

Abstract
Tumor initiation and growth is associated with significant changes in the surrounding tissue. During carcinoma progression, a global stiffening of the extracellular matrix is observed and is interpreted as a signature of aggressive invasive tumors. However, it is still unknown whether this increase in matrix rigidity promotes invasion and whether this effect is constant along the course of invasion. Here we have developed a biomimetic in vitro assay that enabled us to address the question of the importance of tissue rigidity in the chronology of tumor invasion. Using low concentrations of the sugar threose, we can effectively stiffen reconstituted collagen I matrices and control the stiffening in time with no direct effect on residing cells. Our findings demonstrate that, depending on the timing of its stiffening, the extracellular matrix could either inhibit or promote cancer cell invasion and subsequent metastasis: while matrix stiffening after the onset of invasion promotes cancer cell migration and tumor spreading, stiff matrices encapsulate the tumor at an early stage and prevent cancer cell invasion. Our study suggests that adding a temporal dimension in in vitro models to analyze biological processes in four dimensions is necessary to fully capture their complexity.
INTRODUCTION
The tumor microenvironment is characterized by an abnormal synthesis of extracellular matrix (ECM) components and an overall increase of matrix stiffness (Paszek et al., 2005; Levental et al., 2009; Attieh and Vignjevic, 2016). Such severe changes in the microenvironment are integrated at the cellular level. As a result, the transition from a benign to a malignant tumor is not induced by tumor internal signaling alone but results from a dynamic cross-talk between the tumor and its microenvironment (Attieh and Vignjevic, 2016).
In vivo, massive deposition of collagen and enzymatic collagen cross-linking by lysyl oxidase (LOX) are responsible for tumor matrix stiffening (Levental et al., 2009; Barker et al., 2012). However, their impact on tumor progression is not restricted to a mechanical effect. The increase in collagen density also correlates with a higher number of binding sites available and therefore modifies tumor cell adhesion and migration, while LOX also acts as an intracellular factor regulating genes responsible for epithelial–mesenchymal transition (Levental et al., 2009; Barker et al., 2012; Attieh and Vignjevic, 2016).
Although stiff matrices are a signature of aggressive tumors, the causal connection between matrix stiffness and tumor aggressiveness has never been tested. Does matrix stiffening induce cancer cell invasion or is it a consequence of tumor spreading? And is the effect of matrix stiffness on cancer cell invasion constant in time or do cells respond differently to mechanical changes in their environment depending on whether these happen during early or advanced stages of invasion?
To address these questions, in vitro models are valuable tools, as they are less complex than the in vivo microenvironment and offer the ability to decouple the different parameters at play in cancer invasion. Here we show a new approach by which we can modify matrix stiffness alone at different time points during tumor invasion. We use threose, a sugar and natural collagen cross-linker (Kinnunen et al., 2012) that is more effective than the traditional cross-linking agent ribose. As threose does not affect cell autonomous adhesion, migration, and force generation, we can modulate the stiffness of collagen gels without affecting cancer cells embedded in the gel. Our results expose the importance of the temporal dimensions of biological processes and reveal that matrix stiffening inhibits invasion at an early stage but enhances it at later stage.
RESULTS AND DISCUSSION
Threose stiffens collagen more efficiently than ribose
In vitro models of tumor–matrix interactions ideally need to recapitulate the fibrillar structure, rheological properties, and the nature and spatial distribution of ligands of the in vivo ECM to provide biologically relevant insights (Verhulsel et al., 2014). We therefore chose collagen I for our artificial matrix, as it supports the growth of cells in three dimensions while closely mimicking in vivo fibrillar ECMs and reproducing cell–matrix interactions specific to tumors (Wolf et al., 2009; Geraldo et al., 2012; Wolf et al., 2013). Among the different options published in the literature to stiffen collagen (Tanaka et al., 1988; Paszek et al., 2005; Levental et al., 2009), glycation and LOX cross-linking are the only ones also occurring spontaneously in vivo. As LOX activity is known to modulate gene expression, glycation occurring between glucose and collagen better meets our needs for a physiological environment (Tanaka et al., 1988). Ribose is mostly used as a glucose substitute in vitro, but the high concentrations and long incubation time required to significantly stiffen the collagen matrix are presumed to induce a diabetic phenotype in cells (Han et al., 2011).
To overcome this limitation, we tested an alternative collagen cross-linker, threose, which efficiently forms cross-links in bovine articular cartilage (Kinnunen et al., 2012). We incubated collagen gels with threose after polymerization to avoid any interference with fibrillogenesis and minimize potential changes in network architecture. To test whether threose is a more effective cross-linker than ribose, we compared the rheological properties of 2 mg/ml collagen gels alone or supplemented for 48 h either with 1 mM of threose or with 1 or 10 mM of ribose. At concentrations of 1 mM, the elastic modulus of collagen treated with ribose was similar to control, whereas threose treatment induced a 1.8-fold increase in stiffness (Figure 1A). At a concentration of 10 mM, ribose stiffened collagen to the same extent as 1 mM threose (Figure 1A).
FIGURE 1:

Threose stiffens collagen more efficiently than ribose. (A) Elastic moduli obtained with small-amplitude oscillatory rheology for untreated collagen gels, gels treated with 1 or 10 mM of ribose or with 1 mM threose, 48 h after polymerization. Data represent an average over at least three independent measurements. The p values are compared with control condition using Dunnett’s multiple comparison test (*p < 0.05). (B) Measurements of elastic moduli as a function of time over a period of 48 h obtained with small amplitude oscillatory rheology on untreated collagen gels, gels treated with 1 or 10 mM of ribose, or with 1 mM threose. Results are expressed as mean ± SEM obtained as an average over at least three independent measurements. The p values are compared with control condition using Dunnett’s multiple comparison test (****p < 0.0001). (C) Left, maximum intensity projections of reflectance and fluorescence images of collagen networks with or without 1 mM of threose added during 48 h. Scale bar = 10 µm. Right, mesh size measurements of collagen networks. Results are represented as a histogram with mean + SEM for n = 8 positions over n = 3 independent samples. The p values were calculated using an unpaired t test for each separate condition and showed no statistical difference. The mesh size measured with fluorescence microscopy is smaller because confocal reflectance does not allow for visualization of fibers perpendicular to the imaging plane. (D) Mesh size distribution within collagen networks acquired by fluorescence. Results are represented as box and whiskers (minimum to maximum), where each point represents a different region within the matrix, with n > 20 positions over n = 3 independent samples. The p values were calculated using an unpaired t test for each separate condition and showed no statistical difference.
These results suggest that threose is effective at lower concentrations than ribose, which is advantageous, as lower sugar concentrations diminish the likelihood to induce a diabetic phenotype or cause a hypertonic stress to cells.
In addition, monitoring the development of the elastic modulus of collagen gels in time revealed that ribose treatment had a delayed impact on collagen cross-linking compared with threose (Figure 1B). Measurements of the mesh size of the networks by reflectance microscopy validated that the architecture of collagen networks was not affected by threose (Figure 1C). This was confirmed by fluorescence microscopy imaging, although this yielded overall smaller mesh sizes than reflectance imaging because fluorescence microscopy, unlike reflectance imaging, visualizes also fibers orthogonal to the plane of imaging (Figure 1C). Whether in control or threose-treated gels, mesh sizes similarly varied between 2 and 5 µm, indicating that threose did not induce heterogeneous changes in ECM architecture (Figure 1D). Furthermore, we confirmed that the addition of threose did not affect fiber structure by using turbidimetry to compare fiber radii in the absence and presence of threose, which were, respectively, 90 ± 7 and 88 ± 7 nm. Altogether, this characterization shows that a low concentration of threose is effective in changing the stiffness of collagen gels without altering network architecture.
Collagen stiffening before the onset of invasion inhibits cancer cell invasion
We first considered the influence of matrix stiffening at an early stage, when cancer cell invasion has not yet started. To model tumor invasion in vitro, we mixed spheroids of CT26 intestinal adenocarcinoma invasive cells with 2 mg/ml collagen solutions. Once collagen polymerized, gels containing spheroids were incubated in 1 mM of threose for a 24- or 48-h treatment (Figure 2A, Thr_24 h and Thr_48 h). Invasion was quantified 3 d after embedding. The number of cells that invaded out of the spheroid was counted using a three-dimensional automated software. This quantification method generates an invasion index that represents the number of invading cancer cells normalized to the surface area of the spheroid contour. Therefore, we only scored active cell migration and did not discriminate spheroids based on their size (Attieh et al., 2017).
FIGURE 2:

Collagen stiffening before the onset of invasion inhibits cancer cell invasion. (A) Timeline representing the chronology of the experiment. Cancer cell were plated in agarose-coated wells for 3 d to form spheroids in the absence (Control) or presence of threose (Thr_incubation). Spheroids were then embedded in collagen droplets with or without threose. Threose was washed out 24 h (Thr_24 h) and 48 h (Thr_48 h) later. All spheroids were fixed at day 3 postembedding. (B) Maximum intensity projections of cancer cell spheroids at day 3. F-actin (red) and DNA (cyan) were respectively stained with phalloidin-rhodamin and DAPI. Scale bar indicates 100 µm. (C) Quantification of cancer cell invasion. Invasion index is defined as the ratio between the number of invading nuclei of cancer cells and the area of the spheroid contour. Data are expressed as box and whiskers (minimum to maximum) of at least N = 3 separate experiments. The p values are compared with control condition using Newman–Keuls multiple comparison test (*p < 0.05). (D) Quantification of single-cell invasion/migration in three-dimensional collagen gels (left: speed; right: persistence) ± 1 mM of threose, after a 48 h incubation in threose. Persistence was defined as the final cell displacement divided by the length of the trajectory. Results are represented as box and whiskers (minimum to maximum) where each point represents an individual cell. The p value was calculated using Mann–Whitney test for n = 107 cells for control and n = 105 cells for threose over n = 3 separate experiments (***p < 0.001). (E) Quantification of migration (left: speed; right: persistence) of CT26 cancer cells with increasing concentrations of threose treatment plated on glass. Measurements were performed during threose treatment and after wash. Migration persistence was defined as the final cell displacement divided by the length of the trajectory. Results are represented as a histogram with mean + SD for three independent experiments. The p values were calculated using Newman–Keuls multiple comparison test and showed no statistical difference. (F) Quantification of the capacity of control cells to adhere to collagen (control, gray) or to threose-treated collagen (Coll_thr, green) and of threose-treated cells to adhere to control collagen (Cells_thr, blue) or to threose-treated collagen (Cells_thr + coll_thr, red). Measurements were performed at 0, 15 min, 30 min, 1 h, and 2 h. Results are expressed as an XY coordinate with mean ± SD with data representing an average over three independent measurements. The p values are compared with control condition using Dunnett’s multiple comparison test and showed no statistical difference.
Matrix stiffening before the onset of invasion dramatically inhibited cell invasion when collagen was treated with threose compared with control (Figure 2, B and C). Treating collagen with threose for an additional day did not further impede the invasion capacity of cancer cells, as it was already almost completely blocked on a 24 h treatment (Figure 2, B and C). Consistent with these findings, CT26 cancer cells invaded collagen gels with slower speed in the presence of threose when embedded as single cells (Figure 2D). Cell persistence was similar in both conditions, indicating that it was the cells’ capacity to move rather than the directional persistence of their movement that was compromised in a stiffer matrix (Figure 2D).
Inhibition of invasion could be due to an increase in matrix stiffness or to a direct effect of threose on cells. To check whether threose impeded the invasive phenotype of cells, CT26 were preincubated in 1 mM threose for 3 d while spheroids were forming (Figure 2A, Thr_incubation). Invasion was similar for preincubated and control spheroids, indicating that threose did not alter the invasive phenotype of cancer cells (Figure 2, B and C).
We further verified whether threose could alter the molecular machinery driving cell migration by plating cancer cells on glass as an inert substrate that cannot be glycated and treated with increasing concentrations of threose (Figure 2E). Cells migrated with equivalent speed and persistence regardless of threose concentration, suggesting that threose did not affect the migratory phenotype of cancer cells.
Alternatively, threose could decrease adhesion of cells to collagen. We tested the latter hypothesis by plating cells on two-dimensional collagen matrices for increment time points and measuring the percentage of adhered cells after wash (Figure 2F). We found that cell adhesion was unaffected whether collagen (Coll_thr), cells (Cells_Thr), or both (Cells_Thr + Coll_Thr) were treated with threose compared with control, suggesting that threose modified neither collagen adhesion sites nor the adhesion capacity of cancer cells (Figure 2F). We thus concluded that the inhibition of invasion at an early stage was due only to matrix stiffening.
Collagen stiffening before the onset of invasion prevents fiber alignment by cancer cells
Previous reports have shown that prior to invasion, cancer cells contract and align collagen fibers in the direction of their movement (Provenzano et al., 2009; Riching et al., 2014; Kopanska et al., 2016). Once collagen is aligned, cancer cells migrate in a fast and persistent manner, promoting tumor spread (Riching et al., 2014). In in vitro models, collagen fibers are initially aligned parallel to the edge of the tumor (Kopanska et al., 2016). To evaluate whether cells remodeled the matrix, we imaged collagen I fibers surrounding spheroids 3 d postpolymerization and quantified fiber orientation of control and threose-treated gels. Imaging of collagen I revealed that noncross-linked collagen fibers were not parallel to the edge of the tumor and were randomly organized, while fiber alignment parallel to the spheroid was maintained in threose-treated samples (Figure 3A). This observation was validated by the quantification of the orientation of collagen fibers with respect to the spheroid edge (Figure 3B). In threose-treated collagen gels, fibers were mainly parallel to the spheroid, whereas this tendency was less pronounced in control matrices as the proportion of fibers oriented between 0 and 30° was 20% lower (Figure 3B). We thus concluded that 3 d after spheroid embedding, cells started remodeling collagen fibers in control gels but not in threose-treated samples.
FIGURE 3:

Collagen stiffening before the onset of invasion prevents fiber alignment by cancer cells. (A) Two-dimensional slices of cancer cell spheroids in collagen I at day 3. F-actin (red) was stained with phalloidin-rhodamine and collagen was imaged using reflectance (cyan). Right panel, overlaid images of collagen I matrices containing cancer cell spheroids generated using the software CurveAlign (UW–Madison; http://loci.wisc.edu/software/curvealign). Yellow line indicates the edge of the spheroid, and green lines indicate fibers orientation with respect to the closest point on the spheroid edge. Scale bar = 100 µm. (B) Rose plots representing the frequency of distribution of the absolute angles of collagen fibers within the range of 0°–90° with respect to the closest point on the spheroid edge. Data represent the average of 7 < n < 8 spheroids. Fibers parallel to the tumor edge lie in the 0°–30° angles. (C) Left, representative traction force map of a control and a threose-treated cell on collagen-coated polyacrylamide gels with Young’s modulus of 5 kPa. White dotted line represents the cell’s edge. Color code gives the magnitude of traction stress in Pa, which corresponds to forces of pN/µm2. Scale bar = 20 µm. Right, corresponding mean force (strain energy) exerted by CT26 cancer cells. Results are represented as box and whiskers (minimum to maximum), where each point represents an individual cell. The p value was calculated using Mann–Whitney test for n = 27 cells for control and n = 25 cells for threose over three separate experiments and showed no statistical difference.
Impaired matrix remodeling observed in threose-treated hydrogels could be either due to a stiffer matrix or to a direct effect of threose on cell mechanisms regulating contractile forces. To test the latter hypothesis, we performed traction force microscopy on cells plated on polyacrylamide gels containing fluorescent beads. Both control and threose-treated cells exerted comparable traction forces, indicating that threose does not affect traction force generation and that matrix stiffening alone prevented matrix remodeling (Figure 3C).
Altogether these results suggest that matrix remodeling is necessary for cancer cells to migrate out of the spheroid and that a stiffer matrix inhibits this process.
Collagen stiffening after the onset of invasion favors cancer cell invasion
The reduced invasion we observed in a stiffer environment is surprising, as aggressive tumors are typically characterized by high tissue rigidity. However, it is still not known whether the matrix gets stiffer before or after cancer cell invasion (Attieh and Vignjevic, 2016). Matrix stiffening after the onset of invasion might favor cell invasion, as the critical step of matrix remodeling and fiber alignment is then already complete. To test this hypothesis, we embedded spheroids in collagen and monitored how the invasion rate would change when threose was added after invasion started (i.e., from day 3 to day 5 after embedding spheroids in collagen) (Figure 4A, Thr_d3d5). Stiffening after the onset of invasion was compared with early stiffening (Thr_d0d2) and control. Experiments were extended to 7 d after embedding spheroids in collagen to allow for cells to properly invade out of the spheroid. Similarly to the shorter-term experiments, preincubating cancer cells in threose (Figure 4A, Thr_incubation) did not affect their invasion index compared with control, confirming that threose at a concentration of 1 mM does not alter cancer cells’ invasive phenotype even at longer times (Figure 4, B and C).
FIGURE 4:

Collagen stiffening after the onset of invasion favors cancer cell invasion. (A) Timeline representing the chronology of the experiment. Cancer cell spheroids were left to form in agarose-coated wells for 3 d with (Thr_incubation) or without (Control) threose. Spheroids were then embedded in collagen droplets with (Thr_d0d2) or without (Control) threose for 48 h. Three days postembedding, threose was added in the media for 48 h (Thr_d3d5). All spheroids were fixed at day 7 postembedding. (B) Maximum intensity projections of cancer cell spheroids at day 7. F-actin (red) and DNA (cyan) were respectively stained with phalloidin-rhodamin and DAPI. Scale bar indicates 100 µm. (C) Quantification of cancer cell invasion. Invasion index is defined as the ratio between the number of invading nuclei of cancer cells and the area of the spheroid contour. Quantification results are expressed as box and whiskers (minimum to maximum) of N = 3 separate experiments. The p values are compared with control condition using Newman–Keuls multiple comparison test (**p < 0.01). (D) Model, tumor cells are surrounded by the ECM (a). Cancer cells start invading the ECM before the matrix gets remodeled (b) rather than the matrix being remodeled beforehand (b′). As cancer cells invade the matrix, collagen fibers get aligned (c), which sets up positive feedback to enhance invasion.
Surprisingly, cancer cell invasion was increased twofold when the matrix was stiffened after the onset of invasion compared with control (Figure 4, B and C, Thr_d3d5). Moreover, we observed that the decrease of cell invasion observed at day 3 on threose treatment, before the onset of invasion (Figure 2, B and C), vanished when cancer cells were examined at day 7 (Figure 4, B and C, Thr_d0d2): cells showed the same final invasion state as cells invading control gels after 7 d of culture in collagen droplets. This observation indicates that, although matrix stiffening slows down the initiation stage, it enhances cell migration once cells invade the matrix. Given that threose does not interfere with the autonomous migratory phenotype of cancer cells (Figure 2E), we concluded that matrix stiffening enhances cell migration, resulting in extensive invasion at later stages of tumor development.
Altogether, these experiments highlight the importance of the chronology of events in cancer cell invasion. Depending on the stage of the tumor, the changes in the mechanical properties of its microenvironment could differentially influence its aggressiveness. Matrix stiffening before invasion is initiated slows down the transition from noninvasive to invasive tumor stage. On the contrary, stiffening of the matrix after the onset of tumor invasion intensifies tumor invasion. We propose here a model in which, within the chronology of cancer development, cancer cells migrate out of the tumor site, followed by a stiffening of the matrix (Figure 4D).
We have thus uncovered a novel phenotype of invading tumor cells by effectively modulating matrix stiffness in time without altering cancer cells’ autonomous phenotype. We have developed a physiological tumor model composed of spheroids embedded in three-dimensional collagen matrices and selected a natural cross-linker to stiffen collagen. This in vitro model allowed us to directly test the correlation between matrix stiffness and tumor aggressiveness observed in patients (Cox and Erler, 2011; Pickup et al., 2014; Acerbi et al., 2015).
As opposed to other cross-linkers commonly used in the field, threose presents the advantage of effectively stiffening collagen at low concentrations. As proven by our experiments, a strong advantage of using threose is that it does not induce changes in the network architecture in terms of mesh size and fibril diameter, while being effective in changing the stiffness of the matrix. This increase in stiffness is most likely due to a stabilization and an increase in cross-linking of already existing contact points.
Using threose at low concentrations allowed us to modify matrix stiffness while tumor cells were embedded in their physiological three-dimensional matrix, as opposed to studies exposing cancer cells to previously treated and already stiff gels or collagen gels of varying densities that cannot be modulated in time (Paszek et al., 2005; Levental et al., 2009; Provenzano et al., 2009; Krndija et al., 2010). We were thus able to control the temporal onset of matrix stiffening. Although we worked at lower collagen concentration and thereby at lower stiffness than analogous studies using ribose, cells were highly responsive, suggesting that cells are more sensitive to relative changes in matrix rigidity than to its intrinsic value (Bordeleau et al., 2017; Carey et al., 2017).
At an early stage of invasion, cells first remodel the matrix to effectively migrate out of the tumor niche. Contractile forces of cancer cells are required to deform the ECM and lead to tensile radial forces within the matrix that orient the collagen fibers perpendicular to the surface of the tumor (Provenzano et al., 2009; Riching et al., 2014; Kopanska et al., 2016). As there is a mechanical relation between the tensile state of the ECM and invasion, collagen organization at the onset of invasion plays a critical role in the outcome of tumor development. Our results support this hypothesis, as threose-treated matrices are stiffer and therefore more difficult to remodel than control matrices. This is evidenced by the high proportion of collagen fibers oriented parallel to the edge of the tumor 3 d after embedding and the consequent inhibition of cancer cell invasion.
Stiffer matrices are also known to favor a migratory phenotype in cancer cells (Paszek et al., 2005; Levental et al., 2009). This biomechanical process is consistent with the cell response observed when we added threose after the onset of invasion, which led to a twofold increase in invasion. This antagonistic influence of matrix stiffening in the early and late stages of invasion is clearly illustrated by the behavior of cancer cells when collagen is exposed to threose in the early stage: although the transition from noninvasive to invasive phenotype is delayed compared with control, once invasion is initiated, migration is enhanced as cancer cells catch up with control conditions within 4 d of invasion. As CT26 are invasive cancer cells with a mesenchymal-like phenotype, this result is consistent with new findings suggesting that only malignant cancer cells have the ability to adjust to collagen matrices of different densities (Wullkopf et al., 2018).
In conclusion, our in vitro three-dimensional culture model provides a physiologically relevant microenvironment to study the effect of matrix stiffness per se on the transition from benign to malignant tumor. We report here the first data on in vitro collagen cross-linking using threose, which allows us to significantly stiffen collagen matrices without interfering with the phenotype of cancer cells. Furthermore, this study highlights the need to introduce a fourth dimension into cell biological models. Indeed, a static assay where all factors are initially set and remain unchanged could fail to capture the complexity of cancer invasion. The flexibility of our system allowed us to tackle the question of the dynamics of matrix stiffening during tumorigenesis. Our study illustrates that within the chronology of tumor development, neither matrix stiffening nor softening is the most favorable but rather an interchanging state that is used by cancer cells to efficiently invade.
MATERIALS AND METHODS
Rheological measurements
Rheology tests were performed on an Anton Paar Physica MCR 501 rheometer, with a stainless steel 30-mm-diameter and 1° truncation angle cone-plate geometry (CP30-1). The plates were heated to 37° before loading the sample. Water was added to the solvent trap to maintain a moist environment. Twenty minutes after loading the sample, a solution of either pure DMEM or DMEM supplemented with 1–10 mM ribose or 1 mM threose was placed around the geometry and allowed to diffuse in the sample for 48 h. To prevent evaporation during the measurement, a layer of mineral oil was further added around the measuring geometry. During the whole 48 h, the elastic and viscous moduli of the network were probed by applying an oscillatory deformation with 0.5% strain amplitude and a frequency of 0.5 Hz. Results show the values of elastic and viscous moduli after 48 h, averaged over at least three different independent repeats, while the error reported is the standard error of the mean.
Sample preparation
The samples were prepared on ice to prevent early polymerization of collagen. First, collagen was pipetted into an Eppendorf tube and weighed to determine the volume of the final solution. Samples were prepared to have a final collagen concentration of 2 mg/ml, in phosphate buffer saline (PBS) and pH set to 7 with addition of NaOH and topped up to the final volume with DMEM.
Imaging three-dimensional collagen networks
The pure and threose-supplemented collagen networks were imaged in confocal reflectance and confocal fluorescence mode using an inverted Eclipse Ti microscope (Nikon, Tokyo, Japan), a 488-nm Argon laser (Melles Griot, Albuquerque, NM) for illumination, and a 100× objective (Nikon, N.A. 1.49). Z-stacks were acquired 10 μm above the coverslip to avoid surface effects, over a depth of 10 μm, with a step of 0.2 μm and shown as a maximum projection. The networks were allowed to polymerize for 48 h at 37°C with controlled humidity. To visualize fluorescent collagen, we added the collagen binding protein CNA35 (Aper et al., 2014) tagged with green fluorescent protein (pET28a-EGFP-CNA35 was a gift from Maarten Merkx, TU Eindhoven [Addgene plasmid # 61603]) to collagen prior to polymerization in a molar ratio of 20:1.
Mesh size analysis
The mesh size of the networks was determined using an algorithm described in Kaufman et al. (2005). Images were background subtracted and binarized with an Otsu threshold using ImageJ. The resulting image was then analyzed in a custom-written Python routine, which counted the distance between on and off pixels along the rows and columns. The distances obtained were then fitted with an exponential probability distribution, and the mean value (converted from pixels to µm) was taken as the average mesh size. For each condition, we analyzed at least eight randomly sampled images.
Fiber diameter analysis
To analyze the average diameters of the collagen fibers, we performed turbidity measurements with a Lambda 35 dual-beam spectrophotometer (PerkinElmer, Waltham, MA). Samples were polymerized inside a plastic cuvette (UV-Cuvette micro; Plastibrand, Germany) or a quartz cuvette for 48 h before measuring. Subsequently, the optical density of the sample was measured in the wavelength range 650–900 nm. The measured optical density I0 was converted into turbidity τ using the formula
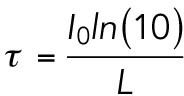 |
where L represents the optical path length. To extract fiber radii and mass-length ratios, we employed the following relation (Yeromonahos et al., 2010):
where A and B are constants corresponding respectively to (88/15)cpπ3ns(dn/dcp)2/NA and (184/154)π2n2s. Here cp represents the collagen concentration, ns the refractive index of the solvent, dn/dcp is the refractive index increment, NA is the Avogadro constant, μ is the mass per length ratio, and a is the fiber radius.
Cell lines
Mouse intestinal cancer cells CT26 were obtained from the American Type Culture Collection. Cells were cultured in DMEM (Life Technologies) supplemented with 10% fetal bovine serum (FBS) (Invitrogen) and 5% CO2. Mycoplasma testing was performed every 2 wk to verify that cells were clear of infections.
Invasion assay
Agarose (Invitrogen) was dissolved in water to a concentration of 0.01 g/ml and boiled. Of the solution, 150 µl was added to wells of a 48-well plate, and agarose was left to polymerize for at least 10 min at room temperature (RT). A solution of 104 cells/ml CT26 cancer cells was made, and 75–100 µl of the solution was added to the wells. The wells were subsequently filled with DMEM supplemented with 10% FBS ± threose (1 mM, T7642, Sigma), and spheroids were left to form for 3–4 d.
Tissue culture plates (30 mm2) were specifically fashioned for the invasion assay: three holes of ∼3–4 mm in diameter were drilled in a plate and widened around the edges using a scalpel.
Rat tail collagen I (2 mg/ml; Corning) was prepared in DMEM, 10X PBS, and 1 M NaOH, to pH = 7, as described under Sample preparation. The solution was kept on ice to avoid collagen polymerization. Spheroids were embedded in 15-µl collagen drops, positioned in the hole of the culture plate. After filling all three holes, the plate was flipped every 30 s for 5 min for the cells to stay in the middle of the collagen drop (preventing sedimentation of spheroids to the glass or to collagen/air interface). Collagen was left to polymerize for an additional 15 min at RT before 3 ml of DMEM supplemented with 5% FBS, 1% antibiotic antimycotic (AA) was added ± 1 mM of threose.
Three-dimensional. immunofluorescence
Spheroids embedded in collagen were fixed using 4% paraformaldehyde in PBS for 30 min at RT. Spheroids were then washed with PBS and permeabilized with 0.1% Triton X-100 in PBS for 30 min at RT. DNA and F-actin were stained using 4′,6-diamidino-2-phenylindole (DAPI) and phalloidin, respectively (Life Technologies). Collagen was imaged using confocal reflection microscopy.
Imaging three-dimensional spheroids
Images were acquired with an inverted AOBS two-photon laser scanning confocal microscope SP8 (Leica) coupled to femtosecond laser Chameleon Vision II (Coherent Inc) using a 25×/1.0NA water-immersion objective. The microscope is equipped with two nondescanned hybrid detectors: NDD1 (500–550 nm) and NDD2 (≥590 nm). Fluorescence channels were recorded simultaneously using the excitation wavelength 980 nm. Collagen was visualized by confocal reflectance microscopy using light at a wavelength of 488 nm and a standard photomultiplier tube detector at a low gain (500 V). Three-dimensional stacks were obtained at a step size of 2-µm intervals. The images were processed with Leica Application Suite (LAS), ImageJ (National Institutes of Health) and Imaris (Bitplane).
Invasion counter software
Quantification of cell invasion from spheroids was performed as already described using a custom semiautomated image analysis program written in Python (Attieh et al., 2017).
Cell adhesion assay
CT26 cells were pretreated for 2 d with 1 mM of threose or no threose in DMEM supplemented with 10% FBS and 1% AA. Collagen (50 µl) (2 mg/ml) was polymerized in 96-well plates and pretreated for 2 d with 1 mM of threose or no threose in DMEM and 1% AA. After 2 d, CT26 cells were stained with CellTracker Red CMTPX Dye (ThermoFisher) and 2 × 105 cells were seeded per well in the 96-well plate. Cells were then incubated at 37°C for 5 min, 15 min, 30 min, 1 h, or 2 h and washed three times to remove nonadherent cells. Fluorescence intensity was measured using a FluoStar fluorescence plate reader (BMG Labtech). Adhesion was represented as the percentage of adherent cells per condition.
Collagen topography measurements
Fibers alignment and their angles with respect to the spheroid edge were measured on images acquired using reflection microscopy with the available software CurveAlign (UW–Madison; http://loci.wisc.edu/software/curvealign) in MatLab. The angles of collagen fibers compared with the spheroid edge were determined for seven to eight slices per condition.
Traction force microscopy
Traction force microscopy was conducted as previously described (Elkhatib et al., 2014).
Glass-bottom cell culture dishes (35 mm; FluoroDish, World Precision Instruments) were silanized with (3-aminopropyl)trimethoxysilane (Sigma-Aldrich) for 15 min and extensively washed with water. The glass surface of the dishes was treated with 0.5% glutaraldehyde (Electron Microscopy sciences) for 30 min and extensively washed with water. Acrylamide 40% (93.3 µl) and 2% bisacrylamide (11 µl) were mixed in PBS solution to a final volume of 500 µl to achieve a Young’s modulus of 5 kPa. For traction force measurements, FluoSphere bead solution (0.2 μm, 580/605 nm; Invitrogen) was added at 2% volume. Polymerization was initiated by addition of 2.5 µl ammonium persulfate (10% wt/vol solution) and 0.25 μl of N,N,N’,N’-tetramethylethane-1,2-diamine. Polyacrylamide solution (12 µl) was rapidly deposited onto the glass-bottom dish and covered with a 18-mm coverslip. After 1 h, PBS was added to the dishes for 10 min, and the coverslips were gently removed under PBS. To allow for collagen coating, the gel surface was activated using Sulfo-SANPAH (1 mg/ml; ThermoFisher) that was photoactivated by ultra-violet (365 nm) for 10 min. After three washes with HEPES and PBS, the gels were coated with monomeric collagen (100 µg/ml) overnight at 4°C. The next day gels were washed three times with PBS. CT26 cells were pretreated for 1 d ± 1 mM threose prior to seeding on the gels. CT26 cells (1 × 104) were plated per gel in DMEM supplemented with 10% FBS, 2% AA, and ± 1 mM threose and cultured on the gels one additional day, thus allowing for a total 2-d incubation ± threose.
To image traction force experiments, we used an Inverted Nikon Ti-E microscope equipped with a scientific complementary metal-oxide semiconductor (sCMOS) 2048 ORCA Flash4.0 V2 camera (Hamamatsu) and a 40× Plan Fluor dry objective (0.75 NA; Nikon). A fluorescent image of the FluoSphere beads and a phase contrast image of the cells were acquired. At the end of the acquisition, cells were detached from the gels using 0.5% Trypsin-EDTA (Life Technologies), and a reference image without cells was recorded. We used a previously described correlation algorithm to extract the bead displacement fields. Traction force was determined using the Fourier transform traction force algorithm, as introduced by Butler et al. (2002). To quantify the force applied by a cell, we measured the strain energy that corresponds to the energy the cell exerts to deform the substrate, which is proportional to the average force applied by a cell.
Two-dimensional. migration
CT26 cells were incubated for 2 d with the following concentrations of threose: 0, 0.5, 1, and 2 mM in DMEM supplemented with 10% FBS and 1% AA. After 2 d, CT26 cells were resuspended and 2500 cells were seeded on uncoated glass-bottom wells of a 96-well plate. After overnight imaging, all the wells were washed and DMEM 10% FBS 1% AA was added, and cell migration was imaged for 8 more hours.
Three-dimensional. migration of single cells
Rat tail collagen I (2 mg/ml; Corning) was prepared in DMEM, 10X PBS, and 1 M NaOH, to pH = 7 as described under Sample preparation. The solution was kept on ice to avoid collagen polymerization. Glass-bottom cell culture dishes (FluoroDish, World Precision Instruments) were coated with poly-l-lysine (Sigma-Aldrich) for 5 min and washed three times with PBS. CT26 cells were resuspended, and 5000 cells were embedded in 40 µl collagen drops that were deposited on the bottom of the culture dishes. Dishes were flipped for 1.5 min for the cells to stay in the center of the collagen drop. Collagen was left to polymerize for an additional 30 min at RT before 2 ml of DMEM supplemented with 10% FBS, 1% AA was added ± 1 mM of threose. Dishes were maintained for 2 d at 37°C, 5% CO2.
Microscopy of CT26 migration and image analysis
Two- and three-dimensional cultures were imaged using an Inverted Nikon Ti-E microscope equipped with a sCMOS 2048 ORCA Flash4.0 V2 camera (Hamamatsu) using the following objectives: Plan Fluor 10x Ph1 DL and Plan Apo VC 20x DIC N2. Images were acquired every 10–20 min for 8–12 h.
A random population of cells was selected and manually tracked in two-dimensional using the MTrackJ plugin (Meijering et al., 2012) in ImageJ (Bitplane). Following tracking of cells, the trajectories were analyzed using custom software written in Python. Migration persistence was defined as the final cell displacement divided by the length of the trajectory.
Statistical analysis
All experiments were performed in a minimum of three independent experiments. All statistical analysis and graphic representations were performed using Prism software.
Acknowledgments
We thank Andrew G. Clark for designing the three-dimensional quantification software, Carlos Perez-Gonzalez for help with the generation of roseplots, Marko Matic for help with quantification of two-dimensional migration data, and Nadia Elkhatib and Jorge Barbazan for help with TFM and cell adhesion. We acknowledge Nikon Imaging Center@CNRS-Institut Curie, PICT-IBiSA@Lhomond supported by the Fondation pour la Recherche Médicale (FRM No. DGE20111123020), the Cancérôple-IdF (No. 2012-2-EML-04-IC-1), and InCA (Cancer National Institute, No. 2011-1-LABEL-IC-4). We also thank La ligue contre le cancer (Y.A. and M.V.), Fondation pour la Recherche Médicale (FRM FDT20170437130), and Ecole Doctorale Frontières du Vivant (F.d.V.)—Programme Bettencourt (R.S.). The work of F.B. and G.H.K. is part of the Industrial Partnership Programme Hybrid Soft Materials that is carried out under an agreement between Unilever Research and Development B.V. and the Netherlands Organisation for Scientific Research (NWO). This work was supported by Labex CelTisBioPhy (Y.A. and M.V.). This article is dedicated to the memory of Maxime Dahan, director of the UMR 168, who recently passed away. As a member of the Labex CelTisPhyBio steering committee, he supported this study through grant funding.
Abbreviations used:
- ECM
extracellular matrix
- GFP
green fluorescent protein
- LOX
lysil oxidase
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E18-01-0068) on October 10, 2018.
REFERENCES
- Acerbi I, Cassereau L, Dean I, Shi Q, Au A, Park C, Chen YY, Liphardt J, Hwang ES, Weaver VM. (2015). Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integr Biol (Camb) , 1120–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aper SJ, van Spreeuwel AC, van Turnhout MC, van der Linden AJ, Pieters PA, van der Zon NL, de la Rambelje SL, Bouten CV, Merkx M. (2014). Colorful protein-based fluorescent probes for collagen imaging. PLoS One , e114983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attieh Y, Clark AG, Grass C, Richon S, Pocard M, Mariani P, Elkhatib N, Betz T, Gurchenkov B, Vignjevic DM. (2017). Cancer-associated fibroblasts lead tumor invasion through integrin-beta3-dependent fibronectin assembly. J Cell Biol , 3509–3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attieh Y, Vignjevic DM. (2016). The hallmarks of CAFs in cancer invasion. Eur J Cell Biol , 493–502. [DOI] [PubMed] [Google Scholar]
- Barker HE, Cox TR, Erler JT. (2012). The rationale for targeting the LOX family in cancer. Nat Rev Cancer , 540–552. [DOI] [PubMed] [Google Scholar]
- Bordeleau F, Mason BN, Lollis EM, Mazzola M, Zanotelli MR, Somasegar S, Califano JP, Montague C, LaValley DJ, Huynh J, et al (2017). Matrix stiffening promotes a tumor vasculature phenotype. Proc Natl Acad Sci USA , 492–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler JP, Tolic-Norrelykke IM, Fabry B, Fredberg JJ. (2002). Traction fields, moments, and strain energy that cells exert on their surroundings. Am J Physiol Cell Physiol , C595–C605. [DOI] [PubMed] [Google Scholar]
- Carey SP, Martin KE, Reinhart-King CA. (2017). Three-dimensional collagen matrix induces a mechanosensitive invasive epithelial phenotype. Sci Rep , 42088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox TR, Erler JT. (2011). Remodeling and homeostasis of the extracellular matrix: implications for fibrotic diseases and cancer. Dis Model Mech , 165–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkhatib N, Neu MB, Zensen C, Schmoller KM, Louvard D, Bausch AR, Betz T, Vignjevic DM. (2014). Fascin plays a role in stress fiber organization and focal adhesion disassembly. Curr Biol , 1492–1499. [DOI] [PubMed] [Google Scholar]
- Geraldo S, Simon A, Elkhatib N, Louvard D, Fetler L, Vignjevic DM. (2012). Do cancer cells have distinct adhesions in 3D collagen matrices and in vivo? Eur J Cell Biol , 930–937. [DOI] [PubMed] [Google Scholar]
- Han C, Lu Y, Wei Y, Liu Y, He R. (2011). D-ribose induces cellular protein glycation and impairs mouse spatial cognition. PLoS One , e24623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman LJ, Brangwynne CP, Kasza KE, Filippidi E, Gordon VD, Deisboeck TS, Weitz DA. (2005). Glioma expansion in collagen I matrices: analyzing collagen concentration-dependent growth and motility patterns. Biophys J , 635–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinnunen J, Kokkonen HT, Kovanen V, Hauta-Kasari M, Vahimaa P, Lammi MJ, Toyras J, Jurvelin JS. (2012). Nondestructive fluorescence-based quantification of threose-induced collagen cross-linking in bovine articular cartilage. J Biomed Opt , 97003. [DOI] [PubMed] [Google Scholar]
- Kopanska KS, Alcheikh Y, Staneva R, Vignjevic D, Betz T. (2016). Tensile forces originating from cancer spheroids facilitate tumor invasion. PLoS One , e0156442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krndija D, Schmid H, Eismann JL, Lother U, Adler G, Oswald F, Seufferlein T, von Wichert G. (2010). Substrate stiffness and the receptor-type tyrosine-protein phosphatase alpha regulate spreading of colon cancer cells through cytoskeletal contractility. Oncogene , 2724–2738. [DOI] [PubMed] [Google Scholar]
- Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, et al (2009). Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell , 891–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijering E, Dzyubachyk O, Smal I. (2012). Methods for cell and particle tracking. Methods Enzymol , 183–200. [DOI] [PubMed] [Google Scholar]
- Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, et al (2005). Tensional homeostasis and the malignant phenotype. Cancer Cell , 241–254. [DOI] [PubMed] [Google Scholar]
- Pickup MW, Mouw JK, Weaver VM. (2014). The extracellular matrix modulates the hallmarks of cancer. EMBO Rep , 1243–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano PP, Inman DR, Eliceiri KW, Keely PJ. (2009). Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK-ERK linkage. Oncogene , 4326–4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riching KM, Cox BL, Salick MR, Pehlke C, Riching AS, Ponik SM, Bass BR, Crone WC, Jiang Y, Weaver AM, et al (2014). 3D collagen alignment limits protrusions to enhance breast cancer cell persistence. Biophys J , 2546–2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Avigad G, Brodsky B, Eikenberry EF. (1988). Glycation induces expansion of the molecular packing of collagen. J Mol Biol , 495–505. [DOI] [PubMed] [Google Scholar]
- Verhulsel M, Vignes M, Descroix S, Malaquin L, Vignjevic DM, Viovy JL. (2014). A review of microfabrication and hydrogel engineering for micro-organs on chips. Biomaterials , 1816–1832. [DOI] [PubMed] [Google Scholar]
- Wolf K, Alexander S, Schacht V, Coussens LM, von Andrian UH, van Rheenen J, Deryugina E, Friedl P. (2009). Collagen-based cell migration models in vitro and in vivo. Semin Cell Dev Biol , 931–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf K, Te Lindert M, Krause M, Alexander S, Te Riet J, Willis AL, Hoffman RM, Figdor CG, Weiss SJ, Friedl P. (2013). Physical limits of cell migration: control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J Cell Biol , 1069–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wullkopf L, West AV, Leijnse N, Cox TR, Madsen CD, Oddershede LB, Erler JT. (2018). Cancer cell ability to mechanically adjust to extracellular matrix stiffness correlates with their invasive potential. Mol Biol Cell , 2378–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeromonahos C, Polack B, Caton F. (2010). Nanostructure of the fibrin clot. Biophys J , 2018–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]