

 A chiral mesoporous hybrid material, containing inserted bis-silylated pyrrolidine building fragments in the framework, for enantioselective Michael addition with high stereocontrol.
A chiral mesoporous hybrid material, containing inserted bis-silylated pyrrolidine building fragments in the framework, for enantioselective Michael addition with high stereocontrol.
Abstract
A new chiral mesoporous hybrid material was synthesized based on pyrrolidine units included in a siliceous framework, HybPyr, and integrated into the organic–inorganic structure, from a specific bis-silylated precursor. A fluoride sol–gel methodology under soft synthesis conditions and in the absence of sophisticated structural directing agents allowed the generation of a mesoporous architecture with a homogeneous distribution of active chiral moieties along the network. The hybrid material was studied by means of different characterization techniques (TGA, NMR and IR spectroscopy, chemical and elemental analyses, TEM, and textural measurements), verifying the stability and integrity of the asymmetric active sites in the solid. The hybrid material, HybPyr, is an excellent asymmetric heterogeneous and recyclable catalyst for enantioselective Michael addition of linear aldehydes to β-nitrostyrene derivatives with high stereocontrol of the reaction products.
1. Introduction
Molecular chirality plays a key role in science and technology. Enantioselective synthesis is a key process in the chemical industry, especially in the field of pharmaceuticals and agrochemicals, and the interest in implementing efficient and economical routes to enantiomerically pure compounds is constantly growing. Thus, the synthesis of optically active intermediates and products as pure enantiomers is a topic of ever-increasing importance for academic research as well as for pharmaceutical development1 and achieving heterogeneous asymmetric catalysis is the most challenging.
Until the last decade, the two main types of efficient asymmetric catalysts were transition metal complexes and enzymes. Indeed, transition metals are considered key to the development of enantioselective catalysts,2 since they are active and enantioselective for a large variety of reactions. Despite their great benefit, there is still room for improvement based on catalyst cost and toxicity that implies rigorous purification techniques to ensure their complete removal from the final target molecules, especially in the case of pharmaceutical uses. Enzymes3 are versatile biocatalysts for a large variety of reactions and exhibit, in biocatalytic transformations, excellent chemo-, regio-, and stereospecificity under mild reaction conditions. Nevertheless, their main drawbacks are long reaction times and low stability under process conditions, which make their recovery and recycling difficult.
Recently, organocatalysts4 have undergone quick and fascinating progress in a short period of time and offered new opportunities in asymmetric synthesis. Organocatalysis is defined as the acceleration of chemical reactions with a sub-stoichiometric amount of an organic compound which does not contain a metal atom. While they exhibit great potential, they also present some limitations such as operation in organic solvent, need of high loadings (10–30 mol%) to accomplish the process in a reasonable reaction time, cost, low stability and difficult separation from reaction medium and recycling. For all, the development of recyclable asymmetric catalysts constitutes important research and, sometimes, industrial goals. Additionally, for easy recovery of catalysts by simple filtration and their possible recycling, the development of solid catalysts makes the implementation of continuous flow processes possible.5 Unfortunately, the number of solid chiral catalysts is small. Different chiral heterogeneous systems have been described, and they are generally based on the immobilization of homogeneous catalytic species on a solid support.6 Nevertheless, ensuring the stability of the catalytic properties in the supported heterogeneous system is a difficult task,7 and a loss of enantioselectivity is often observed during the immobilization process.8 For example, an efficient polymer-supported organocatalyst synthesized from chiral monomers for the Michael addition of ketones to nitroolefins has been prepared by grafting a chiral pyrrolidine monomer;9 metal surfaces were modified by anchoring chiral organic compounds10 and metal–organic frameworks (MOFs), where a chiral tetracarboxylate ligand derived from 1,1′-bi-2-naphthol was incorporated, resulting in active asymmetric Lewis acid catalysts after post-synthesis functionalization with Ti(OiPr)4.11 Very few examples of organosilica hybrid materials incorporating chiral siloxane precursors, such as 1,1′-binaphthalene, (R,R)- or (S,S)-diamino-cyclohexane,12 cinchonidine derivatives,13 prolinamide14 or pyrrolidine-bridged polyhedral oligomeric silsesquioxanes,15 were reported.
Regarding the catalytic topic, we will focus on the use of organocatalysts in the field of asymmetric synthesis. The efficiency and the scope of organocatalysis particularly aminocatalysis have been broadly established since the rediscovery of the proline-catalyzed intermolecular aldol reaction.16 What makes proline different from other aminoacids is its higher effectiveness in aminocatalysis thanks to the pyrrolidine portion which readily reacts with carbonyl compounds to form iminium ions and enamines.17 Chiral secondary amines showed to be effective catalysts for aldol condensation processes by covalent activation of the carbonyl compounds either via nucleophilic enamines or electrophilic iminium ions.18 A wide variety of homogeneous enantioselective catalysts based on pyrrolidine analogues and active to the asymmetric conjugate addition (ACA) have been reported such as pyrrolidine sulfonamide,19 diphenylprolinol silyl ether20 and trans-4-hydroxyprolylamide.21 Other examples of supported polymer-pyrrolidines have been prepared using analogs of Jørgensen catalysts ((R)- and (S)-α,α-bis[3,5-bis(trifluoromethyl)phenyl]-2-pyrrolidinemethanol trimethylsilyl ether) and prolyl-prolinol catalysts.22 They have become of major significance for the stereoselective formation of C–C bonds.
The aim of heterogenization of chiral organocatalysts lies in industrial implementation, the possibility of reducing the costs and facilitation of the recovery, reuse and/or regeneration of the catalysts for several cycles. In this context, the development of new asymmetric hybrid materials offers new opportunities with respect to the mechanical and chemical stability of the materials under reaction process and regeneration conditions. Hybrid materials have been of large interest for the scientific and industrial community because of their potential commercial scope in their applications and because of the challenges presented by their synthesis.
In this work, we report the preparation of a chiral mesoporous hybrid material, containing active pyrrolidine building fragments inserted and stabilized into the framework, based on a methyl-(2S,4R)-4-hydroxypyrrolidine-2-carboxylate bis-silylated derivative, for the enantioselective Michael addition of linear aldehydes to β-nitrostyrene derivatives with high stereocontrol. The asymmetric Michael addition of carbonyl compounds to nitroalkenes is among the most useful synthetic methods for the C–C bond formation and produces nitroaldehydes which constitute key intermediates in organic synthesis, since from nitro groups different functionalities can be created through different organic transformations.23
2. Experimental
2.1. General information
All reagents were purchased from commercial suppliers and used without further purification. Solvents employed in the reactions were purified using a solvent purification system (SPS) MBraun 800. Organic solutions were concentrated under reduced pressure on a Büchi rotary evaporator. The reactions were monitored by GC-FID (Shimadzu, GC Plus ultra 2010) and GC-MS (Shimadzu, GCMS-QP2010 Ultra). 1H and 13C NMR spectra were recorded on a Bruker 300 spectrometer and the chemical shifts are reported in ppm relative to residual proton solvent signals. Data for the 1H NMR spectra are reported as follows: chemical shift (δ, ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, dd = double doublets), coupling constant and integration. Data for 13C NMR spectra are reported in terms of chemical shift (δ, ppm). High performance liquid chromatography (HPLC) was performed with an Agilent Technologies 1220 Infinity Series instrument using a Daicel Chiralpak IC (4.6 × 250 mm). Optical rotations were measured on a Jasco P-2000 polarimeter using the yellow line at 589 nm and [α]20D. C, N, and H contents were determined with a Carlo Erba 1106 elemental analyzer. Thermogravimetric and differential thermal analyses (TGA-DTA) were conducted in an air stream with a Mettler Toledo TGA/SDTA 851E analyzer. Nitrogen adsorption isotherms were measured at 77 K with a Micromeritics ASAP 2010 volumetric adsorption analyzer. Before the measurement, the sample was outgassed for 12 hours at 80 °C. The BET specific surface area24 was calculated from the nitrogen adsorption data in the relative pressure range from 0.04 to 0.2. The total pore volume25 was obtained from the amount of N2 adsorbed at a relative pressure of about 0.99. The external surface area and micropore volume were estimated using the t-plot method in the t range from 3.5 to 5. The pore diameter and the pore size distribution were obtained using the Barrett–Joyner–Halenda (BJH) method26 on the adsorption branch of the isotherms. Solid state MAS-NMR spectra were recorded at room temperature under magic angle spinning (MAS) using a Bruker AV-400 spectrometer. IR spectra of the organic precursors were recorded on KBr disks at room temperature.
2.2. Preparation of disilane (PyrSil)
(2S,4R)-N-1(Benzyl)-4-hydroxyproline-methylester (2)
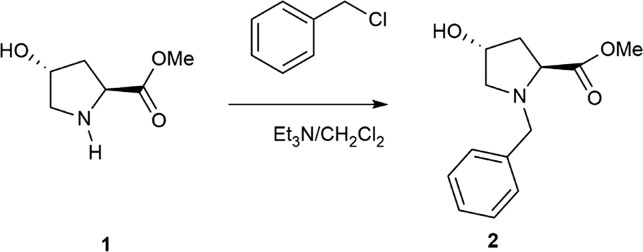
The first step was the protection of the amine group of hydroxyproline compounds. Specifically, (2S,4R)-N-1(benzyl)-4-hydroxyproline-methylester (2) was prepared by following an adapted literature procedure for analogous pyrrolidines.27 Triethylamine (7.6 mL, 55.0 mmol) was added dropwise to a formed suspension of methyl (2S,4R)-4-hydroxypyrrolidine-2-carboxylate hydrochloride (5.0 g, 27.5 mmol) in 23 mL of dry CH2Cl2, and the reaction mixture was continuously stirred. Benzyl chloride (14.8 ml, 118.0 mmol) was added to the mixture in one portion and the formed slurry was stirred at reflux overnight. After reaction, NaOH (solution 2 M) was added until pH = 2. Then, the organic layer was separated and the aqueous phase was extracted three times with dichloromethane (15.0 mL). The combined organic layers were washed with brine and, then, dried over anhydrous sodium sulfate and concentrated to give the crude product, which was purified by silica gel column chromatography (AcOEt/hexane 1/1 as eluent) to afford 2 as an orange yellow liquid (6.1 g, 95%).
[α]D19 = –59 (c = 1, CHCl3). 1H NMR (300 MHz, CDCl3) δ = 7.31 (m, 5H, Ar–H), 4.43 (m, 1H), 3.90 (d, J = 12 Hz, 1H), 3.66 (d, J = 12 Hz, 1H), 3.65 (s, 3H, OCH3), 3.32 (dd, J = 12, 2 Hz, 1H), 2.48 (d, J = 3 Hz, 1H), 2.44 (d, J = 3 Hz, 1H), 2.22 (m, 1H), 2.06 (m, 1H). 13C NMR (75 MHz, CDCl3) δ = 173.9, 138.1, 129.0, 128.3, 127.2, 70.3, 63.6, 61.1, 58.1, 51.7, 39.6.
(3R,5S)-1-Benzyl-5-(hydroxymethyl)pyrrolidin-3-ol (3)
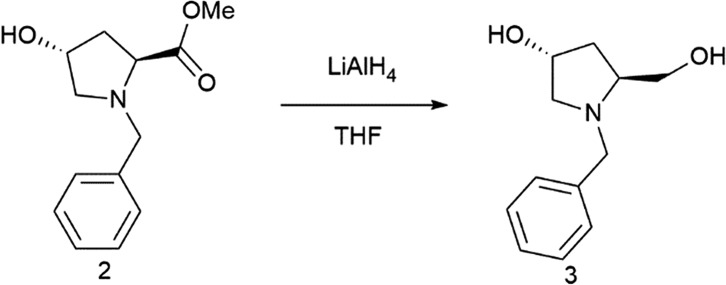
The second step was the reduction of the ester group of compound 2. Then, (3R,5S)-1-benzyl-5-(hydroxymethyl)pyrrolidin-3-ol (3) was synthesized by modifying a reported literature procedure.28 Specifically, a solution of pyrrolidine derivative 2 (9.6 g, 40.8 mmol) in THF (20 ml) was added dropwise to LiAlH4 suspension (2.3 g, 61.2 mmol) in THF (60.0 ml) in a cold bath. When the addition was completed, the mixture was stirred for 30 min. Then, it was heated at reflux for 8 h. The reaction mixture was stirred at room temperature overnight and, using a cold bath, 30.0 mL of water were added, followed by filtration of the salts and extraction with Et2O : AcOEt (1 : 1) (3 × 40.0 mL). The combined organic layers were dried over anhydrous Na2SO4. After solvent removal, product 3 was purified by flash chromatography on a silica gel, using a linear gradient of EtOH in CHCl3. The product was obtained in 91% yield (7.7 g) as a colorless liquid.
[α]D21 = –45 (c = 1, CHCl3). 1H NMR (300 MHz, CDCl3) δ = 7.30 (m, 5H, Ar–H), 4.34 (m, 1H), 4.00 (d, J = 12 Hz 1H), 3.69 (dd, J = 12, 3 Hz, 2H), 3.49 (d, J = 15 Hz, 1H), 3.42 (dd, J = 11, 3 Hz, 1H), 3.26 (dd, J = 12, 6 Hz, 1H), 3.09 (m, 1H), 2.39 (dd, J = 12, 6 Hz 1H), 2.13 (m, 2H), 1.85 (m, 1H). 13C NMR (75 MHz, CDCl3) δ = 138.9, 128.6, 128.4, 127.2, 70.2, 63.3, 62.2, 60.9, 58.6, 37.5.
(3R,5S)-1-Benzyl-5-(8,8-diethoxy-3-oxo-2,9-dioxa-4-aza-8-silaundecyl)pyrrolidin-3-yl(3-(triethoxysilyl)propyl)carbamate (4)
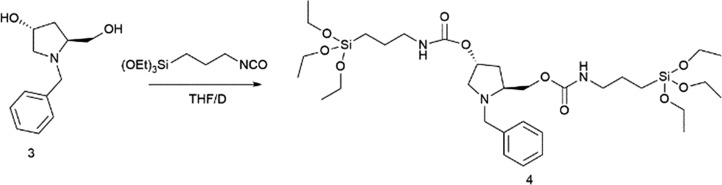
Compound 3 (7.7 g, 37.3 mmol) was added into a 250 mL round-bottom flask and purged under a nitrogen flow. After that, dry THF (62.0 mL) was added. Then, the solution was stirred, and 3-(triethoxysilyl)propylisocyanate (18.4 g, 74.6 mmol) was added dropwise. The mixture was heated at reflux until consumption of the product (∼2 days). After solvent removal under vacuum, the product 4 was obtained in 99% yield (26.1 g) as a yellow oil, without further purification.
[α]D22 = –26 (c = 1, CHCl3). 1H NMR (300 MHz, CDCl3) δ = 7.27 (m, 5H, Ar–H), 4.99 (m, 2H), 4.11 (m, 3H), 3.81 (m, 14H), 3.13 (m, 5H), 2.37 (m, 1H), 1.96 (m, 2H), 1.60 (m, 5H), 1.22 (t, J = 6 Hz, 18H, OCH2CH3), 0.62 (m, 4H, CH2SiO). 13C NMR (75 MHz, CDCl3) δ = 156.4, 156.0, 138.8, 128.8, 128.2, 127.0, 72.7, 66.1, 61.2, 59.6, 58.9, 58.4, 43.4, 43.3, 42.9, 35.9, 23.3, 18.4, 18.3, 7.6.
(3R,5S)-5-(8,8-Diethoxy-3-oxo-2,9-dioxa-4-aza-8-silaundecyl)pyrrolidin-3-yl(3-(triethoxysilyl)propyl)carbamate, PyrSil, (5)
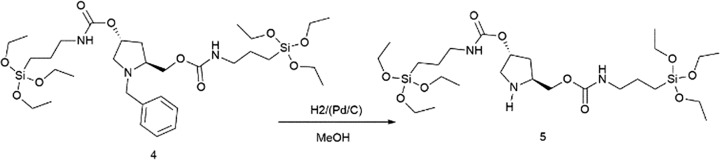
Finally, the amine group of the pyrrolidine backbone was deprotected. For this, compound 4 (5.0 g, 7.1 mmol) was hydrogenated in methanol (15.0 mL) under a pressure of 30 bar in an autoclave, using Pd/C-10% (0.4 g, 3.6 mmol) as a catalyst. When the reaction was completed, the mixture was filtered through Celite and the filtrate was evaporated. Product 5, PyrSil, was obtained in 96% yield (4.2 g) as a yellow oil.
[α]D23 = +12 (c = 1, CHCl3). 1H NMR (300 MHz, CDCl3) δ = 5.10 (m, 2H), 4.09 (m, 2H), 3.80 (q, J = 6 Hz, 12H, OCH2CH3), 3.14 (m, 6H), 2.12 (m, 1H), 1.60 (m, 5H), 1.67–1.53 (m, 4H (d, d′)), 1.21 (t, J = 7 Hz, 18H, OCH2CH3), 1.00 (d, J = 6 Hz, 1H), 0.60 (m, 4H, CH2SiO). 13C NMR (75 MHz, CDCl3) δ = 156.5, 156.0, 67.0, 58.5, 58.4, 58.3, 56.2, 52.7, 50.7, 43.4, 43.0, 35.4, 23.6, 23.3, 18.4, 18.2, 14.6, 7.6, 7.2.
2.3. Synthesis of the chiral hybrid catalyst (HybPyr)
A non-ordered hybrid porous organic–inorganic material, HybPyr, was obtained from a starting mixture of tetramethoxysilane (TMOS) as the silica precursor and an appropriate amount of disilane PyrSil ((R′O)3Si–R–Si(OR′)3), 5, as bridged silsesquioxane (5% mol SiO2 with respect to total mol of SiO2), in methanol. After dissolution of precursors, a water solution of NH4F was added dropwise to the organosilicon alkoxide solution under vigorous stirring. The final reaction mixture has the following molar composition: 1SiO2 : 4MeOH : 4H2O : 0.00313 NH4F where the Si/NH4F and TMOS/disilane ratios were adjusted to 479 and 4, respectively. Hydrolysis and condensation of the silicon precursor were carried out under vigorous stirring in a glass beaker at room temperature. Stirring was continued until gelation occurred. Then, the gel was aged for 24 h at 36 °C and, finally, dried at 150 °C for another 24 h. The solid obtained was a fine brown powder that was exhaustively washed with ethanol and water in consecutive steps to remove the disilane molecules not incorporated into the materials. Finally, the material was dried at 60 °C overnight.
2.4. Catalytic tests
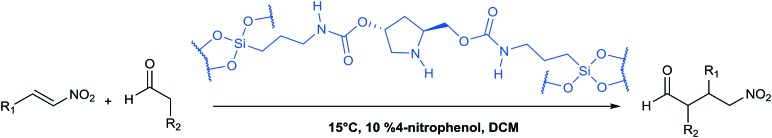
Asymmetric Michael additions: general procedure
The reaction was performed in a 3 mL sealed vial under magnetic stirring. Nitroalkene (0.1 mmol), 10 mol% 4-nitrophenol (0.01 mmol) and 20 mol% catalyst HybPyr (0.02 mmol) were added to aldehyde solution (1.0 mmol) in dry DCM (1 mL). After that, the resulting mixture was stirred at 15 °C for 20 h, and the catalyst HybPyr was recovered by filtration and washed several times with DCM and H2O. The solid catalyst was dried in an oven at 100 °C overnight. The organic extracts were concentrated under reduced pressure. The Michael adducts were obtained as evaporation residues and purified by silica gel column chromatography on TLC plates with hexane : EtOAc = 3 : 1 as the eluent phase. The spectroscopic data of all products obtained were in agreement with the published data.29
The reaction was monitored by GC analysis. The calculations of data for the yield, selectivity and conversion with respect to the limiting reagent (nitrostyrene derivative) were determined from GC analysis (GC-2010-Ultra, Shimadzu, equipped with a FID). The characterization of the Michael adducts was performed by GC-MS and 1H and 13C NMR. The enantiomeric excess (ee), diastereoisomeric ratio (dr) and enantiomeric ratio (er) were determined by HPLC on the purified reaction mixture using a chiral stationary phase (Chiralpak IC column).
3. Results and discussion
3.1. Synthesis and characterization
A mesoporous chiral hybrid material (named HybPyr) was prepared from a suitable bridged silsesquioxane monomer precursor, which contained pyrrolidine-type active fragments as chiral organic linkers, located between reactive siloxane terminal groups (PyrSil). The preparation of this bis-silylated precursor is summarized in Scheme 1. In detail, hydroxypyrrolidine (1) was protected with Cl-Bnz, with ester groups being consecutively reduced to hydroxyls (3) with LiAlH4. Next, silylation of hydroxyl groups was performed through condensation processes with isocyanate siloxane molecules to produce silyl-derivatives (4), containing pyrrolidine-urethane groups as organic bridges. The last step was the deprotection step of pyrrolidine moieties, using Pd/C as a catalyst under H2, to obtain the novel functionalized bis-silylated precursor (5), PyrSil, containing pyrrolidine active units.
Scheme 1. Synthetic pathway to prepare the PyrSil monomer precursor, containing pyrrolidine units between siloxane terminal groups.

The non-ordered mesoporous organic–inorganic hybrid material, HybPyr, was prepared in the presence of the previously obtained PyrSil monomer together with tetramethylorthosilicate (TMOS), as silicon precursors, through a sol–gel synthesis process in methanolic fluoride medium (Scheme 2). This synthesis methodology was based on consecutive hydrolysis and condensation steps at room temperature and neutral pH, with fluoride ions acting as mineralizing agents. The formation of pentacoordinated organosilyl complexes as activated intermediates allowed the rapid gelification process, after a few seconds, by the covalent bonding of siloxane terminal groups.
Scheme 2. Synthesis procedure through a sol–gel process in fluoride medium to obtain the non-ordered mesoporous chiral hybrid material (HybPyr), containing pyrrolidine units in the framework. In the bottom panel, intermediate reactive pentacoordinated siloxanes formed during the synthesis process.

The aging period at 36 °C, followed by a drying process, facilitated the generation of porous organosilicon structures with pyrrolidine chiral moieties isolated into the porous framework. There are few described examples in which this type of chiral organocatalyst is stabilized in the architecture of a robust siliceous material rather than adsorbed or supported onto inorganic matrixes.14
The XRD pattern of the hybrid material HybPyr (not included) did not show diffraction bands and confirmed the formation of the structure without long-order spatial symmetry, as could be expected for the materials obtained through fluoride sol–gel processes in the absence of structural directing agents. This low structuration level was not an obstacle to including pyrrolidine chiral fragments in the network, being ∼5 wt% as estimated from chemical analysis and the associated C/N experimental molar ratio was 4.2, close to the theoretical value for the organic bridge present in the PyrSil monomer precursor (C/N = 4.3). This fact would imply that the derived pyrrolidine building units were introduced into the hybrid solid, maintaining their initial chemical composition.
The organic content (∼18.7 wt%) was also determined by thermogravimetric analysis (TGA) (see Fig. 1), which was higher than that by elemental analysis due to the contribution of hydration water (∼8.4 wt%) and dehydroxylation water (∼5.4 wt%), detected at temperatures lower than 100 °C and higher than 500 °C, respectively, with the latter weight loss being associated with the silanol condensation phenomenon. From the derivative curve (DTA), the main weight loss assigned to the silyl pyrrolidine fragments (∼4.9 wt%) was observed between 200 °C and 400 °C, with the hydrothermal stability of the organic–inorganic porous chiral hybrid material synthesized here being established in this temperature range. It is remarkable that the organic content, regarded only as pyrrolidine building units, was similar for both chemical and thermogravimetric analyses, i.e. around 5 wt%.
Fig. 1. Thermogravimetric curve (TGA) and the corresponding derivative (DTA) of the non-ordered mesoporous chiral hybrid material, HybPyr.

The integrity of the derivative silyl-pyrrolidine building units was confirmed by 13C NMR of the non-ordered mesoporous HybPyr solid, which was able to identify all carbon atoms present in the organic bridge, including those directly bonded to silicon atoms (Fig. 2). This result showed that the organic building units remained intact, as in the starting bis-silylated PyrSil monomer precursor (see Fig. S1 in the ESI†).
Fig. 2. 13C MAS NMR spectrum of the non-ordered mesoporous chiral HybPyr material.

The 13C NMR spectrum confirms that the organic fragments preserved their integrity during the sol–gel synthesis processes, and the 29Si MAS NMR spectra confirm that the organic building units not only remained intact but were also incorporated covalently into the non-ordered porous network bonded to inorganic silica units (Fig. 3). In fact, T-type silicon atoms were detected between –60 and –80 ppm chemical shifts assigned to Si–C species such as T1 (C–Si(OH)2(OSi)), T2 (C–Si(OH)(OSi)2) and T3 (C–Si(OSi)3), together with Q-type silicon atoms of silicon tetrahedral units from the condensed TMOS precursor. This result confirmed that the derivative pyrrolidine units were integrated and stabilized into the framework through effective hydrolysis and condensation between terminal siloxane groups. Additionally, the pure PyrSil monomer precursor showed T-type silicon atoms centered in the chemical shift range of –40––50 ppm that was changed to –60––80 ppm when this organic fragment was incorporated into the framework of the final non-ordered HybPyr solid, with this fact being another confirmation of the effective integration of pyrrolidine moieties into the structure of the hybrid material (see the inset in Fig. 3). Furthermore, the integration of the T and Q signals in the 29Si BD/MAS NMR spectrum allows estimation of the number of functionalized silicon atoms present in the HybPyr material through the T/(T + Q) integrated intensity ratio. The obtained result corroborated that ∼5% of the silicon atoms are functionalized with the derivative pyrrolidine units, with this value being similar to both the amount of bis-silylated precursor used during the synthesis process and the organic content estimated from the chemical and thermogravimetric analyses.
Fig. 3. 29Si MAS NMR spectra of the non-ordered mesoporous hybrid material, HybPyr, and assignment of T- and Q-type silicon atoms: (a) 29Si BD/MAS NMR spectrum and (b) 29Si CP/MAS NMR spectrum. In the inset: 29Si NMR spectrum of the pure bis-silylated precursor, PyrSil.

Infrared spectroscopy results also corroborated the presence and integrity of the pyrrolidine derivative fragments included in the framework of the organic–inorganic hybrid material, HybPyr. The FTIR spectrum of the HybPyr solid shows the bands associated with stretching (ν(–NH–): shoulder at 3260 cm–1) and bending (δ(–NH–): 1559 and 1643 cm–1) vibrations of secondary amines introduced into the organic bridges in the PyrSil monomer precursor, including those due to cyclic aliphatic amines from pyrrolidine groups. The stretching vibration band assigned to ureido-type fragments, ν(–NH–CO–O–), was also located at 1703 cm–1. The –CH2– units due to terminal propyl chains present in the building units were detected through symmetric and asymmetric vibration bands assigned to alkyl groups connected to amino groups (ν(–N–CH2–): 2897 and 2958 cm–1; δ(–N–CH2–): 1383 and 1447 cm–1). Furthermore, a broad band due to the presence of water centered at 3467 cm–1 was clearly observed, overlapping with the band attributed to surface silanol groups (Si–OH), typical of inorganic silicates with structural defects. However, the vibration band at 950 cm–1 was also associated with external silanol groups.
Additionally, in the infrared framework range, a characteristic band located around 790 cm–1 assigned to the Si–C vibration was observed, corroborating the covalent bonds established between tetrahedral silicon units and bis-silylated pyrrolidine moieties. In this range, typical vibration bands (458 and 1080 cm–1) due to the presence of Si–O–Si groups, which were the main structural building units of the organosilicon HybPyr framework, were detected (Fig. 4). Thus, spectroscopic results (NMR and FTIR) evidenced unambiguously the integrity of the organic linker and its effective covalent incorporation into the non-ordered porous hybrid structure.
Fig. 4. FTIR spectrum of non-ordered mesoporous hybrid material, HybPyr.

The textural properties, specific surface area and free porous volume of the organic–inorganic hybrid material, HybPyr, were evaluated from nitrogen adsorption measurements. Specifically, the adsorption isotherm was characteristic of non-ordered porous materials with a marked slope shift at a high relative pressure (P/P0 ∼0.3–0.4), indicating the presence of a large pore diameter in the internal mesopores present in the chiral hybrid architecture (Fig. 5), with a BET surface area and total porous volume of ∼385 m2 g–1 and ∼0.325 cm3 g–1, respectively. The comparison of the textural properties of the non-ordered mesoporous pure silica material is shown in Table 1. It is observed that the presence of pyrrolidine moieties in the framework promoted a reduction of surface area, probably associated with the structural difficulty to homogeneously assemble organic fragments without partially collapsing internal porosity. However, the higher porous contribution in the HybPyr material was due to the formed mesopores, with the generated microporosity being practically inappreciable.
Fig. 5. N2 adsorption isotherms of the non-ordered mesoporous chiral hybrid, HybPyr, and pure silica solids.

Table 1. Textural properties of the porous organic–inorganic hybrid, HybPyr, and pure silica materials.
| Sample | BET surface area (m2 g–1) | External surface (m2 g–1) | Micropore volume (cm3 g–1) | Total pore volume (cm3 g–1) | Mean pore diameter (Å) |
| HybPyr | 386 | 378 | 0.004 | 0.325 | 40 |
| Pure silica | 668 | 547 | 0.053 | 0.473 | 20–30 |
Indeed, the BJH pore size distribution of the non-ordered chiral hybrid material, HybPyr, was characteristic of mesoporous solids prepared through a fluoride sol–gel synthesis route, exhibiting a broad distribution of pores in the mesoporous range (from 17 Å to 300 Å), although with a higher contribution of mesopores with diameters centered at 40 Å (Fig. 6).
Fig. 6. Pore size distributions of the non-ordered mesoporous chiral hybrid, HybPyr, and pure silica solids calculated from the BJH method.

In this case, the large molecular dimensions of PyrSil monomers inserted in the network of the hybrid solid would favor the formation of mesopores with higher internal diameters than in non-ordered pure silica materials without inserted organic building units. This fact was also confirmed from the TEM micrographs, where free mesoporous cavities with a broad internal diameter distribution were observed for the hybrid material containing pyrrolidine units in the framework (Fig. 7).
Fig. 7. TEM micrographs of the non-ordered mesoporous chiral material, HybPyr.

3.2. Enantioselective catalysis
A study of enantioselective Michael addition between aliphatic aldehydes and nitrostyrene derivatives (see Scheme 3) was performed to test the effectiveness and efficiency of the synthesized chiral hybrid material, HybPyr. Amine-catalyzed enantioselective Michael addition of aldehydes to nitroalkenes is an important C–C bond-forming transformation in organic synthesis. Generally, the amine-catalyzed Michael addition of carbonyl compounds to nitroalkenes proceeds via an enamine intermediate. The widely accepted mechanism goes firstly via the formation of the enamine by condensation of a chiral amine and a carbonyl compound. The following enamine addition onto the nitroalkene substrate provides an iminium intermediate. Then, the hydrolysis of the iminium releases the Michael product with regeneration of the asymmetric amine catalyst (Scheme 3). The catalytic cycle can be limited by the last step associated with the amine catalyst released by hydrolysis and thus by its availability. Different studies30 showed the effect of the addition of co-catalysts such as weak acids or bases which boosted considerably the rate of reaction by probably favoring the enamine formation31 and increasing the diastereoselectivity of the Michael reaction, without changing the enantioselectivity.
Scheme 3. Proposed mechanism of chiral amine-catalyzed Michael addition via E-enamine formation.

Herein, we initially studied the enantioselective catalytic performance of the hybrid material HybPyr, containing pyrrolidine-carbamate units in the framework, for the Michael addition of propanal to β-nitrostyrene. Mechanistic insights32 showed that the geometry of enamine would be determined by the catalyst structure, and according to steric hindrance, the E-enamine was thermodynamically favored. The chiral substituent on the catalyst not only controls the geometry of the enamine but also influences the facial selectivity of the Michael addition through steric shielding and/or electronic interaction such as hydrogen bonding established for different aminocatalysts as well as l-proline, tetrazole or thiourea. In this case, hydrogen bonding can be established between the nitro group and carbamate group (Scheme 4). Furthermore, steric shielding could determine the facial selectivity since the bulky substituent group on the catalyst would force the attack from the opposite side. Likewise, for the Michael addition mechanism, the less hindered SiSi transition state via an anti-enamine is usually favored for aldehydes while the ReRe transition state via a syn-enamine is usually preferred for ketones. Both steric shielding and electronic interaction are shown in Scheme 4.
Scheme 4. Electronic interaction and steric shielding transition states with a SiSi approach for propanaldehyde and β-nitrostyrene.

When the chiral Michael addition was performed in the presence of 20 mol% HybPyr catalyst at 30 °C and 0 °C, in the presence of different solvents, the (2R,3S)-2-methyl-4-nitro-3-phenylbutanal (1a) was the main isomer (syn) (Table 2). Based on the enantioselective catalytic results a proposed mechanism is outlined in Scheme 3. According to both steric shielding and electronic interaction, the approach of β-nitrostyrene from the less hindered Si-face of the E-enamine was the most probable.
Table 2. Effect of the solvent on the enantioselective catalytic performance of the HybPyr catalyst.
| Entry | Solvent | T (°C) | t (h) | Yield a (%) | ee b (%) | dr b |
| 1 | n-Hex | 30 | 24 | 99 | 56 | 64 : 36 |
| 2 | Tol | 30 | 24 | 97 | 64 | 70 : 30 |
| 3 | Et2O | 30 | 24 | 95 | 60 | 67 : 33 |
| 4 | DCM | 30 | 12 | 96 | 72 | 83 : 17 |
| 5 | CF3-Tol | 30 | 16 | 97 | 72 | 78 : 22 |
| 6 | CHCl3 | 30 | 20 | 97 | 70 | 74 : 26 |
| 7 | THF | 30 | 24 | 95 | 68 | 74 : 26 |
| 8 | AcOEt | 30 | 72 | 95 | 60 | 68 : 32 |
| 9 | CH3CN | 30 | 24 | 91 | 66 | 74 : 26 |
| 10 | MeOH | 30 | 8 | 99 | 64 | 77 : 23 |
| 11 | EtOH | 30 | 24 | 99 | 58 | 69 : 31 |
| 12 | H2O | 30 | 20 | 81 | 60 | 76 : 24 |
| 13 | Tol : H2O (1 : 3) | 30 | 24 | 93 | 66 | 86 : 14 |
| 14 | Tol : THF (5 : 1) | 30 | 16 | 97 | 66 | 76 : 24 |
| 15 | DCM : H2O (1 : 3) | 30 | 24 | 92 | 66 | 85 : 15 |
| 16 | CHCl3 : IPrOH (9 : 1) | 30 | 16 | 100 | 72 | 79 : 21 |
| 17 | DCM | 0 | 69 | 83 | 74 | 88 : 12 |
| 18 | CHCl3 | 0 | 160 | 41 | 74 | 89 : 11 |
| 19 | THF | 0 | 135 | 94 | 70 | 86 : 16 |
| 20 | iPrOH | 0 | 21 | 82 | 70 | 81 : 19 |
| 21 | MeOH | 0 | 21 | 99 | 72 | 86 : 14 |
| 22 | H2O | 0 | 44 | 69 | 58 | 86 : 14 |
aYield and conversion were determined by GC.
bDetermined by HPLC on the purified reaction mixture, using a chiral stationary phase (Chiralpak IC column). Tol: toluene.
Solvents with different polarities and properties (protonic) as well as mixtures of them were used to maximize the enantioselectivity of the reaction. In all cases, the selectivity towards Michael adducts was >99%. When dichloromethane (DCM), chloroform (CHCl3) or trifluorotoluene (CF3-toluene) were used as solvents, the Michael product was obtained with excellent yields (90–96%) and good enantiomeric excess (ee), 70–72% (Table 2, entries 4–6). However, when other solvents were tested such as toluene, hexane (n-Hex), tetrahydrofuran (THF), diethyl ether (Et2O), acetonitrile (CH3CN), ethyl acetate (AcOEt), ethanol (EtOH), methanol (MeOH) or water, the ee decreased while in all cases high Michael adducts yields were achieved, up to 99%. Additionally, when the reaction temperature was decreased to 0 °C, a slight increase of enantiomeric excess and diastereoisomeric ratio (dr) was observed (Table 2, entries 17, 18 and 21). Following the study and according to these catalytic and enantioselective results, dichloromethane was chosen as the more suitable solvent. Furthermore, when the Michael addition was performed in the presence of the homogenous pyrrolidine precursor (PyrSil), 78% ee was provided after 6 h of reaction with 96% Michael adduct yield. These results showed that the chiral organo-disiloxane, containing pyrrolidine moieties, was successfully incorporated into the hybrid material without loss of its asymmetric catalytic properties.
The effect of the addition of acids or bases as additives or co-catalysts with different strength values (pKa) to the reaction medium was also examined. From Table 3, a clear trend can be observed since the use of additives was advantageous and allowed boosting the reaction rate and achieving higher yields in shorter reaction times. The best results were provided with the addition of NMM (N-methylmorpholine), nitrophenol and K2CO3, the since reaction time was reduced up to 3 times at 30 °C. Moreover, a very slight increase of enantioselectivity, from 76% until 78%, and slight changes in diastereoselectivity were observed, especially when 4-nitrophenol (NO2-PhOH) was used as the additive (Table 3, entries 10–12). At this stage, the evolution of ee% and the Michael adduct yield versus reaction time was examined (Fig. 8), where ee% being observed remained constant independent of the reaction progress. At this point, the heterogeneity of the process was controlled filtering out the catalyst after 1 h of reaction (conversion level: 30.6%) for the Michael addition of propanal to β-nitrostyrene. Then, the process was continued for 4 h, and no change in the yield was registered, ensuring that no active species migrated from the solid to the liquid. Finally, the stability and recyclability of the heterogeneous chiral catalyst were examined. The results showed that the HybPyr catalyst could be reused perfectly for four consecutive cycles without observing substantial activity loss and providing good enantio- and diastereoselectivity (Fig. 9), retaining the initial structuration of the hybrid catalyst (see Fig. S2 and S3 in the ESI†).
Table 3. Effect of the additive use on the enantioselective catalytic performance of the HybPyr catalyst.
| Entry | Additives | mol% | pKa | T °C | Time (h) | Conv.% a | ee% b | dr b |
| 1 | — | — | — | 30 | 12 | 96 | 72 | 83 : 17 |
| 2 | — | — | — | 15 | 21 | 97 | 76 | 86 : 14 |
| 3 | — | — | — | 0 | 69 | 83 | 74 | 88 : 12 |
| 4 | NO2-PhCO2H | 15 | 3.41 | 0 | 30 | 93 | 74 | 81 : 19 |
| 5 | PhCO 2 H | 10 | 4.2 | 30 | 9 | 63 | 70 | 86 : 16 |
| 6 | PhCO2H | 5 | 4.2 | –10 | 64 | 28 | 74 | 91 : 09 |
| 7 | Acetic acid | 5 | 4.75 | –10 | 64 | 25 | 72 | 91 : 09 |
| 8 | NO2-PhOH | 20 | 7.15 | 0 | 48 | 86 | 76 | 87 : 13 |
| 9 | NO 2 -PhOH | 20 | 7.15 | 30 | 4 | 98 | 76 | 84 : 16 |
| 10 | NO2-PhOH | 20 | 7.15 | 15 | 7 | 98 | 78 | 89 : 11 |
| 11 | NO2-PhOH | 10 | 7.15 | 15 | 8 | 94 | 78 | 88 : 12 |
| 12 | NO2-PhOH | 5 | 7.15 | 15 | 12 | 92 | 78 | 88 : 12 |
| 13 | NMM | 20 | 7.61 | 30 | 5 | 99 | 74 | 82 : 18 |
| 14 | NMM | 20 | 7.61 | 15 | 9 | 93 | 76 | 89 : 11 |
| 15 | DMAP | 20 | 9.6 | 30 | 5 | 99 | 68 | 63 : 37 |
| 16 | PhOH | 20 | 9.95 | 30 | 7 | 98 | 76 | 82 : 18 |
| 17 | PhOH | 20 | 9.95 | 15 | 9 | 98 | 76 | 82 : 18 |
| 18 | K2CO3 | 10 | 10.33 | 15 | 6 | 97 | 76 | 89 : 11 |
| 19 | K2CO3 | 5 | 10.33 | 15 | 9 | 95 | 76 | 88 : 12 |
| 20 | K2CO3 | 20 | 10.33 | 0 | 24 | 94 | 74 | 70 : 30 |
aYield and conversion were determined by GC.
bDetermined by HPLC on the purified reaction mixture, using a chiral stationary phase (Chiralpak IC column).
Fig. 8. Evolution of ee% or Michael adduct yield versus time when the Michael addition of propanaldehyde (1 mmol) to β-nitrostyrene (0,1 mmol) was performed in DCM (1 mL), at 15 °C, with 10 mol% nitrophenol and 20 mol% HybPyr catalyst. Selectivity towards Michael adducts was >99%.

Fig. 9. Catalytic performance of the HybPyr material for four consecutive runs for the addition of propanal to β-nitrostyrene (β-nitrostyrene (0,1 mmol), aldehyde (1 mmol), 10 mol% 4-nitrophenol, 1 mL of DCM and 20 mol% HybPyr), 8–10 h. Selectivity towards Michael adducts was >99%.

When the hybrid material, HybPyr, was used to carry out the Michael addition of isobutyraldehyde, a branched aldehyde, onto β-nitrostyrene in DCM, a lower yield and enantiomeric ratio were achieved. In order to improve the outcome of the reaction, different solvents were examined. From Table S1,† we observed that alcohols were the most adequate solvents, especially methanol, since 96% yield of Michael adducts was achieved (entry 14). Nevertheless, the enantiomeric ratio of the reaction was moderate. When DMSO or DMF were used as the solvent, a racemic mixture was obtained, probably because of the competitive adsorption of reactants and high polar solvent over the catalytic surface, limiting the optimal spatial organization via non-covalent interactions between the substrates and the chiral moieties through hydrogen bonds and van der Waals interactions. Additionally, the reaction temperature was varied, without substantial variation in the enantioselectivity (Table S1 (ESI†), entries 16 and 18). A similar trend was reported in the literature, whereby the use of a 2,2′-bipyrrolidine organocatalyst to perform asymmetric Michael addition showed that linear aldehydes provided higher reaction rates and selectivity than branched aldehydes.33
On the other hand, when the Michael reaction was performed with n-alkyl Michael donors such as butyraldehyde and valeraldehyde, under the same reaction conditions, with 20 mol% catalyst at 15 °C in dichloromethane (1 mL), the hybrid material HybPyr exhibited good asymmetric catalytic performance. Likewise, 88 and 95% Michael adduct yields were achieved after 4 and 2 days of reaction with 77 and 76% ee, respectively. These results confirmed the effect of steric hindrance of branched versus linear aldehydes rather than an inductive effect, since these latter higher reaction rate and enantioselectivity were provided.
The beneficial addition of carboxylic acid (25 mol%) allowed us to improve the Michael adduct yields, by reducing the reaction time. The use of strong acids inhibited the reaction (Table S2 (ESI†), entries 4, 9 and 12), while the use of acids with moderate strength (pKa range: 4.2–4.75) increased the reaction rate and gave higher adduct yields while the stereoselectivity of the reaction was unaltered (Table S2 (ESI†), entries 5–8 and 11).
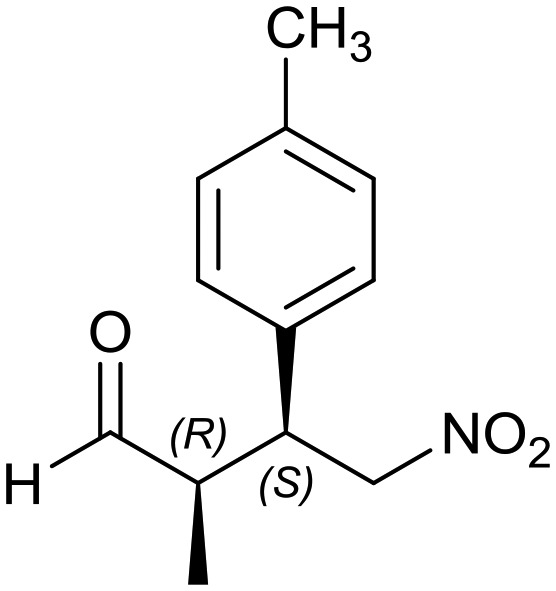
Generalizing the scope of the reaction, the addition of different linear aldehydes to β-nitrostyrene with several aromatic substituent groups was examined. The presence of electron donating (Me, OMe) or withdrawing (Br, CF3) groups was analyzed, in order to evaluate the performance of the hybrid material HybPyr as an asymmetric heterogeneous catalyst. The Michael addition was then performed with 20 mol% catalyst, at 15 °C, in dichloromethane. As we expected, when withdrawing groups were present in the para position, an increase of reaction rate (shorter reaction time) was observed (Table 4, 6b, 7b and 8b). Meanwhile, when a donating group in the para position or withdrawing group in the meta position was present, a decrease of reactivity was noticed (Table 4, 6d, 7d, 8d, 6c, 7c, 8c, 6e, 7e and 8e). In all cases, high Michael adduct yields were achieved, and the diastereoselectivity and enantioselectivity were also high, up to 90% and 75%, respectively, indicating that changes of the electronic properties of the nitroaromatic compound did not affect the stereocontrol and the course of reaction. In conclusion, the high potential of the pyrrolidine-carbamate-type hybrid catalyst (HybPyr) for addition of linear aldehydes to β-nitrostyrene derivatives with high enantioselectivity was confirmed.
Table 4. Scope of asymmetric and catalytic performance of the HybPyr catalyst.
| |||||
| Substrate | R1 = Ph | R1 = 4-BrPh | R1 = 4-MePh | R1 = 2-CF3Ph | R1 = 4-MeOPh |
| R2 = Me |
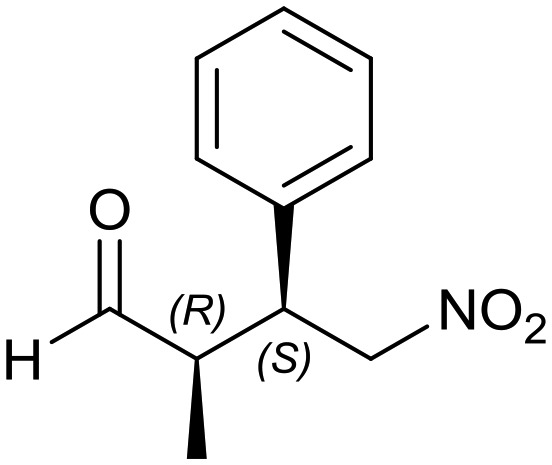 6a: yield = 94% (8 h)
6a: yield = 94% (8 h) |
 6b: yield = 98% (8 h)
6b: yield = 98% (8 h) |
 6c: yield = 99% (14 h)
6c: yield = 99% (14 h) |
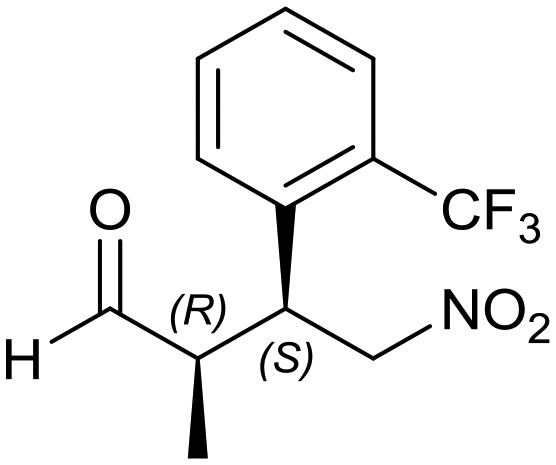 6d: yield = 94% (56 h)
6d: yield = 94% (56 h) |
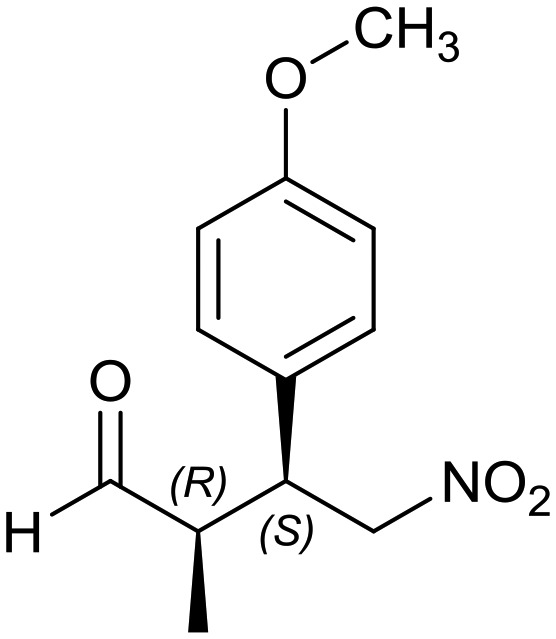 6e: yield = 95% (24 h)
6e: yield = 95% (24 h) |
| dr = 89 : 11/ee = 78% | dr = 89 : 11/ee = 78% | dr = 90 : 10/ee = 80% | dr = 88 : 12/ee = 76% | dr = 89 : 11/ee = 78% | |
| syn/anti = 88 : 12 | syn/anti = 89 : 11 | syn/anti = 88 : 12 | syn/anti = 89 : 11 | syn/anti = 87 : 13 | |
| R2 = Et |
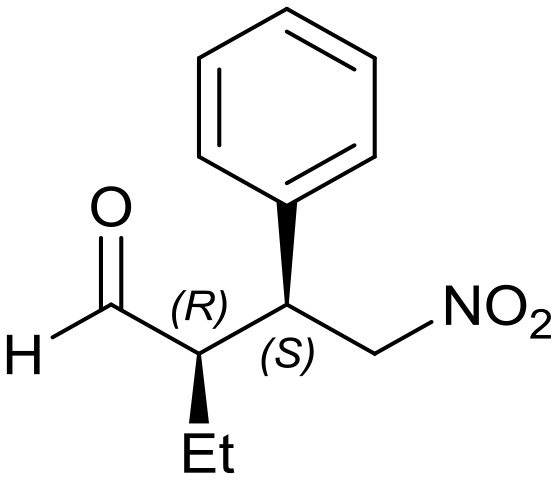 7a: yield = 99% (10 h)
7a: yield = 99% (10 h) |
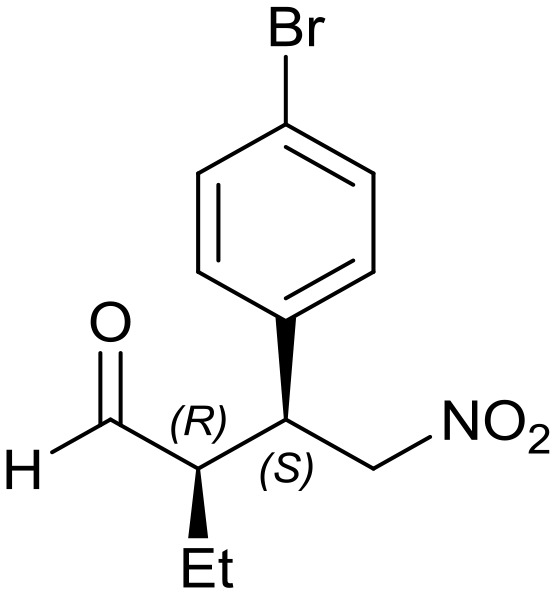 7b: yield = 100% (10 h)
7b: yield = 100% (10 h) |
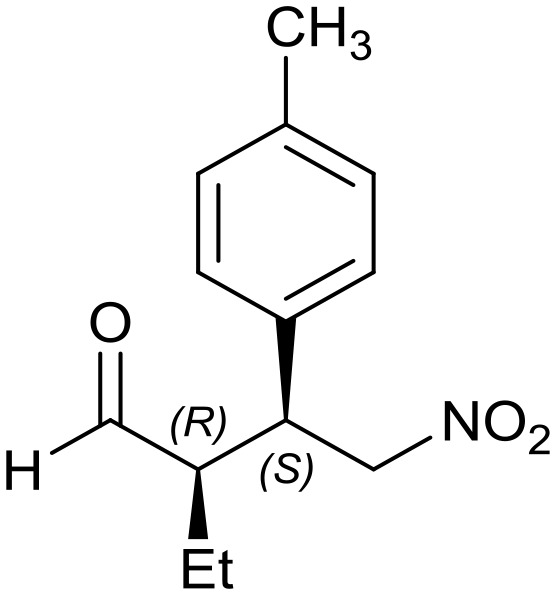 7c: yield = 99% (14 h)
7c: yield = 99% (14 h) |
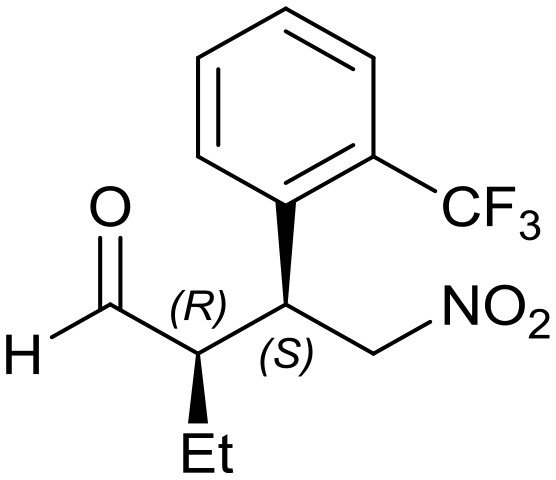 7d: yield = 94% (45 h)
7d: yield = 94% (45 h) |
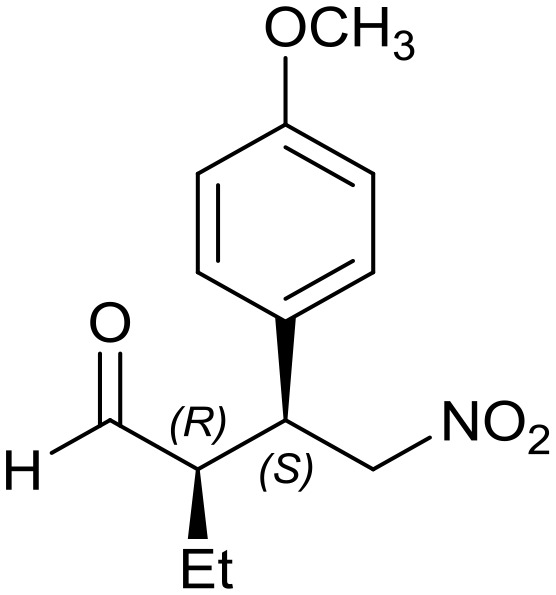 7e: yield = 99% (45 h)
7e: yield = 99% (45 h) |
| dr = 91 : 9/ee = 82% | dr = 91 : 9/ee = 82% | dr = 90 : 10/ee = 80% | dr = 91 : 9/ee = 80% | dr = 89 : 11/ee = 78% | |
| syn/anti = 92 : 8 | syn/anti = 89 : 11 | syn/anti = 89 : 11 | syn/anti = 92 : 8 | syn/anti = 87 : 13 | |
| R2 = Pr |
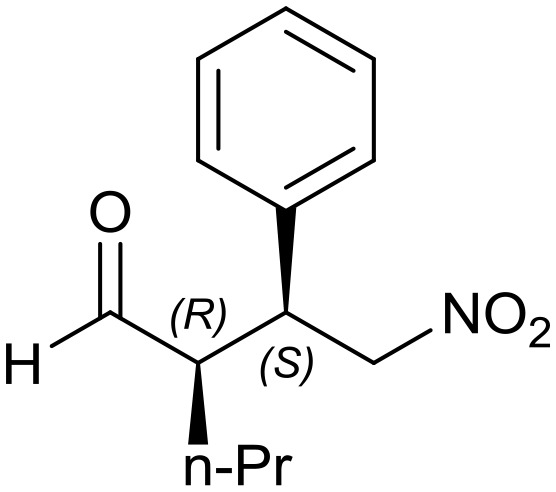 8a: yield = 97% (12 h)
8a: yield = 97% (12 h) |
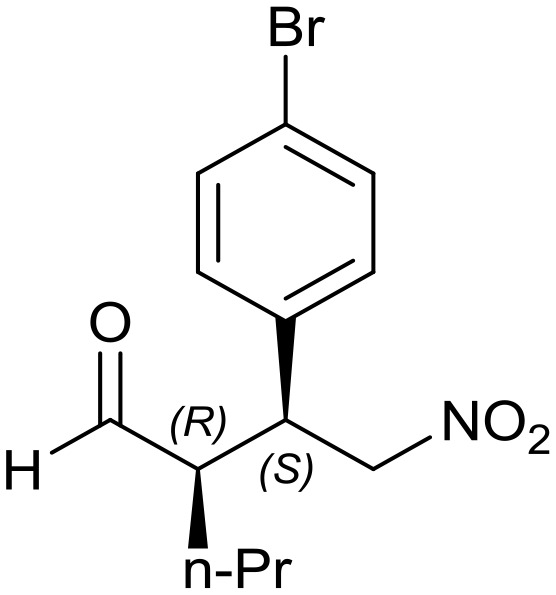 8b: yield = 97% (7 h)
8b: yield = 97% (7 h) |
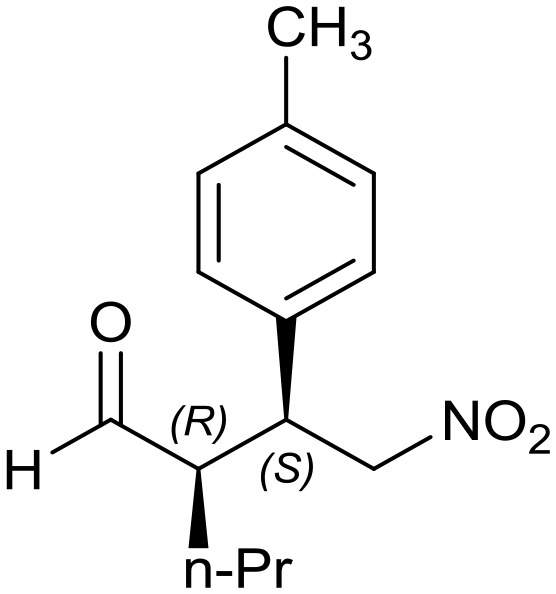 8c: yield = 94% (15 h)
8c: yield = 94% (15 h) |
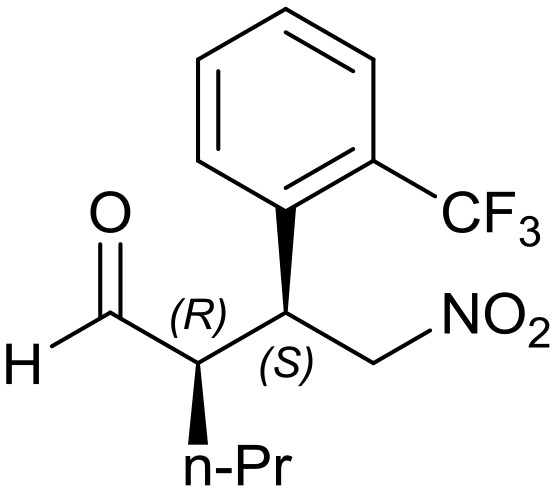 8d: yield = 98% (40 h)
8d: yield = 98% (40 h) |
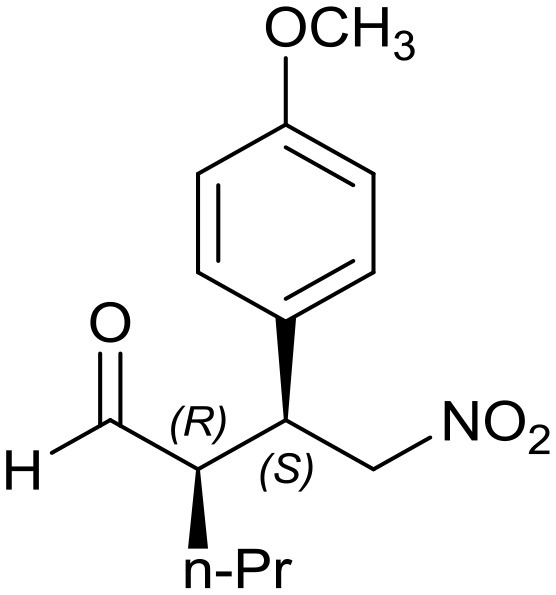 8e: yield = 93% (29 h)
8e: yield = 93% (29 h) |
| dr = 90 : 10/ee = 80% | dr = 91 : 9/ee = 82% | dr = 91 : 9/ee = 82% | dr = 91 : 9/ee = 82% | dr = 88 : 12/ee = 76% | |
| syn/anti = 87 : 13 | syn/anti = 95 : 5 | syn/anti = 91 : 9 | syn/anti = 92 : 8 | syn/anti = 87 : 13 | |
Definitively, this study shows the successful incorporation and stabilization of pyrrolidine carbamate disilane into the mesoporous siliceous framework together with the high potential of the hybrid material, HybPyr, as an effective and efficient asymmetric catalyst to perform the Michael addition of linear aldehydes onto nitro aromatic compounds, overcoming traditional problems of chiral organocatalytic moieties embedded into organic polymers in which the porous confinement effect is not possible and random distribution of chiral active sites is also observed.34
4. Conclusions
In summary, we have synthesized an innovative chiral mesoporous hybrid material, HybPyr, based on a bis-silylated chiral derivative of pyrrolidine, using a fluoride sol–gel route in the absence of structural directing agents and under soft synthesis conditions. The effective incorporation and stabilization of these organic units were confirmed by different characterization techniques (elemental analysis, TGA, NMR, FTIR, and TEM). The chiral material was successfully used as an asymmetric catalyst to perform the Michael addition between a wide range of linear aldehydes to nitroalkenes offering direct access to highly functionalized products through a rapid and atom-economical way and, at the same time, with excellent yields as well as high enantioselectivity. The hybrid catalyst was stable, easily separable and can be reused several times. We believe that the solid chiral hybrid catalyst prepared in this work may provide new synthetic possibilities for multicomponent transformations, with the consequent preparation of chiral and more sophisticated demanded products and commodities.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
The authors are grateful for financial support from the Spanish Government by MAT2014-52085-C2-1-P, MAT2017-82288-C2-1-P and Severo Ochoa Excellence Program SEV-2016-0683. S. Ll. thanks predoctoral fellowships from MINECO for economical support (BES-2015-072627). The authors thank the MULTY2HYCAT (EU-Horizon 2020 funded project under grant agreement no. 720783). The European Union is also acknowledged by ERC-AdG-2014-671093-SynCatMatch.
Footnotes
†Electronic supplementary information (ESI) available: Tables related to the effects of the solvent and additive addition on the enantioselective catalytic performance of the HybPyr catalyst. NMR spectra of the bis-silylated precursor and previous intermediates together with spectra of hybrid catalysts after successive catalytic cycles. Racemic and chiral HPLC chromatograms for Michael adducts. See DOI: 10.1039/c8cy01650j
References
- Gawley R. E. and Aub J., Principles of Asymmetric Synthesis, Elsevier, 2012. [Google Scholar]
- Hultzsch K. C., Giri R., Shi B. F., Engle K. M., Maugel N., Yu J. Q. Adv. Synth. Catal. Chem. Soc. Rev. 2005;2009;34738:367. 3242. [Google Scholar]
- Machajewski T. D., Wong C. H. Angew. Chem., Int. Ed. 2000;39:1352. doi: 10.1002/(sici)1521-3773(20000417)39:8<1352::aid-anie1352>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Dalko P. I., Moisan L., Houk K. N., List B., Notz W., Tanaka F., Barbas C. F. Angew. Chem., Int. Ed. Acc. Chem. Res. Acc. Chem. Res. 2004;2004;2004;433737:5138. 487, 580. [Google Scholar]
- Chiroli V., Benaglia M., Puglisi A., Porta R., Jumdeb R. P., Mandoli A. Green Chem. 2014;16:2798. [Google Scholar]
- Sheldon R. A. and Bekkum van H., Fine Chemicals through Heterogeneous Catalysis, Wiley-VCH Verlag GmbH, Weinheim, 2001. [Google Scholar]
- Rase H. F., Handbook of Commercial Catalysts: Heterogeneous Catalysts, CRC Press, New York, 2000. [Google Scholar]
- Hallman K., Moberg C., Clarke R. J., Shannon I. J., Bigi F., Moroni L., Maggi R., Sartori G. Tetrahedron: Asymmetry. Chem. Commun. Chem. Commun. 2001;2001;2002;12:1475. 1936, 716. [Google Scholar]
- Alza E., Cambeiro X. C., Jimeno C., Pericas M. A. Org. Lett. 2007;9:3717. doi: 10.1021/ol071366k. [DOI] [PubMed] [Google Scholar]
- Mallat T., Orglmeister E., Baiker A., Humblot V., Haq S., Muryn C., Hofer W. A., Raval R. Chem. Rev. J. Am. Chem. Soc. 2007;2002;107124:4863. 503. doi: 10.1021/cr0683663. [DOI] [PubMed] [Google Scholar]
- Ma L., Falkowski J. M., Abney C., Lin W. Nat. Chem. 2010;2:838. doi: 10.1038/nchem.738. [DOI] [PubMed] [Google Scholar]
- Moreau J., Vellutini L., Man M., Bied C., Yang Q., Han D., Yang H., Li C. J. Am. Chem. Soc. Chem. – Asian J. 2001;2008;1233:1509. 1214. [Google Scholar]
- Corma A., Iborra S., Rodríguez I., Iglesias M., Sánchez F. Catal. Lett. 2002;82:237. [Google Scholar]
- Monge-Marcet A., Pleixats R., Cattoën X., Wong Chi Man M., Alonso D. A., Nájera C. New J. Chem. 2011;35:2766. [Google Scholar]
- Luanphaisarnnont T., Hanprasit S., Somjit V., Ervithayasuporn V. Catal. Lett. 2018;148:779. [Google Scholar]
- List B., Lerner R. A., Barbas C. F. J. Am. Chem. Soc. 2000;122:2395. [Google Scholar]
- Hickmott P. W. Tetrahedron. 1982;38:1975. [Google Scholar]
- Lelais G., MacMillan D. W. C., List B., Palomo C., Mielgo A. Aldrichimica Acta. Chem. Commun. Angew. Chem., Int. Ed. 2006;2006;2006;3945:79. 819, 7876. [Google Scholar]
- Wang W., Wang J., Li H., Wang W., Wang J., Li H. Angew. Chem. Angew. Chem., Int. Ed. 2005;2005;11744:1393. 1369. [Google Scholar]
- Hayashi Y., Gotoh H., Hayashi T., Shoji M., Hayashi Y., Gotoh H., Hayashi T., Shoji M. Angew. Chem. Angew. Chem., Int. Ed. 2005;2005;11744:4284. 4212. [Google Scholar]
- Palomo C., Vera S., Mielgo A., Gomez-Bengoa E., Palomo C., Vera S., Mielgo A., Gomez-Bengoa E. Angew. Chem. Angew. Chem., Int. Ed. 2006;2006;11845:6130. 5984. [Google Scholar]
- Atodiresei I., Vila C., Rueping M., Giacalone F., Gruttadauria M., Agrigento P., Campisciano V., Noto R. ACS Catal. Catal. Commun. 2015;2011;516:6241. 75. [Google Scholar]
- Berner B. J., Tedeschi L., Enders D. Eur. J. Org. Chem. 2002;12:1877. [Google Scholar]
- Gregg S. J. and Sing K. S. W., Adsorption, Surface Area and Porosity, Academic Press, 2nd edn, 1982. [Google Scholar]
- Sing K. S. W., Everett D. H., Haul R. A. W., Moscou L., Pierotti R. A., Rouquerol J., Siemieniewska T. Pure Appl. Chem. 1985;57:603. [Google Scholar]
- Barrett E. P., Joyner L. G., Halenda P. P. J. Am. Chem. Soc. 1951;73:373. [Google Scholar]
- Gray D., Concellón C., Gallagher T., Orliac A., Routier J., Burgat Charvillon F., Sauer W. H. B., Bombrun A., Kulkarni S. S., Gómez Pardo D., Cossy J. J. Org. Chem. Chem. – Eur. J. 2004;2014;6920:4849. 3813. doi: 10.1021/jo049562h. [DOI] [PubMed] [Google Scholar]
- Kovačková S., Dračínský M., Rejman D., Heindl C., Hübner H., Gmeiner P. Tetrahedron. Tetrahedron: Asymmetry. 2011;2003;6714:1485. 3153. [Google Scholar]
- Betancort J. M., Barbas C. F., Alexakis A., Andrey O., Andrey O., Alexakis A., Tomassini A., Bernardinelli G., Wang W., Wang J., Li H., Wang W., Wang J., Li H., Barros M. T., Faísca Phillips A. M., Lu D., Gong Y., Wang W., Wang W. H., Abe T., Wang X. B., Kodama K., Hirose T., Zhang G. Y., Sagamanova I., Rodríguez-Escrich C., Molnár I. G., Sayalero S., Gilmour R., Pericàs M. A. Org. Lett. Org. Lett. Adv. Synth. Catal. Angew. Chem. Angew. Chem., Int. Ed. Eur. J. Org. Chem. Adv. Synth. Catal. Tetrahedron: Asymmetry. ACS Catal. 2001;2002;2004;2005;2005;2007;2010;2010;2015;34346117441352215:3737. 3611, 1147, 1393, 1369, 178, 644, 2925, 6241. [Google Scholar]
- Patora-Komisarskaa K., Benohouda M., Ishikawaa H., Seebach D., Hayashi Y. Helv. Chim. Acta. 2011;94:719. [Google Scholar]
- Rantanen T., Shiffers I., Zani L. Angew. Chem., Int. Ed. 2005;44:1758. doi: 10.1002/anie.200500154. [DOI] [PubMed] [Google Scholar]
- Sulzer-Mossé S., Alexakis A. Chem. Commun. 2007:3123. doi: 10.1039/b701216k. [DOI] [PubMed] [Google Scholar]
- Alexakis A., Andrey O. Org. Lett. 2002;4:3611. doi: 10.1021/ol026543q. [DOI] [PubMed] [Google Scholar]
- Li P., Wang L., Wang M., Zhang Y., Androvic L., Drabina P., Svodova M., Sedlak M., Omar E. M., Dhungana K., Headley A. D., Rahman M. B. A., Ni B., Zhang Q., Dhungana K., Headley A. D. Eur. J. Org. Chem. Tetrahedron: Asymmetry. Lett. Org. Chem. Org. Lett. 2008;2015;2011;2009;727811:1157. 782, 170, 1037. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.