental Table S1. Cultures were established from frozen aliquots by adding 1 ml of thawed cells to 10 ml of 0.2× High Nutrient Media (Supplemental Table S1). After the cells reached a density of 104 cells/ml, the culture was split 1:2 into 1× High Nutrient Media with a constant volume of 0.24 ml/cm2. After this initial split (denoted as day 0), cells were passaged in 1× High Nutrient Media according to the following schedule: 1:4 dilution on day 1, 1:8 dilution on day 2, 1:16 on day 3. Subsequently cells were passaged every day at a 1:24 dilution or every other day as a 1:48 dilution of cells. To induce the development of multicellular rosette colonies (Figures 1C and 4, E–G; Supplemental Figures S4B and S9B), outer membrane vesicles purified from the supernatant of Algoriphagus machiponginensis cultures were added to cultures of S. rosetta feeding on E. pacifica (Alegado et al., 2011; Levin et al., 2014; Woznica et al., 2016).
On the basis of the recommendation from Lonza to use a medium with a low calcium concentration for transfecting mammalian cells, we searched for a seawater recipe with a lower concentration of calcium than the routinely used artificial seawater made from Tropic of Marin sea salts (Tropic of Marin, Wartenberg, Germany; Levin and King, 2013), which has a calcium concentration of 9.1 mM at a salinity of 35 g/kg (Atkinson and Bingman, 1998). The AK seawater formulation (Supplemental Table S1) has been used to culture marine algae (Hallegraeff et al., 2004) and dinoflagellates (Skelton et al., 2009) and has a calcium concentration of 2.7 mM. We found that S. rosetta grows more rapidly in 1× High Nutrient Media prepared in AK seawater rather than seawater prepared with Tropic of Marin sea salts (Supplemental Figure S2A). Therefore, we switched to a growth medium based on AK seawater for routine culturing. After optimizing the nucleofection protocol, we demonstrated that growing S. rosetta in AK seawater also resulted in higher transfection efficiencies (Supplemental Figure S2B) than growing S. rosetta in seawater prepared with Tropic of Marin sea salts.
Reporter plasmid design and molecular cloning
Supplemental Dataset S1 lists the complete inventory of engineered plasmids with a summary of primers, cloning methods, and annotations for constructing each plasmid. Complete plasmid sequences and plasmids have also been deposited at Addgene (www.addgene.org/Nicole_King). Below is a brief summary of considerations for designing plasmids, and a more detailed description of standard molecular cloning methods for engineering plasmids can be found in the Supplemental Information.
Cloning regulatory regions from S. rosetta genes.
Because we had no previous knowledge about the architecture of choanoflagellate regulatory regions, we aimed to clone as many as 1000 base pairs upstream and downstream of targeted open reading frames, as these fragments are slightly larger than the mean intergenic distance of 885 base pairs (Sebe-Pedros et al., 2017). Of necessity, the cloned intergenic sequences reported here were shorter to avoid repetitive CA and GT sequences that were present before the putative promoter and after the 3′-UTR, respectively. To increase the specificity of primers, we designed the primers to anneal to regions with a GC content ≤50%, as the S. rosetta genome is 56% GC. Ultimately, the cloned regions that encompass the promoter and the 5′-UTR ranged in size from 550 to 1095 base pairs and those encompassing the 3′UTR ranged from 200 to 807 base pairs.
Synthetic gene design.
Synthetic reporter genes (nanoluc and the genes encoding diverse fluorescent proteins listed in Supplemental File 1) were codon-optimized to match the codon usage of the set of highly expressed intronless genes listed in Supplemental Figure S1, as codon usage can be biased for highly expressed genes (Hiraoka et al., 2009). A codon usage table (Supplemental Dataset S2) was generated from the coding sequences of highly expressed intronless genes (Supplemental Dataset S2A) and from all coding sequences (Supplemental Dataset S2B) using the “cusp” tool in Emboss (Rice et al., 2000). The codon usage table was then used to generate a codon-optimized DNA sequence for each target protein sequence with the “backtranseq” tool in Emboss. The DNA sequences were further edited by making synonymous substitutions with less-frequently-used codons to change restriction enzyme sites and to remove repetitive sequences. Finally, sequences were added to the ends of these designed genes for cloning with restriction enzymes or Gibson assembly. The engineered reporter gene sequences are available through Addgene (Supplemental Dataset S1; www.addgene.org/Nicole_King).
Subcellular marker design.
Dataset S3 provides the amino acid sequences for all of the subcellular markers reported in Figure 3. To ensure that the fluorescent protein tag for each marker would not interfere with the functions of proteins or peptides that determine localization, some of the constructs were engineered to have a flexible linker sequence (SGGSGGS) separating the fluorescent protein and the localization signals.
Optimized transfection protocol
The protocol is summarized in Figure 2 and detailed protocols for reagent preparation and transfection are available at protocols.io at the following link: www.protocols.io/groups/king-lab.
Culture.
Two days prior to transfection, a culture flask (Falcon; Corning Life Sciences, Oneonta, NY; Cat. No. 353144) was seeded with S. rosetta at a density of 5000 cells/ml in 200 ml of 1× High Nutrient Medium. The culture was supplemented with 2 mg of frozen E. pacifica by resuspending a 10-mg pellet of flash-frozen E. pacifica in 1 ml of media and then adding 200 µl of the resuspended pellet to the culture of S. rosetta.
Washing.
After 36–48 h of growth, bacteria were washed away from S. rosetta cells through three consecutive rounds of centrifugation and resuspension in sterile AK seawater. The culture flask was vigorously shaken for 30 s to homogenize the 200 ml culture that was seeded 2 d before (see above) and then transferred to 50-ml conical tubes and spun for 5 min at 2000 × g and 22°C. The supernatant was removed with a serological pipette, and residual media were removed with a fine-tip transfer pipette. The cell pellets were resuspended in a total volume of 100 ml of AK seawater, vigorously shaken in their conical tubes for 30 s, and then centrifuged for 5 min at 2200 × g and 22°C. The supernatant was removed as before. Each cell pellet was resuspended in 50 ml of AK seawater, vigorously shaken for 30 s, and centrifuged for 5 min at 2400 × g and 22°C. After the supernatant was removed, the cells were resuspended in a total volume of 100 µl of AK seawater. A 100-fold dilution of cells fixed with a 100-fold dilution of 37% (wt/vol) formaldehyde was counted on a Luna-FL automated cell counter (Logos Biosystems, Anyang, Korea; Cat. No. L20001) and the remaining cells were diluted to a final concentration of 5 × 107 choanoflagellate cells/ml. The resuspended cells were divided into 100-µl aliquots with 5 × 106 cells per aliquot to immediately prime cells in the next step. A 200-ml culture typically yields 6–8 aliquots of cells.
Prime.
After bacteria were washed away, each aliquot of S. rosetta cells was incubated in priming buffer to remove the extracellular material coating the cell. The 100-µl aliquots, which contained 5 × 106 cells, were centrifuged for 5 min at 800 × g and at room temperature. The supernatant was removed with a fine-tip micropipette. Cells were resuspended in 100 µl of priming buffer (40 mM HEPES-KOH, pH 7.5; 34 mM lithium citrate; 50 mM l-cysteine; 15% [wt/vol] PEG 8000; and 1 µM papain [Millipore Sigma, St. Louis, MO; Cat. No. P3125-100MG]) and then incubated for 30 min. Priming was quenched by adding 2 µl of 50-mg/ml bovine serum albumin-fraction V (Thermo Fisher Scientific, Waltham, MA; Cat. No. BP1600-100) and then centrifuged for 5 min at 1250 ×g and 22°C with the centrifuge brake set to a “soft” setting. The supernatant was removed with a fine-tip micropipette, and the cells were resuspended in 25 µl of SF Buffer (Lonza, Basel, Switzerland; Cat. No. V4SC-2960).
Nucleofection.
Each transfection reaction was prepared by adding 2 µl of “primed” cells resuspended in SF buffer to a mixture of 14 µl of SF buffer; 2 µl of 20 µg/µl pUC19; 1 µl of 250 mM ATP, pH 7.5; 1 µl of 100 mg/ml sodium heparin; and ≤7 µl of reporter DNA. (Note that higher volumes of nucleofection lead to lower transfection frequencies; thus, reporter DNA should be as concentrated as possible, not exceeding 7 µl. Also, see Note about titrating reporter plasmids.) The transfection reaction was transferred to one well of a 96-well nucleofection plate (Lonza; Cat. No. V4SC-2960) or a 16-well strip (Lonza; Cat. No. V4XC-2032). The nucleofection plate was placed in a 96-well shuttle device (Lonza; Cat. No. AAM-1001S) or X-unit (Lonza; Cat. No. AAF-1002F) connected to a Nucleofector 4D core unit (Lonza; Cat. No. AAF-1002B), and the CM156 pulse was applied to each well.
Rest and recovery.
Immediately after pulsation, 100 µl of ice-cold recovery buffer (10 mM HEPES-KOH, pH 7.5; 0.9 M sorbitol; 8% [wt/vol] PEG 8000) was added to the cells. Recovery buffer was gently mixed with the transfected cells by firmly tapping the side of the plate and then incubating the samples for 5 min. The whole volume of the transfection reaction plus the recovery buffer was transferred to 1 ml of 1× High Nutrient Medium in a 12-well plate. After the cells recovered for 1 h, 5 µl of a 10-mg frozen E. pacifica pellet resuspended in medium (see above) was added to each well. The cells were grown for 24–48 h before being assayed for luminescence or fluorescence.
Note about establishing transfection in nonmodel microeukaryotes.
Establishing a transfection protocol for S. rosetta required adapting several different transfection procedures for a variety of eukaryotic cells to meet the unique requirements of S. rosetta. While the specific details for transfecting S. rosetta may not be readily applicable to other organisms, the general considerations and the process for optimization that led to the development of the transfection protocol described here could inform efforts to transfect other microeukaryotes. Therefore, we have included a summary in the Supplemental Information (see the text and Supplemental Figures S10 and S11) of the initial development and optimization of the aforementioned protocol.
Nanoluc reporter assay
To measure relative transfection efficiency resulting from different transfection protocols and promoters, we performed luciferase assays on lysates of transfected cells. Cells transfected with 2.5 µg of nanoluc reporter plasmids were pelleted by centrifuging for 10 min at 4200 × g and 4°C. The supernatant was removed and the cells were resuspended in 50 µl of NanoGlo buffer (Promega, Madison, WI; Cat. No. N1110) and then transferred to a well of a white, opaque 96-well plate (Greiner Bio-one, Monroe, NC; Cat. No.655083). Luminescence was immediately recorded on a Spectramax L Microplate Reader (Molecular Devices, San Jose, CA) with 1 min dark adaption and 10 s dwell time with the photomultiplier gain set to photon-counting mode.
Based on standard definitions from analytical chemistry (Harris, 2007), the detection limit was set to three SDs above the background signal so that any signal above the detection limit would have less than a 1% chance of arising from random error. The limit of detection was calculated in two different ways. First, the y-axis intercept and SD were calculated from a standard curve (Harris, 2007) fitted to a serial dilution of nanoluc versus luciferase activity (Supplemental Figure S3A). To decrease the bias toward higher luciferase values, the standard curve was fitted with the objective
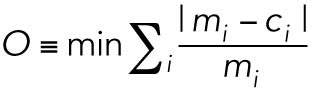 |
where m is the measured luciferase value for a given data point i and c is the calculated luciferase value. Second, the detection limit was also determined as three SDs above the mean of eight replicate luciferase measurements of cells transfected without any reporter plasmid, which resulted in the same calculated detection limit.
Reproducibility in luciferase assays was assessed by performing at least two independent experiments on separate days with different preparations of “primed” cells; data presented in Figure 2 and Supplemental Figures S2, S3, S10, and S11 represent one of the independent experiments. Within each experiment from the same preparation of primed cells, replicate measurements were performed by setting up three to five independent transfections for each condition (shown as black dots); bar graphs in Figure 2 and Supplemental Figures S2, S3, S10, and S11 show the mean values of the five independent transfections, with error bars showing the SD. Before performing statistical tests that relied on a normal distribution, luciferase data were transformed to a lognormal distribution by taking the base-10 logarithm of luciferase values, as gene expression data from luciferase assays display a lognormal distribution (Muranaka et al., 2013).
Flow cytometry
To measure the percentage of cells expressing each of the different transgenes under different transfection conditions, we used flow cytometry. Cells were transfected with 10 µg of mWasabi or 10 µg of TagRFP-T reporter plasmids for flow cytometry because these fluorophores produced the highest fluorescence signals upon illumination with the 488 and 561 nm lasers, respectively. To prepare cells for flow cytometry, cultures from 10–12 transfections were pooled 24 h after transfection and centrifuged for 15 min at 3600 × g and 4°C. The supernatant was removed with a fine-tip transfer pipette to avoid disturbing the pellet. The pelleted cells were resuspended in 500 µl of 0.22-µm filtered AK seawater and then filtered through a 40-µm filter. Cell populations were then analyzed using a LSRFortessa cell analyzer (BD Biosciences, San Jose, CA) flow cytometer (Figure 2E) or a FACSAria Fusion (BD Biosciences) fluorescence-activated cell sorter (Supplemental Figure S6B).
Because a large number of bacteria were present in the cultures, S. rosetta cells were gated based on the area of forward-scattering signal versus the area of the side-scattering signal and the area of the forward-scattering signal versus the height of the forward-scattering signals. To differentiate transfected cells from untransfected cells, the fluorescence signal was measured using lasers and filters for the fluorophores FITC (green fluorescence) and PE (red fluorescence); untransfected cells form a population along the y = x line of these plots, and the population of transfected cells is skewed along one axis that corresponds to the fluorophore. The transfected cells were gated to exclude >99.99% of untransfected cells as determined from a negative control reaction that was transfected without a fluorescent reporter (Figure 2E, left panel).
Live-cell imaging
An important benefit of transgenics is the ability to visualize protein localization and cell architecture in living cells. To this end, we have established improved protocols for live-cell imaging in S. rosetta. Glass-bottomed dishes (World Precision Instruments, Sarasota, FL; Cat. No. FD35-100) were prepared for live-cell microscopy by corona-treating the glass for 10 s. Afterward, 300 µl of 0.1 mg/ml poly-d-lysine (Millipore Sigma; Cat. No. P6407-5MG) was applied to the glass cover (18 µl/cm2), incubated for 10 min at room temperature, and then removed. Excess poly-d-lysine was washed away from the glass surface with three rinses of 500 µl artificial seawater.
Cells transfected with 5 µg of each fluorescent reporter were prepared for microscopy by centrifuging 1–2 ml of transfected cells for 10 min at 3600 × g and 4°C. After centrifugation, the supernatant was removed and the cell pellet was resuspended in 200 µl of 4/5 Tropic of Marin artificial seawater with 100 mM LiCl. Lithium chloride slows flagellar beating, as in spermatozoa (Brokaw, 1987; Gibbons and Gibbons, 2013), to decrease the movement of cells during imaging. The resuspended cells were pipetted on top of the poly-d-lysine–coated glass-bottomed dish and adsorbed onto the surface for 10 min. Last, 200 µl of 20% (wt/vol) Ficoll 400 dissolved in 4/5 Tropic of Marin artificial seawater with 100 mM LiCl was pipetted drop by drop on top of the cells. The addition of Ficoll decreases flagellar movement by increasing the viscosity of the medium (Pate and Brokaw, 1980; Wilson et al., 2015) without significantly changing the osmolarity or refractive index of the sample (GE Healthcare Lifesciences, Pittsburgh, PA; Cat. No. 17030010).
Confocal microscopy was performed on a Zeiss Axio Observer LSM 880 with an Airyscan detector and a 63x/NA1.40 Plan-Apochromatic oil immersion objective (Carl Zeiss AG, Oberkochen, Germany). The mTFP1 and mCherry fluorophores were selected for two-color imaging due to their high photostability and minimal spectral overlap. Confocal stacks were acquired in superresolution mode using ILEX line scanning and twofold averaging and the following settings: 40 × 40 nm pixel size, 93 nm z-step, 0.9–1.0 µs/pixel dwell time, 850 gain, 458-nm laser operating at 5% laser power, 561-nm laser operating at 3% laser power, 458-/561-nm multiple beam splitter, and 495–550-nm bandpass/570-nm longpass filter. Images were initially processed using the automated Airyscan algorithm (Zeiss) and then reprocessed by setting the Airyscan threshold 0.5 units higher than the value reported from automated Airyscan processing. The stacks were further processed by correcting for signal decay, background, and flicker in Zen Blue (Zeiss). Last, FIJI (Schindelin et al., 2012) was used to apply a gamma factor to each channel and subtract the background using a 100-pixel radius.
Epifluorescence and differential interference contrast images were recorded using a Zeiss Axio Observer.Z1/7 Widefield microscope with a Hamamatsu Orca-Flash 4.0 LT CMOS Digital Camera (Hamamatsu Photonics, Hamamatsu City, Japan) and 40×/NA 1.1 LD C-Apochromatic water immersion, 63×/NA1.40 Plan-Apochromatic oil immersion, or 100× NA 1.40 Plan-Apochromatic oil immersion objectives (Zeiss). Green fluorescent proteins were imaged with a 38 HE filter set and red fluorescent proteins with a 43 HE filter set. Images were processed by applying a gamma factor and background-subtracting fluorescence channels in FIJI.
Note about titrating reporter plasmids.
A titration of fluorescent reporter plasmids showed that 10 µg of total reporter plasmids best balanced transfection efficiency, brightness, and a faithful indication of subcellular architecture. We caution that high plasmid concentrations can result in the overexpression of fluorescent markers, leading to aberrant localization of the marker and gross changes in cell morphology. Such artifacts can be avoided by performing a titration to determine the best concentration of plasmid and recording images from cells with a range of fluorescence intensities that result from any transfection. One of the best markers for assessing optimal reporter plasmid concentrations is the tubulin marker because of its distinct localization, which can be benchmarked with immunofluorescence.
Immunofluorescence staining and imaging
Immunofluorescence was performed as previously described (Woznica et al., 2016) with modifications to better preserve features of the cytoskeleton. Cells (2 ml) were concentrated by centrifugation for 10 min at 2750 × g and 4°C. The cells were resuspended in 400 µl of artificial seawater and applied to poly-l-lysine–coated coverslips (Corning Life Sciences; Cat. No.354085) placed at the bottom of each well of a 24-well cell culture dish. After the cells were allowed to settle on the coverslip for 30 min, 150 µl of the cell solution was gently removed from the side of the dish. It is crucial to leave a small layer of buffer on top of cells to preserve the cell morphology; hence 250 µl of liquid was left in the well. All of the subsequent washes and incubations during the staining procedure were performed by adding and removing 200 µl of the indicated buffer.
Cells were fixed in two stages. First, the coverslip was washed once with 6% acetone in cytoskeleton buffer (10 mM MES, pH 6.1; 138 KCl, 3 mM MgCl2; 2 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N’,N’-tetraacetic acid [EGTA]; 675 mM sucrose), which better preserves the actin cytoskeleton (Symons and Mitchison, 1991; Cramer and Mitchison, 1995), and then incubated for 10 min at room temperature after a second application of the acetone solution. Subsequently, the coverslip was washed once with 4% formaldehyde diluted in cytoskeleton buffer and then incubated for 15 min at room temperature after a second application of the formaldehyde solution. Last, the coverslip was gently washed three times with cytoskeleton buffer.
Cells were permeabilized by washing the coverslip once with permeabilization buffer (100 mM PIPES, pH 6.95; 2 mM EGTA; 1 mM MgCl2; 1% [wt/vol] bovine serum albumin (BSA)-fraction V; 0.3% [vol/vol] Triton X-100) and then incubated for 30 min upon a second addition of permeabilization buffer. After the permeabilization buffer was removed, the coverslip was washed once with primary antibody, 50 ng/ml mouse E7 anti-tubulin antibody (Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA; Cat. No. AB_2315513) diluted in permeabilization buffer, and then incubated for 1 h in a second application of primary antibody. The coverslip was gently washed twice in permeabilization buffer. Next, the coverslip was washed once with secondary antibody, 8 ng/ml donkey anti-mouse immunoglobulin G–AlexaFluor568 (Thermo Fisher Scientific; Cat. No. A10037) diluted in permeabilization buffer, and then incubated for 1 h after a second application of secondary antibody. Afterward, the coverslip was washed once in permeabilization buffer and then three times with PEM (100 mM PIPES-KOH, pH 6.95; 2 mM EGTA; 1 mM MgCl2). The coverslip was washed once with 10 µg/ml Hoechst 33342 (Thermo Fisher Scientific; Cat. No. H3570) and 4 U/ml Phalloidin-AlexaFluor488 (Thermo Fisher Scientific; Cat. No. A12379) in PEM and then incubated for 30 min with a second application of Hoechst33342/Phalloidin. Finally, the coverslip was washed once in PEM.
To prepare a slide for mounting, 10 µl of Pro-Long Diamond (Thermo Fisher Scientific; Cat. No. P36970) was added to a slide. The coverslip was gently removed from the well with forceps, excess buffer was blotted from the side with a piece of filter paper, and the coverslip was gently placed on the drop of Pro-Long diamond. The mounting media cured overnight before visualization.
Images were acquired on a Zeiss LSM 880 Airyscan confocal microscope with a 63× objective (as described for live cell imaging) by frame scanning in the superresolution mode with the following settings: 35 × 35 nm pixel size; 80 nm z-step; 0.64 µs/pixel dwell time; 561-nm laser operating at 1.5% power with a 488-/561-nm beam splitter, a 420–480-/495–620-nm bandpass filter, and a gain of 750; 488-nm laser operating at 1.5% power with a 488-/561-nm beam splitter, a 420–480-/495–550-nm bandpass filter, and a gain of 750; and 405nm laser operating at 1.5% power with a 405 nm beam splitter, a 420–480-/495–550-nm bandpass filter, and a gain of 775.
Supplementary Material
Acknowledgments
Laura Wetzel, Monika Sigg, Hannah Elzinga, Lily Helfrich, and Reef Aldayafleh helped with experiments and reagent preparation. Corey Allard, as part of the Marine Biological Laboratory’s Physiology Course, helped with early tests of priming conditions. Kent McDonald generously provided a transmission electron micrograph from samples prepared by Pawel Burkhardt. We thank these people for providing access and support for scientific instruments: Russell Vance and lab for use of their luminometer, Hector Nolla and Alma Valeros in the Flow Cytometry Facility, and the UC Berkeley DNA Sequencing Facility. The following individuals generously donated reagents and provided technical support: Brad Hook and Dee Czarniecki from Promega, Ethan Brooks from Lonza, and Colleen Manning from Zeiss. We appreciate scientific discussions and advice from these individuals: David Schaffer, Sabrina Sun, Jorge Ortiz, Niren Murthy, Tara DeBoer, Fyodor Urnov, Matt Welch and lab, Rebecca Heald and lab, Abby Dernburg and lab, and Amy Gladfelter. We thank members of the King lab for helpful discussions, research support, and comments on the manuscript, especially Arielle Woznica, Kayley Hake, and Ben Larson. We also thank the following people for providing comments on the manuscript: Candace Britton, Pawel Burkhardt, Matt Daugherty, Galo Garcia, Tera Levin, Kristin Patrick, and Dan Richter. D.S.B. is supported as a Simons Foundation Postdoctoral Fellow of the Jane Coffin Childs Memorial Fund for Biomedical Research. This work was funded in part by a grant from the Gordon and Betty Moore Foundation’s Marine Microbiology Initiative for establishing Emerging Model Systems.
Abbreviations used:
- act
actin
- CoxIV
cytochrome C oxidase, subunit IV
- efl
elongation factor L
- ER
endoplasmic reticulum
- G-domain
guanosine triphosphate–binding domain
- H3
histone H3
- HDEL
peptide sequence of His-Asp-Glu-Leu
- SV40
simian virus 40
- tub
α-tubulin
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E18-08-0514) on October 3, 2018.
REFERENCES
- Abedin M, King N. (2008). The premetazoan ancestry of cadherins. Science , 946–948. [DOI] [PubMed] [Google Scholar]
- Adam JC, Pringle JR, Peifer M. (2000). Evidence for functional differentiation among Drosophila septins in cytokinesis and cellularization. Mol Biol Cell , 3123–3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ai HW, Henderson JN, Remington SJ, Campbell RE. (2006). Directed evolution of a monomeric, bright and photostable version of Clavularia cyan fluorescent protein: structural characterization and applications in fluorescence imaging. Biochem J , 531–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ai HW, Olenych SG, Wong P, Davidson MW, Campbell RE. (2008). Hue-shifted monomeric variants of Clavularia cyan fluorescent protein: identification of the molecular determinants of color and applications in fluorescence imaging. BMC Biol , 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alegado RA, Brown LW, Cao S, Dermenjian RK, Zuzow R, Fairclough SR, Clardy J, King N. (2012). A bacterial sulfonolipid triggers multicellular development in the closest living relatives of animals. Elife , e00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alegado RA, Ferriera S, Nusbaum C, Young SK, Zeng Q, Imamovic A, Fairclough SR, King N. (2011). Complete genome sequence of Algoriphagus sp. PR1, bacterial prey of a colony-forming choanoflagellate. J Bacteriol , 1485–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson MJ, Bingman C. (1998). Elemental composition of commercial seasalts. J Aquariculture Aquatic Sciences , 39–43. [Google Scholar]
- Barral Y, Mermall V, Mooseker MS, Snyder M. (2000). Compartmentalization of the cell cortex by septins is required for maintenance of cell polarity in yeast. Mol Cell , 841–851. [DOI] [PubMed] [Google Scholar]
- Berepiki A, Read ND. (2013). Septins are important for cell polarity, septation and asexual spore formation in Neurospora crassa and show different patterns of localisation at germ tube tips. PLoS One , e63843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertin A, McMurray MA, Grob P, Park SS, Garcia G, Patanwala I, Ng HL, Alber T, Thorner J, Nogales E. (2008). Saccharomyces cerevisiae septins: supramolecular organization of heterooligomers and the mechanism of filament assembly. Proc Natl Acad Sci USA , 8274–8279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertin A, McMurray MA, Thai L, Garcia G, Votin V, Grob P, Allyn T, Thorner J, Nogales E. (2010). Phosphatidylinositol-4,5-bisphosphate promotes budding yeast septin filament assembly and organization. J Mol Biol , 711–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen JR, Hwang D, Bai X, Roy D, Spiliotis ET. (2011). Septin GTPases spatially guide microtubule organization and plus end dynamics in polarizing epithelia. J Cell Biol , 187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges AA, Jentzsch MS, Oakes PW, Occhipinti P, Gladfelter AS. (2016). Micron-scale plasma membrane curvature is recognized by the septin cytoskeleton. J Cell Biol , 23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges AA, Zhang H, Mehta SB, Occhipinti P, Tani T, Gladfelter AS. (2014). Septin assemblies form by diffusion-driven annealing on membranes. Proc Natl Acad Sci USA , 2146–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brokaw CJ. (1987). A lithium-sensitive regulator of sperm flagellar oscillation is activated by cAMP-dependent phosphorylation. J Cell Biol , 1789–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet T, King N. (2017). The origin of animal multicellularity and cell differentiation. Dev Cell , 124–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger G, Forget L, Zhu Y, Gray MW, Lang BF. (2003). Unique mitochondrial genome architecture in unicellular relatives of animals. Proc Natl Acad Sci USA , 892–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhardt P, Gronborg M, McDonald K, Sulur T, Wang Q, King N. (2014). Evolutionary insights into premetazoan functions of the neuronal protein homer. Mol Biol Evol , 2342–2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon KS, Woods BL, Crutchley JM, Gladfelter AS. (2018). An amphipathic helix enables septins to sense micron-scale membrane curvature. bioRxiv 379982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caro F, Miller MG, DeRisi JL. (2012). Plate-based transfection and culturing technique for genetic manipulation of Plasmodium falciparum. Malar J , 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casamayor A, Snyder M. (2003). Molecular dissection of a yeast septin: distinct domains are required for septin interaction, localization, and function. Mol Cell Biol , 2762–2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cid VJ, Adamikova L, Cenamor R, Molina M, Sanchez M, Nombela C. (1998). Cell integrity and morphogenesis in a budding yeast septin mutant. Microbiology , 3463–3474. [DOI] [PubMed] [Google Scholar]
- Cramer LP, Mitchison TJ. (1995). Myosin is involved in postmitotic cell spreading. J Cell Biol , 179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayel MJ, Alegado RA, Fairclough SR, Levin TC, Nichols SA, McDonald K, King N. (2011). Cell differentiation and morphogenesis in the colony-forming choanoflagellate Salpingoeca rosetta. Dev Biol , 73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayel MJ, King N. (2014). Prey capture and phagocytosis in the choanoflagellate Salpingoeca rosetta. PLoS One , e95577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairclough SR, Chen Z, Kramer E, Zeng Q, Young S, Robertson HM, Begovic E, Richter DJ, Russ C, Westbrook MJ, et al. (2013). Premetazoan genome evolution and the regulation of cell differentiation in the choanoflagellate Salpingoeca rosetta. Genome Biol , r15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairclough SR, Dayel MJ, King N. (2010). Multicellular development in a choanoflagellate. Curr Biol , 875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fares H, Peifer M, Pringle JR. (1995). Localization and possible functions of Drosophila septins. Mol Biol Cell , 1843–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford SK, Pringle JR. (1991). Cellular morphogenesis in the Saccharomyces cerevisiae cell cycle: localization of the CDC11 gene product and the timing of events at the budding site. Dev Genet , 281–292. [DOI] [PubMed] [Google Scholar]
- Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. (2011). ER tubules mark sites of mitochondrial division. Science , 358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia G, Bertin A, Li Z, Song Y, McMurray MA, Thorner J, Nogales E. (2011). Subunit-dependent modulation of septin assembly: budding yeast septin Shs1 promotes ring and gauze formation. J Cell Biol , 993–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons BH, Gibbons IR. (2013). Lithium reversibly inhibits microtubule-based motility in sperm flagella. Nature , 560–562. [DOI] [PubMed] [Google Scholar]
- Gresch O, Engel FB, Nesic D, Tran TT, England HM, Hickman ES, Korner I, Gan L, Chen S, Castro-Obregon S, et al. (2004). New non-viral method for gene transfer into primary cells. Methods , 151–163. [DOI] [PubMed] [Google Scholar]
- Haarer BK, Pringle JR. (1987). Immunofluorescence localization of the Saccharomyces cerevisiae CDC12 gene product to the vicinity of the 10-nm filaments in the mother-bud neck. Mol Cell Biol , 3678–3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall MP, Unch J, Binkowski BF, Valley MP, Butler BL, Wood MG, Otto P, Zimmerman K, Vidugiris G, Machleidt T, et al. (2012). Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem Biol , 1848–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallegraeff GM, Anderson DM, Cembella AD. (2004). Manual on Harmful Marine Microalgae, Paris, France: United Nations Educational, Scientific and Cultural Organization. [Google Scholar]
- Hamm A, Krott N, Breibach I, Blindt R, Bosserhoff AK. (2002). Efficient transfection method for primary cells. Tissue Eng , 235–245. [DOI] [PubMed] [Google Scholar]
- Harris DC. (2007). Quantitative Chemical Analysis, New York: Freeman. [Google Scholar]
- Hartwell LH. (1971). Genetic control of the cell division cycle in yeast. IV. Genes controlling bud emergence and cytokinesis. Exp Cell Res , 265–276. [DOI] [PubMed] [Google Scholar]
- Helfer H, Gladfelter AS. (2006). AgSwe1p regulates mitosis in response to morphogenesis and nutrients in multinucleated Ashbya gossypii cells. Mol Biol Cell , 4494–4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraoka Y, Kawamata K, Haraguchi T, Chikashige Y. (2009). Codon usage bias is correlated with gene expression levels in the fission yeast Schizosaccharomyces pombe. Genes Cells , 499–509. [DOI] [PubMed] [Google Scholar]
- Hoi H, Howe ES, Ding Y, Zhang W, Baird MA, Sell BR, Allen JR, Davidson MW, Campbell RE. (2013). An engineered monomeric Zoanthus sp. yellow fluorescent protein. Chem Biol , 1296–1304. [DOI] [PubMed] [Google Scholar]
- Hu Q, Milenkovic L, Jin H, Scott MP, Nachury MV, Spiliotis ET, Nelson WJ. (2010). A septin diffusion barrier at the base of the primary cilium maintains ciliary membrane protein distribution. Science , 436–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YW, Yan M, Collins RF, Diciccio JE, Grinstein S, Trimble WS. (2008). Mammalian septins are required for phagosome formation. Mol Biol Cell , 1717–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O’Shea EK. (2003). Global analysis of protein localization in budding yeast. Nature , 686–691. [DOI] [PubMed] [Google Scholar]
- James-Clark H. (1868). On the spongiae ciliatae as infusoria flagellata: or observations on the structure, animality and relationship of Leucosolenia botryoides Bowerbank. Annals and Magazine of Natural History , 250–264. [Google Scholar]
- Janse CJ, Franke-Fayard B, Mair GR, Ramesar J, Thiel C, Engelmann S, Matuschewski K, van Gemert GJ, Sauerwein RW, Waters AP. (2006a). High efficiency transfection of Plasmodium berghei facilitates novel selection procedures. Mol Biochem Parasitol , 60–70. [DOI] [PubMed] [Google Scholar]
- Janse CJ, Ramesar J, Waters AP. (2006b). High-efficiency transfection and drug selection of genetically transformed blood stages of the rodent malaria parasite Plasmodium berghei. Nat Protoc , 346–356. [DOI] [PubMed] [Google Scholar]
- Kalderon D, Roberts BL, Richardson WD, Smith AE. (1984). A short amino acid sequence able to specify nuclear location. Cell , 499–509. [DOI] [PubMed] [Google Scholar]
- Kanda T, Sullivan KF, Wahl GM. (1998). Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr Biol , 377–385. [DOI] [PubMed] [Google Scholar]
- Karpov SA, Leadbeater BSC. (1998). Cytoskeleton structure and composition in choanoflagellates. J Eukaryot Microbiol , 361–367. [Google Scholar]
- Kawai S, Hashimoto W, Murata K. (2010). Transformation of Saccharomyces cerevisiae and other fungi: methods and possible underlying mechanism. Bioeng Bugs , 395–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WS. (1871). Affinities of the sponges. Nature , 184. [Google Scholar]
- Kim HB, Haarer BK, Pringle JR. (1991). Cellular morphogenesis in the Saccharomyces cerevisiae cell cycle: localization of the CDC3 gene product and the timing of events at the budding site. J Cell Biol , 535–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Cooper JA. (2018). Septins regulate junctional integrity of endothelial monolayers. Mol Biol Cell , 1693–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SK, Shindo A, Park TJ, Oh EC, Ghosh S, Gray RS, Lewis RA, Johnson CA, Attie-Bittach T, Katsanis N, et al. (2010). Planar cell polarity acts through septins to control collective cell movement and ciliogenesis. Science , 1337–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King N. (2004). The unicellular ancestry of animal development. Dev Cell , 313–325. [DOI] [PubMed] [Google Scholar]
- King N, Hittinger CT, Carroll SB. (2003). Evolution of key cell signaling and adhesion protein families predates animal origins. Science , 361–363. [DOI] [PubMed] [Google Scholar]
- King N, Westbrook MJ, Young SL, Kuo A, Abedin M, Chapman J, Fairclough S, Hellsten U, Isogai Y, Letunic I, et al. (2008). The genome of the choanoflagellate Monosiga brevicollis and the origin of metazoans. Nature , 783–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King N, Young SL, Abedin M, Carr M, Leadbeater BS. (2009). Visualizing the subcellular localization of actin, beta-tubulin, and DNA in Monosiga brevicollis. Cold Spring Harb Protoc , pdb.prot5150. [DOI] [PubMed] [Google Scholar]
- Kremer BE, Haystead T, Macara IG. (2005). Mammalian septins regulate microtubule stability through interaction with the microtubule-binding protein MAP4. Mol Biol Cell , 4648–4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusch J, Meyer A, Snyder MP, Barral Y. (2002). Microtubule capture by the cleavage apparatus is required for proper spindle positioning in yeast. Genes Dev , 1627–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang BF, O’Kelly C, Nerad T, Gray MW, Burger G. (2002). The closest unicellular relatives of animals. Curr Biol , 1773–1778. [DOI] [PubMed] [Google Scholar]
- Leadbeater BSC. (2015). The Choanoflagellates: Evolution, Biology and Ecology, Cambridge, UK: Cambridge University Press. [Google Scholar]
- Levin TC, Greaney AJ, Wetzel L, King N. (2014). The rosetteless gene controls development in the choanoflagellate S. rosetta. Elife , e04070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin TC, King N. (2013). Evidence for sex and recombination in the choanoflagellate Salpingoeca rosetta. Curr Biol , 2176–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Neundorf I, Nitsche F. (2018). First efficient transfection in choanoflagellates using cell-penetrating peptides. bioRxiv 260190. [Google Scholar]
- Manning G, Young SL, Miller WT, Zhai Y. (2008). The protist, Monosiga brevicollis, has a tyrosine kinase signaling network more elaborate and diverse than found in any known metazoan. Proc Natl Acad Sci USA , 9674–9679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrakis M, Azou-Gros Y, Tsai FC, Alvarado J, Bertin A, Iv F, Kress A, Brasselet S, Koenderink GH, Lecuit T. (2014). Septins promote F-actin ring formation by crosslinking actin filaments into curved bundles. Nat Cell Biol , 322–334. [DOI] [PubMed] [Google Scholar]
- McMurray MA, Bertin A, Garcia G, Lam L, Nogales E, Thorner J. (2011). Septin filament formation is essential in budding yeast. Dev Cell , 540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meads T, Schroer TA. (1995). Polarity and nucleation of microtubules in polarized epithelial cells. Cell Motil Cytoskeleton , 273–288. [DOI] [PubMed] [Google Scholar]
- Meseroll RA, Howard L, Gladfelter AS. (2012). Septin ring size scaling and dynamics require the coiled-coil region of Shs1p. Mol Biol Cell , 3391–3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muranaka T, Kubota S, Oyama T. (2013). A single-cell bioluminescence imaging system for monitoring cellular gene expression in a plant body. Plant Cell Physiol , 2085–2093. [DOI] [PubMed] [Google Scholar]
- Nedashkovskaya OI, Kim SB, Vancanneyt M, Lysenko AM, Shin DS, Park MS, Lee KH, Jung WJ, Kalinovskaya NI, Mikhailov VV, et al. (2006). Echinicola pacifica gen. nov., sp. nov., a novel flexibacterium isolated from the sea urchin Strongylocentrotus intermedius. Int J Syst Evol Microbiol , 953–958. [DOI] [PubMed] [Google Scholar]
- Neufeld TP, Rubin GM. (1994). The Drosophilapeanut gene is required for cytokinesis and encodes a protein similar to yeast putative bud neck filament proteins. Cell , 371–379. [DOI] [PubMed] [Google Scholar]
- Nishihama R, Onishi M, Pringle JR. (2011). New insights into the phylogenetic distribution and evolutionary origins of the septins. Biol Chem , 681–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunnari J, Marshall WF, Straight A, Murray A, Sedat JW, Walter P. (1997). Mitochondrial transmission during mating in Saccharomyces cerevisiae is determined by mitochondrial fusion and fission and the intramitochondrial segregation of mitochondrial DNA. Mol Biol Cell , 1233–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill RS, Clark DV. (2013). The Drosophila melanogaster septin gene Sep2 has a redundant function with the retrogene Sep5 in imaginal cell proliferation but is essential for oogenesis. Genome , 753–758. [DOI] [PubMed] [Google Scholar]
- Pan F, Malmberg RL, Momany M. (2007). Analysis of septins across kingdoms reveals orthology and new motifs. BMC Evol Biol , 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parfrey LW, Lahr DJ, Knoll AH, Katz LA. (2011). Estimating the timing of early eukaryotic diversification with multigene molecular clocks. Proc Natl Acad Sci USA , 13624–13629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park TJ, Kim SK, Wallingford JB. (2015). The planar cell polarity effector protein Wdpcp (Fritz) controls epithelial cell cortex dynamics via septins and actomyosin. Biochem Biophys Res Commun , 562–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra-Acero H, Ros-Rocher N, Perez-Posada A, Koz˙yczkowska A, Sánchez-Pons N, Nakata A, Suga H, Najle SR, Ruiz-Trillo I. (2018). Transfection of Capsaspora owczarzaki, a close unicellular relative of animals. Development , dev162107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pate EF, Brokaw CJ. (1980). Movement of spermatozoa in viscous environments. J Exp Biol , 395–397. [DOI] [PubMed] [Google Scholar]
- Pedelacq JD, Cabantous S, Tran T, Terwilliger TC, Waldo GS. (2006). Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol , 79–88. [DOI] [PubMed] [Google Scholar]
- Reid TS, Terry KL, Casey PJ, Beese LS. (2004). Crystallographic analysis of CaaX prenyltransferases complexed with substrates defines rules of protein substrate selectivity. J Mol Biol , 417–433. [DOI] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A. (2000). EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet , 276–277. [DOI] [PubMed] [Google Scholar]
- Richter D, Fozouni P, Eisen M, King N. (2018). Gene family innovation, conservation and loss on the animal stem lineage. eLife , e34226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, Bradke F, Jenne D, Holak TA, Werb Z, et al. (2008). Lifeact: a versatile marker to visualize F-actin. Nat Methods , 605–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Boulan E, Macara IG. (2014). Organization and execution of the epithelial polarity programme. Nat Rev Mol Cell Biol , 225–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rols MP, Teissie J. (1989). Ionic-strength modulation of electrically induced permeabilization and associated fusion of mammalian cells. Eur J Biochem , 109–115. [DOI] [PubMed] [Google Scholar]
- Rols MP, Teissie J. (1990). Modulation of electrically induced permeabilization and fusion of Chinese hamster ovary cells by osmotic pressure. Biochemistry , 4561–4567. [DOI] [PubMed] [Google Scholar]
- Ruiz-Trillo I, Roger AJ, Burger G, Gray MW, Lang BF. (2008). A phylogenomic investigation into the origin of metazoa. Mol Biol Evol , 664–672. [DOI] [PubMed] [Google Scholar]
- Schiestl RH, Gietz RD. (1989). High efficiency transformation of intact yeast cells using single stranded nucleic acids as a carrier. Curr Genet , 339–346. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods , 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebe-Pedros A, Burkhardt P, Sanchez-Pons N, Fairclough SR, Lang BF, King N, Ruiz-Trillo I. (2013). Insights into the origin of metazoan filopodia and microvilli. Mol Biol Evol , 2013–2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebe-Pedros A, Degnan BM, Ruiz-Trillo I. (2017). The origin of Metazoa: a unicellular perspective. Nat Rev Genet , 498–512. [DOI] [PubMed] [Google Scholar]
- Sellin ME, Holmfeldt P, Stenmark S, Gullberg M. (2011). Microtubules support a disk-like septin arrangement at the plasma membrane of mammalian cells. Mol Biol Cell , 4588–4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. (2004). Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol , 1567–1572. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Lambert GG, Chammas A, Ni Y, Cranfill PJ, Baird MA, Sell BR, Allen JR, Day RN, Israelsson M, et al. (2013). A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat Methods , 407–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Lin MZ, McKeown MR, Steinbach PA, Hazelwood KL, Davidson MW, Tsien RY. (2008). Improving the photostability of bright monomeric orange and red fluorescent proteins. Nat Methods , 545–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman-Gavrila RV, Hales KG, Wilde A. (2008). Anillin-mediated targeting of peanut to pseudocleavage furrows is regulated by the GTPase Ran. Mol Biol Cell , 3735–3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirajuddin M, Farkasovsky M, Hauer F, Kuhlmann D, Macara IG, Weyand M, Stark H, Wittinghofer A. (2007). Structural insight into filament formation by mammalian septins. Nature , 311–315. [DOI] [PubMed] [Google Scholar]
- Skelton HM, Burkholder JM, Parrow MW. (2009). Axenic culture of the heterotrophic dinoflagellate Pfiesteria shumwayae in a semi-defined medium. J Eukaryot Microbiol , 73–82. [DOI] [PubMed] [Google Scholar]
- Spiliotis ET. (2010). Regulation of microtubule organization and functions by septin GTPases. Cytoskeleton (Hoboken) , 339–345. [DOI] [PubMed] [Google Scholar]
- Spiliotis ET, Gladfelter AS. (2012). Spatial guidance of cell asymmetry: septin GTPases show the way. Traffic , 195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiliotis ET, Hunt SJ, Hu Q, Kinoshita M, Nelson WJ. (2008). Epithelial polarity requires septin coupling of vesicle transport to polyglutamylated microtubules. J Cell Biol , 295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stajich JE, Berbee ML, Blackwell M, Hibbett DS, James TY, Spatafora JW, Taylor JW. (2009). The fungi. Curr Biol , 840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straight AF, Marshall WF, Sedat JW, Murray AW. (1997). Mitosis in living budding yeast: anaphase A but no metaphase plate. Science , 574–578. [DOI] [PubMed] [Google Scholar]
- Suga H, Dacre M, de Mendoza A, Shalchian-Tabrizi K, Manning G, Ruiz-Trillo I. (2012). Genomic survey of premetazoans shows deep conservation of cytoplasmic tyrosine kinases and multiple radiations of receptor tyrosine kinases. Sci Signal , ra35. [DOI] [PubMed] [Google Scholar]
- Suga H, Ruiz-Trillo I. (2013). Development of ichthyosporeans sheds light on the origin of metazoan multicellularity. Dev Biol , 284–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symons MH, Mitchison TJ. (1991). Control of actin polymerization in live and permeabilized fibroblasts. J Cell Biol , 503–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takizawa PA, DeRisi JL, Wilhelm JE, Vale RD. (2000). Plasma membrane compartmentalization in yeast by messenger RNA transport and a septin diffusion barrier. Science , 341–344. [DOI] [PubMed] [Google Scholar]
- Tanaka-Takiguchi Y, Kinoshita M, Takiguchi K. (2009). Septin-mediated uniform bracing of phospholipid membranes. Curr Biol , 140–145. [DOI] [PubMed] [Google Scholar]
- Tooley AJ, Gilden J, Jacobelli J, Beemiller P, Trimble WS, Kinoshita M, Krummel MF. (2009). Amoeboid T lymphocytes require the septin cytoskeleton for cortical integrity and persistent motility. Nat Cell Biol , 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinayak S, Pawlowic MC, Sateriale A, Brooks CF, Studstill CJ, Bar-Peled Y, Cipriano MJ, Striepen B. (2015). Genetic modification of the diarrhoeal pathogen Cryptosporidium parvum. Nature , 477–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Casey PJ. (2016). Protein prenylation: unique fats make their mark on biology. Nat Rev Mol Cell Biol , 110–122. [DOI] [PubMed] [Google Scholar]
- Wilson KS, Gonzalez O, Dutcher SK, Bayly PV. (2015). Dynein-deficient flagella respond to increased viscosity with contrasting changes in power and recovery strokes. Cytoskeleton (Hoboken) , 477–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woznica A, Cantley AM, Beemelmanns C, Freinkman E, Clardy J, King N. (2016). Bacterial lipids activate, synergize, and inhibit a developmental switch in choanoflagellates. Proc Natl Acad Sci USA , 7894–7899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woznica A, Gerdt JP, Hulett RE, Clardy J, King N. (2017). Mating in the closest living relatives of animals is induced by a bacterial chondroitinase. Cell , 1183.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang TT, Cheng L, Kain SR. (1996). Optimized codon usage and chromophore mutations provide enhanced sensitivity with the green fluorescent protein. Nucleic Acids Res , 4592–4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young SL, Diolaiti D, Conacci-Sorrell M, Ruiz-Trillo I, Eisenman RN, King N. (2011). Premetazoan ancestry of the Myc–Max network. Mol Biol Evol , 2961–2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.