

Abstract
Acute kidney injury is a medical condition characterized by kidney damage with a rapid decline of renal function, which is associated with high mortality and morbidity. Recent research has further established an intimate relationship between acute kidney injury and chronic kidney disease. Perturbations of kidney cells in acute kidney injury result in the accumulation of unfolded and misfolded proteins in the endoplasmic reticulum, leading to unfolded protein response or endoplasmic reticulum stress. In this review, we analyze the role and regulation of endoplasmic reticulum stress in acute kidney injury triggered by renal ischemia-reperfusion and cisplatin nephrotoxicity. The balance between the two major components of unfolded protein response, the adaptive pathway and the apoptotic pathway, plays a critical role in determining the cell fate in endoplasmic reticulum stress. The adaptive pathway is evoked to attenuate translation, induce chaperones, maintain protein homeostasis, and promote cell survival. Prolonged endoplasmic reticulum stress activates the apoptotic pathway, resulting in the elimination of dysfunctional cells. Therefore, regulating ER stress in kidney cells may provide a therapeutic target in acute kidney injury.
Keywords: Endoplasmic reticulum stress, Acute kidney injury, Ischemia-reperfusion injury, Cisplatin, Autophagy, Apoptosis
Introduction
The endoplasmic reticulum (ER) is an intracellular organelle that plays an important role in protein homeostasis (also referred as proteostasis), including protein synthesis, folding, modification, and degradation (1). Under pathological conditions, cells may lose the proteostasis resulting in the accumulation of unfolded and misfolded proteins in the ER, which triggers the unfolded protein response (UPR) or ER stress (2). There are three main ER stress sensors, including inositol-requiring protein 1(IRE1), protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK), and activating transcription factor 6 (ATF6) (3, 4). Under normal conditions, these sensors are inactive by binding to the ER-resident chaperone BiP/GRP78 (immunoglobulin heavy chain binding protein/78 kDa glucose-regulated protein) in the ER lumen (5, 6). However, when the cells are under ER stress, an exquisite network of mechanisms is activated to determine cell fate (2). If the UPR pathway (also called adaptive pathway under this situation) successfully solves this crisis by attenuating translation to reduce protein stress, inducing chaperonins to increase the ER folding capacity (3), and degrading these abnormal proteins via ER-associated degradation (ERAD) or autophagy (7), the cells survive. But if the pathogenic stimuli are too severe or prolonged, the deranged proteostasis cannot be rectified, then apoptotic UPR pathway ensues (8).
When the UPR fails to protect the disturbed cell, ER stress becomes one of the main pathogenic factors in various human diseases, such as neurodegenerative diseases, liver disease, diabetes, and cardiovascular diseases (1, 3, 9). In kidneys, ER stress has been implicated in the development and progression of a variety of renal diseases (10). Acute kidney injury (AKI) is a major renal disease associated with high morbidity and mortality (11, 12). In clinical settings, AKI is mainly caused by ischemia-reperfusion, nephrotoxicity, and sepsis (13). AKI is characterized by the rapid decline of kidney function which can also result in a long-term loss of renal function (14). Recent work has further established the inter-relationship between AKI and CKD (15–17). ER stress has been implicated in the pathogenesis of AKI induced by renal ischemia-reperfusion injury (IRI) and nephrotoxicity (18). In this review, we briefly describe ER stress and then focus on ER stress in the acute injury phase of AKI. We will also analyze the emerging evidence for the involvement of ER stress in kidney repair and renal fibrosis following AKI.
Three main branches in UPR and ER stress
UPR is a homeostatic network that responds to the changes in protein folding within a cell (19). There are three main branches in UPR to transduce the status information of ER protein folding to cytosol and nucleus, forming an exquisite adaptive mechanism (20) (Figure 1).
Figure 1:
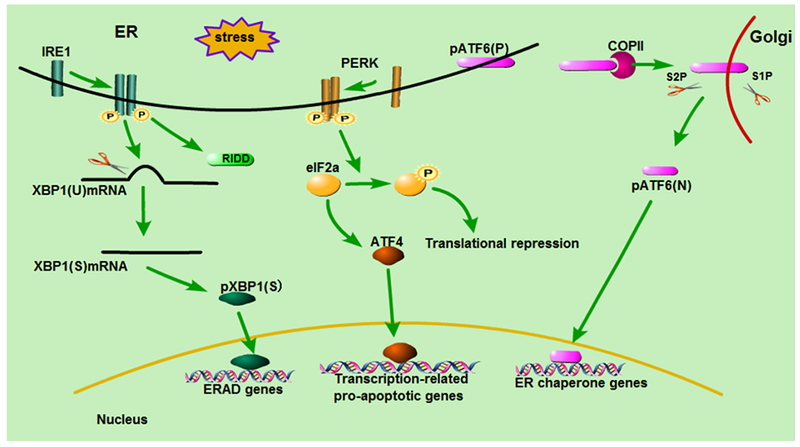
Main signaling pathways in UPR/ER stress. The UPR has three main signaling branches. IRE1 pathway catalyzes the splicing of X-box-binding protein 1 (XBP1) mRNA and cleaves special mRNAs by regulated IRE1-dependent decay (RIDD). PERK phosphorylates and inactivates eukaryotic translation initiator factor 2α (eIF2α) resulting in the repression of protein translation and the transcription of apoptotic genes. ATF6 is transported to the Golgi apparatus and cleaved by the Site-1 and -2 Proteases (SiP1, S2P). Then it migrates to the nucleus and activates the transcription of chaperone genes.
IRE1 pathway:
IRE1 pathway is known as the most highly conserved mechanism of the UPR (21). In the presence of ER stress, IRE1 dimerizes and autotransphosphorylates to trigger its intrinsic kinase and endoribonuclease activities (6), catalyzing the splicing of X-box-binding protein 1 (XBP1) mRNA or cleaving special mRNAs for degradation by regulated IRE1-dependent decay (RIDD) (22). XBP1, as a potent transcription activator, upregulates the transcription of a subset of UPR genes that promote ERAD to eliminate misfolded proteins and augment the protein folding capacity by increasing the translation of ER chaperones (23) (Figure 1). However, under severe stress, phosphorylated IRE1 may also recruit TNF receptor-associated adapter protein 2 (TRAF2), and signals to the apoptosis signaling-regulating kinase 1 (ASK1), causing the activation of Jun N-terminal kinase (JNK) signal pathway to induce apoptosis (24, 25). In addition, IRE1–TRAF2 complex may directly activate caspase-12 mediated apoptotic pathway (26).
PERK pathway:
PERK possesses protein serine/threonine kinase activity and is activated after disassociation from the intraluminal chaperone BiP/GRP78 (1, 27). Upon activation, PERK phosphorylates and inactivates eukaryotic initiator factor 2α (eIF2α), resulting in the reduction of global protein synthesis to alleviate cellular protein load and re-built homeostasis (Figure 1). In addition, phospho-eIF2α activates ATF4, which promotes the transcription of multiple ER stress-associated genes to enhance ER stress response, including cAMP response elements (CRE) and C/EBP–homologous protein (CHOP) (28–30) (Figure 1).
ATF6 pathway:
ATF6 has two Golgi localization sequences, but it resides at the ER membrane under basal conditions. Upon ER stress, ATF6 translocates to the Golgi apparatus with the assistance of the coat protein II (COPII) complex, where it is sequentially cleaved by site-1 and -2 proteases (S1P, S2P)) (31) (Figure 1). Consequently, a 50kDa cytosolic domain fragment of ATF6 (ATF6f) is released and transferred to the nucleus where it binds to ATF/CRE and ER stress response element (ERSE) and activates the transcription of ER chaperone genes, including the genes encoding ERAD components and XBP1 (22, 32) (Figure 1).
ER stress in ischemic AKI
Renal ischemia-reperfusion injury (IRI) is a major cause of AKI associated with multiple clinical situations, such as renal vascular obstruction, resuscitation from cardiac arrest, and kidney transplantation (33). Several studies have demonstrated that renal IRI induces ER stress in renal tubule epithelial cells. For example, Montie et al. (34) detected an evident increase of eIF2α phosphorylation (phospho-eIF2α), accompanied by the phosphorylation of PERK in tubular epithelial cells in mice after cardiac arrest-induced ischemia and reperfusion. In immunohistochemistry, phospho-eIF2α staining was mostly in the corticomedullary junction, the kidney region most susceptible to renal IRI. Additionally, BiP/GRP78, a key element in UPR, was activated in mice following 35 min of bilateral renal ischemia and reperfusion, and similar to phospho-eIF2α, BiP/GRP78 was mostly located in the corticomedullary junction area (35).
The mechanism underlying ER stress in AKI remains poorly understood. Some studies focused on ER molecular chaperones, such as BiP/GRP78 and GRP94, which bind to the misfolded or unfolded proteins in the ER lumen under the condition of cell stress. In vitro, pretreatment with ER molecular chaperones reduced ATP depletion-induced injury in Madin-Darby canine kidney (MDCK) cells (36). Moreover, chemical UPR inducers were shown to selectively induce BiP/GRP78 and ameliorate renal IRI in vivo (37–39). Of note, recent studies have demonstrated a role of autophagy in ER stress-related protective effects in renal IRI. In 2015, Chandrika et al. showed that tunicamycin (ER stress inducer) induced UPR and autophagy in renal tubular cells in vitro and in kidneys in mice (40). In this study, inhibition of autophagy increased tubular cell death in vitro and worsened ischemic AKI in mice, indicating a protective role of autophagy under these conditions (40). In support, Ling and colleagues demonstrated that the exogenous H2S alleviated renal IRI by up-regulating ER stress-induced autophagy in rats (41). While these studies suggest a protective role of UPR/ER stress in renal IRI, there is also evidence for the opposite. In this regard, a recent study (42) reported that intermedin (IMD) (a new calcitonin/calcitonin gene-related peptide family) suppressed BiP/GRP78, CHOP, and caspase-12 in renal IRI. Moreover, overexpression of IMD significantly reduced apoptosis during hypoxia/reoxygenation in renal epithelial cells and markedly improved renal function after IRI in rats, suggesting a pathogenic role of ER stress in renal IRI (42). The exact cause of the discrepancy between this study and the previous studies remains unclear, but it is in line with the “double-edged sword” concept about ER stress. In other words, mild to moderate ER stress simulates a cytoprotective UPR, whereas apoptotic pathways may be activated when the stress is too severe or excessive. Noh et al. (35) focused on the function of CHOP, which is an important participant in ER stress-associated apoptosis in renal IRI. Compared with wild-type mice, CHOP-knockout mice suffered significantly less degrees of renal IRI, supporting a pathogenic role of CHOP-mediated apoptosis under this condition (35). Thus, it is critically important to control and monitor the level of ER stress when considering UPR/ER stress-targeted preventative and/or therapeutic strategies for AKI in renal IRI.
Accumulating evidence has shown that patients who recovered from AKI may suffer from chronic kidney disease (CKD) years later (16, 17, 43, 44). A critial mechanism mediating the progression of AKI to CKD is maladaptive repair in renal tubules leading to interstitial fibrosis. ER stress has been implicated in renal interstitial fibrosis. For example, in the rat model of unilateral ureteral obstruction (UUO), ER stress-associated apoptosis mediated by CHOP was shown to play a role in the development of renal fibrosis (45). Interestingly, ER stress at the early stage of AKI may be beneficial in terms of the development of renal fibrosis. In a retrospective study (46), Fan et al. recently showed the induction of multiple ER stress markers in the renal biopsies of AKI patients, including Reticulon-1A (RTN1A), a key mediator of ER stress. Interestingly, the induction of RTN1A correlated positively with the severity of AKI. In mice, RN1A knockdown in renal tubules attenuated ER stress and apoptosis in nephrotoxic AKI and also suppressed subsequent development of renal fibrosis. Inversely, RTN1A overexpression in renal tubules exacerbated early renal injury and renal fibrosis in mice (46). In another study, Gai et al. suggested that pharmacological activation of Farnesoid X receptor could ameliorate ischemic AKI and prevent subsequent renal fibrosis and the progression to chronic kidney disease by suppressing ER stress and oxidative stress (47). It is noteworthy that, in these studies, inhibition of ER stress reduced both acute injury and late development of renal pathologies. Therefore, the role of ER stress specifically in AKI to CKD transition remains to be clarified.
ER stress in cisplatin-induced AKI
Cisplatin is a widely used chemotherapeutic drug in clinics for various malignancies. However, over one-quarter of patients who receive a single dose of cisplatin develop renal problems especially AKI, which is the major usage-limiting side effect of cisplatin (48). The underlying molecular mechanism of cisplatin-induced AKI involves multiple factors, among which is ER stress and associated apoptosis (48, 49). ER stress markers (such as XBP, BiP/GRP78) were shown to increase in kidney tissues after cisplatin injection in rats (50). Moreover, elevated ER stress and reduced Akt activity were observed in kidney tissues of hyperhomocysteinemia (HHcy) mice after cisplatin injection. HHcy mice developed more severe renal injury after cisplatin injection and IRI as indicated by more severe renal tubular damage and higher serum creatinine (51). On the other hand, preconditioning of ER stress afforded protective effects against cisplatin-induced nephrotoxicity in vitro (52).
Apoptosis in renal tubules is one of the main features of cisplatin-induced AKI. Cisplatin-induced renal cell apoptosis was associated with the activation of calpain and caspase-12 (53, 54), which predominantly reside in ER and mediate ER stress-associated cell death. Several studies have suggested the involvement of ER stress in cisplatin-induced apoptosis in kidney cells and tissues. For example, in 2005, Liu and Baliga demonstrated a role of caspase-12 in cisplatin-induced apoptosis in LLC-PK1 renal tubular epithelial cells (54). More recently, Chen et al. showed that blockade of KCa3.1 potassium channels protected against cisplatin-induced AKI by attenuating the mitochondrial as well as ER stress pathways of apoptosis (55). Therefore, depending on the cellular context and experimental condition, ER stress may play an important role in cisplatin-induced nephrotoxicity or AKI.
Recent clinical evidence of ER stress in AKI.
While the relationship between ER stress and AKI has been suggested by both in vitro and in vivo experiments, the clinical evidence from human patients remains sketchy. Nonetheless, three recent studies have shed lights in this area. As discussed above, Fan and colleagues recently showed the induction of ER stress markers in renal biopsies of AKI patients, which correlated with the severity of AKI (46). Travernier et al. also reported that the level of urinary angiogenin correlated with worse renal function in human kidney transplants and may be a risk predictor of renal allograft failure (56). Of note, urinary angiogenin was mainly produced by renal tubular cells upon the activation of transcription factor XBP1 in ER stress, suggesting the occurrence of ER stress in this clinical setting (56). In addition, cysteine-rich with EGF-like domains 2 (CRELD2) was recently shown as a sensitive urinary biomarker for detecting ER stress in various kidney diseases including ischemic AKI. Notably, in pediatric patients undergoing cardiac surgery, a strong association between urinary levels of CRELD2 within postoperative 6 hours with severe AKI after surgery. In addition, urinary CRELD2 levels were shown to significantly increase in patients with autosomal dominant tubulointerstitial kidney disease caused by UMOD (uromodulin gene) mutations, a prototypical tubular ER stress disease (57). Thus, there is emerging clinical evidence for ER stress in AKI and related kidney diseases in human patients. Moreover, the release of ER stress-related factors into urine may serve as promising biomarkers for AKI.
ER stress-associated autophagy in kidneys
Macroautophagy (referred as autophagy) is a cellular homeostatic mechanism that plays a critical role in a wide range of cell physiological and pathophysiological conditions (58–60). During autophagy, autophagosomes sequester parts of the cytoplasm including protein aggregates, misfolded proteins and defective organelles, and then fuse with lysosomes for degradation (61). In this process, the basic materials are released for reuse in the biosynthetic pathway to maintain cellular homeostasis (62). Autophagosome formation is initiated by the UNC-51-like kinase (ULK) complex, which is regulated negatively by mechanistic target of rapamycin complex 1 (mTORC1) and positively by 5ʹ AMP-activated protein kinase (AMPK) (63).
Emerging evidence has revealed a crosstalk between autophagy and ER stress (64). ER stress/UPR is known to activate autophagy, which, in addition to ERAD, acts as a mechanism for clearing or degrading misfolded proteins(8, 65–68). Moreover, autophagy can selectively engulf and degrade dysfunctional ER to maintain the structural and functional homeostasis of the cell. An early study of yeasts indicated that ER stress triggered autophagy in an autophagy-related gene (Atg) dependent manner (64). ER stress may promote autophagy by inducing the deactivation of mTOR (69). It has also been reported that the activation of the PERK/eIF2α pathway in ER stress contributes to polyglutamine-induced LC3 conversion, an important step in autophagy (70). In a model of Crohn’s disease or chronic intestinal inflammation, Adolph and colleagues (71) demonstrated that blockade of UPR or autophagy in intestinal epithelial cells led to each other’s compensatory engagement, whereas blockade of both resulted in a severe inflammatory disease. Moreover, induction of autophagy ameliorated ER stress-induced intestinal inflammation as well as cell death. At the molecular level, ATG16L1 (autophagy-related 16–like 1) was suggested to inhibit IRE1α in ER stress/UPR (71). Thus, there is a fascinating interaction between ER stress/UPR and autophagy under disease conditions. ER stress induces autophagy to remove protein aggregates and probably damaged parts of ER to re-establish homeostasis (8).
Autophagy is activated in AKI for kidney protection (72–75). The connection between autophagy and ER has also been suggested in kidney cells and tissues (67, 76). For example, in 2009, Kawakami et al. reported that treatment with classic ER stress inducers (tunicamycin, brefeldin A) led to a significant increase in LC3 II (biochemical hallmark of autophagy) in renal proximal tubular cells, suggesting autophagy activation by ER stress (77). It is generally accepted that, during ER stress, autophagy is an adaptive mechanism for cell survival. As a result, it was shown that inhibition of autophagy deteriorated cell function and accelerated cell death (78). Interestingly, Dong et al. showed that mTOR was rapidly activated during ER stress in renal tubular cells, and inhibition of mTOR by rapamycin increased cell viability under this condition (79). Although not examined, inhibition of mTOR in this study is expected to activate autophagy, which may be responsible for the protective effect of rapamycin during ER stress. Based on experimental testing, Kapuy et al. further proposed a mathematical model to describe the relationship between mTOR, autophagy, and apoptosis during UPR/ER stress (80). In this model, under low ER stress, autophagy is activated overcoming the suppression by mTOR and prevents apoptosis to promote cell survival. On the contrary, in response to severe ER stress, autophagy may be blocked due to mTOR activation, and apoptosis ensues (80).
ER stress-associated apoptosis in kidneys
As alluded above, when cells fail to maintain the protein homeostasis via the adaptive UPR pathway under excessive or severe ER stress, apoptosis is triggered (81). Several mechanisms are known to contribute to this process under ER stress (Figure 2). The first is CHOP-mediated pathway (30). CHOP, a bZIP transcription factor, induces the expression of pro-apoptotic genes (e.g. Bax and Bak) and suppresses the transcription of anti-apoptotic Bcl2, causing apoptosis (82). Notably, CHOP can be induced via all three major ER stress pathways, including PERK, ATF6, and IRE1 (82). The second mechanism of ER stress-associated apoptosis is specifically initiated by IRE1 (83). In this pathway, IRE1 recruits tumor necrosis factor receptor-associated factor 2 (TRAF2) leading to the activation of the TRAF2-ASK-JNK pathway of apoptosis (84). The third mechanism involves Ca2+-mediated signaling, where ER stress leads to the release of Ca2+ from ER to mitochondria resulting in the activation caspase-4/caspase-12) for apoptosis (85–87). ER stress-mediated apoptosis eliminates irreversibly deranged cells, which is an important factor in the pathogenesis of kidney diseases (88). In both mouse and rat models of renal IRI, pharmacological inhibitors of ER stress (i.e. Tauroursodeoxycholic acid-TUDCA) suppressed tubular cell death and kidney tissue damage providing protective effects against AKI. Thus, targeting ER stress may offer therapeutic opportunities for the prevent and treatment of AKI.
Figure 2: Adaptive and pro-apoptotic pathways activated in ER stress.

When the stress is mild, the adaptive pathway is activated to repress translation to reduce protein load, activate chaperone expression to increase the ER folding capacity, and degrade abnormal proteins via ER-associated degradation (ERAD) quality control or autophagy. When the stress is too long or too severe, the cell capacity of proteostasis is overwhelmed and apoptotic pathways are activated. The first apoptotic pathway is mediated by CHOP, which induces the expression of pro-apoptotic genes (e.g. Bax and Bak) and suppresses the transcription of anti-apoptotic Bcl2, causing apoptosis. The second pathway involves the activation of TRAF2 via IRE1, which results in sequential activation of ASK1 and JNK to trigger apoptosis. The third mechanism involves Ca2+ released from ER that accumulates in mitochondria resulting in cytochrome c release and caspase activation.
Targeting ER stress for therapy of AKI.
As describe above, ER stress is involved in AKI and probably also in its progression to CKD. Under these conditions, the balance between the adaptive and apoptotic pathways of UPR determines the fate of tubular cells and the prognosis of the disease (89). In view of this, therapeutic strategies have to consider whether ER stress/UPR in a specific disease condition is mainly adaptive for cell survival or injurious for cell death.
On the one hand, strategies enhancing the adaptive pathway may increase renal cell survival, ameliorate kidney injury, and promote kidney repair. In this aspect, preconditioning targeting ER stress has been proved cytoprotective in AKI. Eyrou and Cribb showed that preconditioning with ER stress inducers (tunicamycin, thapsigargin, oxidized dithiothreitol) had rotective effects on the cytotoxicity of several clinically relevant drugs in multiple kidney cell lines (52). Prachasilchai et al. further demonstrated that pretreatment with thapsigargin resulted in the amelioration of ischemic AKI as indicated by both serum creatinine measurement and histopathological analysis (90). In addition, overexpression of ER chaperone proteins, such as ORP150, showed significant protective effects against ischemic AKI (91). In rats, pre-treatment with trans-4,5-dihydroxy-1,2-dithiane induced ER stress and protected kidneys from nephrotoxic injury (92, 93). These studies indicate that ER stress preconditioning or prior induction may afford protective effects against subsequent AKI, supporting the prophylactic use of ER stress inducers (at low doses) in expected conditions of AKI.
On the other hand, when the ER stress is too severe, the pro-apoptotic pathway of UPR becomes dominant. Under such conditions, it is important to block ER stress for preventing cell death and alleviating organ damage and failure. In 2012, Gao et al. demonstrated the inhibitory effect of tauroursodeoxycholic acid (TUDCA, a bile acid derivative with chaperone properties) on ER stress during ischemic AKI and, importantly, TUDCA protected against renal tubule damage (38). In 2014, Carlisle et al. reported that 4-PBA (pharmacologic inhibitor of ER stress) partially protected the kidney from Tunicamycin-induced AKI by repressing ER stress-induced CHOP expression(94).
Conclusions
There is emerging evidence for the involvement of UPR/ER stress in AKI induced by ischemia-reperfusion and nephrotoxicity. In these disease conditions, the signaling cascade of ER stress that aims at re-establishing microenvironment homeostasis is quite complex and deserves further delineation. Functionally, it is generally understood that under mild-moderate ER stress, the cell may activate UPR to restore the proteostasis to promote cell survival and strengthen the cellular ability to endure further stress. However, once the ER stress is excessive, the adaptive capacity of UPR is overwhelmed, resulting in the activation of the suicidal program of apoptosis to eliminate the irreversibly damaged cells (Figure 2). While this thinking is logical, it is apparently a simplistic view. First, there is no distinct demarcation point between the signaling networks of the adaptive and apoptotic pathways. Moreover, it is important to understand the mechanism underlying the cross-talk between these two intertwined pathways. Another area of future investigation is the dynamic crosstalk between ER and other organelles (mitochondria, Golgi, nucleus), which forms an interlaced signaling network to influence the response of a cell to ER stress. Therapeutically, as presented in this review there is evidence for the kidney protective effect of some chemical compounds targeting the key ER stress modulators. Especially, ER stress preconditioning and chemical chaperones may improve the capacity of protein folding and promote kidney cell survival in AKI. However, these observations need to be verified and extended to other relevant models. Finally, very limited information is currently available from clinical studies. Therefore, there is a pressing need to investigate ER stress in samples from AKI patients and, hopefully, conduct clinical trials to test the effect of ER stress modulating chemicals.
Key Messages:
Perturbations of kidney cells in acute kidney injury result in the accumulation of unfolded and misfolded proteins in ER, leading to unfolded protein response (UPR) or ER stress.
The balance between the adaptive pathway and the apoptotic pathway of UPR plays a critical role in determining the cell fate in ER stress.
Modulation of ER stress in kidney cells may provide a therapeutic strategy for acute kidney injury.
Acknowledgments:
The study was supported in part by grants from National Natural Science Foundation of China (81720108008, 81430017), the National Institutes of Health (2R01DK058831, 1R01 DK087843) of USA, and Department of Veterans Administration (5I01BX000319) of USA.
Footnotes
Conflicts of Interest: The authors declare no conflict of interest.
References
- 1.Bravo R, Parra V, Gatica D, Rodriguez AE, Torrealba N, Paredes F, et al. Endoplasmic reticulum and the unfolded protein response: dynamics and metabolic integration. International review of cell and molecular biology. 2013;301:215–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Inagi R, Ishimoto Y, Nangaku M. Proteostasis in endoplasmic reticulum--new mechanisms in kidney disease. Nature reviews Nephrology. 2014;10(7):369–78. [DOI] [PubMed] [Google Scholar]
- 3.Dara L, Ji C, Kaplowitz N. The contribution of endoplasmic reticulum stress to liver diseases. Hepatology. 2011;53(5):1752–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334(6059):1081–6. [DOI] [PubMed] [Google Scholar]
- 5.Sommer T, Jarosch E. BiP binding keeps ATF6 at bay. Developmental cell. 2002;3(1):1–2. [DOI] [PubMed] [Google Scholar]
- 6.Prischi F, Nowak PR, Carrara M, Ali MM. Phosphoregulation of Ire1 RNase splicing activity. Nature communications. 2014;5:3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Credle JJ, Finer-Moore JS, Papa FR, Stroud RM, Walter P. On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(52):18773–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hetz C The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nature reviews Molecular cell biology. 2012;13(2):89–102. [DOI] [PubMed] [Google Scholar]
- 9.Schonthal AH. Endoplasmic reticulum stress: its role in disease and novel prospects for therapy. Scientifica. 2012;2012:857516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taniguchi M, Yoshida H. Endoplasmic reticulum stress in kidney function and disease. Curr Opin Nephrol Hypertens. 2015;24(4):345–50. [DOI] [PubMed] [Google Scholar]
- 11.Togel F, Westenfelder C. Recent advances in the understanding of acute kidney injury. F1000prime reports. 2014;6:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang C, Dong Z. Epigenetic regulation in acute kidney injury: new light in a dark area. Kidney international. 2015;88(4):665–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Basile DP, Anderson MD, Sutton TA. Pathophysiology of acute kidney injury. Comprehensive Physiology. 2012;2(2):1303–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yaklin KM. Acute kidney injury: an overview of pathophysiology and treatments. Nephrology nursing journal : journal of the American Nephrology Nurses’ Association. 2011;38(1):13–8; quiz 9. [PubMed] [Google Scholar]
- 15.Yang L, Humphreys BD, Bonventre JV. Pathophysiology of acute kidney injury to chronic kidney disease: maladaptive repair. Contributions to nephrology. 2011;174:149–55. [DOI] [PubMed] [Google Scholar]
- 16.He L, Wei Q, Liu J, Yi M, Liu Y, Liu H, et al. AKI on CKD: heightened injury, suppressed repair, and the underlying mechanisms. Kidney Int. 2017;92(5):1071–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Venkatachalam MA, Weinberg JM, Kriz W, Bidani AK. Failed Tubule Recovery, AKI-CKD Transition, and Kidney Disease Progression. J Am Soc Nephrol. 2015;26(8):1765–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inagi R Endoplasmic reticulum stress as a progression factor for kidney injury. Current opinion in pharmacology. 2010;10(2):156–65. [DOI] [PubMed] [Google Scholar]
- 19.Kozutsumi Y, Segal M, Normington K, Gething MJ, Sambrook J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature. 1988;332(6163):462–4. [DOI] [PubMed] [Google Scholar]
- 20.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annual review of biochemistry. 2005;74:739–89. [DOI] [PubMed] [Google Scholar]
- 21.Kimata Y, Kohno K. Endoplasmic reticulum stress-sensing mechanisms in yeast and mammalian cells. Current opinion in cell biology. 2011;23(2):135–42. [DOI] [PubMed] [Google Scholar]
- 22.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107(7):881–91. [DOI] [PubMed] [Google Scholar]
- 23.Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415(6867):92–6. [DOI] [PubMed] [Google Scholar]
- 24.Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, et al. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes & development. 2002;16(11):1345–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu X, Zhang J, Sun H, Jiang C, Dong Y, Shan Q, et al. Ubiquitination of inositol-requiring enzyme 1 (IRE1) by the E3 ligase CHIP mediates the IRE1/TRAF2/JNK pathway. The Journal of biological chemistry. 2014;289(44):30567–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tong Q, Wu L, Jiang T, Ou Z, Zhang Y, Zhu D. Inhibition of endoplasmic reticulum stress-activated IRE1alpha-TRAF2-caspase-12 apoptotic pathway is involved in the neuroprotective effects of telmisartan in the rotenone rat model of Parkinson’s disease. European journal of pharmacology. 2016;776:106–15. [DOI] [PubMed] [Google Scholar]
- 27.Ishiwata-Kimata Y, Promlek T, Kohno K, Kimata Y. BiP-bound and nonclustered mode of Ire1 evokes a weak but sustained unfolded protein response. Genes Cells. 2013;18(4):288–301. [DOI] [PubMed] [Google Scholar]
- 28.Ohoka N, Yoshii S, Hattori T, Onozaki K, Hayashi H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 2005;24(6):1243–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hiramatsu N, Messah C, Han J, LaVail MM, Kaufman RJ, Lin JH. Translational and posttranslational regulation of XIAP by eIF2alpha and ATF4 promotes ER stress-induced cell death during the unfolded protein response. Mol Biol Cell. 2014;25(9):1411–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18(24):3066–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen J, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Developmental cell. 2002;3(1):99–111. [DOI] [PubMed] [Google Scholar]
- 32.Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Molecular biology of the cell. 1999;10(11):3787–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bastin AJ, Ostermann M, Slack AJ, Diller GP, Finney SJ, Evans TW. Acute kidney injury after cardiac surgery according to Risk/Injury/Failure/Loss/End-stage, Acute Kidney Injury Network, and Kidney Disease: Improving Global Outcomes classifications. Journal of critical care. 2013;28(4):389–96. [DOI] [PubMed] [Google Scholar]
- 34.Montie HL, Kayali F, Haezebrouck AJ, Rossi NF, Degracia DJ. Renal ischemia and reperfusion activates the eIF 2 alpha kinase PERK. Biochimica et biophysica acta. 2005;1741(3):314–24. [DOI] [PubMed] [Google Scholar]
- 35.Noh MR, Kim JI, Han SJ, Lee TJ, Park KM. C/EBP homologous protein (CHOP) gene deficiency attenuates renal ischemia/reperfusion injury in mice. Biochim Biophys Acta. 2015;1852(9):1895–901. [DOI] [PubMed] [Google Scholar]
- 36.Bush KT, George SK, Zhang PL, Nigam SK. Pretreatment with inducers of ER molecular chaperones protects epithelial cells subjected to ATP depletion. The American journal of physiology. 1999;277(2 Pt 2):F211–8. [DOI] [PubMed] [Google Scholar]
- 37.Prachasilchai W, Sonoda H, Yokota-Ikeda N, Ito K, Kudo T, Imaizumi K, et al. The protective effect of a newly developed molecular chaperone-inducer against mouse ischemic acute kidney injury. Journal of pharmacological sciences. 2009;109(2):311–4. [DOI] [PubMed] [Google Scholar]
- 38.Gao X, Fu L, Xiao M, Xu C, Sun L, Zhang T, et al. The nephroprotective effect of tauroursodeoxycholic acid on ischaemia/reperfusion-induced acute kidney injury by inhibiting endoplasmic reticulum stress. Basic Clin Pharmacol Toxicol. 2012;111(1):14–23. [DOI] [PubMed] [Google Scholar]
- 39.Gupta S, Li S, Abedin MJ, Noppakun K, Wang L, Kaur T, et al. Prevention of acute kidney injury by tauroursodeoxycholic acid in rat and cell culture models. PLoS One. 2012;7(11):e48950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chandrika BB, Yang C, Ou Y, Feng X, Muhoza D, Holmes AF, et al. Endoplasmic Reticulum Stress-Induced Autophagy Provides Cytoprotection from Chemical Hypoxia and Oxidant Injury and Ameliorates Renal Ischemia-Reperfusion Injury. PloS one. 2015;10(10):e0140025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ling Q, Yu X, Wang T, Wang SG, Ye ZQ, Liu JH. Roles of the Exogenous H2S-Mediated SR-A Signaling Pathway in Renal Ischemia/Reperfusion Injury in Regulating Endoplasmic Reticulum Stress-Induced Autophagy in a Rat Model. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology. 2017;41(6):2461–74. [DOI] [PubMed] [Google Scholar]
- 42.Wang Y, Tian J, Qiao X, Su X, Mi Y, Zhang R, et al. Intermedin protects against renal ischemia-reperfusion injury by inhibiting endoplasmic reticulum stress. BMC nephrology. 2015;16(1):169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Basile DP, Bonventre JV, Mehta R, Nangaku M, Unwin R, Rosner MH, et al. Progression after AKI: Understanding Maladaptive Repair Processes to Predict and Identify Therapeutic Treatments. J Am Soc Nephrol. 2016;27(3):687–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Humphreys BD, Cantaluppi V, Portilla D, Singbartl K, Yang L, Rosner MH, et al. Targeting Endogenous Repair Pathways after AKI. J Am Soc Nephrol. 2016;27(4):990–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chiang CK, Hsu SP, Wu CT, Huang JW, Cheng HT, Chang YW, et al. Endoplasmic reticulum stress implicated in the development of renal fibrosis. Mol Med. 2011;17(11–12):1295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fan Y, Xiao W, Lee K, Salem F, Wen J, He L, et al. Inhibition of Reticulon-1A-Mediated Endoplasmic Reticulum Stress in Early AKI Attenuates Renal Fibrosis Development. Journal of the American Society of Nephrology : JASN. 2017;28(7):2007–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gai Z, Chu L, Xu Z, Song X, Sun D, Kullak-Ublick GA. Farnesoid X receptor activation protects the kidney from ischemia-reperfusion damage. Scientific reports. 2017;7(1):9815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pabla N, Dong Z. Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney Int. 2008;73(9):994–1007. [DOI] [PubMed] [Google Scholar]
- 49.Yan M, Tang C, Ma Z, Huang S, Dong Z. DNA damage response in nephrotoxic and ischemic kidney injury. Toxicol Appl Pharmacol. 2016;313:104–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peyrou M, Hanna PE, Cribb AE. Cisplatin, gentamicin, and p-aminophenol induce markers of endoplasmic reticulum stress in the rat kidneys. Toxicological sciences : an official journal of the Society of Toxicology. 2007;99(1):346–53. [DOI] [PubMed] [Google Scholar]
- 51.Long Y, Zhen X, Zhu F, Hu Z, Lei W, Li S, et al. Hyperhomocysteinemia Exacerbates Cisplatin-induced Acute Kidney Injury. International journal of biological sciences. 2017;13(2):219–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Peyrou M, Cribb AE. Effect of endoplasmic reticulum stress preconditioning on cytotoxicity of clinically relevant nephrotoxins in renal cell lines. Toxicology in vitro : an international journal published in association with BIBRA. 2007;21(5):878–86. [DOI] [PubMed] [Google Scholar]
- 53.Mandic A, Hansson J, Linder S, Shoshan MC. Cisplatin induces endoplasmic reticulum stress and nucleus-independent apoptotic signaling. The Journal of biological chemistry. 2003;278(11):9100–6. [DOI] [PubMed] [Google Scholar]
- 54.Liu H, Baliga R. Endoplasmic reticulum stress-associated caspase 12 mediates cisplatin-induced LLC-PK1 cell apoptosis. J Am Soc Nephrol. 2005;16(7):1985–92. [DOI] [PubMed] [Google Scholar]
- 55.Chen CL, Liao JW, Hu OY, Pao LH. Blockade of KCa3.1 potassium channels protects against cisplatin-induced acute kidney injury. Archives of toxicology. 2016;90(9):2249–60. [DOI] [PubMed] [Google Scholar]
- 56.Tavernier Q, Mami I, Rabant M, Karras A, Laurent-Puig P, Chevet E, et al. Urinary Angiogenin Reflects the Magnitude of Kidney Injury at the Infrahistologic Level. Journal of the American Society of Nephrology : JASN. 2017;28(2):678–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim Y, Park SJ, Manson SR, Molina CA, Kidd K, Thiessen-Philbrook H, et al. Elevated urinary CRELD2 is associated with endoplasmic reticulum stress-mediated kidney disease. JCI insight. 2017;2(23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hale AN, Ledbetter DJ, Gawriluk TR, Rucker EB 3rd. Autophagy: regulation and role in development. Autophagy. 2013;9(7):951–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ryter SW, Choi AM. Autophagy: An Integral Component of the Mammalian Stress Response. J Biochem Pharmacol Res. 2013;1(3):176–88. [PMC free article] [PubMed] [Google Scholar]
- 60.Fougeray S, Pallet N. Mechanisms and biological functions of autophagy in diseased and ageing kidneys. Nat Rev Nephrol. 2015;11(1):34–45. [DOI] [PubMed] [Google Scholar]
- 61.He L, Livingston MJ, Dong Z. Autophagy in acute kidney injury and repair. Nephron Clinical practice. 2014;127(1-4):56–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Takabatake Y, Kimura T, Takahashi A, Isaka Y. Autophagy and the kidney: health and disease. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2014;29(9):1639–47. [DOI] [PubMed] [Google Scholar]
- 63.Cybulsky AV. Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nature reviews Nephrology. 2017;13(11):681–96. [DOI] [PubMed] [Google Scholar]
- 64.Yorimitsu T, Nair U, Yang Z, Klionsky DJ. Endoplasmic reticulum stress triggers autophagy. The Journal of biological chemistry. 2006;281(40):30299–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hoyer-Hansen M, Jaattela M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell death and differentiation. 2007;14(9):1576–82. [DOI] [PubMed] [Google Scholar]
- 66.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annual review of genetics. 2009;43:67–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cybulsky AV. The intersecting roles of endoplasmic reticulum stress, ubiquitin-proteasome system, and autophagy in the pathogenesis of proteinuric kidney disease. Kidney Int. 2013;84(1):25–33. [DOI] [PubMed] [Google Scholar]
- 68.Rashid HO, Yadav RK, Kim HR, Chae HJ. ER stress: Autophagy induction, inhibition and selection. Autophagy. 2015;11(11):1956–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Qin L, Wang Z, Tao L, Wang Y. ER stress negatively regulates AKT/TSC/mTOR pathway to enhance autophagy. Autophagy. 2010;6(2):239–47. [DOI] [PubMed] [Google Scholar]
- 70.Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, et al. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell death and differentiation. 2007;14(2):230–9. [DOI] [PubMed] [Google Scholar]
- 71.Adolph TE, Tomczak MF, Niederreiter L, Ko HJ, Bock J, Martinez-Naves E, et al. Paneth cells as a site of origin for intestinal inflammation. Nature. 2013;503(7475):272–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tang C, Han H, Yan M, Zhu S, Liu J, Liu Z, et al. PINK1-PRKN/PARK2 pathway of mitophagy is activated to protect against Renal ischemia-reperfusion injury. Autophagy. 2018:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang D, Pan J, Xiang X, Liu Y, Dong G, Livingston MJ, et al. Protein Kinase Cdelta Suppresses Autophagy to Induce Kidney Cell Apoptosis in Cisplatin Nephrotoxicity. J Am Soc Nephrol. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu J, Livingston MJ, Dong G, Tang C, Su Y, Wu G, et al. Histone deacetylase inhibitors protect against cisplatin-induced acute kidney injury by activating autophagy in proximal tubular cells. Cell Death Dis. 2018;9(3):322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Livingston MJ, Dong Z. Autophagy in acute kidney injury. Semin Nephrol. 2014;34(1):17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Oliva Trejo JA, Asanuma K, Kim EH, Takagi-Akiba M, Nonaka K, Hidaka T, et al. Transient increase in proteinuria, poly-ubiquitylated proteins and ER stress markers in podocyte-specific autophagy-deficient mice following unilateral nephrectomy. Biochemical and biophysical research communications. 2014;446(4):1190–6. [DOI] [PubMed] [Google Scholar]
- 77.Kawakami T, Inagi R, Takano H, Sato S, Ingelfinger JR, Fujita T, et al. Endoplasmic reticulum stress induces autophagy in renal proximal tubular cells. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2009;24(9):2665–72. [DOI] [PubMed] [Google Scholar]
- 78.Rovetta F, Stacchiotti A, Consiglio A, Cadei M, Grigolato PG, Lavazza A, et al. ER signaling regulation drives the switch between autophagy and apoptosis in NRK-52E cells exposed to cisplatin. Experimental cell research. 2012;318(3):238–50. [DOI] [PubMed] [Google Scholar]
- 79.Dong G, Liu Y, Zhang L, Huang S, Ding HF, Dong Z. mTOR contributes to ER stress and associated apoptosis in renal tubular cells. American journal of physiology Renal physiology. 2015;308(3):F267–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kapuy O, Vinod PK, Banhegyi G. mTOR inhibition increases cell viability via autophagy induction during endoplasmic reticulum stress - An experimental and modeling study. FEBS open bio. 2014;4:704–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhao L, Ackerman SL. Endoplasmic reticulum stress in health and disease. Current opinion in cell biology. 2006;18(4):444–52. [DOI] [PubMed] [Google Scholar]
- 82.Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell death and differentiation. 2004;11(4):381–9. [DOI] [PubMed] [Google Scholar]
- 83.Hetz C, Bernasconi P, Fisher J, Lee AH, Bassik MC, Antonsson B, et al. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha. Science. 2006;312(5773):572–6. [DOI] [PubMed] [Google Scholar]
- 84.Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287(5453):664–6. [DOI] [PubMed] [Google Scholar]
- 85.Matsuzaki S, Hiratsuka T, Kuwahara R, Katayama T, Tohyama M. Caspase-4 is partially cleaved by calpain via the impairment of Ca2+ homeostasis under the ER stress. Neurochemistry international. 2010;56(2):352–6. [DOI] [PubMed] [Google Scholar]
- 86.Binet F, Chiasson S, Girard D. Evidence that endoplasmic reticulum (ER) stress and caspase-4 activation occur in human neutrophils. Biochemical and biophysical research communications. 2010;391(1):18–23. [DOI] [PubMed] [Google Scholar]
- 87.Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403(6765):98–103. [DOI] [PubMed] [Google Scholar]
- 88.Iurlaro R, Munoz Pinedo C. Cell death induced by endoplasmic reticulum stress. The FEBS journal. 2015. [DOI] [PubMed] [Google Scholar]
- 89.Pluquet O, Pourtier A, Abbadie C. The unfolded protein response and cellular senescence. A review in the theme: cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. American journal of physiology Cell physiology. 2015;308(6):C415–25. [DOI] [PubMed] [Google Scholar]
- 90.Prachasilchai W, Sonoda H, Yokota-Ikeda N, Oshikawa S, Aikawa C, Uchida K, et al. A protective role of unfolded protein response in mouse ischemic acute kidney injury. European journal of pharmacology. 2008;592(1–3):138–45. [DOI] [PubMed] [Google Scholar]
- 91.Bando Y, Tsukamoto Y, Katayama T, Ozawa K, Kitao Y, Hori O, et al. ORP150/HSP12A protects renal tubular epithelium from ischemia-induced cell death. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2004;18(12):1401–3. [DOI] [PubMed] [Google Scholar]
- 92.Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science. 2005;307(5711):935–9. [DOI] [PubMed] [Google Scholar]
- 93.Asmellash S, Stevens JL, Ichimura T. Modulating the endoplasmic reticulum stress response with trans-4,5-dihydroxy-1,2-dithiane prevents chemically induced renal injury in vivo. Toxicological sciences : an official journal of the Society of Toxicology. 2005;88(2):576–84. [DOI] [PubMed] [Google Scholar]
- 94.Carlisle RE, Brimble E, Werner KE, Cruz GL, Ask K, Ingram AJ, et al. 4-Phenylbutyrate inhibits tunicamycin-induced acute kidney injury via CHOP/GADD153 repression. PloS one. 2014;9(1):e84663. [DOI] [PMC free article] [PubMed] [Google Scholar]