

ABSTRACT
Aberrant expression of noncoding RNAs plays a critical role during tumorigenesis. To uncover novel functions of long non-coding RNA (lncRNA) in lung adenocarcinoma, we used a microarray-based screen identifying LINC00673 with elevated expression in matched tumor versus normal tissue. We report that loss of LINC00673 is sufficient to trigger cellular senescence, a tumor suppressive mechanism associated with permanent cell cycle arrest, both in lung cancer and normal cells in a p53-dependent manner. LINC00673-depleted cells fail to efficiently transit from G1- to S-phase. Using a quantitative proteomics approach, we confirm the modulation of senescence-associated genes as a result of LINC00673 knockdown. In addition, we uncover that depletion of p53 in normal and tumor cells is sufficient to overcome LINC00673-mediated cell cycle arrest and cellular senescence. Furthermore, we report that overexpression of LINC00673 reduces p53 translation and contributes to the bypass of Ras-induced senescence. In summary, our findings highlight LINC00673 as a crucial regulator of proliferation and cellular senescence in lung cancer.
KEYWORDS: LINC00673, long noncoding RNA, lung cancer, p53, senescence
Introduction
Lung cancer accounts for most of the cancer-related deaths worldwide and is associated with a poor prognosis [1]. The group of non-small cell lung cancer (NSCLC) represents 85% of diagnosed lung neoplasms with lung adenocarcinoma (ADC) being the most abundant subtype [2]. Comprehensive analyses revealed that over 80% of the human genome is transcribed and the class of long noncoding RNAs (lncRNA) accounts for a significant proportion of the noncoding transcriptome [3,4]. LncRNAs are generally defined to be longer than 200 nucleotides with little or no protein-coding potential [5]. In-depth analyses of the epigenetic, genomic and transcriptional landscape of human cancers revealed that many lncRNAs exhibit cancer type-specific aberrant expression [6], thus presenting a source of novel markers and alternative target molecules for cancer therapy [7]. LncRNAs play critical roles in the onset and progression of cancer [8–10], and various studies have confirmed their impact on epigenetic, transcriptional as well as post-transcriptional gene regulation [5]. To comprehensively understand lung cancer-promoting pathways and uncover potential novel druggable targets for cancer therapy, multiple screens for lncRNAs were conducted utilizing lung cancer specimens [9]. However, to date only few lncRNAs have been thoroughly characterized.
In this study, we identify the altered expression of 479 ncRNAs in lung ADC patient samples compared to normal tissue using a comparative microarray profiling approach. We characterize the most upregulated lncRNA LINC00673 in functional detail and define its role in the regulation of cellular senescence, a state of stable cell cycle arrest which is recognized as an effective tumor suppressor mechanism [11].
Results
LINC00673 is upregulated in lung adenocarcinoma
To identify novel dysregulated ncRNAs, 27 early-stage lung ADC and adjacent non-malignant lung samples derived from patients, were subjected to a microarray-based expression profiling. 479 ncRNAs were differentially expressed between tumor and normal tissues with a significant and minimum twofold change (Fig. 1A, Suppl. Fig. S1, Suppl. Table S1). As a control for the validity of our microarray hybridization and analysis, we identified sex-specific expression patterns between normal or malignant lung tissues from male versus female patients (Suppl. Fig. S2A,B). Six mRNAs and one lncRNA, namely XIST, were independently identified as significantly differentially expressed between male and female patients (adjusted P < 0.05 (FDR), fold change FC ≥ 2). Notably, the six male-specific transcripts were all derived from the Y chromosome while the one female-specific transcript was XIST, the lncRNA involved in inactivation of the second X chromosome in female cells. The results were identical when either normal or tumor tissues were analyzed separately demonstrating the reliability of the identified transcript signature. In agreement with previous reports, the majority of detected ncRNAs was less abundant compared to mRNAs [3,12] (Suppl. Fig. S3). The ncRNA LINC00673 was the most significantly and most highly upregulated ncRNA in lung ADC using our comparative microarray analysis (Fig. 1B). We validated our microarray results by RT-qPCR in two independent patient cohorts (Fig. 1C, Suppl. Fig. S4) and identified LINC00673 expression in multiple cancer cell lines derived from different tumors (Suppl. Fig. S5A). In addition, we used the TANRIC platform [13] to uncover the enhanced expression of LINC00673 in various other malignancies (Suppl. Fig. S5B) supporting a potentially oncogenic role for this lncRNA. LINC00673 expression did not correlate with patient survival, lung cancer stage or the smoking behavior in TCGA lung ADC (Suppl. Fig. S6A–C) suggesting a low prognostic and predictive value for lung ADC.
Figure 1.
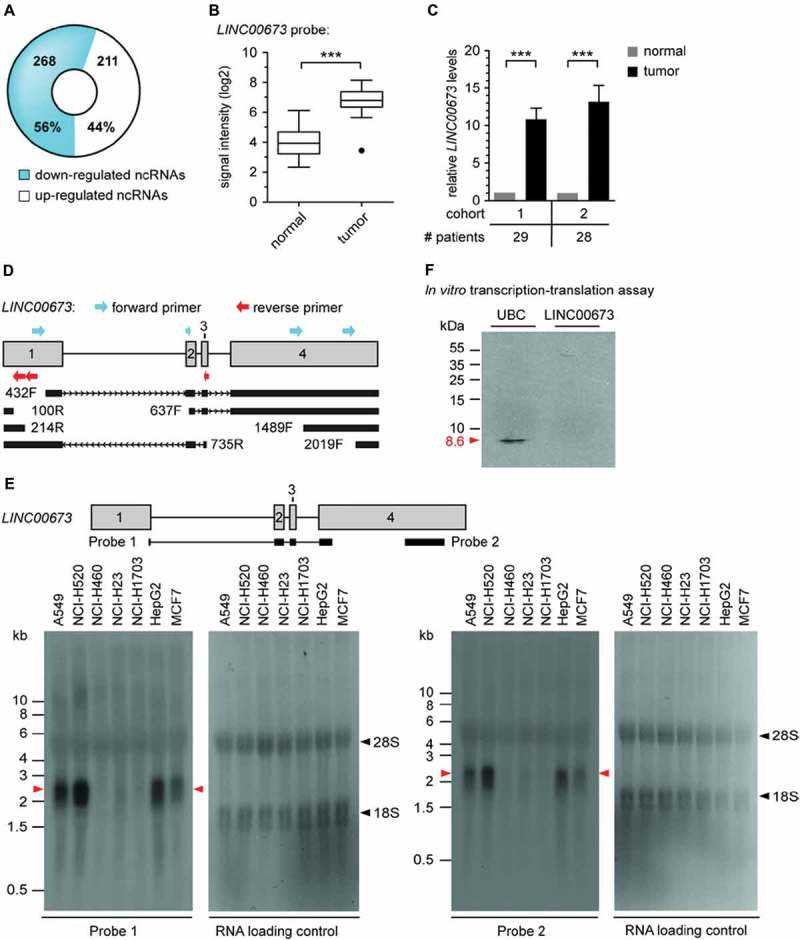
Identification, validation and characterization of LINC00673 lncRNA. (A) Comparative microarray analysis identified 479 significantly deregulated ncRNAs in non-matched lung ADC vs. normal lung patient samples (n = 27, corrected P < 0.05, FC ≥ 2). (B) LINC00673 lncRNA expression was significantly enhanced in lung ADC (n = 27; absolute FC between tumor and normal samples is shown, with ***, P < 0.001). (C) Elevated LINC00673 levels were validated by RT-qPCR analysis in two patient cohorts (cohort 1 overlapping with the microarray cohort) comprising matched tumor and normal samples. PPIA was used as reference gene and the mean ratios of tumor/normal (T/N) + SEM are shown. Statistical significance was determined with paired Student t test, with ***, P < 0.001. (D) Schematic overview of detected transcripts matching LINC00673 reference sequence by RACE in A549 cDNA. The first nucleotide of each RACE primer is indicated (F: forward primer, R: reverse primer). (E) Northern Blot analysis of LINC00673 in different cell lines. 18S and 28S rRNA bands are indicated by black arrows. (F) In vitro transcription-translation assay based on DNA templates. The incorporation of [35S] methionine into proteins was detected by autoradiography, and ubiquitin C (UBC) served as positive control. One representative experiment is shown (n = 3).
Molecular characterization of LINC00673
We next established the full-length sequence of the LINC00673 transcript by rapid amplification of cDNA ends (RACE) in the lung cancer cell line A549 (Fig. 1D) and by Northern blot (Fig. 1E). The transcript comprised of 4 exons and a length of roughly 2.3 kb coinciding with the database sequence of LINC00673 (NR_036488). Notably, the detected Northern blot signal intensities for LINC00673 positively correlated with the determined relative expression of LINC00673 using RT-qPCR in selected cell lines (Suppl. Fig. S5A). We further examined the coding potential of LINC00673 by conducting a search for putative open reading frames (ORF) using the NCBI ORF Finder tool. A total of six putative ORFs were identified, of which five matched the canonical AUG start codon (data not shown). None of the predicted peptides were identified in the deposited tandem mass spectrometry data provided by PeptideAtlas [14]. Further supporting the noncoding nature of LINC00673, an in vitro translation approach failed to generate detectable proteins from a DNA template, while the ubiquitin C (UBC) control protein was efficiently produced (Fig. 1F).
The determination of the subcellular localization of a lncRNA might provide critical information about its biological functions [15]. Hence, cellular fractionation experiments of A549 cells revealed an enrichment of LINC00673 in the cytoplasm (Fig. 2A), which was further confirmed by RNA FISH (fluorescence in situ hybridization; Fig. 2B). Moreover, we examined the half-life of LINC00673 by measuring the relative abundance of transcripts in actinomycin D-treated A549 cells by RT-qPCR (Fig. 2C). LINC00673 displayed a short half-life of <2 h, which is characteristic of known regulatory RNAs [16]. The stable housekeeping mRNA PPIA [16] and the rapidly processed 45S pre-rRNA were used as controls.
Figure 2.
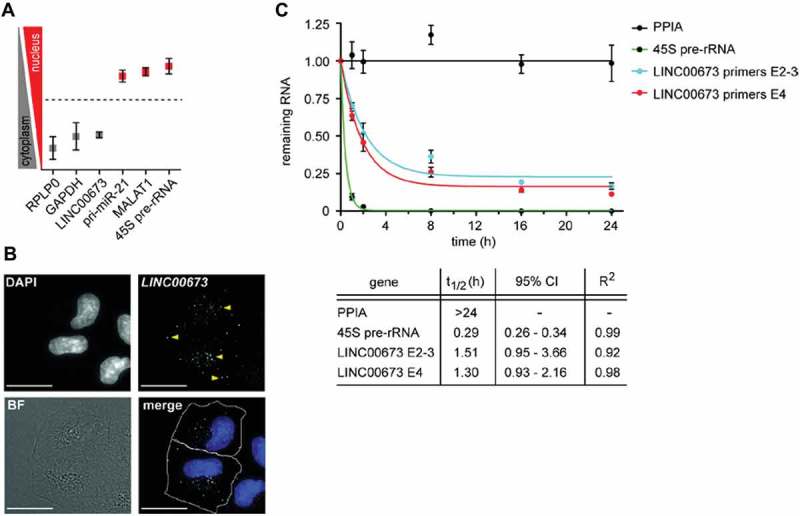
LINC00673 is a cytoplasmic lncRNA with a short half-life. (A) Cellular fractionation of A549 cells and subsequent RT-qPCR (represented as nuclear/cytosolic (log2) ratios) indicated an enrichment of LINC00673 in the cytoplasmic fraction. Data represent the mean ± SEM (n = 3). (B) RNA FISH using custom Stellaris (LGC Biosearch Technologies) probes for LINC00673 revealed a cytoplasmic localization. Yellow arrows indicate lncRNA signal, and the scale bars represent 20 μm. BF: brightfield. (C) RNA stability was determined by actinomycin D (10 µg/ml) treatment of A549 cells for up to 24 h and the RNA levels were measured by RT-qPCR. LINC00673 expression was detected with two different primer pairs spanning exon 2–3 (E2-3) and located within exon 4 (E4), respectively. Data represent the mean ± SEM (n = 3), t1/2: half-life, CI: confidence interval. Data was fitted to a non-linear least squares regression curve (one phase decay) with GraphPad Prism 5.
LINC00673 is a regulator of cell proliferation
As a first step to approach the possible function of LINC00673 in lung cancer, we sought to analyze commonalities with genes whose expression correlated with LINC00673 lncRNA in the TCGA lung ADC set using the TANRIC platform. Subsequent functional profiling of positively correlated genes with g:Profiler [17] revealed a striking enrichment of E2F-regulated genes (transcription factor binding sites are retrieved from TRANSFAC database, Suppl. Fig. S7A). In addition, levels of LINC00673 and E2F1 protein significantly correlated in different lung fibroblast and lung cancer cell lines (Suppl. Fig. S7B,C,D, Suppl. Table S2). Implicating LINC00673 in the same pathway by guilt-by-association, the putative LINC00673 promoter region itself was recognized by E2F1 according to publically available ChIP-Seq data (Suppl. Fig. S7E). To investigate whether E2F1, an important regulator of cell cycle progression [18], also regulated the expression of LINC00673, we took advantage of the human lung cancer cell line NCI-H1299 and the human embryonic lung fibroblasts WI-38 stably expressing conditionally active E2F1, namely ER-E2F1 [19]. The activation of ER-E2F1 by 4-hydroxytamoxifen (OHT) significantly increased LINC00673 expression in both cell lines (Suppl. Fig. S7F). In support of a direct regulation of LINC00673 levels, the activation of ectopic E2F1 in the presence of the protein synthesis inhibitor cycloheximide (CHX) still led to the induction of LINC00673 (Suppl. Fig. S7G). To investigate whether LINC00673 levels were also regulated during the cell cycle, IMR-90 normal immortalized human lung fibroblast cells as well as A549 lung adenocarcinoma cells were arrested in G0 by serum starvation and transcript levels measured after releasing the cells from the cell cycle block. In the normal fibroblasts, E2F1 and its known target gene MCM6 [20] were induced, while LINC00673 levels were not regulated in a cell cycle-dependent manner (Suppl. Fig. S7H). In the lung cancer cells, LINC00673, E2F1 and MCM6 RNA levels correlated well with each other showing a decrease upon serum starvation and an increase upon release from starvation (Suppl. Fig. S7I). We concluded that endogenous E2F1 did not regulate LINC00673 during normal cell cycle progression, but that aberrant E2F1 activity in transformed cells lead to elevated LINC00673 levels. Given the correlation of LINC00673 with E2F-regulated cell cycle-associated genes at steady state levels, we sought to investigate whether LINC00673 itself was a crucial regulator of cell proliferation.
To explore the cellular functions of LINC00673, we conducted knockdown studies by applying two different knockdown tools, namely LNA longRNA GapmeRs (Exiqon) and siPOOLs (siTOOLs Biotech). The latter comprise a well-defined, custom-designed pool of 30 siRNAs targeting the gene of interest and were previously shown to eliminate off-target effects that are frequently observed with single siRNAs [21]. Knockdown efficiency of LINC00673 in the lung cancer cell lines A549 and Calu-3 was confirmed using either GapmeRs or the siPOOL (Suppl. Fig. S8A,B). Depletion of LINC00673 using either GapmeRs or the siPOOL significantly decreased A549 cell number (Fig. 3A). This effect was not caused by an increased apoptotic response since no significantly enhanced activation of the cellular caspases 3 and 7 was observed (Fig. 3B). To investigate whether the decrease in cell number was a result of reduced cell proliferation, we quantified the incorporation of BrdU 24 h and 48 h after LINC00673 knockdown, respectively. The GapmeR- and siPOOL-mediated knockdown of LINC00673 both significantly decreased cell proliferation of A549 and Calu-3 cells (Fig. 3C). Notably, LINC00673 knockdown also reduced cell proliferation of the normal human lung fibroblasts WI-38 and IMR-90 (Fig. 3D). The described observations were consistent with GapmeR- and siPOOL-mediated knockdown of LINC00673 in lung cancer cells. Discrepancies in the strength of monitored effects were mainly observed between the two GapmeRs and pointed towards possible off-target effects despite comparable knockdown efficiencies (Fig. 3A). For this reason, siPOOL-mediated knockdown of LINC00673 was used in all subsequent experiments.
Figure 3.
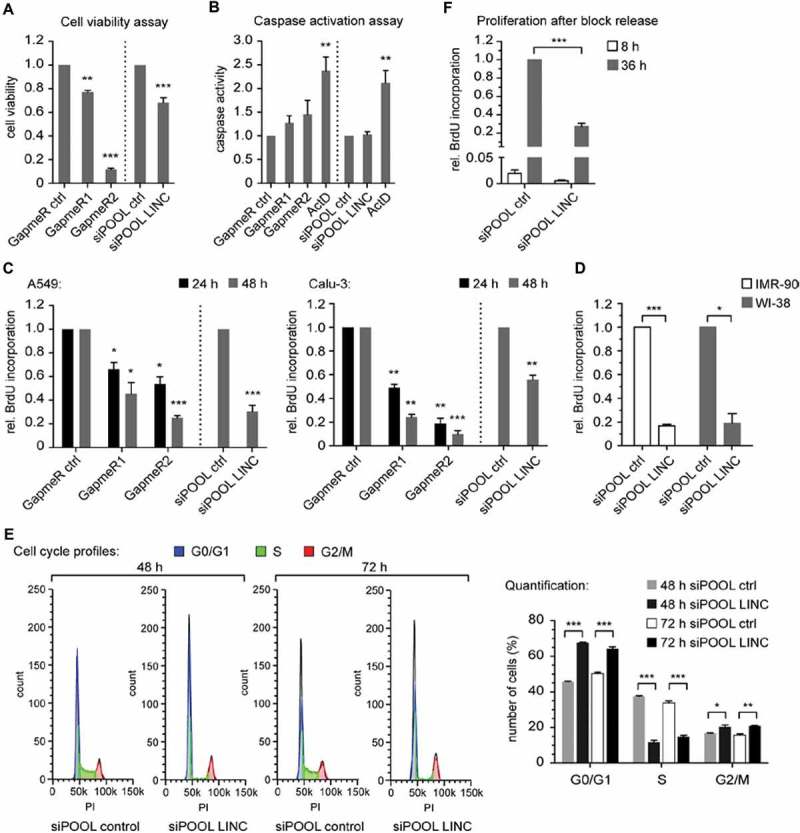
LINC00673 reduces cell proliferation and induces a cell cycle arrest. (A) CellTiter-Glo Luminescent Cell Viability Assay (Promega) was performed 48 h after LINC00673 knockdown in A549 cells with GapmeRs (60 nM; n = 3) and siPOOLs (10 nM; n = 4), respectively. (B) Caspase-Glo 3/7 luminescent assay (Promega) was performed 24 h after LINC00673 knockdown in A549 cells (final concentration of GapmeRs and siPOOLs as in A; n = 6 for GapmeR-mediated knockdown and n = 4 for siPOOL-mediated knockdown experiments). Treatment with actinomycin D (ActD; 5 µg/ml) served as a positive control for apoptosis induction. (C) BrdU Cell Proliferation ELISA Kit (Roche) was used to quantify cell proliferation 24 h and 48 h after LINC00673 knockdown (final concentrations as in A; n = 3 for GapmeR-mediated knockdown in A549 and Calu-3; n = 5 for siPOOL-mediated knockdown in A549 and n = 3 for Calu-3). (D) BrdU Cell Proliferation ELISA Kit was used to quantify cell proliferation 48 h after siPOOL-mediated knockdown (final concentration of 1 nM) of LINC00673 in IMR-90 and WI-38 cells (n = 3). (E) Cell cycle profiles of A549 cells were analyzed by FACS after 48 h and 72 h of LINC00673 knockdown. Quantification of cells with the cell cycle function of FlowJo (V10) and the cell cycle profiles of one representative experiment are shown (n = 3). (F) IMR-90 cells were serum starved for 24 h and subsequently transfected with 0.3 nM siPOOLs. Cells were released by adding complete medium supplemented with 10% FBS at 72 h after knockdown and BrdU incorporation was measured 8 h and 36 h following block release (n = 4). In A-F the mean + SEM is shown and the statistical significance was determined per two-sided unpaired Student t test, with *, P < 0.05; **, P < 0.01; ***, P < 0.001.
The reduction of LINC00673 levels in A549 cells was accompanied by a prominent increase of cells in G0/G1-phases and a concomitant decrease of cells in S-phase (Fig. 3E). Moreover, we found that LINC00673-depleted IMR-90 cells arrested in G0/G1 were unable to re-enter the cell cycle efficiently and consequently displayed a strong reduction in proliferation after serum stimulation (Fig. 3F). We confirmed an efficient knockdown of LINC00673 in starved IMR-90 cells and additionally were able to show a diminished induction of E2F1 and MCM6 upon cell cycle re-entry as compared to the siPOOL control condition (Suppl. Fig. S8C). In accordance with our experimental data, we uncovered a highly significant enrichment of LINC00673-correlated genes in the gene ontology terms of mitotic cell cycle and cell cycle phase transition (Suppl. Fig. S9). Thus, loss of LINC00673 severely impaired cell cycle progression.
LINC00673 depletion triggers cellular senescence
LINC00673 knockdown caused a change of cellular morphology, namely increased cell size and adaptation of a flat cell morphology in both A549 lung cancer cells and IMR-90 normal lung fibroblast cells (Suppl. Fig. S10). These alterations are morphological hallmarks of cellular senescence [22], a state of stable proliferative arrest.
In response to various stimuli, cellular senescence is controlled by the tumor suppressor pathways p16INK4a/pRb and p53 [22,23]. Noteworthy, disruption of the pRb and the p53 pathways is frequent in NSCLC [24–26]. The experimental cell models used here reflect these different genetic backgrounds. A549 cells present a homozygous deletion of the p16INK4a/p14ARF locus, whereas IMR-90 lung fibroblasts employ functional pRb and p53 pathways.
Numerous senescence markers have been proposed and their occurrence partially depends on cell and tissue types [27]. We chose to first analyze the accumulation of senescence-associated β-galactosidase (SA-β-Gal) [28] and the decreased expression of E2F1 target genes [29–32] as indicators of senescence. We confirmed a strong increase of SA-β-Gal positive A549 and IMR-90 cells 4 days after LINC00673 knockdown (Fig. 4A,B). Doxorubicin treatment of cells served as a positive control for SA-β-Gal stainings. Next, we investigated the expression of E2F-regulated genes and crucial cell cycle regulators following LINC00673 knockdown [20]. Significant reduction of selected genes became evident at the transcript level in A549 cells as soon as 48 h following depletion (Fig. 4C), an early timepoint undergoing cell cycle arrest (Fig. 3E). Furthermore, we noted the elevated expression of genes characteristic for the senescence-associated secretory phenotype (SASP) [33] in A549 cells (Fig. 4D) and IMR-90 cells (Suppl. Fig. S11A, B).
Figure 4.
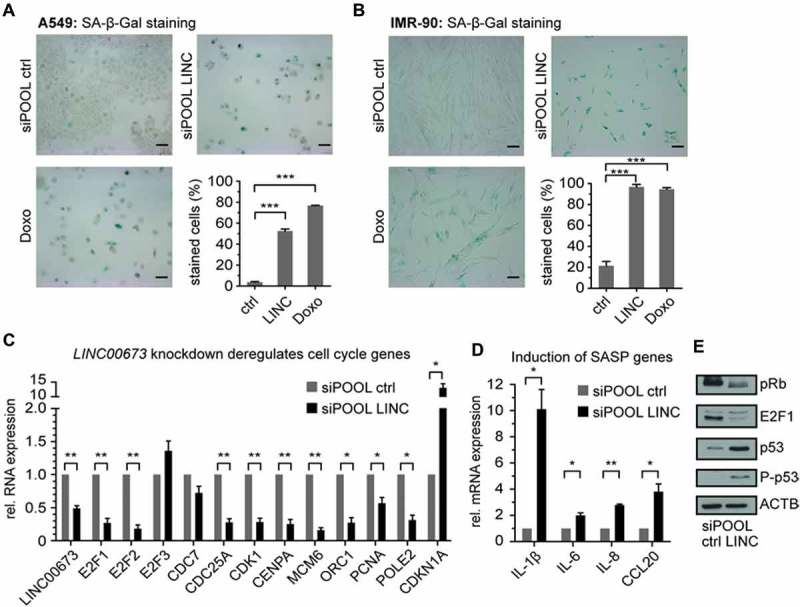
LINC00673 depletion induces a senescence-like phenotype. (A) SA-β-Gal accumulated in A549 cells 4 days after LINC00673 knockdown with siPOOLs (3 nM). As a positive control for staining, cells were treated with 200 nM doxorubicin. At least 200 cells were counted per condition (n = 4). The scale bar represents 100 µm. (B) SA-β-Gal staining of IMR-90 cells as in A (n = 3). (C) Cell cycle and E2F1-regulated genes were determined by RT-qPCR at 48 h after LINC00673 knockdown with siPOOLs (3 nM) in A549 cells. GAPDH was used as reference gene (n = 3). (D) SASP gene expression in A549 cells was determined by RT-qPCR as in C (n = 3). (E) Western Blot analysis 48 h after LINC00673 knockdown with siPOOLs (3 nM) in A549 cells. Specific antibodies were used for indicated proteins. One representative Western Blot is shown (n = 3). All data is shown as mean + SEM and the statistical significance was determined per two-sided unpaired Student t test, with *, P < 0.05; **, P < 0.01; ***, P < 0.001.
The senescence-associated cell cycle exit was previously linked to the formation of facultative heterochromatin foci at E2F-responsive promoters, a process that is mediated by pRb [32]. IMR-90 cells contain a functional p16INK4a/pRb pathway and therefore displayed enhanced formation of H3K9me3-positive foci, a surrogate marker of the senescence-associated heterochromatin foci (SAHF) phenotype (Suppl. Fig. S12). Also, LINC00673 depletion-mediated senescence was not primarily caused by persistent DNA damage since we were unable to detect the accumulation of γ-H2AX foci 4 days after LINC00673 knockdown [34–36] (Suppl. Fig. S12).
Depletion of E2F1 in cancer cells was previously linked to the induction of senescence independent of the pRb and p53 status of the cell line [37]. We also noted a decrease of E2F1 transcript and protein levels upon LINC00673 knockdown in A549 cells (Fig. 4C,E). Simultaneously, the accumulation and activation of p53 and the elevated expression of p21 underlined the potential contribution of the p53 pathway in establishing the LINC00673 depletion-mediated senescence phenotype (Fig. 4E). Although we were unable to detect persistent DNA damage foci in senescent cells, the occurrence of phospho-p53S15 suggested that the DNA damage signaling pathway contributed, at least partially, to the onset of the phenotype [34–36]. In addition, we noted an increase in hypophosphorylated pRb in A549 cells (Fig. 4E), which was in agreement with a previous report on DNA damage-induced senescence in PC-3 prostate cancer cells [37]. In summary, we concluded that the LINC00673 depletion-mediated senescence phenotype might engage the p53 pathway but did not require p16INK4a activity and was not a direct cause of persistent DNA damage.
LINC00673 depletion-induced senescence relies on the p53 pathway
We sought to analyze in more detail whether the cell cycle arrest upon LINC00673 depletion is a consequence of p53 or pRb pathway activation. For this purpose, we quantified the ability of A549 and IMR-90 cells to proliferate following simultaneous knockdown of LINC00673, p53 and/or pRb. The loss of p53 was sufficient to rescue the proliferation defect in both A549 and IMR-90 cells (Fig. 5A,B). In contrast, pRb depletion did not restore cell proliferation. The ability to proliferate positively correlated with E2F1 levels, which could only be restored and induced upon p53 depletion in A549 and IMR-90 cells, respectively (Fig. 5C,D). Active p53 protein was necessary to establish the cell cycle arrest as monitored by the specific accumulation of p53 and the increase in CDKN1A transcript levels (Fig. 5C,D, Suppl. Fig. S13). We confirmed that the knockdowns were efficient in all conditions both on RNA and protein levels (Fig. 5C,D, Suppl. Fig. S13). Finally, the siPOOL-mediated depletion of p53 was sufficient to overcome senescence triggered by LINC00673 depletion in A549 cells (Fig. 5E).
Figure 5.
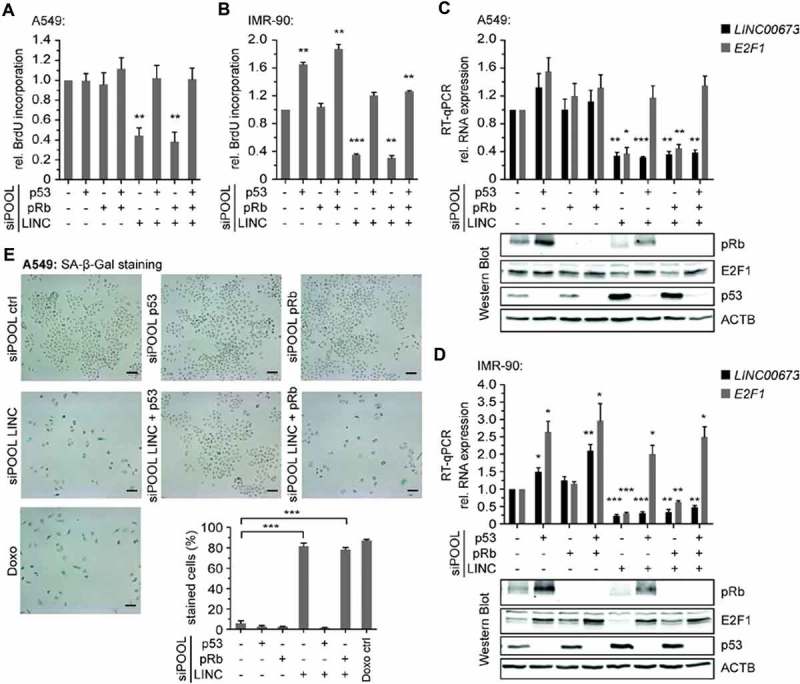
LINC00673-mediated senescence is p53-dependent. (A) BrdU Cell Proliferation ELISA Kit was used to quantify cell proliferation 48 h after knockdown of p53, pRb and LINC00673 in A549 (n = 4). The reactions contained siPOOL negative control (12 nM) or a mix of 1 nM siPOOL p53, pRb and 10 nM siPOOL LINC00673 supplemented with siPOOL negative control for a final concentration of 12 nM. (B) BrdU incorporation in IMR-90 cells was measured as in A. The reactions contained siPOOL negative control (0.9 nM) or a mix of 0.3 nM siPOOL p53, pRb and LINC00673 supplemented with siPOOL negative control for a final concentration of 0.9 nM. (C) Relative RNA levels were determined by RT-qPCR at 48 h after knockdown in A549 cells (n = 3). The reactions contained siPOOL negative control (5 nM) or a mix of 1 nM siPOOL p53, pRb and 3 nM siPOOL LINC00673 supplemented with siPOOL negative control for a final concentration of 5 nM. GAPDH was used as reference gene. One representative Western Blot is shown (n = 3). (D) RT-qPCR as in C using IMR-90 cells (n = 4). The siPOOL concentrations were as described in B. One representative Western Blot is shown (n = 4). (E) SA-β-Gal accumulated in A549 cells 4 days after knockdown. The reactions contained siPOOL negative control (4 nM) or a mix of 3 nM siPOOL LINC00673 and 1 nM siPOOL p53 or pRb supplemented with siPOOL negative control for a final concentration of 4 nM (n = 4, scale bar = 100 µm). In A-F, the mean + SEM is shown. The statistical significance was determined per two-sided unpaired Student t test, with *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Since activating mutations of Ras [38] and elevated expression of LINC00673 (Fig. 1) constitute frequent events in lung cancer, we determined whether ectopic expression of LINC00673 can contribute to the bypass of Ras-induced senescence. In human lung fibroblasts, activated Ras engages both the p53 and pRb pathways, and these two pathways need to be inactivated in order to bypass Ras-induced senescence [39,40]. While the sole expression of LINC00673 was not sufficient to bypass Ras-induced senescence, combined expression of LINC00673 and inactivation of the pRb pathway using E7 from the human papillomavirus (HPV) type 16 significantly abrogated the senescence response (Suppl. Fig. S14). On the other hand, expression of LINC00673 in p53-inactivated cells using the HPV E6 oncoprotein had no impact on senescence entry upon Ras expression (Suppl. Fig. S14). These results suggested that elevated levels of LINC00673 contribute to the bypass of Ras-induced cellular senescence by inactivating the p53 pathway, and could thus play a critical role during tumorigenesis.
To verify our findings and monitor alterations in protein expression at an early timepoint, namely 48 h following LINC00673 knockdown in A549 cells, we chose an unbiased quantitative mass spectrometry approach using SILAC. Thereby, we identified a total of 20 up- and 12 downregulated proteins in comparison to the siPOOL control condition (biological duplicate, t test with P < 0.05, fold change >1.5, Fig. 6A, Suppl. Table S3, Suppl. Fig. S15). We validated our results by RT-qPCR and confirmed that the majority of the modulated proteins displayed altered mRNA levels (Suppl. Fig. S16A,B). Interestingly, 50 % (10 out of 20) of the identified upregulated proteins were direct targets of p53, while 47 % (15 out of 32) of all identified proteins were previously associated with cellular senescence (Suppl. Table S4). Specifically, we identified the upregulation of the p53 targets and known senescence regulators PAI-1 [41] and DDB2 [42]. Among the proteins with reduced abundances, we detected UHRF1 and PRC1. While UHRF1 is known to negatively regulate the tumor suppressors p16INK4A, hMLH1, p21, pRb and PML [43], reduced PRC1 levels were previously observed in oncogenic Ras-induced and replicative senescence [44]. We found that p53 knockdown was able to at least partially restore the gene expression of selected SILAC hits (Suppl. Fig. S17). Overall, the deregulation of identified genes is in line with the early induction of a senescence phenotype in A549 cells.
Figure 6.
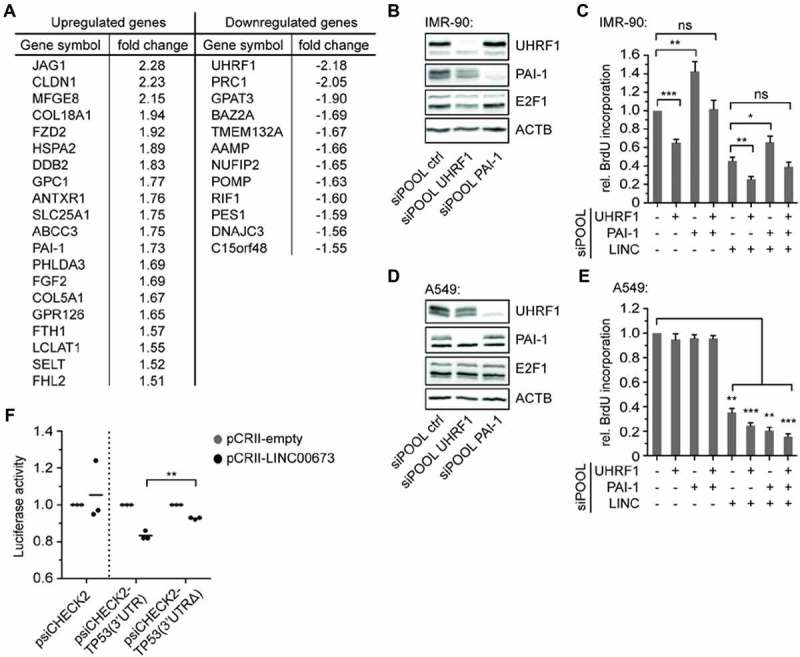
LINC00673 acts by regulating p53 translation. (A) A549 cells were adapted to SILAC medium and total protein lysates were prepared at 48 h following siPOOL-mediated knockdown of LINC00673. The lysates were analyzed by mass spectrometry and proteins that displayed an absolute fold change >1.5 as compared to the siPOOL control are listed (n = 2, t test with P < 0.05). (B) Protein levels were determined with specific antibodies 48 h after UHRF1 and PAI-1 knockdown with siPOOLs (final concentration of 0.3 nM) in IMR-90 cells. One representative Western Blot is shown (n = 3). (C) Cell proliferation was quantified by BrdU incorporation at 48 h after knockdown of UHRF1, PAI-1 and LINC00673 in IMR-90 (n = 7). The reactions contained siPOOL negative control (0.9 nM) or a mix of 0.3 nM siPOOL UHRF1, PAI-1 supplemented with siPOOL negative control for a final concentration of 0.9 nM. (D) Protein expression was monitored at 48 h after UHRF1 and PAI-1 knockdown with siPOOLs (final concentration of 1 nM) in A549 cells. One representative Western Blot is shown (n = 3). (E) Cell proliferation was quantified in A549 cells as described in C (n = 3). The reactions contained siPOOL negative control (12 nM) or a mix of 1 nM siPOOL UHRF1, PAI-1 supplemented with siPOOL negative control for a final concentration of 12 nM. (F) Luciferase assays were performed in quadruplicates for each sample at 48 h after Hek293 transfection of a 1:5 ratio of psiCHECK2 and pCRII plasmids (n = 3). In A-E, the mean + SEM is shown. The statistical significance was determined per two-sided unpaired Student t test, with *, P < 0.05; **, P < 0.01; ***, P < 0.001.
We further analyzed whether the knockdown of UHRF1 would be sufficient to reduce cell proliferation of IMR-90 normal human lung fibroblasts. We confirmed an efficient knockdown by siPOOL both on protein and mRNA level (Fig. 6B, Suppl. Fig. S18A), and noted a reduction in BrdU incorporation (Fig. 6C). This effect was further significantly enhanced by simultaneous LINC00673 knockdown suggesting a relevant role for UHRF1 in cell cycle progression in IMR-90 cells. Since PAI-1 is a critical downstream target of p53 for the induction of replicative senescence in primary fibroblasts [41], we investigated the influence of PAI-1 knockdown on IMR-90 cell proliferation. Indeed, the efficient reduction of PAI-1 levels by siPOOL (Fig. 6B, Suppl. Fig. S18A) increased BrdU incorporation and partially rescued the reduction of cell proliferation by LINC00673 knockdown in IMR-90 cells (Fig. 6C). In contrast to our observations in IMR-90 cells, neither UHRF1 nor PAI-1 knockdown significantly affected A549 cell proliferation (Fig. 6D,E, Suppl. Fig. S18B). Noteworthy, neither UHRF1 nor PAI-1 knockdown significantly altered LINC00673 levels, and UHRF1 knockdown reduced E2F1 levels only in IMR-90 cells (Suppl. Fig. S18A, B). Together, these findings indicate that LINC00673 depletion-mediated senescence, especially in lung cancer cells, is not established by a single downstream effector but rather by a more complex network of cellular responses depending on the activation of the p53 pathway.
In a recent study, direct interactions between the 7SL RNA and the 3ʹ-untranslated region (UTR) of TP53 mRNA were linked to reduced p53 translation [45]. We noted that LINC00673 contained an Alu sequence (nucleotides 902–1217) displaying reverse complementarity to the TP53 3ʹUTR. We therefore hypothesized that LINC00673 may negatively regulate p53 translation by directly interacting with the TP53 3ʹUTR, using a similar mechanism as the 7SL RNA. In analogy to a previous study by Abdelmohsen et al. [45], we cloned the TP53 3ʹUTR (nucleotides 1421–2591) into the psiCHECK2 dual luciferase vector [psiCHECK2-TP53(3ʹUTR)]. Additionally, we generated a psiCHECK2-TP53(3ʹUTRΔ) plasmid lacking the LINC00673 interaction region. In Hek293 cells co-transfected with pCRII-LINC00673 and psiCHECK2-TP53(3ʹUTR), luciferase activity was significantly reduced as compared to the psiCHECK2-TP53(3ʹUTRΔ) (Fig. 6F). As a control, luciferase activity was not significantly altered after co-transfection of empty psiCHECK2 plasmid with pCRII and pCRII-LINC00673, respectively (Fig. 6F). In summary, our data indicate that the enhanced expression of LINC00673 in lung cancer promotes cell proliferation by negatively regulating p53 expression and thereby confers resistance to senescence, which is a potent anti-tumor barrier (Fig. 7).
Figure 7.
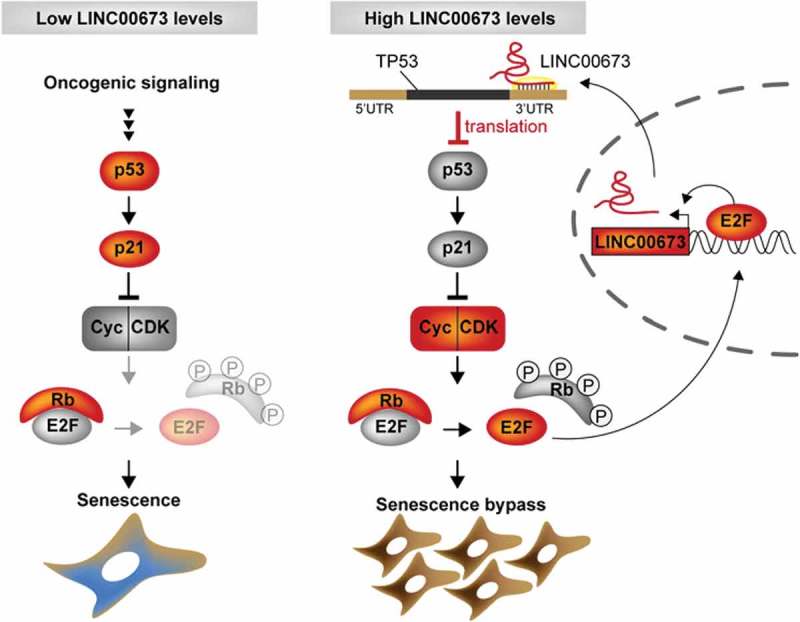
Schematic model of LINC00673-mediated senescence bypass in lung ADC. Constitutive mitogenic signaling e.g. by oncogenic Ras triggers p53 accumulation and activation resulting in cell cycle arrest and cellular senescence. In lung cells overexpressing oncogenic LINC00673, TP53 translation is reduced by direct interactions between the TP53 3ʹUTR and LINC00673. Consequently, p53-mediated cell cycle arrest is abrogated leading to bypass of cellular senescence.
Discussion
In this study, we show the elevated expression of lncRNA LINC00673 or linc00673 in lung ADC patient samples compared to normal surrounding tissue, which is in agreement with previous reports in NSCLC [46–48]. Elevated LINC00673 levels were also described in melanoma [49], tongue squamous cell carcinoma [50] and gastric cancer [51,52], and found in other types of human cancers including breast, liver and thyroid cancer (Suppl. Fig. S5B).
Along with our data, recent studies about LINC00673 function in lung cancer agree on a role in cell proliferation but not apoptosis [47,48]. More precisely, our results indicate that LINC00673 depletion provokes a strong G1-phase arrest culminating in cellular senescence in lung cancer cells. One central property of tumor cells, cell cycle progression, is controlled by E2F transcription factors [18]. The multifunctional transcription factor E2F1 participates in the timely regulation of replication genes to promote G1/S transition, and has previously been shown to regulate the expression of various lncRNAs [19,53–56]. We provide evidence that E2F1 could elevate LINC00673 levels in tumor cells. Interestingly, E2F1 was attributed a role in senescence regulation [57,58]. In cancer cell lines, E2F1 depletion was sufficient to promote cellular senescence [37] and consequently, E2F1-responsive genes were downregulated [20,31,59]. With regard to LINC00673, we show that the depletion-mediated G1-phase arrest is also accompanied by reduced E2F1 and E2F-target levels.
Cellular senescence is a potent barrier against tumorigenesis in vivo [60–63], and hence, the discovery of novel regulators of senescence provides alternative cancer treatment options for the future [58]. Insights into the functional role of nuclear lncRNAs in the induction and maintenance of senescence were gained from studies that were mainly conducted in human fibroblast cells, and underlined the importance of nuclear lncRNAs in regulating senescence-associated gene signatures [64–66]. We show that a large fraction of LINC00673 transcripts is localized in the cytoplasm, which is in line with recent reports in lung, pancreatic and gastric cancer [47,52,67]. Shi et al. proposed that LINC00673 controls lung cancer cell proliferation by directly interacting with the H3K4 histone demethylase LSD1 and epigenetically silencing NCALD [47]. In our study, LINC00673 knockdown does not cause extensive SAHF formation in lung cancer cell lines, which argues against a major role in epigenetic gene silencing in cellular senescence. The execution of the LINC00673 depletion-mediated cellular senescence program does not require pRb action in cancer cells. Since p53 has been shown to induce cellular senescence by selectively cooperating with Rb2/p130 [68], Rb2 contribution in p16INK4a-proficient cells should also be investigated in further studies. However, our data clearly shows the importance of the p53-p21 pathway in LINC00673 depletion-mediated cellular senescence as well as Ras-induced senescence.
We hypothesized that, by virtue of its cytosolic localization, LINC00673 could act as a post-transcriptional regulator of senescence-inducing target genes. With regard to the mechanism, the ncRNAs HULC, PTENP1 and linc-MD1 were proposed to act as competing endogenous RNAs (ceRNA) that regulate their target genes post-transcriptionally by competing for the same set of miRNAs [69]. Recently, LINC00673 has been shown to promote TGF-β-induced EMT in NSCLC by sponging miR-150-5p and thereby enabling ZEB1 accumulation [48]. Other lncRNAs were implicated in the regulation of mRNA stability [70,71], decay [72,73] or mRNA translation [74–76]. Altogether only few senescence-associated cytoplasmic lncRNAs have been described to date [66]. Interestingly, reduction of 7SL RNA caused senescence by promoting p53 translation [45]. In analogy, we demonstrate a reduction in p53 translation by direct interaction between LINC00673 (nucleotides 902–1217) and TP53 3ʹ-UTR. The regulatory function of LINC00673 is reinforced by its short half-life. We identified a number of additional dysregulated proteins following LINC00673 depletion using a proteomics approach. Further studies are necessary to more precisely define further factors involved in p53 translational suppression, and investigate whether the proposed mechanism of post-transcriptional gene regulation by LINC00673 can be extended to other proteins. Nuclear LINC00673 could additionally engage in epigenetic regulation of target genes by associating with LSD1 and EZH2 in lung cancer [47,77]. In gastric cancer, a similar mode of action for oncogenic LINC00673 was described, where direct interactions with LSD1 and EZH2 promoted repression of KLF2 and LATS2 gene expression [51], and LINC00673 association with EZH2 and DNMT1 epigenetically silenced the tumor suppressor KLF4 [52]. In a comprehensive study, Zheng et al. uncovered that the G > A variation at rs11655237 in LINC00673 conferred susceptibility to pancreatic ductal adenocarcinoma in Han Chinese by creating a binding site for miR-1231 interfering with LINC00673-assisted PTPN11 degradation [67].
In pancreatic cancer, LINC00673 transcript levels were reduced [67] indicating that LINC00673 can exert oncogenic or tumor suppressive functions depending on the investigated tissue or cell type. Therefore, it is expected that LINC00673 employs various different modes of action across different types of cancer. We suggest that enhanced LINC00673 expression in early lung ADC contributes to lung tumorigenesis by reducing p53 protein levels and bypassing cellular senescence. In mouse xenograft models, shRNA-mediated LINC00673 knockdown significantly reduced tumor growth [47,51,77] emphasizing that LINC00673 could be a non-protein-coding key regulator of cell proliferation. Reducing its levels may delay cancer progression of early lung ADC, thereby positively influencing patient survival.
Materials & methods
RACE, in vitro translation, RNA FISH, cell fractionation and RNA stability
For 5ʹ- and 3ʹ-RACE analyses, the SMARTer RACE cDNA Amplication Kit (Clontech Takara Bio, Mountain View, CA, USA) was used according to the manufacturer’s instructions. For first-strand cDNA synthesis, DNase I-treated total RNA from A549 cells was used. The gene-specific primers are summarized in Suppl. Table S7. The in vitro transcription-translation assay was based on rabbit reticulocyte extract using the TNT T7 Quick for PCR DNA kit (Promega) as described by the manufacturer. Briefly, 800 ng of gel-purified PCR products were mixed with 40 μl of TNT T7 PCR Quick Master Mix and 3 μl of [35S] methionine (Perkin Elmer) in a final volume of 50 μl and the reactions were incubated for 90 min at 30°C. For SDS-PAGE, 10 μl of each reaction were mixed with 5 μl of SDS-Laemmli sample buffer and heated for 1 min at 90°C. The proteins were separated on a 20% SDS gel, fixed for 30 min (50% methanol and 10% acetic acid) then vacuum-dried for 1 h at 80°C. Signals were monitored by autoradiography. The primers used to generate PCR templates from plasmid DNA are listed in Suppl. Table S8. The RNA FISH probes were designed by and purchased from Stellaris (LGC Biosearch Technologies, Novato, CA, USA). For hybridization, 75 nM of probes (labeled with Quasar-670) were used and staining was performed as recommended by the manufacturer. As a negative control, the coverslips were hybridized with buffer only. The RNA FISH pictures were taken with an Olympus Cell^R microscope utilizing z-stacks and a 60x objective. All pictures of the same experiment were processed with the same settings in ImageJ, thereby using the unstained cells as a negative control to avoid signal artifacts. The cellular fractionation was carried out as previously described [78]. To estimate the half-life of LINC00673, A549 cells were seeded into 6-wells to reach 80% confluence one day later. For treatment, cell culture media containing actinomycin D (10 µg/ml; Sigma-Aldrich) or the equivalent volume of DMSO (solvent control) were added to the cells. The cells were lysed in TRIzol at 0, 1, 2, 8, 16 and 24 h after treatment and the relative abundance of transcripts was determined by RT-qPCR. The respective 0 h values were used as reference and the ratios of actinomycin D-treated and DMSO solvent controls were calculated for each timepoint. The half-lives were determined by fitting the data with a non-linear least squares regression (one phase decay) with GraphPad Prism 5.
Cellular senescence assay
5 x 104 A549 cells and 1 × 105 IMR-90 cells were grown on 60 mm dishes and the senescence-associated β-galactosidase (SA-β-Gal) activity was detected 4 days after reverse transfection. As a positive control, A549 cells were treated with 200 nM doxorubicin (Calbiochem, Merck) 12 hrs after seeding while IMR-90 cells were treated with 1 µM doxorubicin for 2 hrs at 24 hrs after seeding. Cells were washed with PBS and fixed with 0.5% glutaraldehyde in PBS for 15 min at room temperature. Then, cells were washed twice with PBS supplemented with 1 mM MgCl2 (pH 6.0) for 5 min on a rocker. 2 ml X-Gal staining solution (PBS containing 1 mM MgCl2, 5 mM potassium hexacyanoferrate (III), 5 mM potassium hexacyanoferrate (II) trihydrate, 1 mg/ml X-Gal (5-bromo-4-chloro-3-indolyl-beta-D-galacto-pyranoside), pH 6.0) were added and the dishes were incubated overnight at 37°C. Next day, the cells were washed three times with distilled water and microscopy pictures were taken with the Zeiss Cell Observer using a 10x objective. For analyses, at least 200 cells were counted per condition in 3–4 independent experiments.
Funding Statement
Research in the Diederichs lab is supported by the Deutsche Forschungsgemeinschaft (DFG Di 1421/7-1) and the RNA@DKFZ Cross Program Topic.
Acknowledgments
The patient tissues were provided by the Lung Biobank Heidelberg, member of the Biomaterial Bank Heidelberg (BMBH), and the biobank platform of the German Center for Lung Research (DZL). We would like to thank Vladimir Benes and Tomi Ivacevic (EMBL, Heidelberg, Germany) for providing the infrastructure and support with the microarray analysis; Johanna Schott (DKFZ-ZMBH Alliance, Heidelberg, Germany) for the re-annotation of the microarray; Stefanie Grund for the cellular fractionation; Hans Johansson (LGC Biosearch Technologies) for the design of the FISH probes; the DKFZ Light Microscopy Core Facility, the ZMBH Flow Cytometry & FACS Core Facility (Heidelberg, Germany) for the technical support, the ZMBH Mass Spectrometry Core Facility for the sample preparation, technical support and data analysis. This work is part of the PhD thesis of A.R.
Author contributions
S.D. conceived the microarray study and performed the analysis. A.W., P.A.S., T.M., M.M., H.Z. and H.H. carried out the primary tumor analysis and patient RNA isolation. M.P.S. performed the microarrays. A.R. and K.B. and S.D. designed and analyzed the experiments. A.R., K.B. and M.G. performed the experiments. O.B. and D.G. carried out experiments in ER-E2F1 cells. M.R., K.B. and F.A.M designed, performed and analyzed retroviral infection experiments. F.A.M. provided valuable suggestions. A.R. wrote the manuscript.
Data availability statement
The microarray data generated within this study are available at the NCBI Gene Expression Omnibus (GEO) GSE113852.
Disclosure statement
S.D. is a co-owner of siTOOLs Biotech GmbH, Planegg/Martinsried, Germany.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Torre LA, Bray F, Siegel RL, et al. Global cancer statistics, 2012. CA Cancer J Clin. 2015. March;65(2):87–108. [DOI] [PubMed] [Google Scholar]
- [2].Langer CJ, Besse B, Gualberto A, et al. The evolving role of histology in the management of advanced non-small-cell lung cancer. J Clin Oncol. 2010. December 20;28(36):5311–5320. [DOI] [PubMed] [Google Scholar]
- [3].Djebali S, Davis CA, Merkel A, et al. Landscape of transcription in human cells. Nature. 2012. September 6;489(7414):101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hangauer MJ, Vaughn IW, McManus MT.. Pervasive transcription of the human genome produces thousands of previously unidentified long intergenic noncoding RNAs. PLoS Genet. 2013. June;9(6):e1003569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet. 2009. March;10(3):155–159. [DOI] [PubMed] [Google Scholar]
- [6].Yan X, Hu Z, Feng Y, et al. Comprehensive Genomic Characterization of Long Non-coding RNAs across Human Cancers. Cancer Cell. 2015. October 12;28(4):529–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chen J, Wang R, Zhang K, et al. Long non-coding RNAs in non-small cell lung cancer as biomarkers and therapeutic targets. J Cell Mol Med. 2014. December;18(12):2425–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Gutschner T, Diederichs S. The hallmarks of cancer: a long non-coding RNA point of view. RNA Biol. 2012. June;9(6):703–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Roth A, Diederichs S. Long noncoding RNAs in lung cancer. Curr Top Microbiol Immunol. 2016;394:57–110. [DOI] [PubMed] [Google Scholar]
- [10].Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011. September 16;43(6):904–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013;75:685–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Cabili MN, Trapnell C, Goff L, et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011. September 15;25(18):1915–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Li J, Han L, Roebuck P, et al. TANRIC: an interactive open platform to explore the function of lncRNAs in cancer. Cancer Res. 2015. September 15;75(18):3728–3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Desiere F, Deutsch EW, King NL, et al. The PeptideAtlas project. Nucleic Acids Res. 2006. January 1;34(Database issue):D655–D658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zhang K, Shi ZM, Chang YN, et al. The ways of action of long non-coding RNAs in cytoplasm and nucleus. Gene. 2014. August 15;547(1):1–9. [DOI] [PubMed] [Google Scholar]
- [16].Tani H, Mizutani R, Salam KA, et al. Genome-wide determination of RNA stability reveals hundreds of short-lived noncoding transcripts in mammals. Genome Res. 2012. May;22(5):947–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Reimand J, Kull M, Peterson H, et al. g:profiler–aweb-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res. 2007. July;35(Web Server issue):W193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Polager S, Ginsberg D. E2F - at the crossroads of life and death. Trends Cell Biol. 2008. November;18(11):528–535. [DOI] [PubMed] [Google Scholar]
- [19].Feldstein O, Nizri T, Doniger T, et al. The long non-coding RNA ERIC is regulated by E2F and modulates the cellular response to DNA damage. Mol Cancer. 2013;12(1):131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Vernier M, Bourdeau V, Gaumont-Leclerc MF, et al. Regulation of E2Fs and senescence by PML nuclear bodies. Genes Dev. 2011. January 1;25(1):41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hannus M, Beitzinger M, Engelmann JC, et al. siPools: highly complex but accurately defined siRNA pools eliminate off-target effects. Nucleic Acids Res. 2014. July;42(12):8049–8061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kuilman T, Michaloglou C, Mooi WJ, et al. The essence of senescence. Genes Dev. 2010. November 15;24(22):2463–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Campisi J, d’Adda Di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007. September;8(9):729–740. [DOI] [PubMed] [Google Scholar]
- [24].Tanaka H, Fujii Y, Hirabayashi H, et al. Disruption of the RB pathway and cell-proliferative activity in non-small-cell lung cancers. Int J Cancer J Inter Du Cancer. 1998. April 17;79(2):111–115. [DOI] [PubMed] [Google Scholar]
- [25].Kashiwabara K, Oyama T, Sano T, et al. Correlation between methylation status of the p16/CDKN2 gene and the expression of p16 and Rb proteins in primary non-small cell lung cancers. Int J Cancer J Inter Du Cancer. 1998. June 19;79(3):215–220. [DOI] [PubMed] [Google Scholar]
- [26].Mogi A, Kuwano H. TP53 mutations in nonsmall cell lung cancer. J Biomed Biotechnol. 2011;2011:583929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Althubiti M, Lezina L, Carrera S, et al. Characterization of novel markers of senescence and their prognostic potential in cancer. Cell Death Dis. 2014;5:e1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Dimri GP, Lee X, Basile G, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995. September 26;92(20):9363–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Frolov MV, Dyson NJ. Molecular mechanisms of E2F-dependent activation and pRB-mediated repression. J Cell Sci. 2004. May 1;117(Pt 11):2173–2181. [DOI] [PubMed] [Google Scholar]
- [30].Chicas A, Wang X, Zhang C, et al. Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. Cancer Cell. 2010. April 13;17(4):376–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Chen T, Xue L, Niu J, et al. The retinoblastoma protein selectively represses E2F1 targets via a TAAC DNA element during cellular senescence. J Biol Chem. 2012. October 26;287(44):37540–37551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Narita M, Nunez S, Heard E, et al. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003. June 13;113(6):703–716. [DOI] [PubMed] [Google Scholar]
- [33].Coppe JP, Desprez PY, Krtolica A, et al. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Bartkova J, Rezaei N, Liontos M, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006. November 30;444(7119):633–637. [DOI] [PubMed] [Google Scholar]
- [35].Di Micco R, Fumagalli M, Cicalese A, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006. November 30;444(7119):638–642. [DOI] [PubMed] [Google Scholar]
- [36].Mallette FA, Gaumont-Leclerc MF, Ferbeyre G. The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes Dev. 2007. January 1;21(1):43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Park C, Lee I, Kang WK. E2F-1 is a critical modulator of cellular senescence in human cancer. Int J Mol Med. 2006. May;17(5):715–720. [PubMed] [Google Scholar]
- [38].Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007. April;7(4):295–308. [DOI] [PubMed] [Google Scholar]
- [39].Serrano M, Lin AW, McCurrach ME, et al. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997. March 7;88(5):593–602. [DOI] [PubMed] [Google Scholar]
- [40].Mallette FA, Goumard S, Gaumont-Leclerc MF, et al. Human fibroblasts require the Rb family of tumor suppressors, but not p53, for PML-induced senescence. Oncogene. 2004. January 8;23(1):91–99. [DOI] [PubMed] [Google Scholar]
- [41].Kortlever RM, Higgins PJ, Bernards R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat Cell Biol. 2006. 8;Aug(8):877–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Roy N, Stoyanova T, Dominguez-Brauer C, et al. DDB2, an essential mediator of premature senescence. Mol Cell Biol. 2010. June;30(11):2681–2692. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [43].Guan D, Factor D, Liu Y, et al. The epigenetic regulator UHRF1 promotes ubiquitination-mediated degradation of the tumor-suppressor protein promyelocytic leukemia protein. Oncogene. 2013. August 15;32(33):3819–3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Mason DX, Jackson TJ, Lin AW. Molecular signature of oncogenic ras-induced senescence. Oncogene. 2004. December 9;23(57):9238–9246. [DOI] [PubMed] [Google Scholar]
- [45].Abdelmohsen K, Panda AC, Kang MJ, et al. 7SL RNA represses p53 translation by competing with HuR. Nucleic Acids Res. 2014. September;42(15):10099–10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].White NM, Cabanski CR, Silva-Fisher JM, et al. Transcriptome sequencing reveals altered long intergenic non-coding RNAs in lung cancer. Genome Biol. 2014;15(8):429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Shi X, Ma C, Zhu Q, et al. Upregulation of long intergenic noncoding RNA 00673 promotes tumor proliferation via LSD1 interaction and repression of NCALD in non-small-cell lung cancer. Oncotarget. 2016. May 3;7(18):25558–25575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lu W, Zhang H, Niu Y, et al. Long non-coding RNA linc00673 regulated non-small cell lung cancer proliferation, migration, invasion and epithelial mesenchymal transition by sponging miR-150-5p. Mol Cancer. 2017. July 11;16(1):118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Schmidt K, Joyce CE, Buquicchio F, et al. The lncRNA SLNCR1 mediates melanoma invasion through a conserved SRA1-like region. Cell Rep. 2016. May 31;15(9):2025–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Yu J, Liu Y, Gong Z, et al. Overexpression long non-coding RNA LINC00673 is associated with poor prognosis and promotes invasion and metastasis in tongue squamous cell carcinoma. Oncotarget. 2017. March 7;8(10):16621–16632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Huang M, Hou J, Wang Y, et al. Long noncoding RNA LINC00673 Is activated by SP1 and exerts oncogenic properties by interacting with LSD1 and EZH2 in gastric cancer. Mol ther. 2017. April 5;25(4):1014–1026. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [52].Ba MC, Long H, Cui SZ, et al. Long noncoding RNA LINC00673 epigenetically suppresses KLF4 by interacting with EZH2 and DNMT1 in gastric cancer. Oncotarget. 2017. November 10;8(56):95542–95553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Berteaux N, Lottin S, Monte D, et al. H19 mRNA-like noncoding RNA promotes breast cancer cell proliferation through positive control by E2F1. J Biol Chem. 2005. August 19;280(33):29625–29636. [DOI] [PubMed] [Google Scholar]
- [54].Wan G, Mathur R, Hu X, et al. Long non-coding RNA ANRIL (CDKN2B-AS) is induced by the ATM-E2F1 signaling pathway. Cell Signal. 2013. May;25(5):1086–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Bida O, Gidoni M, Ideses D, et al. A novel mitosis-associated lncRNA, MA-linc1, is required for cell cycle progression and sensitizes cancer cells to Paclitaxel. Oncotarget. 2015. September 29;6(29):27880–27890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Zhang E, Yin D, Han L, et al. E2F1-induced upregulation of long noncoding RNA LINC00668 predicts a poor prognosis of gastric cancer and promotes cell proliferation through epigenetically silencing of CKIs. Oncotarget. 2016. Apr 26;7(17):23212–23226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Polager S, Ginsberg D. p53 and E2f: partners in life and death. Nat Rev Cancer. 2009. October;9(10):738–748. [DOI] [PubMed] [Google Scholar]
- [58].Laine A, Westermarck J. Molecular pathways: harnessing E2F1 regulation for prosenescence therapy in p53-defective cancer cells. Clin Cancer Res off J Am Assoc Cancer Res. 2014. July 15;20(14):3644–3650. [DOI] [PubMed] [Google Scholar]
- [59].Young AP, Nagarajan R, Longmore GD. Mechanisms of transcriptional regulation by Rb-E2F segregate by biological pathway. Oncogene. 2003. October 16;22(46):7209–7217. [DOI] [PubMed] [Google Scholar]
- [60].Braig M, Lee S, Loddenkemper C, et al. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005. August 4;436(7051):660–665. [DOI] [PubMed] [Google Scholar]
- [61].Chen Z, Trotman LC, Shaffer D, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005. August 4;436(7051):725–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Collado M, Gil J, Efeyan A, et al. Tumour biology: senescence in premalignant tumours. Nature. 2005. August 4;436(7051):642. [DOI] [PubMed] [Google Scholar]
- [63].Michaloglou C, Vredeveld LC, Soengas MS, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005. August 4;436(7051):720–724. [DOI] [PubMed] [Google Scholar]
- [64].Lazorthes S, Vallot C, Briois S, et al. A vlincRNA participates in senescence maintenance by relieving H2AZ-mediated repression at the INK4 locus. Nat Commun. 2015;6:5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Abdelmohsen K, Gorospe M. Noncoding RNA control of cellular senescence. Wiley Interdiscip Rev RNA. 2015. November;6(6):615–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Montes M, Lund AH. Emerging roles of lncRNAs in senescence. FEBS J. 2016. July;283(13):2414–2426. [DOI] [PubMed] [Google Scholar]
- [67].Zheng J, Huang X, Tan W, et al. Pancreatic cancer risk variant in LINC00673 creates a miR-1231 binding site and interferes with PTPN11 degradation. Nat Genet. 2016. May 23;48:747–757. [DOI] [PubMed] [Google Scholar]
- [68].Kapic A, Helmbold H, Reimer R, et al. Cooperation between p53 and p130(Rb2) in induction of cellular senescence. Cell Death Differ. 2006. February;13(2):324–334. [DOI] [PubMed] [Google Scholar]
- [69].Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014. January 16;505(7483):344–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Faghihi MA, Modarresi F, Khalil AM, et al. Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretase. Nat Med. 2008. July;14(7):723–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Kretz M, Siprashvili Z, Chu C, et al. Control of somatic tissue differentiation by the long non-coding RNA TINCR. Nature. 2013. January 10;493(7431):231–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Gong C, Maquat LE. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3ʹ UTRs via Alu elements. Nature. 2011. February 10;470(7333):284–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Liu X, Li D, Zhang W, et al. Long non-coding RNA gadd7 interacts with TDP-43 and regulates Cdk6 mRNA decay. Embo J. 2012. November 28;31(23):4415–4427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Carrieri C, Cimatti L, Biagioli M, et al. Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature. 2012. November 15;491(7424):454–457. [DOI] [PubMed] [Google Scholar]
- [75].Yoon JH, Abdelmohsen K, Srikantan S, et al. LincRNA-p21 suppresses target mRNA translation. Mol Cell. 2012. August 24;47(4):648–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Gumireddy K, Li A, Yan J, et al. Identification of a long non-coding RNA-associated RNP complex regulating metastasis at the translational step. Embo J. 2013. October 16;32(20):2672–2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Ma C, Wu G, Zhu Q, et al. Long intergenic noncoding RNA 00673 promotes non-small-cell lung cancer metastasis by binding with EZH2 and causing epigenetic silencing of HOXA5. Oncotarget. 2017. May 16;8(20):32696–32705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Grund SE, Polycarpou-Schwarz M, Luo C, et al. Rare Drosha splice variants are deficient in microRNA processing but do not affect general microRNA expression in cancer cells. Neoplasia. 2012. March;14(3):238–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The microarray data generated within this study are available at the NCBI Gene Expression Omnibus (GEO) GSE113852.