

ABSTRACT
Long noncoding RNAs (lncRNAs) are emerging as critical mediators of various biological processes in the immune system. The current data showed that the lncRNA Malat1 is highly expressed in T cell subsets, but the function of Malat1 in T cell remains unclear. In this study, we detected the T cell development and both CD8+ and CD4+ T cell response to LCMV infection using Malat1−/- mice model. To our surprise, there were no significant defects in thymocytes at different developmental stages and the peripheral T cell pool with ablation of Malat1. During LCMV infection, Malat1−/- mice exhibited normal effector and memory CD8+ T cells as well as TFH cells differentiation. Our results indicated that Malat1 is not essential for T cell development and T cell-mediated antiviral response though it expresses at very high level in different T cell populations.
KEYWORDS: lncRNA, Malat1, T cell development, effector CD8+ T cells, memory CD8+ T cells, TFH cells
Introduction
Long noncoding RNAs (lncRNAs) are non-protein coding transcripts, which coordinate diverse aspects of cell and tissue development [1]. It is well documented that lncRNAs regulate the development of cardiomyocytes, stem cells, epithelial cells, erythrocytes, and adipocyte [2]. A large number of studies have indicated lncRNAs also regulate the development and differentiation of several immune cell lineages, such as myeloid cells and dendritic cell development [3,4]. Previous works from different labs have characterized that distinct T cell subsets express unique profiles of lncRNAs at different stages of development [5–8]. The lncRNA Malat1 (metastasis-associated lung adenocarcinoma transcript 1) is a nuclear localized RNA, which is a highly conserved transcript that is involved in alternative splicing [9,10]. The expression of Malat1 is correlated with tumorigenesis and metastasis in multiple myeloma and solid tumors, suggesting its universal role in cancer [11]. Ma et al. reported that Malat1 plays a critical role in regulating proliferation and maintaining undifferentiated status of early-stage hematopoietic cells [12]. To our knowledge, although Malat1 plays essential role in many kinds of cancer cells and emerging roles in immune system, its role in T cell development and T cell-mediated immune response remains largely unknown.
T cells, the essential regulators of cellular immunity, are produced in thymus and undergo a series of well-documented differentiation steps. Pluripotent precursors derived from hematopoietic stem cells in the bone marrow migrate to the thymus, which initiate and sustain T cell development at CD4−CD8− double-negative (DN) stage [13,14]. The surface expression of CD44 and CD25 characterizes the four major DN cell subsets: CD44+CD25− (DN1), CD44+CD25+ (DN2), CD44−CD25+ (DN3), and CD44−CD25− (DN4) cells. When cells proceed to differentiate from DN2 to DN4 stages, the pre-TCR is expressed, which is comprised of the non-rearranging pre-TCR α-chain and a rearranged TCR β-chain [15]. Successful pre-TCR expression leads to DN4 cells transition to CD4+CD8+ double-positive (DP) thymocytes and replacement of the pre-TCR α-chain with a newly rearranged TCR α-chain, which yields a complete αβ TCR [16]. The αβ-TCR+ DP thymocytes undergo the processes of positive and negative selection based on their relative ability to interact with thymic selecting ligands [17]. The positive-selected DP cells first differentiate into CD4+CD8lo intermediate (IM) thymocytes, which then give rise to mature major histocompatibility complex (MHC) class II–restricted CD4+ or MHC class I–restricted CD8+ single-positive (SP) T cells. After that, mature T cells emigrate and join the peripheral lymphocyte pool.
T cells play vital roles in controlling the adaptive immune response. As they not only control a multitude of immune responses directly, but also regulate B cell immune responses. CD4-bearing T cells are associated with helper functions and CD8-bearing T cells are associated with cytotoxicity. CD4+ T cells produce functional T helper cells that are tailored to their respective roles in host defense. Follicular helper T (TFH) cells, a unique subset of T helper cells, are the specialized providers of B cell help, and are essential for germinal center (GC) formation and the development of high-affinity antibodies-secreted plasma cells and memory B cells [18,19]. TFH cell differentiation is a multistage, multifactorial process [20]. Priming by dendritic cells triggers upregulation of ICOS on CD4+ T cells, which is required for the expression of Bcl-6, a master regulator of TFH cells differentiation [21–23]. Bcl-6 in turn induces chemokine receptor CXCR5 expression [18], which allows the early TFH cells migrate to the border of the B cell follicular and undergo further differentiation. At the T cell-B cell border, early TFH cells interact with activated B cells presenting cognate antigen, results in the early TFH cells providing help to B cells. Meanwhile, cognate help from B cells drives the full development of TFH cells. Within GC, TFH cells continue to provide help to the B cells, facilitating the establishment of GC reaction and promoting the GC B cells differentiate into long-lived plasma cells and memory B cells. Reciprocal signals provided by the B cells are also crucial for sustaining the TFH cells [24].
Cytotoxic effector CD8+ T cells are an essential component of the immune system against various intracellular pathogens including viruses and intracellular bacterial. Upon encountering antigen, antigen‐presenting cells (APCs) initiate responses from rare, antigen-specific CD8+ T cells. The activated pathogen‐specific CD8+ T cells embark on a proliferative expansion in numbers and differentiate into primary effector populations. These effector CD8+ T cells acquire the ability to produce interferon-γ (IFN-γ), lesser extent tumor-necrosis factor (TNF), and the ability to perform cytolysis to manifest antimicrobial functions. Most of the effector cells succumb to apoptosis during contraction phase, and only a small portion of surviving CD8+ T cells initiate the memory pool and differentiate to long-lived memory cells, capable of providing strengthened protection against same pathogens [25–27]. Memory CD8+ T cells are heterogeneous, consisting of at least four distinct subsets: effector memory T cells (TEM), central memory T cells (TCM), tissue-resident memory T cells (TRM), and stem memory T cells [28]. At effector phage, CD8+ T cells downregulate the expression of CD127 (also known as IL-7Rα) and CD62L, but generally are KLRG1hi. The differentiation of effector cells to memory CD8+ T cells is accompanied with CD127 upregulation and KLRG1 downregulation. TEM cells and TCM cells have different expression pattern of homing molecules, chiefs among these are CCR7 and CD62L [29]. TEM cells and TCM cells are phenotypically and functionally vary. TCM cells usually reside in secondary lymphoid organs and have greater proliferation potential, whereas TEM cells constitutively exert effector functions such as cytotoxicity [30,31].
Here we set out to investigate the function of Malat1 in T cell development and T cell-mediated immune response. To our surprise, despite Malat1 is highly expressed in T cell subsets, the role of Malat1 in T cell development and T cell-mediated antiviral response is nonessential, which suggests Malat1 may not be a master regulator in T cells.
Results
To identify new regulators of T cell development, we analyzed published RNA-seq data from T cell at the different developmental stages (GSE109125). Comparing with several well-known T cell specific genes (Bcl11b, Id2, Lef1, Notch1, Rag1, Tcf3, Tcf7 and Tox), we found the relative expression of the lncRNA Malat1 was especially higher in T cell subsets (Figure 1(a)). In addition, Malat1 was identified to play key roles in tumorigenesis, so that we chose this gene for further investigation in order to characterize its function in T cell development and response to infection. We isolated different T cell subsets and subjected them to quantitative RT-PCR by three pairs of primers located at 5ʹ-region, middle of gene body and 3ʹ-region, respectively. Malat1 was detected in all populations of T cells at very high level, with peaking in DP thymocytes (Figure 1(b)).
Figure 1.
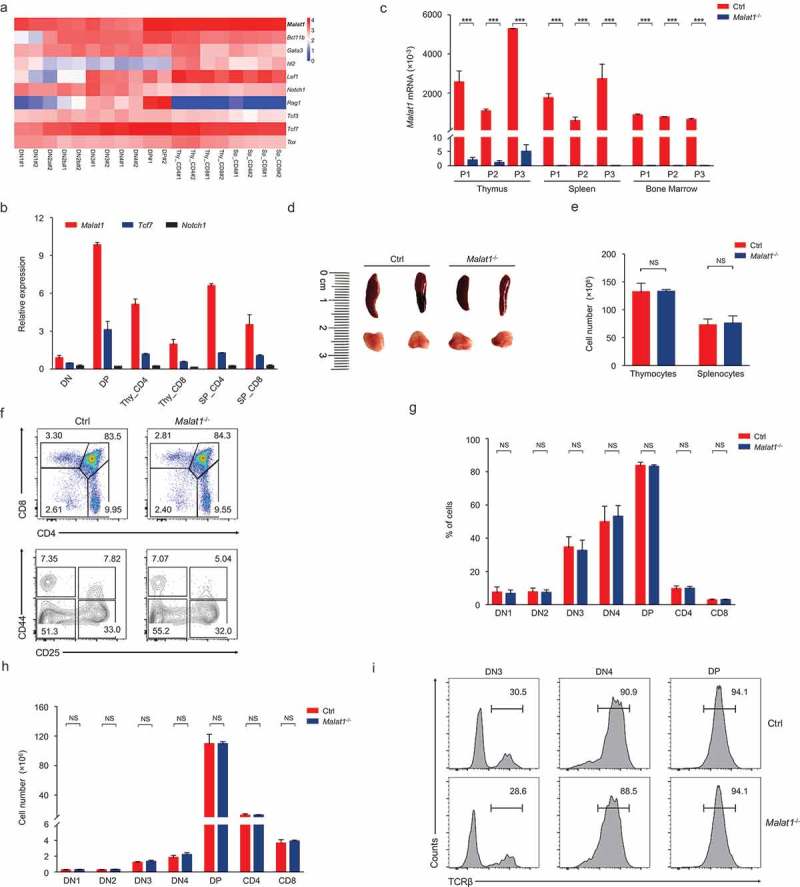
Malat1 is not required for early T cell development. (a) Expression of Malat1 and key transcription factors of T cell development in different T cell subsets from published RNA-seq. (b) Analysis of Malat1 expression in thymocytes and peripheral T cells by quantitative RT-PCR. Relative gene expression levels in each sample were normalized to Hprt1. (c) Analysis of Malat1 expression in thymus, spleen and bone marrow from wild-type (Ctrl) and Malat1−/- mice. Relative gene expression levels in each sample were normalized to Hprt1. (d-e) The size (d) and cellularity (e) of thymus and spleen from wild-type (Ctrl) and Malat1−/- mice. (f) Surface staining of CD4 and CD8 on thymocytes and CD25 and CD44 on DN thymocytes. (g-h) The percentages (g) and numbers (h) of thymocytes in f. (i) Expression of TCRβ in DN3, DN4 and DP thymocytes. Data represent mean ± s.d.
To address the functional importance of Malat1 in the T cell lineage, we investigated the T cell development in Malat1−/- mice. Quantitative RT-PCR results indicated that the expression of Malat1 was significantly decreased in the thymus, spleen and bone marrow from Malat1−/- mice (Figure 1(c)) as well as all subsets of thymocytes and peripheral T cells (Data not shown). These results confirmed that Malat1 gene was completely inactivated in Malat1−/- mice. Mice with a null Malat1 allele were viable and fertile and showed no obvious signs of immune deficiency syndrome. The size and cellularity of thymi and spleens were similar in Malat1−/- mice and their littermate control mice (Figure 1(d, e)). The frequency and numbers of DN, CD4 SP, CD8 SP, and DP thymocytes were similar in Malat1−/- and their littermate control mice. The percentages and numbers of DN1-DN4 subsets based on expression of CD44 and CD25 were showed no difference between Malat1−/- mice and their littermate control mice (Figure 1(f-h)). TCRβ gene rearrangements occurs in DN3 transition to DN4 and DP stage, namely V(D)J recombination, and β-selection [16]. Intracellular staining of TCRβ in DN3, DN4 and DP thymocytes indicated Malat1−/- mice have no defects in TCRβ expression (Figure 1(i)). These result suggested Malat1 is not essential for maturation of T cell from the DN to SP stage.
Next we explored whether Malat1 is essential for negative and positive selection and/or lineage commitment of αβ thymocytes. The frequency of surface TCRβhi subsets was similar in mice of both genotypes (Figure 2(a, b)). Downregulation of the expression of CD24 and CD69 states the intrathymic positively selected TCRβhi thymocytes maturation [32]. We found that the maturation of CD24+CD69+ subsets to CD24−CD69− cells was not blockaded in Malat1−/- mice (Figure 2(c, d)). CD24+CD69+TCRβhi cells contains post-selection DP thymocytes and CD4+CD8lo intermediate thymocytes, which are the precursors of immature CD4+ or CD8+ SP thymocytes [17]. We observed the numbers of DP, CD4+, CD8+ SP and CD4+CD8lo thymocytes were not altered in Malat1-deficiency mice (Figure 2(e, f)). CD24−CD69−TCRβhi cells only contains mature thymocytes [17], and we noted ablation of Malat1 did not affect the frequency and numbers of mature thymocytes (Figure 2(g, h)). Collectively, these results indicated that Malat1 is not required for late T cell development and lineage commitment. Given Malat1 is not essential for T cell development, we questioned whether deficiency in Malat1 changed the populations of peripheral T cells. We noted peripheral CD4+ and CD8+ T cells were similar in Malat1−/- mice and their littermate control mice (Figure 2(i, j)).
Figure 2.
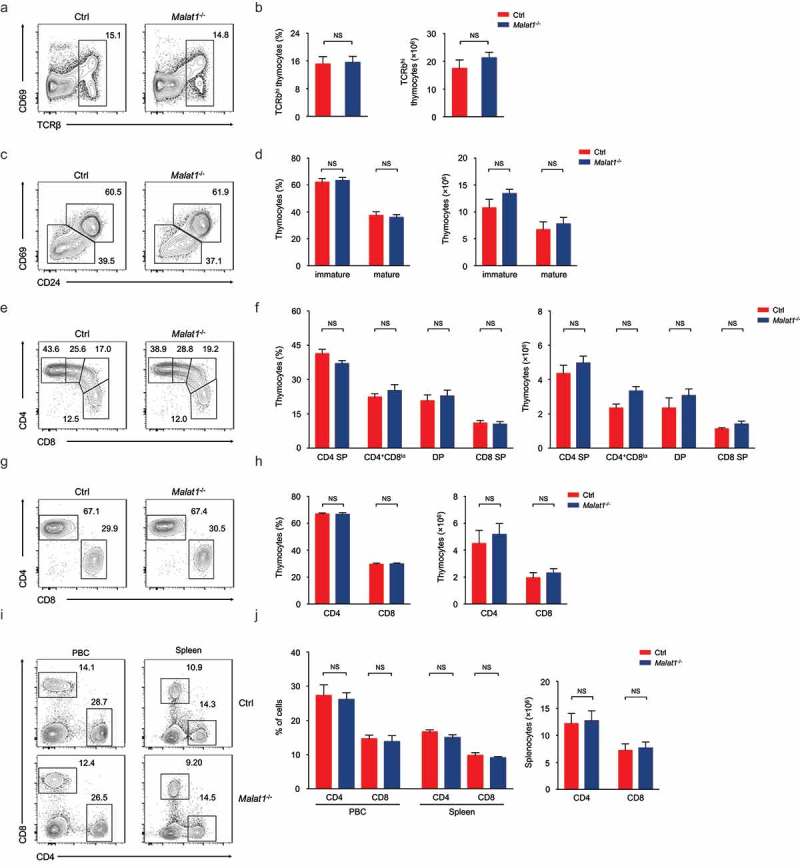
Ablation of Malat1 does not alter late T cell development and migration. (a-b) Flow cytometry analysis of post-selection TCRβhi thymocytes. The percentages (left) and numbers (right) of post-selection TCRβhi thymocytes are shown in b. (c-d) Flow cytometry analysis of mature (CD24−CD69−) thymocytes (bottom left) and immature (CD24+CD69+) thymocytes (top right) gated on post-selection TCRβhi thymocytes. The frequency (left) and numbers (right) of immature and mature subsets are shown in d. (e-f) Flow cytometry analysis of CD4+ SP, CD4+CD8lo, DP and CD8+ SP subsets gated on immature CD24+CD69+ TCRβhi thymocytes. The percentages (left) and numbers (right) of indicated subsets are shown in f. (g-h) Flow cytometry analysis of CD4+ and CD8+ subsets gated on mature CD24−CD69− TCRβhi thymocytes. The frequency (left) and numbers (right) of CD4+ and CD8+ subsets are shown in h. (i-j) Flow cytometry analysis of CD4+ and CD8+ T cells in peripheral blood cells (left column) and spleen (right column). The percentages (left) and numbers (right) of peripheral T cells are shown in j. Data represent mean ± s.d.
Due to CD4+ and CD8+ T cells are essential mediators of cellular immune response. We next investigated whether Malat1 plays an important role in T cell-mediated antiviral immune. The mice were infected with lymphocytic choriomeningitis virus (LCMV), and the TFH cells differentiation as well as effector and memory CD8+ T cells were analyzed. We found that ablation of Malat1 did not alter CD4+ T cells pool (Figure 3(a, b)) at 8 days after infection. Analysis of CD44hiCD62L− activated CD4+ T cells revealed that the percentages and numbers of SLAMloCXCR5+ TFH cells, PD-1hiCXCR5+ GC TFH cells and Bcl-6hiCXCR5+ GC TFH were similar in Malat1−/- mice compared with that of littermate control mice (Figure 3(c-e)). Moreover, Malat1−/- mice exhibited similar frequency and numbers of GL7+Fas+ GC B cells and B220−CD138+ plasma cells and equivalent antigen-specific Ig secretion with that of littermate control mice (Figure 3(f-h)). In summary, these data indicated that Malat1 is dispensable for TFH cells differentiation and B cell helping functions.
Figure 3.
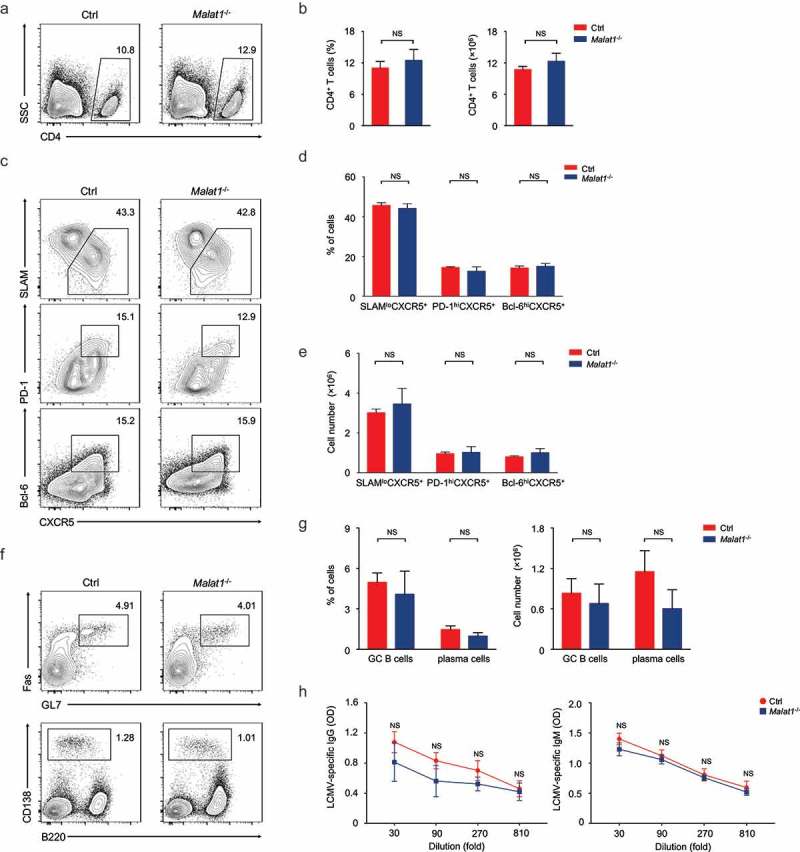
Malat1 is dispensable for TFH differentiation. (a-b) Flow cytometry analysis of CD4+ T cell pool at 8 days after infection. The percentage (left) and number (right) of effector CD4+ T cells are shown in b. (c) Flow cytometry analysis of SLAM− CXCR5+ TFH cells (top), PD-1hiCXCR5+ GC TFH cells (middle), and Bcl-6hiCXCR5+ GC TFH cells (bottom) at day 8 after LCMV infection. (d-e) The percentages (d) and numbers (e) of TFH cells in c. (f) Analysis of GC B cells (top) and plasma cells (bottom) in spleen by flow cytometry. (g) The percentages (left) and numbers (right) of GC B and plasma cells in f. (h) Analysis of LCMV-specific IgG (left) and IgM (right) in the sera after LCMV infection. Data represent mean ± s.d.
We next examined effector differentiation of CD8+ T cells on day 8 after infection. We found normal CD8+ T cells expansion in Malat1−/- mice compared with that in wild-type mice (Figure 4(a, b)). Malat1−/- effector cells exhibited similar downregulation of CD62L and CD127, and similar upregulation of CD44 and KLRG1 (Figure 4(c-e)). Furthermore, Malat1−/- effector cells showed equivalent secretion of interferon-γ (IFN-γ) and similar tumor necrosis factor-α (TNF-α) and Granzyme B production compared with that of littermate control mice (Figure 4(f-h)). We also noted that the expression of Eomes and T-bet were similar between Malat1−/- and their littermate control mice (Figure 4(i)). Collectively, our results demonstrated that Malat1 is not essential for effector CD8+ T cells formation and expansion to response to LCMV infection.
Figure 4.
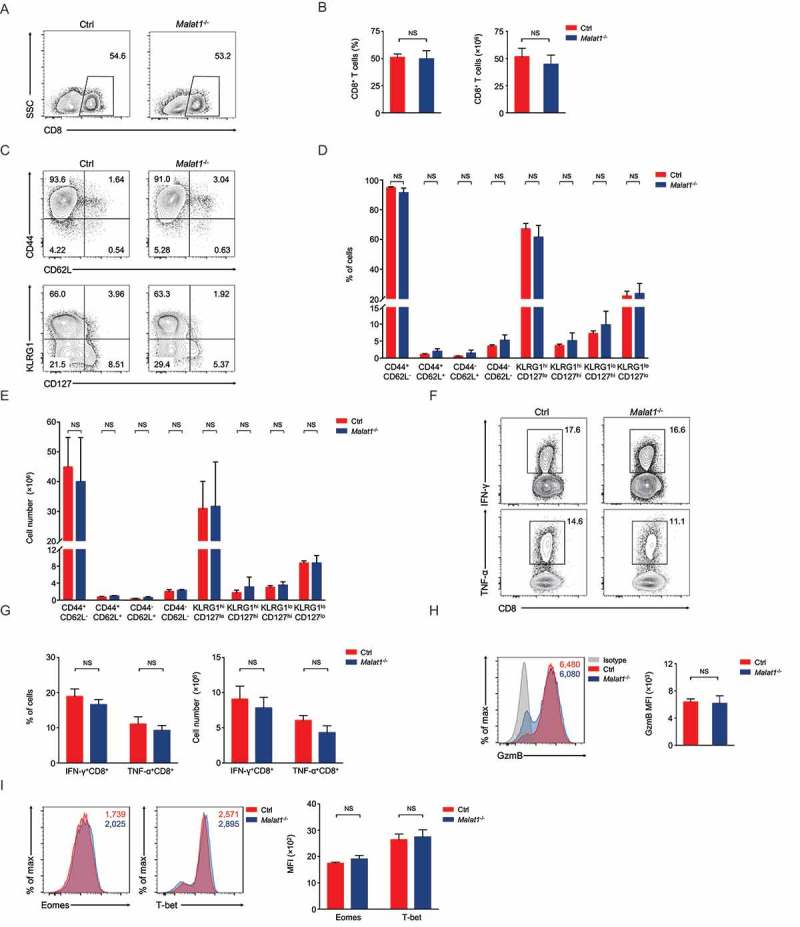
Malat1 is not essential for effector differentiation of CD8+ T cell. (a) Analysis of CD8+ T cells expansion at 8 days after infection. (b) The percentage (left) and number (right) of effector CD8+ T cells in a. (c) Analysis of surface expression of CD44, CD62L, KLRG1 and CD127 from day 8 effector T cells. (d-e) The percentages (d) and numbers (e) of effector T cells in c. (f) Production of effector molecules IFN-γ and TNF-α by effector T cells. (g) The percentages (left) and numbers (right) of IFN-γ+CD8+ and TNF-α+CD8+ T cells in f. (h) Expression of Granzyme B (GzmB) from day 8 effector T cells. (i) Intracellular staining of Eomes and T-bet expression in CD8+ T cells 8 days post infection. Data represent mean ± s.d.
Giving its dispensable role in effector differentiation of CD8+ T cells, we questioned whether deficiency of Malat1 altered the memory T cells formation. Analysis of memory CD8+ T cells at 72 days after infection indicated that the Malat1 deficiency did not affect memory CD8+ T cells pool formation (Figure 5(a, b)). We also noted that the percentages and numbers of CD44+CD62L+TCM and CD44+CD62L−TEM were similar in Malat1−/- mice compared with that of littermate control mice (Figure 5(c, d)). Moreover, the expression of CCR7, CD127 and KLRG1 was considerable in Malat1−/- and wild-type memory T cells (Figure 5(e)). Upon peptide stimulation in vitro, Malat1−/- and wild-type memory CD8+ T cells were both capable of secreting IFN-γ, TNF-α and producing Granzyme B (Figure 5(f-h)). Furthermore, we noted that the protein expression of key transcription factors T-bet and Eomes were not altered in Malat1−/- memory CD8+ T cells compared with that in wild-type memory CD8+ T cells (Figure 5(i)). Taken together, these results revealed that Malat1 is unnecessary for memory CD8+ T cells formation.
Figure 5.
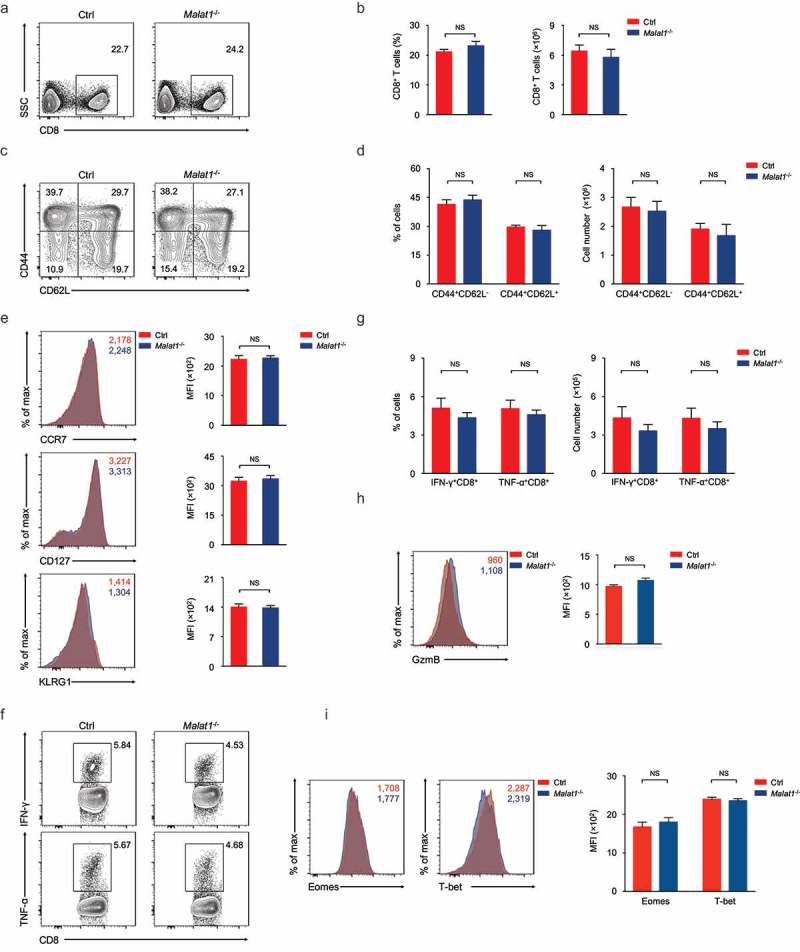
Ablation of Malat1 exhibited normal memory CD8+ T cell pool formation. (a) Analysis of memory CD8+ T cells pool at 72 days after infection. (b) The percentage (left) and number (right) of effector CD8+ T cells in a. (c) Analysis of TEM (CD44+CD62L−) and TCM (CD44+CD62L+) gated on CD8+ T cells. (d) The percentages (left) and numbers (right) of TEM (CD44+CD62L−) and TCM (CD44+CD62L+) in c. (e) Analysis of the expression of CCR7 (top), CD127 (middle), and KLRG1 (bottom) by memory CD8+ T cells. (f) Analysis of the secretion of IFN-γ (top) and TNF-α (bottom) by memory CD8+ T cells. (g) The percentages (left) and numbers (right) of IFN-γ+CD8+ and TNF-α+CD8+ T cells in f. (h) Analysis of the production of Granzyme B by memory CD8+ T cells. (i) Analysis of the expression of Eomes (left) and T-bet (right) by memory CD8+ T cells. Data represent mean ± s.d.
Discussion
Recent studies have reported that lncRNAs are essential for the differentiation and activation of immune cells, and they may be critical determinants of various biological processes. The differentiation of CD4+ T cells into T helper cell subsets is vital for the initiation of adaptive immune responses, and several studies have indicated that distinct T cell subsets express unique profiles of lncRNAs. lncRNA linc-MAF-4 controls the differentiation of TH1 cells by suppress expression of the MAF, a TH2 cells-associated transcription factor. The genomic regions of linc-MAF-4 and MAF form a long-distance interaction, and linc-MAF-4 recruits chromatin modifiers LSD1 and EZH2 to deposit H3K27me3 marks at the promoter of MAF to silence its expression in TH1 cells [8]. In TH2 cells, lncRNA LincR-Ccr2-5′AS, together with Gata3, is a ‘master’ component of a regulatory circuit in gene expression specific to the TH2 cells and is essential for the migration of TH2 cells [6]. Besides CD4+ T cells, lncRNAs also involve in CD8+ T cell immune response [33]. CD244 signaling induces the expression of lncRNA-CD244 and mediates the repression of IFN-γ/TNF-α expression in CD8+ T cells. Similar to linc-MAF-4, lncRNA-CD244 recruits EZH2 to the promoters of IFNG and TNF, and mediates the deposition of repressive chromatin marks.
The lncRNA Malat1 has been reported that it can regulates alternative splicing of pre-mRNAs by modulating SR splicing factor phosphorylation [10]. More importantly, the emerging evidences indicated Malat1 plays an important role in cancers and metastasis [34,35]. Malat1-deficiency cancer cells showed impaired cell migration and defective in metastatic tumor nodules formation, which indicated Malat1 is a critical regulator of the metastasis of cancer cells [36]. Numerous studies have also reported Malat1 functions in lung cancer, liver cancer, breast cancer, bladder cancer, and osteosarcoma [34,37–39]. The existing data reveal that Malat1 is higher expressed in various immune cell subsets, but its roles in immune cells are rarely reported. It should be a significant work to identify the function and mechanism of Malat1 in T cells.
In this study, we identified the different developmental stages in T cell using Malat1−/- mice, but no significant phenotypes were observed. Subsequently, we detected the effector and memory CD8+ T cells and TFH cell response to LCMV infection, but no defects were detected after Malat1−/- ablation. Taken together, the lncRNA Malat1-deficiency mice have normal T cell development and peripheral T cells, and Malat1 is not required for effector and memory CD8+ T cell formation and function as well as TFH cell differentiation. Our study demonstrated Malat1 is not a key regulator in T cell no matter how high expression level it is. Undoubtedly, these data will contribute to a better understanding of lncRNA Malat1 as well as lncRNA in regulating T cell development and antiviral response.
Materials and methods
Mice
Malat1−/- mice were from Texas A&M Institute for Genomic Medicine (TIGM). All animals were on a fully C57BL/6J background. 6- to 10-week-old mice were used in this study, and both male and female mice were included without randomization or ‘blinding’. Mice were bred and housed in specific pathogen-free conditions in accordance with the Institutional Animal Care and Use Committee of China Agricultural University.
LCMV infection
LCMV-Armstrong strain was grown on BHK-21 cells (ATCC, Manassas, VA, USA) and titers were determined as described before [40]. Age and sex matched wild-type mice and Malat1−/- mice were infected by 2 × 105 plaque-forming units LCMV-Armstrong strain intraperitoneally.
Flow cytometry and cell sorting
Single cell suspensions were prepared from bone marrow, spleen, or/and thymus and used for flow cytometry analysis or cell sorting. Surface staining was performed in FACS Buffer (PBS supplemented with 2% fetal bovine serum). The following fluorescence-labeled monoclonal antibodies were used: anti-CD19 (1D3), anti-CD25 (PC61.5), anti-CD279 (J43), anti-CD4 (RM4-5), anti-CD44 (IM7), anti-CD45R (RA3-6B2), anti- CD62L (MEL-14), anti-CD8a (53–6.7), anti-GL7 (GL7), anti-Granzyme B (GB11), anti-KLRG1 (2F1), anti-TCRβ (H57-597), anti-TNF-α (MP6-XT22) (from ebiosciences, CA, USA); anti-CD138 (281–2), anti-CD95 (Jo2), anti-IFN-γ (XMG1.2) (from BD Biosciences, San Jose, CA, USA). CXCR5 staining was done using purified anti-CXCR5 (2G8, BD Biosciences, San Jose, CA, USA) for 1 h, followed by biotin-conjugated goat anti-rat IgG (Cat # 112–066-143, Jackson Immunoresearch, West Grove, PA, USA) for 30 min, and then by fluorescence-conjugated streptavidin at 4 °C for 30 min in PBS supplemented with 2% normal mouse serum, 2% FCS, and 0.5% BSA. Staining for Bcl-6 (K112-91, BD Biosciences, San Jose, CA, USA) was performed with Foxp3/Transcription Factor Staining Buffer Set following the manufacturer’s instructions. Data were acquired on a FACSVerse (BD Biosciences, San Jose, CA, USA) and were analyzed with FlowJo software (Treestar, Ashland, Oregon, USA). All cell sorting experiments were carried out on a FACSAria II sorter (BD Biosciences, San Jose, CA, USA).
Functional characterization of antigen-specific CD8+ T cells
For functional characterization of antigen-specific CD8+ T cells, splenocytes were stimulated with 1 μM of GP33–41 (KAVYNFATC), NP396–404 (FQPQNGQLI) for 5 hours in the presence of GolgiStop and GlogiPlug (BD Biosciences, San Jose, CA, USA). The stimulated cells were then surfaced stained, fixed and permeabilized using BD Cytofix/Cytoperm and BD Perm/Wash solutions (BD Biosciences, San Jose, CA, USA), and intracellularly stained for IFN-γ, IL-2, TNF-α following the manufacturer’s instructions.
Quantitative RT-PCR
Cells were sorted and subsequently lysed in TRIzol reagent (Life Technologies, Carlsbad, CA, USA). Total RNA was extracted followed by cDNA synthesis with FastQuant RT Kit (Tiangen, Beijing, China). cDNA was analyzed for expression of various genes with the SYBR Green SuperReal PreMix (Tiangen, Beijing, China) on a CFX96 Real-Time System (Bio-Rad, Hercules, CA, USA). The following primers were used: for Gapdh, 5ʹ- ACTCCACTCACGGCAAATTCA-3ʹ and 5ʹ-GGCCTCACCCCATTTGATG-3ʹ; for Hprt1, 5ʹ- GCGTCGTGATTAGCGATGATG-3ʹ and 5ʹ-CTCGAGCAAGTCTTTCAGTCC-3ʹ; for Malat1 (P1), 5ʹ- AGAGTGAGTTCCAGGACAGC-3ʹ and 5ʹ-GCTTCCTACTACCTGTGCCT-3ʹ; for Malat1 (P2), 5ʹ- CATCTCGGAGCAGGAAACAG-3ʹ and 5ʹ-CCATCATGAAAGCCCATCGG-3ʹ for Malat1 (P3), 5ʹ-TGAGGACAACAGGTGAACGA-3ʹ and 5ʹ-GGCTCCGCTGTCCTACATTA-3ʹ; for Tcf7, 5ʹ-CCCTTCCTGCGGATATAGAC-3ʹ and 5ʹ-GGTACACCAGATCCCAGCAT-3ʹ; and for Notch1, 5ʹ- CCCTTGCTCTGCCTAACGC-3ʹ and 5ʹ-GGAGTCCTGGCATCGTTGG-3ʹ.
Statistical analysis
Statistical analysis was conducted with Prism 7.0 (GraphPad, La Jolla, CA, USA). An unpaired Student’s t-test with a 95% confidence interval was used for calculation of P value. NS not significant, * P < 0.05, ** P < 0.01 and *** P < 0.001.
Funding Statement
This study was supported in part by grants National Key Research and Development Program of China (2017YFA0104401), National Natural Scientific Foundation of China (Grant Numbers: 31571522, 31630038 & 31422037) to Dr. Shuyang Yu.
Disclosure statement
No potential conflict of interest was reported by the authors.
Author Contributions
Y.Y., W.G., J.C., P.G., G.Y., Juanjuan L., Jingjing L. and M.Y. performed the experiments; F.W. analyzed the high-throughput data and assisted the Figures processing; T.Z., Y.K. and X.M. provided scientific insights and helped the overall study; X.M. revised the manuscript; Y.Y. and S.Y. analyzed the data and wrote the paper; and S.Y. conceived of the project and supervised the whole study.
References
- [1].Hu W, Alvarez-Dominguez JR, Lodish HF.. Regulation of mammalian cell differentiation by long non-coding RNAs. EMBO Rep. 2012;13(11):971–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Atianand MK, Caffrey DR, Fitzgerald KA. Immunobiology of Long Noncoding RNAs. Annu Rev Immunol. 2017;35:177–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kotzin JJ, Spencer SP, McCright SJ, et al. The long non-coding RNA Morrbid regulates Bim and short-lived myeloid cell lifespan. Nature. 2016;537(7619):239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wang P, Xue Y, Han Y, et al. The STAT3-binding long noncoding RNA lnc-DC controls human dendritic cell differentiation. Science. 2014;344(6181):310–313. [DOI] [PubMed] [Google Scholar]
- [5].Gomez JA, Wapinski OL, Yang YW, et al. The NeST long ncRNA controls microbial susceptibility and epigenetic activation of the interferon-γ locus.. Cell. 2013;152(4):743–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hu G, Tang Q, Sharma S, et al. Expression and regulation of intergenic long noncoding RNAs during T cell development and differentiation. Nat Immunol. 2013;14(11):1190–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Turner M, Galloway A, Vigorito E. Noncoding RNA and its associated proteins as regulatory elements of the immune system. Nat Immunol. 2014;15(6):484–491. [DOI] [PubMed] [Google Scholar]
- [8].Ranzani V, Rossetti G, Panzeri I, et al. The long intergenic noncoding RNA landscape of human lymphocytes highlights the regulation of T cell differentiation by linc-MAF-4. Nat Immunol. 2015;16(3):318–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Huarte M. The emerging role of lncRNAs in cancer. Nat Med. 2015;21(11):1253–1261. [DOI] [PubMed] [Google Scholar]
- [10].Tripathi V, Ellis JD, Shen Z, et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell. 2010;39(6):925–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yoshimoto R, Mayeda A, Yoshida M, et al. MALAT1 long non-coding RNA in cancer. Biochim Biophys Acta. 2016;1859(1):192–199. [DOI] [PubMed] [Google Scholar]
- [12].Ma X-Y, Wang J-H, Wang J-L, et al. Malat1 as an evolutionarily conserved lncRNA, plays a positive role in regulating proliferation and maintaining undifferentiated status of early-stage hematopoietic cells. Bmc Genomics. 2015;16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Rothenberg EV, Moore JE, Yui MA. Launching the T-cell-lineage developmental programme. Nat Rev Immunol. 2008;8(1):9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Issuree PD, Ng CP, Littman DR. Heritable Gene Regulation in the CD4: CD8T Cell Lineage Choice. Front Immunol. 2017;8:291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].von Boehmer H, Fehling HJ. Structure and function of the pre-T cell receptor. Annu Rev Immunol. 1997;15:433–452. [DOI] [PubMed] [Google Scholar]
- [16].Germain RN. T-cell development and the CD4-CD8 lineage decision. Nat Rev Immunol. 2002;2(5):309–322. [DOI] [PubMed] [Google Scholar]
- [17].Kappes DJ, He X, He X. CD4-CD8 lineage commitment: an inside view. Nat Immunol. 2005;6(8):761–766. [DOI] [PubMed] [Google Scholar]
- [18].Crotty S. Follicular helper CD4 T cells TFH. Annu Rev Immunol. 2011;29:621–663. [DOI] [PubMed] [Google Scholar]
- [19].Jogdand GM, Mohanty S, Devadas S. Regulators of Tfh cell differentiation. Front Immunol. 2016;7:520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity. 2014;41(4):529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Johnston RJ, Poholek AC, DiToro D, et al. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science. 2009;325(5943):1006–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Nurieva RI, Chung Y, Martinez GJ, et al. Bcl6 mediates the development of T follicular helper cells. Science. 2009;325(5943):1001–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Yu D, Rao S, Tsai LM, et al. The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity. 2009;31(3):457–468. [DOI] [PubMed] [Google Scholar]
- [24].Ma CS, Deenick EK, Batten M, et al. The origins, function, and regulation of T follicular helper cells. J Exp Med. 2012;209(7):1241–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Harty JT, Badovinac VP. Shaping and reshaping CD8+ T-cell memory. Nat Rev Immunol. 2008;8(2):107–119. [DOI] [PubMed] [Google Scholar]
- [26].Zhou X, Yu S, Zhao D-M, et al. Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity. 2010;33(2):229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kim EH, Suresh M. Role of PI3K/Akt signaling in memory CD8 T cell differentiation. Front Immunol. 2013;4:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Chang JT, Wherry EJ, Goldrath AW. Molecular regulation of effector and memory T cell differentiation. Nat Immunol. 2014;15(12):1104–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lefrancois L, Marzo AL. The descent of memory T-cell subsets. Nat Rev Immunol. 2006;6(8):618–623. [DOI] [PubMed] [Google Scholar]
- [30].Masopust D, Vezys V, Marzo AL, et al. Preferential localization of effector memory cells in nonlymphoid tissue. Science. 2001;291(5512):2413–2417. [DOI] [PubMed] [Google Scholar]
- [31].Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol. 2012;12(11):749–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Steinke FC, Yu S, Zhou X, et al. TCF-1 and LEF-1 act upstream of Th-POK to promote the CD4(+) T cell fate and interact with Runx3 to silence Cd4 in CD8(+) T cells.. Nat Immunol. 2014;15(7):646–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wang Y, Zhong H, Xie X, et al. Long noncoding RNA derived from CD244 signaling epigenetically controls CD8+ T-cell immune responses in tuberculosis infection. Proc Natl Acad Sci U S A. 2015;112(29):E3883–E3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Gutschner T, Hammerle M, Diederichs S. MALAT1 - a paradigm for long noncoding RNA function in cancer. J Mol Med (Berl). 2013;91(7):791–801. [DOI] [PubMed] [Google Scholar]
- [35].Diederichs S. The four dimensions of noncoding RNA conservation. Trends Genet. 2014;30(4):121–123. [DOI] [PubMed] [Google Scholar]
- [36].Gutschner T, Hämmerle M, Eissmann M, et al. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013;73(3):1180–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Luo J-H, Ren B, Keryanov S, et al. Transcriptomic and genomic analysis of human hepatocellular carcinomas and hepatoblastomas. Hepatology. 2006;44(4):1012–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ellis MJ, Ding L, Shen D, et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. 2012;486(7403):353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ying L, Chen Q, Wang Y, et al. Upregulated MALAT-1 contributes to bladder cancer cell migration by inducing epithelial-to-mesenchymal transition. Mol Biosyst. 2012;8(9):2289–2294. [DOI] [PubMed] [Google Scholar]
- [40].Welsh RM, Seedhom MO. Lymphocytic choriomeningitis virus (LCMV): propagation, quantitation, and storage. Curr Protoc Microbiol. 2008;Chapter 15:Unit 15A 1. [DOI] [PMC free article] [PubMed] [Google Scholar]