

ABSTRACT
Myeloid cell leukemia-1 (Mcl −1) is one of the most frequently amplified genes in cancer, and its overexpression is associated with poor prognosis and drug resistance. As a member of the Bcl-2 family it is involved in the control of the mitochondrial (intrinsic) cell death pathway. Alternative splicing of the (Mcl-1) gene results in the expression of two functionally distinct proteins, the anti-apoptotic Mcl-1L (exon 2 included) and the pro-apoptotic Mcl-1S (exon 2 skipped). Our data shows that transfecting siRNAs that target hnRNP K and the hnRNP F/H family result in a switch in splicing towards the pro-apoptotic Mcl-1S. Specific binding sites for these and other Mcl-1 splicing factors were investigated and identified by RNA immunoprecipitation and through construction of a Mcl-1 minigene construct. Moreover, this study shows up to a 30 fold change in the levels of Mcl-1S can be achieved through double and triple knockdowns of the most significant RNA binding proteins involved in Mcl-1 splicing, as well as activation of the mitochondrial cell death pathway. Targeting the splicing process of Mcl-1 along with other apoptotic regulators provides an exciting new therapeutic target in cancer cells, and may provide a way to overcome therapy resistance.
KEYWORDS: Apoptosis, Mcl −1, hnRNPs, SRSF1, alternative splicing
Introduction
Evading apoptosis is one of the key hallmarks of cancer and is important for its initiation and development as well as in resistance to therapy[1]. Apoptosis can be triggered by a number of different pathways, but the mitochondrial (intrinsic) cell death pathway is commonly dysregulation in cancer. The activation of this pathway is controlled by the Bcl2 protein family [2–4]. This family contains both pro-apoptotic proteins (Bak, Bax, Bim and Puma) as well as anti-apoptotic proteins (Bcl-2, Bcl-XL and Mcl-1L) and it is the overall balance between the two groups which determines whether apoptosis is triggered.
Due to their central role in controlling the mitochondrial apoptotic pathway, a number of the Bcl-2 family members have been implicated in cancer development and drug resistance [2,3,5,6]. Indeed the anti-apoptotic Mcl-1 and Bcl-X have been shown to be frequently amplified across a range of different human cancers, with the amplification of Mcl-1 occurring in more than 10% of cancers[7]. Additionally, increased expression of Mcl-1 protein has been observed in a wide range of different cancers [8–10], including breast cancer where it is associated with a higher tumour grade and a poor prognosis[11].
There is also evidence to suggest that Mcl-1 may be involved in the development of therapy resistance, as elevated Mcl-1 levels have been found in resistant cells. This includes chemotherapy-resistant triple negative breast cancer cells where Mcl-1 was found to be the second most amplified gene, with amplification in 54% of the samples tested[12]. As well as in antiestrogen-resistant breast cancer cells where there were increased levels of Mcl-1 protein[13]. A number of studies have also demonstrated that sensitivity of these drug resistant cells can be restored upon depletion of Mcl-1.
Along with gene amplifications and transcriptional regulation of the Bcl-2 family, alternative splicing of Bcl-2 family members can also control activation of apoptosis, by producing spliced variants with differing functions. The alternative splicing of Bcl-X in particular has been widely studied. Bcl-X produces two protein variants, the main transcript encodes for the anti-apoptotic Bcl-XL, whereas an alternatively spliced transcript encodes for the shorter pro-apoptotic Bcl-XS variant. Numerous splicing regulators have been identified which are involved in Bcl-X splice site selection [14–18]. Although historically less well studied than the Bcl-X splicing event, Mcl-1 also undergoes alternative splicing to produce functionally different proteins [19,20]. The full length transcript of Mcl-1 encodes for Mcl-1L, which is the main anti-apoptotic variant. The other variant, Mcl-1S, is produced when the second exon of Mcl-1 is alternatively spliced out. This splicing event results in a shift in the reading frame and the loss of all downstream domains, resulting in a pro-apoptotic BH-3 only protein being produced. Mcl-1S exerts its pro-apoptotic activity through specifically binding to anti-apoptotic Mcl-1L, rather than through any other members of the Bcl-2 family[21]. A switch in Mcl-1 splicing towards the anti-apoptotic Mcl-1L variant has been shown to occur in both breast and ovarian cancer [22,23]. Recently, some studies have started to look at the regulation of this alternative splicing event and have identified some splicing factors involved. These include two members of the SR protein family, SRSF1 and SRSF5, as well as RBM4 [24,25].
A wide range of RNA-binding proteins have been implicated in regulating alternative splicing but most of the well-studied splicing factors are members of the SR or hnRNP family of RNA binding proteins. As previously mentioned the role of the SR family in regulating Mcl-1 splicing has been investigated, but currently little is known about the involvement of the hnRNP family in this splicing process. Therefore, in this study we wanted to investigate the role of members of the hnRNP family in the regulation of the Mcl-1 splicing event. From an initial screen of the family members, hnRNP K and hnRNP F/H subfamily were identified as key regulators of Mcl-1 splicing. Loss of these hnRNPs along with SRSF1 through RNA interference produced significant switches in Mcl-1 splicing and induced apoptosis. Therefore these splicing factors along with their identified binding site represent an exciting new therapeutic target.
Results
To identify potential hnRNP splicing regulators we screened cDNA collected from a previous hnRNP siRNA study for Mcl-1 splicing changes. This study had reduced the levels of a number of different hnRNP family members (hnRNP A/B, hnRNP A2/B1, hnRNP D, hnRNP F, hnRNP H1, hnRNP I (PTB) and hnRNP K) by siRNA transfection in SKBR3 cells. Splicing changes were detected by conventional PCR with Mcl-1 primers which can detect both isoforms, and showed changes in Mcl-1 splicing for cells transfected with hnRNP H1, hnRNP K and hnRNP I siRNAs (data not shown).
The results from this initial screen were then confirmed in two breast cancer cell lines, MCF-7 and MDA-MD-231 cells. MCF-7 cells are an ER positive cell line which were selected due to their high expression of Mcl-1L[24]. MDA-MD-231 cells were selected as they are a triple negative breast cancer cell line, and this subset of breast cancer has been demonstrated to be particularly sensitive to Mcl-1 depletion[26]. Along with siRNAs targeting the hnRNPs identified in the initial screen, siRNAs targeting hnRNP F were also included, as hnRNP H1 and F are very closely related family members and can bind the same sequences within the RNA [27,28]. Double knockdowns of both hnRNP H1 and F were also performed as these two closely related proteins may also be acting redundantly[29]. Knockdown of both hnRNP H1 and F was achieved by transfection of two separate siRNA targeting hnRNP F and hnRNP H1, and by using a single siRNA designed against a conserved region of hnRNP H1 and F. Efficient knockdown of all the mRNA targets was achieved by 48 hours including the double knockdowns of hnRNP H1 and F (Fig. 1A). Reductions to protein levels of the hnRNP F, hnRNP H1 and hnRNP K was determined to have occurred by 72 hours (Fig. S1). The hnRNP H1 antibody used to detect hnRNP F and H1, also detected additional bands at 37kDa which correspond to the size of hnRNP H3. These additional bands believed to be hnRNP H3 are upregulated in response to hnRNP F and H1 knockdown. Unfortunately proteins levels of hnRNP I were not detectable by western blotting in these cell lines.
Changes in Mcl-1 splicing were assessed by splice specific TaqMan assays for Mcl-1L and Mcl-1S 72 hours after cells were transfected. This time point was selected as reductions to hnRNP protein levels had been demonstrated to have occurred at this point. Reduction of hnRNP K produced increased levels of Mcl-1S compared with Mcl-1L in both cell lines (Fig. 1B and D). Knockdown of hnRNP H1 also resulted in an increase in the short form over the longer isoform in the MCF-7 cell line, although this was not observed in the MDA-MD-231 cell line. Despite its similarities with hnRNP H1 reduction of hnRNP F on its own had no effect on the splicing of Mcl-1. Some redundancy does appear to occur between the two proteins as the double knockdown of hnRNP F and H1 produced a much greater switch in splicing than knockdown of hnRNP H1 alone. A switch in splicing was also observed in the MCF-7 cell line after treatment with the hnRNP I siRNAs where levels of Mcl-1S were reduced compared to Mcl-1L (Fig. 1C).
Figure 1.
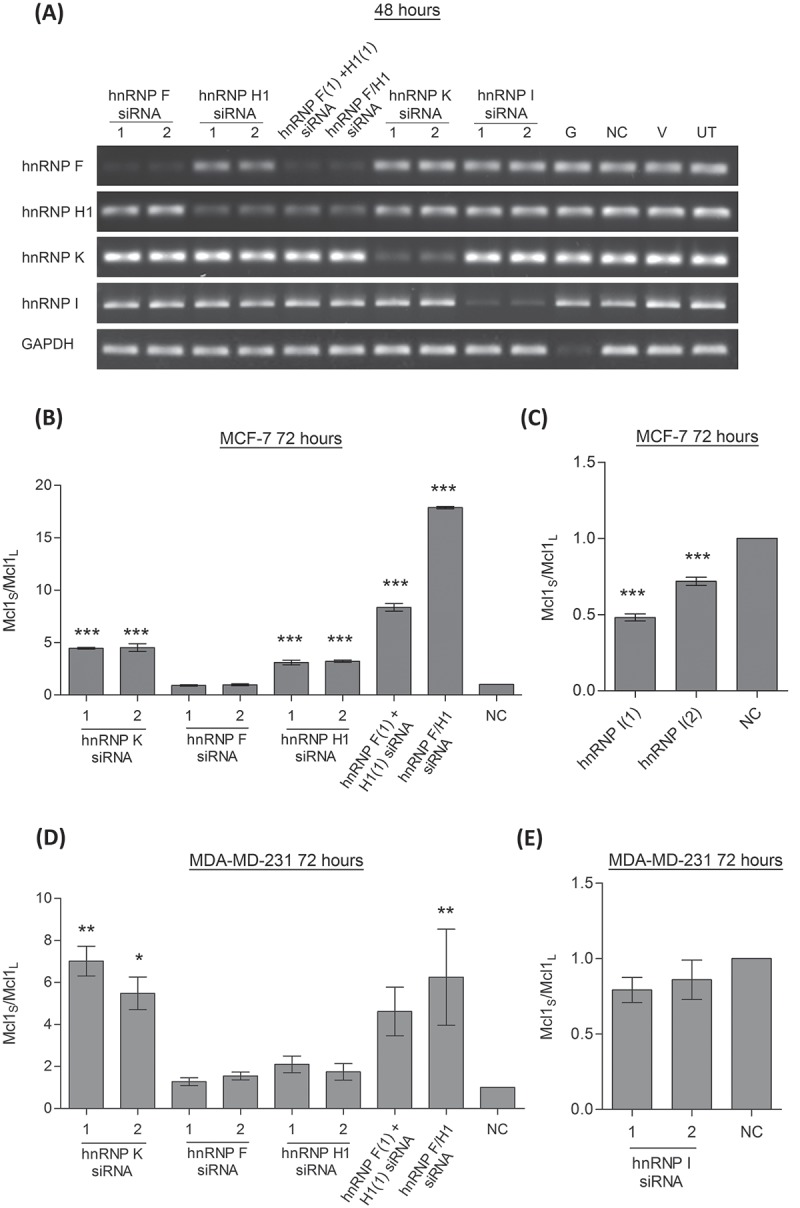
Effect of hnRNP siRNAs on Mcl-1 splicing. (A) RNA interference was used to reduce the levels hnRNP F, H1, K and I in MCF7 cells. Combined knockdown of hnRNP F and H1 was also achieved by either combined transfection of two siRNAs (hnRNP F(1) and H1(1) siRNAs) or by using a single siRNA against a conserved region (hnRNP F/H1 siRNA). Four controls comprising a positive transfection control (GAPDH siRNA (G)), a negative scrambled sequence control siRNA (NC), a vehicle only control (V) and untreated cells (UT) were also used. Knockdown of hnRNPs was assessed at mRNA level at 48 hour using classical PCR. Alternative splicing events in MCF7 cells (B and C) and MDA-MD-231 cells (D and E) were measured using splice specific primers by qPCR at 72 hours post-transfection (mean (n = 3) ± SEM). * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001.
The hnRNPs identified clearly affect the splicing of Mcl-1 but as there is a lot of cross regulation amongst splicing factors, further investigations were required to demonstrate a direct interaction. Firstly, the effect of the reduction of these specific hnRNPs on the mRNA levels of known Mcl-1 splicing regulators was investigated by conventional PCR. This showed no changes in the mRNA levels of SRSF1, SRSF5 and RBM4 after hnRNP siRNA treatment (Fig. S1). Secondly, RNA immunoprecipitations were performed to demonstrate direct binding of the hnRNPs to Mcl-1 pre-mRNA. Fig. 2 shows that hnRNP H1 and hnRNP K antibodies were able to pull down Mcl-1 pre-mRNA with a fold enrichment of 15.75 and 5.47 respectively. Although the hnRNP H1 antibody was raised against hnRNP H1 it is also able to detect other family members by western blotting (Fig. S1)[30]. Therefore the RNA immunoprecipitation with this antibody demonstrates that the hnRNP F/H family bind to Mcl-1 pre-mRNA rather than hnRNP H1 specifically. The hnRNP I antibody on the other hand was unable to precipitate Mcl-1 pre-mRNA and therefore it is unlikely that it is directly binding to Mcl-1.
Figure 2.
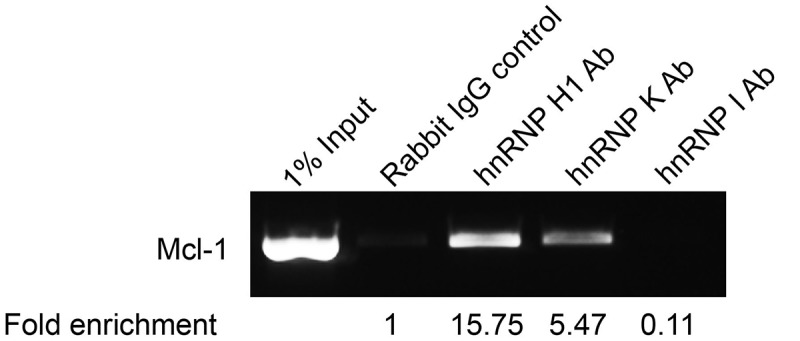
Binding of hnRNPs to Mcl-1 transcript. Binding of splicing factors to endogenous Mcl-1 pre-mRNA was determined by RNA immunoprecipitation using antibodies that target hnRNP H1, hnRNP K and hnRNP I. Fold enrichment of target mRNA was determined by qPCR.
To identify specific binding sites for the Mcl-1 splicing factors so far identified a Mcl-1 minigene was constructed. The minigene covers the end of exon 1 to the beginning of exon 3 and includes the entire second exon and both introns (Fig. 3A). Potential binding sites for hnRNP K and the hnRNP F/H family, as well as for SRSF1 (a key Mcl-1 splicing factor [24]), were identified by splicing factor binding site prediction programs (SpliceAid (http://www.introni.it/splicing.html) and ESE Finder [31,32]). These programs predicted a SRSF1 binding site towards the end of exon 2 (Fig. S2), and site-directed mutagenesis was employed to mutate it so that it could no longer be bound by SRSF1. Care was also taken not to create additional binding sites with this mutation and therefore the mutated sequences were checked again by the splicing factor binding site prediction programs (Fig. S2). Transfection of the MCF-7 cells with the wild-type and the SRSF1 mutated minigenes showed that the loss of this binding site resulted in a significant shift in splicing towards the shorter isoform (Fig. 3B).
Figure 3.
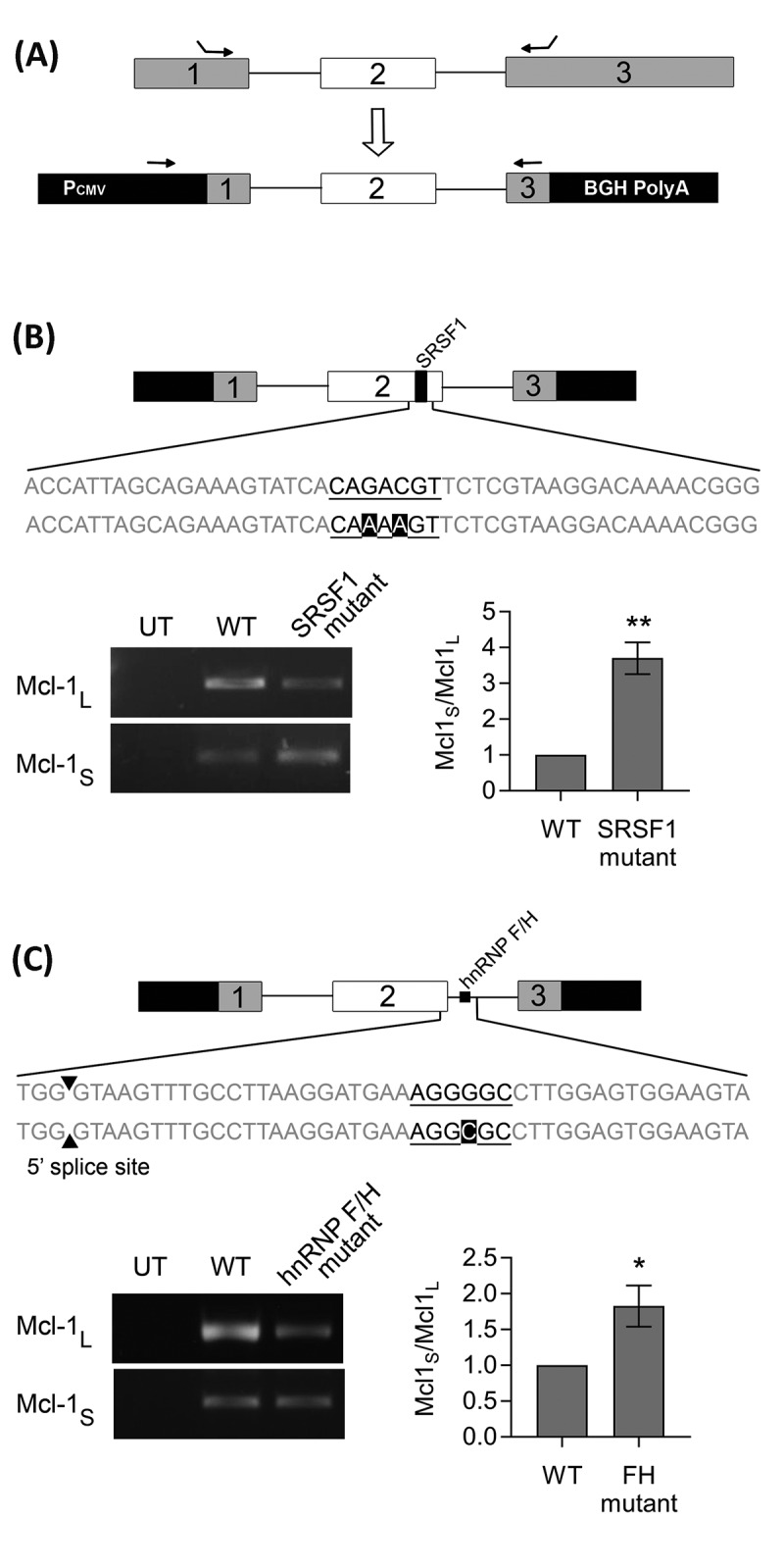
Identifying splicing factor binding sites using a Mcl-1 minigene. (A) Construction of the Mcl-1 minigene containing the last 115bp of exon 1, intron 1, exon 2, intron 2 and the first 157bp of exon 3. Location of minigene specific primers are indicated by arrows. Site directed mutagenesis was performed on the predicted binding sites for SRSF1 (B) and hnRNP F/H (C) within the Mcl-1 minigene. Changes to the splicing pattern were determined using the minigene specific primers and classical PCR. Bands were quantified by capillary electrophoresis (mean (n = 3) ± SEM). * P ≤ 0.05, ** P ≤ 0.01.
Predicted binding sites for hnRNP F and H1 were much more numerous. The hnRNP F/H family are able to act as both enhancers and silencers of splicing usually dependent on the location of their binding sites. In the case of Mcl-1 they are acting as splicing enhancers, therefore their binding sites (G tracts) are likely to be found between 10 and 70 bp downstream of the 5ʹ splice site [29,33]. A potential hnRNP F/H binding site within this region was identified within Mcl-1 (Fig. S3). Mutation of this site within the Mcl-1 minigene resulted in a significant switch in splicing, increasing levels of the shorter isoform compared to the wild-type Mcl-1 minigene control (Fig. 3C). It is possible that these mutations are also affecting the stability of the RNA. Particularly for the hnRNP F/H mutant where the switch mainly occurs as a result of decreased levels of Mcl-1L, but as this mutation is within the intron it should effect both variants equally and there is no corresponding decrease in Mcl-1S.
Four potential hnRNP K binding sites within the Mcl-1 minigene were also identified (Fig. S4). These were also each in turn mutated to abolish the predicted binding of hnRNP K, however none of the mutations produced a significant switch in the splicing of the minigene (Fig. S4).
To try and create greater switches in Mcl-1 splicing, combined knockdowns of some of the key Mcl-1 splicing factors were performed. This included siRNAs against hnRNP K, SRSF1 and the siRNA targeting the conserved region of hnRNP H1 and F. Cotransfecting two separate siRNAs did not appear to reduce the efficiency of the individual siRNAs (Fig. 4A, B)). When the levels of the two spliced variants were measured after the double knockdowns, all of the combinations produced extremely large switches in splicing (Fig. 4B). As with the single siRNA transfections the largest changes in Mcl-1 splicing were observed in the MCF-7 cell line. Although all siRNA combinations produced large switches in splicing, the knockdown of hnRNP F/H1 and hnRNP K together produced the greatest change in both cell lines.
Figure 4.
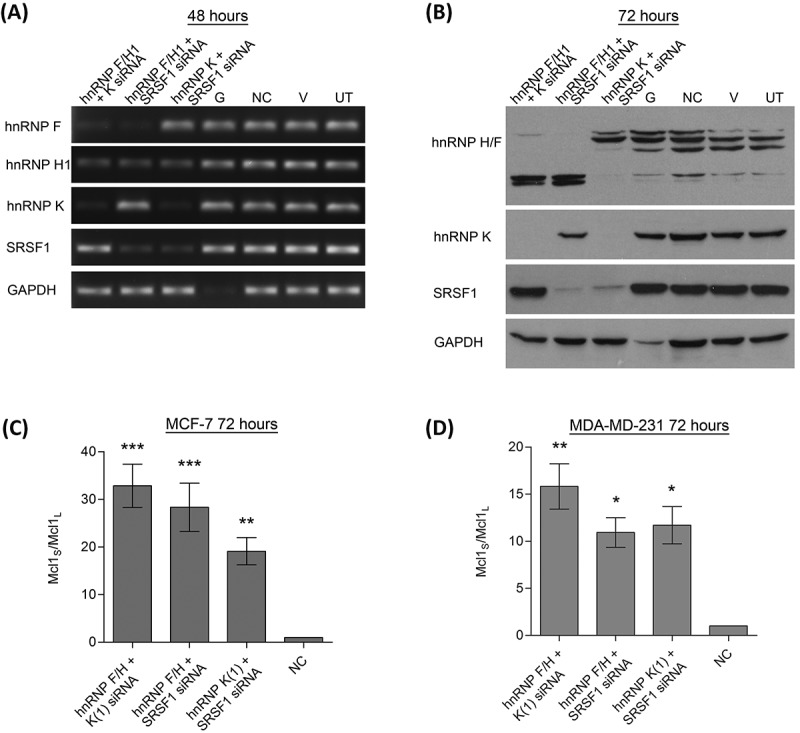
Double knockdowns of key Mcl-1 splicing factors. Cells were transfected with two separate siRNAs to achieve double knockdowns of key splicing factors (hnRNP K(1), hnRNPF/H1 and SRSF1 siRNA). Four controls comprising a positive transfection control (GAPDH siRNA (G)), a negative scrambled sequence control siRNA (NC), a vehicle only control (V) and untreated cells (UT) were also used. Knockdown of hnRNPs in MCF7 cells was assessed at 48 hours using classical PCR (A) and at 72 hours by western blot (B). Alternative splicing of Mcl-1 was measured in MCF7 (C) and MDA-MD-231 (D) cells using splice specific primers by qPCR at 72 hours post-transfection (mean (n = 3) ± SEM). * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001.
It should be noted that it has previously been observed that knockdown of SRSF1 also reduces the levels of hnRNP H1[30]. This was indeed observed to be the case with the combined knockdown of hnRNP K and SRSF1 siRNAs which also reduced the levels of hnRNP H1. Despite this, the double knockdown of SRSF1 and hnRNP F/H1 produced a much greater switch than individual knockdowns indicating that the role of SRSF1 in Mcl-1 splicing is not just mediated through hnRNP H1 reductions.
To assess whether these dual knockdowns can have a functional effect on the two breast cancer cell lines, cell counts were performed to assess cell numbers and viability. In the MCF-7 cell line the combined knockdown of hnRNP F/H1 and hnRNP K produced a significant reduction in cell viability (Fig. 5A), this was also the combination of siRNAs that produced the greatest change in Mcl-1 splicing. In comparison the MDA-MD-231 cells showed much greater sensitivity to reductions in the splicing factors, with a greater reduction in cell viability after knockdown of hnRNP F/H1 and hnRNP K (Fig. 5B). Additionally, MDA-MD-231 cells were particularly sensitive to the combined knockdown of hnRNP K and SRSF1, despite this combination not producing the greatest switch in Mcl-1 splicing. To determine whether the reduced viability was a result of the activation of the mitochondrial (intrinsic) cell death pathway, caspase 9 activity was measured, as caspase 9 is specifically activated by this pathway. Significant increases in caspase 9 activity were observed in the MDA-MD-231 cells with the combined knockdown of hnRNP F/H1 and hnRNP K, and with hnRNP K and SRSF1, mirroring the reductions observed in cell viability.
Figure 5.
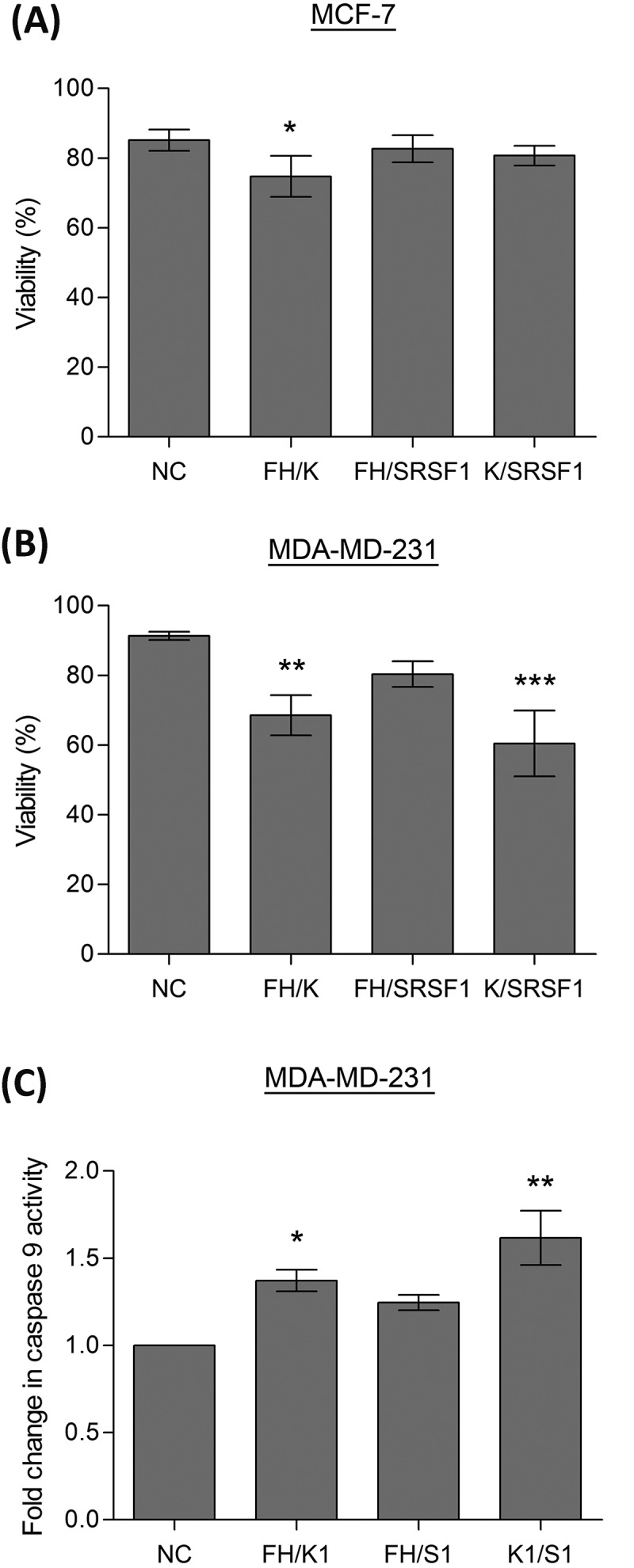
Induction of apoptosis by splicing factors. Cells were transfected with two separate siRNAs to achieve double knockdowns of key splicing factors (hnRNP K(1), hnRNPF/H1 and SRSF1 siRNA), a negative scrambled sequence control siRNA (NC) was used as the control. Cell viability of MCF7 (A) and MDA-MD-231 (B) cells was assessed by cell counts and trypan blue exclusion (mean (n = 3) ± SEM). (C) Caspase 9 activity was determined in the MDA-MD-231 cells 72 hours after transfection with siRNAs. Enzymatic activity of caspase 9 is expressed as fold induction compared to the control (negative control siRNA) and given as mean (n = 3) ± SEM. * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001.
Discussion
Mcl-1 has recently emerged as a key survival and resistance factor involved in a cancer cells evasion of apoptosis. Therefore it is important to explore ways to target Mcl-1 therapeutically, such as switching its splicing to reverse its anti-apoptotic function. This study identifies for the first time members of the hnRNP family of RNA binding proteins that are involved in the regulation of the Mcl-1 splicing event. Both hnRNP K and members of the hnRNP F/H family were identified as splicing enhancers of Mcl-1 promoting the inclusion exon 2 and the production of the anti-apoptotic Mcl-1L.
The involvement of hnRNP K in the production of the anti-apoptotic Mcl-1L variant is consistent with its emerging role in cancer development. HnRNP K has been shown to be overexpressed in a variety of different cancer tissues, including lung, colorectal, prostate, oral squamous cell and nasopharyngeal cancer [34–38]. Overexpression of hnRNP K is also frequently correlated with increased tumour grade and poor prognosis [35–38], including in breast cancer where increased expression of hnRNP K was observed in Grade III cancer compared to Grade II cancer[39].
HnRNP K has also been implicated in the regulation of apoptosis, with several studies showing that depletion of hnRNP K leads to the induction of apoptosis [40–42]. Some potential apoptotic targets of hnRNP K have been previously identified, and include FLIP [42] and XIAP [40], which are two anti-apoptotic proteins which are transcriptionally activated by hnRNP K. Additionally, hnRNP K has been shown to be an important regulator of alternative splicing in apoptotic genes[43]. In a study by Venables et al [43] the effect of depletion of hnRNP family members on alternative splicing events in apoptotic genes was assessed by PCR. HnRNP K emerged as the most influential hnRNP protein, with more than 40% of the alternative splicing events affected by its depletion. Although the alternative splicing of Mcl-1 was not assessed in that study, the results presented here mean Mcl-1 can now be added to the list of hnRNP K apoptotic targets.
The hnRNP FH family were also shown to be involved in the production of the anti-apoptotic Mcl-1L variant, but the role of this family in the overall development of tumorigenesis is less clear. There is some evidence that hnRNP H1 is upregulated in certain tumours such as hepatocellular carcinoma, glioma, gastric carcinoma, head and neck carcinomas and carcinoma of the pancreas [44–46]. High levels of hnRNP H has also been linked to a splicing oncogenic switch involved in the development of malignant gliomas[45]. Despite these observations another study by Hope et al [47] has shown that strong nuclear hnRNP H expression is associated with increased survival. Overexpression of hnRNP F in tumours on the other hand has been observed less frequently, with only gastric carcinoma showing an increase in expression[44].
The hnRNP FH family have also been implicated in the alternative splicing of numerous apoptotic genes. Although the resulting effect on the apoptotic process is unclear as they are able to promote the production of anti-apoptotic variants including a-raf [46] and MADD [45] as well as pro-apoptotic variants such as Bcl-xS [16]. Despite these contradictory effects, downregulation of hnRNP H1 has been demonstrated to induce apoptosis in some carcinoma cell lines [45,46], and although some other apoptotic targets have been identified, Mcl-1 splicing may also contribute to this phenotype.
The role of hnRNP K and the hnRNP FH family in Mcl-1 splicing appears to be by a direct mechanism as both show direct binding to Mcl-1 pre-mRNA via RNA immunoprecipitation. A specific binding site for the hnRNP FH family was identified using the Mcl-1 minigene construct. The site, which is predicted to be bound by both hnRNP H1 and hnRNP F, acted as an intronic splicing enhancer site, and its location just 24bp from the 5ʹ splice site is typical for a G tract involved in enhancing spicing [29,33]. Despite the clear switch in splicing of Mcl-1 after knockdown of hnRNP K and strong binding of hnRNP K to Mcl-1 pre-mRNA, a binding site within the Mcl-1 minigene was not found. Mutation of the four predicted binding sites had no effect on the splicing, but all four sites had low scores for binding affinity from SpliceAid. Further analysis of the Mcl-1 gene using SpliceAid did identify a number of high scoring predicted binding sites for hnRNP K towards the end of exon 3. These sites though lie outside the region contained in the Mcl-1 minigene construct. Alternatively, the binding site of hnRNP K may lie within the Mcl-1 minigene construct but consist of a novel sequence not currently in the SpliceAid database.
The proto-oncogene SRSF1 has previously been identified as a key regulator of Mcl-1 alternative splicing, and been shown to bind to Mcl-1 pre-mRNA [17,24]. Here we identified a specific binding site for SRSF1 within the second exon of Mcl-1. Therefore this appears to be a classic exon splicing enhancer site close to the 3ʹ splice site which is bound by SRSF1. Identification of these binding sites opens up the possibility in the future of designing therapeutic splice switching oligonucleotides to target them.
The data presented here also highlights the advantage of targeting multiple splicing factors when trying to alter the splicing pattern of genes. The alternative splicing process is often controlled by multiple splicing factors which can sometimes act redundantly. By targeting multiple splicing factors agreater switch in splicing was observed which may potentially be more therapeutically relevant. Specifically the combined knockdowns of hnRNP F/H and K which produced the biggest switch in Mcl-1 splicing also significantly reduced the viability of both cell lines. Previously, antisense oligonucleotides have induced apoptosis is a similar manner by altering the splicing of Mcl-1 [48,49]. Although the switch in Mcl-1 splicing was greater in the MCF7 cells the observed reductions in viability were less than with the MDA-MD-231 cells. This observation may reflect the underlying levels of Mcl-1L within the cell lines, with MCF7 cells having high levels of Mcl-1L which would have to be overcome to trigger apoptosis. It should be noted though that these reductions in viability were observed despite there being no additional apoptotic stimulus applied to the cells.
One unexpected result from the combined siRNA transfections was that knockdown of both SRSF1 and hnRNP K produced the greatest induction of apoptosis within the MDA-MD-231 cell line. This was not observed in the MCF7 cells where there was no significant reduction. A possible explanation for this anomaly may be the relative importance of the other Bcl-2 family members within individual cell lines. Some redundancy has been shown to occur with the Bcl-2 family both among pro-apoptotic as well as anti-apoptotic proteins. This redundancy is thought to account for some of the resistance observed to BH3 mimetics which target specific Bcl-2 anti-apoptotic proteins[2]. In particular there is growing evidence that Mcl-1 and Bcl-xL can act redundantly in cancer cells, with some cells requiring reductions in both of them to fully induce apoptosis [50,51]. In addition to affecting the splicing of Mcl-1, the splicing factors used for the double knockdowns have all previously been shown to affect the splicing of Bcl-x [14,16–18]. Both hnRNP K and SRSF1 have been demonstrated to repress the production of the pro-apoptotic Bcl-xS. Therefore, reducing the levels of these proteins will result in a switch towards the anti-apoptotic variants of both Mcl-1 and Bcl-x. The hnRNP F/H family on the other hand enhances splicing at the Bcl-xS splice site, increasing production of the pro-apoptotic variant. So when levels of hnRNP F and H1 are reduced, although there is a switch in splicing towards the pro-apoptotic Mcl-1 variant there is also a corresponding switch in Bcl-x splicing towards the anti-apoptotic Bcl-xL. Therefore, the MDA-MD-231 cell line may be more dependent on Bcl-xL for cell survival than the MCF7 cell line, and it is the combined switch in splicing of both Mcl-1 and Bcl-x that produces such a big induction of apoptosis.
Another possibility is that other apoptotic factors are also affected by reductions in these RNA binding proteins and are contributing to the induction of apoptosis. Previous studies have shown that SRSF1 can also affect the splicing of other apoptotic factors including the pro-apoptotic Bcl-2 protein Bim and caspase 2 [17,52]. In all of these cases SRSF1 promotes the production of more anti-apoptotic variants at the expense of pro-apoptotic variants. This implicates SRSF1 as a more general regulator of apoptosis induction. As discussed previously, hnRNP K has also been implicated in the control of a variety of different apoptotic factors. Therefore these two splicing factors may prove to be key therapeutic targets for the induction of apoptosis.
In summary this study is the first to identify hnRNP F/H and K as regulators of Mcl-1 alternative splicing. Furthermore, targeting multiple splicing factors proved to be more effective at switching Mcl-1 splicing and triggering apoptosis. In the future targeting the splicing factors and binding sites identified in this study and by others may prove an effective way of triggering apoptosis, and overcoming therapy resistance in cancer cells.
Materials and methods
Cell culture
MCF-7 and MDA-MD-231 breast cancer cells were maintained in DMEM medium (Sigma-Aldrich) supplemented with 10% Foetal Calf Serum (Sigma-Aldrich), 8 mM L-glutamine and 1 unit/ml Penicillin/Streptomycin (Sigma-Aldrich) and incubated at 37°C with 5% CO2.
Transfections
MCF-7 and MDA-MD-231 cells were transfected with Silencer® Select siRNA (Ambion; Table 1), Silencer® Negative Control siRNA (Ambion), Silencer® Select GAPDH Positive Control siRNA (Ambion) using INTERFERin® Transfection Agent (Polyplus transfection™) according to the manufacturer’s instructions. At least two siRNA oligonucleotides were used for each splicing factor. Following optimisation cells were seeded at 4 × 104 cells/well in 24 well plates, or 2 × 105 cells/well in 6 well plates. After 24 hours cells were transfected with 5nM siRNA with 4 µl/ml of INTERFERin® transfection agent. Cells and siRNA were incubated together at 37°C in an atmosphere of 5% CO2. RNA was collected 48 and 72 hours post-transfection.
Table 1.
siRNA sequences.
| Target gene | Sense (5ʹ->3ʹ) | Antisense (5ʹ->3ʹ) |
|---|---|---|
| hnRNP H1 (1) | GAAGCAUACUGGUCCAAAUtt | AUUUGGACCAGUAUGCUUCaa |
| hnRNP H1 (2) | GAGUAACUGGUGAAGCAGAtt | UCUGCUUCACCAGUUACUCtg |
| hnRNP F (1) | GCAGCACAGAUAUAUAGAAtt | UUCUAUAUAUCUGUGCUGCat |
| hnRNP F (2) | CGACCGAGAACGACAUUUAtt | UAAAUGUCGUUCUCGGUCGct |
| hnRNP F/H | GGAAGAAAUUGUUCAGUUCtt | GAACUGAACAAUUUCUUCCtt |
| hnRNP K (1) | CGAUGAAACCUAUGAUUAUtt | AUAAUCAUAGGUUUCAUCGta |
| hnRNP K (2) | GAGCUUCGAUCAAAAUUGAtt | UCAAUUUUGAUCGAAGCUCcc |
| hnRNP I (1) | CAAAGCCUCUUUAUUCUUUtt | AAAGAAUAAAGAGGCUUUGgg |
| hnRNP I (2) | GCAUCACGCUCUCGAAGCAtt | UGCUUCGAGAGCGUGAUGCgg |
| SRSF1 | GGAUAACACUAAGUUUAGAtt | UCUAAACUUAGUGUUAUCCag |
FuGENE® HD transfection reagent (Promega) was used according to manufacturer’s instructions to transfect the Mcl-1 minigene plasmids into MCF-7 cells. Briefly, 1.5 × 105 cells/well were seeded into a 12 well plate. After 24 hours each well was transfected with 0.5 µg plasmid DNA and 1.5 µl of FuGENE® HD transfection reagent. Cells and siRNA were incubated together at 37°C in an atmosphere of 5% CO2. RNA was collected 24 hours after transfection.
RNA isolation and PCR
Total RNA was extracted with RNeasy spin columns (Qiagen) and treated with DNase I (Qiagen) following manufacturer’s instructions. One µg of total RNA was reverse transcribed using oligo (dT) primers and Superscript III Reverse Transcriptase (RT) (Invitrogen, Life Technologies) following manufacturer’s instructions. PCRs were performed with the cDNA, specific primers (Table 2) and PCR master mix (Promega). The cDNA products were separated by electrophoresis on 1.5% (w/v) agarose gels and visualised under UV following ethidium bromide staining. Bands were quantified by capillary gel electrophoresis (Qiaxcel, Qiagen).
Table 2.
PCR primers.
| Target gene | Forward primer (5ʹ->3ʹ) | Reverse primer (5ʹ->3ʹ) | Cycle number |
|---|---|---|---|
| hnRNP H1 | TCCTGACACGGCCAATGATG | TTGGCACGATTTCCAACCCT | 26 |
| hnRNP F | GAGGGAGGTGAAGGCTTTGT | CGGCCCCATCATGAATCGT | 25 |
| hnRNP K | GTGCACAGCCTTATGATCCC | ACCAGGACGCATTCTGTCAA | 25 |
| hnRNP I | GGTACAAAGCGGGGATCTGA | GGGGTCACCGAGGTGTAGTA | 25 |
| SRSF1 | GCCCCGCAGGGAACAACGAT | CGTCTCGCGGGTCCTCGAAC | 26 |
| GAPDH | CTGCCGTCTAGAAAAACC | CCAAATTCGTTGTCATACC | 20 |
| Mcl-1 | GGACACAAAGCCAATGGGCAGGT | GCAAAAGCCAGCAGCACATTCCTGA | |
| Mcl-1 minigene | TAGGGAGACCCAAGCTGGCTA | AGCAGCACATTCCTGATGCC | 20/30 |
| RBM4 | GCCATTCCGGTTGTAGGCT | GCGGACGCTTCTGACAAAAC | 30 |
| SRSF5 | TTGCGGATGCACACCGACCT | TCCGGCTCCTGCTCCTGCTT | 24 |
Real-time PCR was performed on cDNA using inventoried TaqMan® assays (Applied Biosystems) and TaqMan® Universal Master Mix II (Applied Biosystems). Taqman assays were selected that were specific for each of the splice variants of Mcl-1 as well as for the endogenous control GAPDH. The assay was performed in quadruplet and the PCR amplification was performed using the OneStepPlus real-time PCR system (Applied Biosystems). All experiments were performed in triplicate with a minimum of three independent experiments.
Protein extraction and Western Blotting
MCF7 cells were washed in ice cold PBS and then collected in RIPA buffer (25 mM Tris-HCl, pH 7.6, 150 mM NaCl, 1% NP-40, 0.1% sodium deoxycholate and 0.1% SDS) supplemented with Protease Inhibitors (Sigma-Aldrich). The cell lysate was passed through a 25 gauge needle, aliquoted and stored at −20°C. Following protein quantification 30µg of protein was heat denatured at 95°C for 5 minutes with SDS loading buffer (0.125M Tris-HCl pH6.8, 2% SDS, 10% glycerol, 10% (v/v) β-mercaptoethanol, 0.1% (v/w) bromophenol blue).
Protein lysates were separated using 12% SDS-polyacrylamide gel electrophoresis (PAGE) gels and transferred electrophoretically onto PVDF membranes. After blocking with 10% dried milk powder in 1x PBS membranes were probed with either rabbit anti-GAPDH (1:1000, Santa Cruz), rabbit anti-hnRNP H1 (1:1000, GeneTex) or rabbit anti-hnRNP K (1:1000, GeneTex) diluted in PBS plus 1% milk and incubated overnight at 4°C. After washing in PBS the appropriate HRP-conjugated secondary antibodies (Dako) diluted in PBS were added. ECL (Pierce) was used for protein detection. RNA Immunoprecipitation
MCF-7 cells were washed in ice cold PBS and then collected in lysis buffer (10 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.5% NP-40, 1% Triton X-100) containing protease inhibitors (Sigma-Aldrich) and RNase OUT (Invitrogen). Equal amounts of cell lysate were then incubated overnight at 4°C with 2 µg of either rabbit anti-hnRNP H1 antibody (GeneTex), rabbit anti-hnRNP I antibody (GeneTex) rabbit anti-hnRNP K antibody (GeneTex) and rabbit IgG control (Santa Cruz). Cell lysates and antibodies were then incubated with Dynabeads Protein G (Invitrogen) for a further hour at 4°C. Beads were then washed five times with lysis buffer before the immunoprecipitated RNA was collected in Trizol reagent (Invitrogen) according to manufacturer’s instructions. The RT-PCR was performed as before but half of the RNA obtained from the immunoprecipitation was used in each reaction. Fold enrichment of target mRNA was determined after normalisation to the input and rabbit IgG controls.
Mcl-1 Minigene Plasmid Construction
The Mcl-1 minigene was generated from human genomic DNA using specific Mcl-1 PCR primers containing specific restriction sites (Fig. 4A): forward primer 5′-TCTAGAGGCGCCAAGGACACAAA-3′ (XbaI), reverse primer 5′-AAGCTTGGTGGTTGGTTAAAAGTCAACTATTG-3′ (HindIII). These primers amplified the last 115 bp of exon 1, intron 1, exon 2, intron 2 and the first 157 bp of exon 3, using Platinum® Taq DNA Polymerase High Fidelity (Invitrogen). The PCR product was cloned into the TOPO®-TA cloning vector (Invitrogen) and sequenced. The fragment was then subcloned into pcDNA3.1 by restriction digestion and ligation using T4 DNA ligase (Invitrogen). Potential splicing factor binding sites within the Mcl-1 minigene construct were mutated using the Q5 site-directed mutagenesis kit (New England BioLabs) with forward primer 5′-AAGTATCACAAAAGTTCTCGTAAGG-3′, and reverse primer 5′-TCTGCTAATGGTTCGATG-3′ for SRSF1 binding site, and forward primer 5′-GGATGAAAGGCGCCTTGGAGTG-3′, and reverse primer 5′-TTAAGGCAAACTTACCCAG-3′ for hnRNP F/H binding site.
Cell Counts
Cells were collected 72 hours after siRNA transfection and resuspended in serum free media. Equal volumes of cell suspension and trypan blue and cell counts performed with the LUNA-II™ Automated Cell Counter.
Caspase 9 Assay
Caspase 9 activity was determined by a colorimetric assay according to the manufacturer’s protocol (R&D Systems). In brief, cells were collected and counted 72 hours after siRNAs transfection and then lysed with the supplied Lysis Buffer (25 μL per 1 × 106 cells). 50 μL of cell lysate was incubated with the supplied reaction buffer and caspase-9 substrate (LEHD-pNA) at 37 °C for 2 hours. The reaction was measured on a microplate reader using a wavelength of 405 nm. The results are expressed as the fold increase in caspase activity over that of NC siRNA treated cells.
Funding Statement
This work was supported by a grant from the Royal Victoria Infirmary Breast Cancer Research Appeal [BH162604].
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplementary data can be accessed here.
References
- [1].Hanahan D, Weinberg R.. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. [DOI] [PubMed] [Google Scholar]
- [2].Delbridge AR, Strasser A. The BCL-2 protein family, BH3-mimetics and cancer therapy. Cell Death Differ. 2015;22:1071–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Correia C, Lee SH, Meng XW, et al. Emerging understanding of Bcl-2 biology: implications for neoplastic progression and treatment. Biochim Biophys Acta. 2015;1853:1658–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Czabotar PE, Lessene G, Strasser A, et al. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15:49–63. [DOI] [PubMed] [Google Scholar]
- [5].Delbridge AR, Grabow S, Strasser A, et al. Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nat Rev Cancer. 2016;16:99–109. [DOI] [PubMed] [Google Scholar]
- [6].Williams MM, Cook RS. Bcl-2 family proteins in breast development and cancer: could Mcl-1 targeting overcome therapeutic resistance? Oncotarget. 2015;6:3519–3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Beroukhim R, Mermel C, Porter D, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cho-Vega J, Rassidakis G, Admirand J, et al. MCL-1 expression in B-cell non-Hodgkin’s lymphomas. Hum Pathol. 2004;35:1095–1100. [DOI] [PubMed] [Google Scholar]
- [9].Aichberger K, Mayerhofer M, Krauth M, et al. Identification of mcl-1 as a BCR/ABL-dependent target in chronic myeloid leukemia (CML): evidence for cooperative antileukemic effects of imatinib and mcl-1 antisense oligonucleotides. Blood. 2005;105:3303–3311. [DOI] [PubMed] [Google Scholar]
- [10].Sieghart W, Losert D, Strommer S, et al. Mcl-1 overexpression in hepatocellular carcinoma: a potential target for antisense therapy. J Hepatol. 2006;44:151–157. [DOI] [PubMed] [Google Scholar]
- [11].Ding Q, He X, Xia W, et al. Myeloid cell leukemia-1 inversely correlates with glycogen synthase kinase-3beta activity and associates with poor prognosis in human breast cancer. Cancer Res. 2007;67:4564–4571. [DOI] [PubMed] [Google Scholar]
- [12].Balko JM, Giltnane JM, Wang K, et al. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov. 2014;4:232–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Thrane S, Pedersen AM, Thomsen MB, et al. A kinase inhibitor screen identifies Mcl-1 and Aurora kinase A as novel treatment targets in antiestrogen-resistant breast cancer cells. Oncogene. 2015;34:4199–4210. [DOI] [PubMed] [Google Scholar]
- [14].Shketa LCA, Toutant J, Rendos H, et al. Regulation of alternative splicing and the case of Bcl-x. Pak J Biochem Mol Biol. 2015;42:27–38. [Google Scholar]
- [15].Cloutier P, Toutant J, Shkreta L, et al. Antagonistic effects of the SRp30c protein and cryptic 5ʹ splice sites on the alternative splicing of the apoptotic regulator Bcl-x. J Biol Chem. 2008;283:21315–21324. [DOI] [PubMed] [Google Scholar]
- [16].Garneau D, Revil T, Fisette JF, et al. Heterogeneous nuclear ribonucleoprotein F/H proteins modulate the alternative splicing of the apoptotic mediator Bcl-x. J Biol Chem. 2005;280:22641–22650. [DOI] [PubMed] [Google Scholar]
- [17].Moore M, Wang Q, Kennedy C, et al. An alternative splicing network links cell-cycle control to apoptosis. Cell. 2010;142:625–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Revil T, Pelletier J, Toutant J, et al. Heterogeneous nuclear ribonucleoprotein K represses the production of pro-apoptotic Bcl-xS splice isoform. J Biol Chem. 2009;284:21458–21467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bae J, Leo C, Hsu S, et al. MCL-1S, a splicing variant of the antiapoptotic BCL-2 family member MCL-1, encodes a proapoptotic protein possessing only the BH3 domain. J Biol Chem. 2000;275:25255–25261. [DOI] [PubMed] [Google Scholar]
- [20].Bingle C, Craig R, Swales B, et al. Exon skipping in Mcl-1 results in a bcl-2 homology domain 3 only gene product that promotes cell death. J Biol Chem. 2000;275:22136–22146. [DOI] [PubMed] [Google Scholar]
- [21].Stewart M, Fire E, Keating A, et al. The MCL-1 BH3 helix is an exclusive MCL-1 inhibitor and apoptosis sensitizer. Nat Chem Biol. 2010;6:595–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Venables J, Klinck R, Bramard A, et al. Identification of alternative splicing markers for breast cancer. Cancer Res. 2008;68:9525–9531. [DOI] [PubMed] [Google Scholar]
- [23].Klinck R, Bramard A, Inkel L, et al. Multiple alternative splicing markers for ovarian cancer. Cancer Res. 2008;68:657–663. [DOI] [PubMed] [Google Scholar]
- [24].Gautrey HL, Tyson-Capper AJ. Regulation of Mcl-1 by SRSF1 and SRSF5 in cancer cells. PLoS One. 2012;7:e51497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lin JC, Lin CY, Tarn WY, et al. Elevated SRPK1 lessens apoptosis in breast cancer cells through RBM4-regulated splicing events. Rna. 2014;20:1621–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Goodwin CMRO, Olejniczak ET, Fesik SW. Myeloid cell leukemia-1 is an important apoptotic survival factor in triple-negative breast cancer. Cell Death Differ. 2015;22(12):2098–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Caputi M, Zahler AM. Determination of the RNA binding specificity of the heterogeneous nuclear ribonucleoprotein (hnRNP) H/H’/F/2H9 family. J Biol Chem. 2001;276:43850–43859. [DOI] [PubMed] [Google Scholar]
- [28].Huelga SC, Vu AQ, Arnold JD, et al. Integrative genome-wide analysis reveals cooperative regulation of alternative splicing by hnRNP proteins. Cell Rep. 2012;1:167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wang E, Aslanzadeh V, Papa F, et al. Global profiling of alternative splicing events and gene expression regulated by hnRNPH/F. PLoS ONE. 2012;7:e51266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Gautrey H, Jackson C, Dittrich AL, et al. SRSF3 and hnRNP H1 regulate a splicing hotspot of HER2 in breast cancer cells. RNA Biol. 2015;12:1139–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Cartegni L, Wang J, Zhu Z, et al. ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003;31:3568–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Smith PJ, Zhang C, Wang J, et al. An increased specificity score matrix for the prediction of SF2/ASF-specific exonic splicing enhancers. Hum Mol Genet. 2006;15:2490–2508. [DOI] [PubMed] [Google Scholar]
- [33].Xiao X, Wang Z, Jang M, et al. Splice site strength-dependent activity and genetic buffering by poly-G runs. Nat Struct Mol Biol. 2009;16:1094–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Pino I, Pio R, Toledo G, et al. Altered patterns of expression of members of the heterogeneous nuclear ribonucleoprotein (hnRNP) family in lung cancer. Lung Cancer. 2003;41:131–143. [DOI] [PubMed] [Google Scholar]
- [35].Carpenter B, McKay M, Dundas SR, et al. Heterogeneous nuclear ribonucleoprotein K is over expressed, aberrantly localised and is associated with poor prognosis in colorectal cancer. Br J Cancer. 2006;95:921–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Barboro P, Repaci E, Rubagotti A, et al. Heterogeneous nuclear ribonucleoprotein K: altered pattern of expression associated with diagnosis and prognosis of prostate cancer. Br J Cancer. 2009;100:1608–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Matta A, Tripathi SC, DeSouza LV, et al. Heterogeneous ribonucleoprotein K is a marker of oral leukoplakia and correlates with poor prognosis of squamous cell carcinoma. Int J Cancer J Inter Du Cancer. 2009;125:1398–1406. [DOI] [PubMed] [Google Scholar]
- [38].Chen LC, Hsueh C, Tsang NM, et al. Heterogeneous ribonucleoprotein k and thymidine phosphorylase are independent prognostic and therapeutic markers for nasopharyngeal carcinoma. Clin Cancer Res off J Am Assoc Cancer Res. 2008;14:3807–3813. [Google Scholar]
- [39].Mandal M, Vadlamudi R, Nguyen D, et al. Growth factors regulate heterogeneous nuclear ribonucleoprotein K expression and function. J Biol Chem. 2001;276:9699–9704. [DOI] [PubMed] [Google Scholar]
- [40].Xiao Z, Ko HL, Goh EH, et al. hnRNP K suppresses apoptosis independent of p53 status by maintaining high levels of endogenous caspase inhibitors. Carcinogenesis. 2013;34:1458–1467. [DOI] [PubMed] [Google Scholar]
- [41].Gao FH, Wu YL, Zhao M, et al. Protein kinase C-delta mediates down-regulation of heterogeneous nuclear ribonucleoprotein K protein: involvement in apoptosis induction. Exp Cell Res. 2009;315:3250–3258. [DOI] [PubMed] [Google Scholar]
- [42].Chen LC, Chung IC, Hsueh C, et al. The antiapoptotic protein, FLIP, is regulated by heterogeneous nuclear ribonucleoprotein K and correlates with poor overall survival of nasopharyngeal carcinoma patients. Cell Death Differ. 2010;17:1463–1473. [DOI] [PubMed] [Google Scholar]
- [43].Venables J, Koh C, Froehlich U, et al. Multiple and specific mRNA processing targets for the major human hnRNP proteins. Mol Cell Biol. 2008;28:6033–6043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Honore B, Baandrup U, Vorum H. Heterogeneous nuclear ribonucleoproteins F and H/H’ show differential expression in normal and selected cancer tissues. Exp Cell Res. 2004;294:199–209. [DOI] [PubMed] [Google Scholar]
- [45].LeFave CV, Squatrito M, Vorlova S, et al. Splicing factor hnRNPH drives an oncogenic splicing switch in gliomas. Embo J. 2011;30:4084–4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Rauch J, O’Neill E, Mack B, et al. Heterogeneous nuclear ribonucleoprotein H blocks MST2-mediated apoptosis in cancer cells by regulating A-Raf transcription. Cancer Res. 2010;70:1679–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Hope NR, Murray GI. The expression profile of RNA-binding proteins in primary and metastatic colorectal cancer: relationship of heterogeneous nuclear ribonucleoproteins with prognosis. Hum Pathol. 2011;42:393–402. [DOI] [PubMed] [Google Scholar]
- [48].Shieh J, Liu K, Huang S, et al. Modification of alternative splicing of Mcl-1 pre-mRNA using antisense morpholino oligonucleotides induces apoptosis in basal cell carcinoma cells. J Invest Dermatol. 2009;129:2497–2506. [DOI] [PubMed] [Google Scholar]
- [49].Kim D, Kim J, Park M, et al. Modulation of biological processes in the nucleus by delivery of DNA oligonucleotides conjugated with gold nanoparticles. Biomaterials. 2011;32:2593–2604. [DOI] [PubMed] [Google Scholar]
- [50].Wei G, Margolin AA, Haery L, et al. Chemical genomics identifies small-molecule MCL1 repressors and BCL-xL as a predictor of MCL1 dependency. Cancer Cell. 2012;21:547–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Xiao Y, Nimmer P, Sheppard GS, et al. MCL-1 is a key determinant of breast cancer cell survival: validation of MCL-1 dependency utilizing a highly selective small molecule inhibitor. Mol Cancer Ther. 2015;14:1837–1847. [DOI] [PubMed] [Google Scholar]
- [52].Anczuków O, Rosenberg A, Akerman M, et al. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat Struct Mol Biol. 2012;19:220–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.