

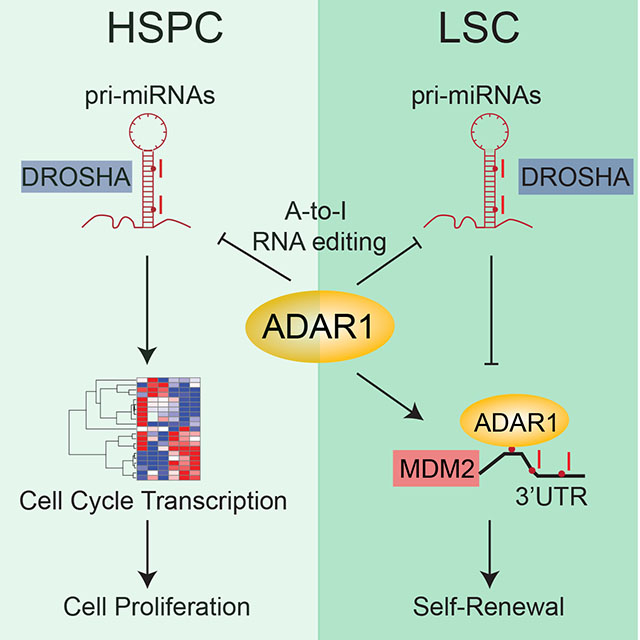
Summary
Adenosine deaminase associated with RNA1 (ADAR1) deregulation contributes to therapeutic resistance in many malignancies. Here we show that ADAR1-induced hyper-editing in normal human hematopoietic progenitors impairs miR-26a maturation, which represses CDKN1A expression indirectly via EZH2 thereby accelerating cell cycle transit. However, in blast crisis chronic myeloid leukemia progenitors, loss of EZH2 expression and increased CDKN1A oppose cell cycle transit. Moreover, A-to-I editing of both the MDM2 regulatory miRNA and its binding site within the 3’UTR region stabilize MDM2 transcripts thereby enhancing BC progenitor propagation. These data reveal a dual mechanism governing malignant transformation of progenitors that is predicated on hyper-editing of cell cycle regulatory miRNAs and the 3’UTR binding site of tumor suppressor miRNAs.
Keywords: Epitranscriptome, RNA hyper-editing, ADAR1, microRNAs, 3’ UTR, progenitors, cell cycle, leukemia
Graphical Abstract
INTRODUCTION
Post-transcriptional RNA modifications, referred to as epitranscriptomic alterations, include methylation, splicing and editing (Jiang et al., 2017) and contribute to mammalian gene expression diversity. While processing of nascent RNA, including 5’ processing (capping), 3’ processing (cleavage and polyadenylation), RNA methylation and adenosine to inosine (A-to-I) editing have been studied primarily in mouse models, a recent study shows striking species and tissue specificity of RNA editing by adenosine deaminase associated with RNA 1 and 2 (ADAR1 and ADAR2)(Tan et al., 2017). As essential components of the innate immune system, ADARs evolved to protect tissues from retroviral infection as well as to guide tissue repair and regeneration in response to injury, frequently in the context of inflammatory cytokines(Liddicoat et al., 2015; Mannion et al., 2014). ADAR-mediated deamination of adenosine to inosine (A-to-I), which is subsequently read as a guanosine (G), rewrites (editing in non-coding regions), recodes (editing in protein coding regions), and rewires (editing-induced changes in topology) transcripts (Tan et al., 2017; Zipeto et al., 2015). Compelling murine studies demonstrate that A-to-I RNA editing is vital for normal hematopoietic stem and progenitor cell (HSPC) maintenance (Hartner et al., 2009; Wang et al., 2000). However, a recently published comparative RNA-sequencing (RNA-seq) study demonstrated that A-to-I editing in mammals occurs most frequently in the context of primate-specific repetitive elements and in a tissue specific manner thereby underscoring the importance of studying ADAR1 function in human cells (Tan et al., 2017).
Humans have three forms of ADAR, including ADAR1, ADAR2 and ADAR3. While ADAR3 appears to inhibit ADAR2 editing within coding regions, ADAR1 edits primarily within double stranded RNA (dsRNA) loops formed by inverted primate-specific Alu repetitive elements (Jiang et al., 2017; Tan et al., 2017; Zipeto et al., 2015). Approximately, 11% of the human genome is composed of Alu elements (one million copies) and 90% of ADAR-mediated editing occurs within regions containing these transcribed repetitive elements (Batzer and Deininger, 2002; Jurka and Smith, 1988). Thus, the epitranscriptomic impact of ADAR1 must be considered in the context of human tissues and likely in a cell-type and context-specific manner. While ADAR1 editase activity has been strongly linked to murine HSPC maintenance, the role of ADAR1-mediated A-to-I editing in normal human HSPC cell cycle transit and maintenance has not been extensively studied. Recently, we showed that overexpression of ADAR1p150, a cytoplasmic isoform of ADAR1, enhances self-renewal gene expression in a deaminase dependent manner in human cord blood HSPC (Zipeto et al., 2016). These data suggest that ADAR1 may play a key role in normal human HSPC maintenance.
Deregulation of ADAR1 has emerged as a dominant driver of cancer progression and therapeutic resistance (Chen et al., 2013; Han et al., 2015; Jiang et al., 2017). Cumulative whole transcriptome RNA-seq analyses have uncovered inflammatory cytokine networks that activate ADAR during relapse or progression of lobular breast (Shah et al., 2009), hepatocellular (Qi et al., 2014) and esophageal squamous cell (Qin et al., 2014) carcinomas and in the most highly studied myeloproliferative neoplasm (MPN), chronic myeloid leukemia (CML) (Jiang et al., 2013). During blast crisis (BC) transformation of chronic phase (CP) CML, increased A-to-I RNA editing of self-renewal transcripts is triggered by inflammatory cytokine activation of JAK/STAT signaling (Zipeto et al., 2016). Thus, a mechanistic understanding of the cell type and context specific effects of ADAR1 activation on cell cycle transit and self-renewal would inform therapeutic strategies aimed at predicting and preventing malignant RNA editing that fuels cancer stem cell (CSC) generation as well as further refining ADAR-mediated RNA repair strategies (Cox et al., 2017).
RESULTS
ADAR1 activation induces hematopoietic stem and progenitor (HSPC) cell cycle transit
To gain insight into the function of ADAR1 in normal HSPC, normal cord blood CD34+ cells were lentivirally transduced with the wild-type (WT) ADAR1 or ADAR1E921A, an editing defective mutant, labeled with GFP. Overexpression of ADAR1 WT induced proliferation of both stem (CD34+38−Lin−) and progenitor (CD34+38+Lin−) populations and increased Ki67 expression (Figures 1A-1E and S1A-S1C). Moreover, DiR-labeled GFP+ ADAR1-WT expressing HSPCs quickly lost DiR signal, indicative of accelerated cell cycle transit (Figures 1F-G and S1D). Interestingly, ADAR1 increased B cell (CD19+) expansion, which concurs with the recent finding that ADAR1 is required for B-cell lineage development (Marcu-Malina et al., 2016) (Figures S1E-F). Moreover, q-RT-PCR array and RNA-seq analyses revealed that overexpression of ADAR1 WT significantly altered KEGG cell cycle regulatory transcript expression (Figures 1H-J and S1G; Table S1). Differential expression of certain cell cycle transcripts was observed with ADAR1 WT but not ADAR1E921A, suggesting these cell cycle transcript changes were A-to-I RNA editing dependent (Zipeto et al., 2016) (Figures 1K and S1H). Interestingly, expression of CDKN1A, a cyclin-dependent kinase inhibitor that induces quiescence in response to DNA damage, was reduced following ADAR1 WT overexpression and is the central hub for cell cycle regulation by ADAR1 WT (Figures 1L-M). To ascertain the effect of ADAR1 in HSPC cell cycle regulation, we also performed ADAR1 knockdown in cord blood HSPCs with a shRNA (Jiang et al., 2013; Zipeto et al., 2016) (Figures 1N-P and S1I). As expected, knockdown of ADAR1 displayed a reversal of ADAR1 overexpression phenotypes, including an increased quiescent G0 population as well as increased CDKN1A expression (Figures 1N-P and S1I). In contrast to ADAR1 overexpression (Zipeto et al., 2016), shRNA-mediated ADAR1 knockdown in HSPC reduced the percentage of replated colonies in in vitro replating assay (Figure 1P). Previous studies have shown that the replating capacity correlates closely serial transplantation potential (Crews et al., 2016; Zipeto et al., 2016). In addition, these ADAR1 shRNA knockdown human HSPC replating data are compatible with a previous report showing that conditional ADAR1 deletion in murine HSPC impairs long-term multi-lineage reconstituting potential underscoring the importance of ADAR1 for normal HSPC maintenance (Hartner et al., 2009).
Figure 1. ADAR1 regulates cell cycle in normal hematopoiesis.
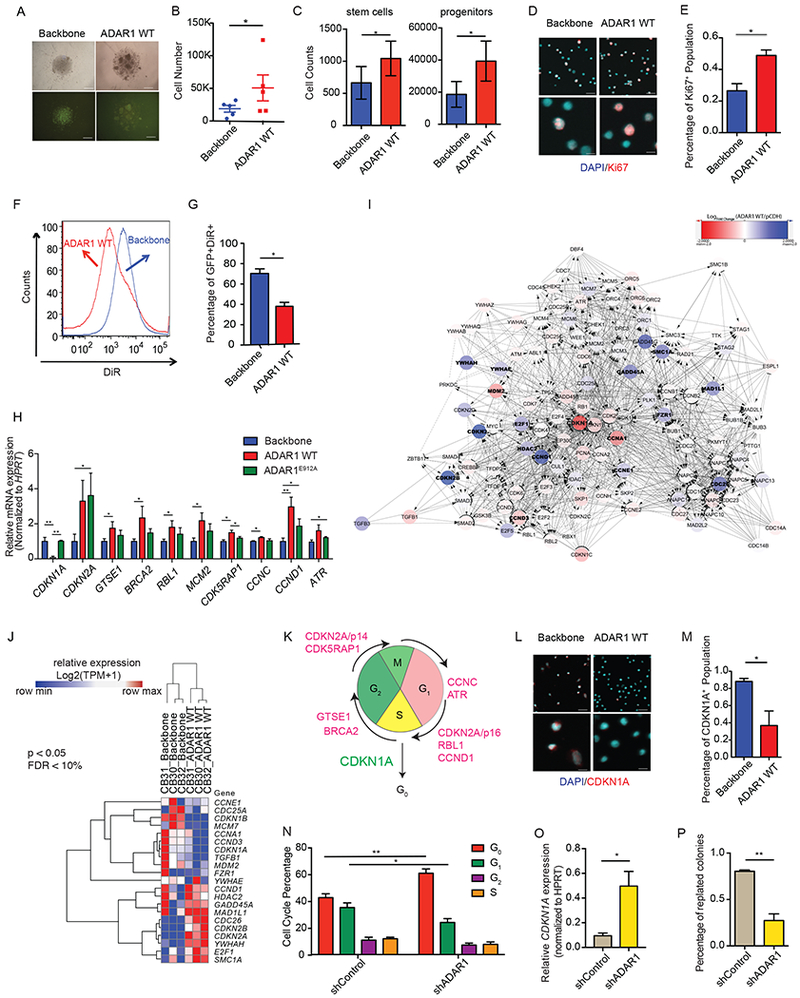
(A) Representative picture of ADAR1-WT or lentiviral backbone transduced cord blood CD34+ cells. (B and C) Cell counts of total cell number (B), stem cells (CD34+CD38−Lin−), and progenitors (CD34+CD38+Lin−) (C) following ADAR1 WT overexpression in cord blood CD34+ cells (n=3). (D and E) Representative image of Ki67 immunofluorescent staining in ADAR1 WT-expressing cord blood CD34+ cells (D) and the corresponding calculation of Ki67+ cells (n=3). (F and G) Representative flow cytometry of DiR tracing in cord blood CD34+ cells transduced with backbone control or ADAR1 WT (F) and the corresponding calculation of GFP+DiR+ cells (G) (n=3). (H) Significant differential expressed cell cycle transcripts were determined by qRT-PCR array of 84 transcripts on cord blood HSPC (n=5) transduced with ADAR1 WT, ADAR1E912A, or lentiviral vector control. (I) Cytoscape analysis of differentially expressed transcripts of KEGG Cell Cycle Pathway in ADAR1 WT-transduced cord blood (n=3) versus lentiviral vector control (n=3) by whole transcriptome RNA-seq. (J) RNA-seq quantification on ADAR1 WT-transduced cord blood (n=3) and lentiviral vector control (n=3) for genes corresponding to the KEGG Cell Cycle Pathway visualized in a heatmap (p<0.05, FDR <10%). (K) Representative image of ADAR1-mediated differentially expression targets in cell cycle stages. (L and M). Representative images (L) and quantification (M) of immunofluorescent staining of CDKN1A protein in ADAR1 WT-expressing CD34+ cells (n=3). (N) Cell cycle analysis in cord blood CD34+ HSPC transduced with shRNA targeting ADAR1 (shADAR1) or control shRNA (shControl) as measured by flow cytometry of Ki-67 and 7AAD (n=4). (O). CDKN1A expression measured by qRT-PCR in cord blood CD34+ HSPC (n=3). (P) Percentage of replated colonies in cord blood CD34+ HSPC transduced with shADAR1 or shControl (n=3). All graphs show mean with SEM and statistical analysis was calculated using the Student’s t-test. *p<0.05, **p<0.005, ***p<0.0005. See also Figure S1 and Table S1.
ADAR1 pri-miRNA editing regulates progenitor cell cycle transit
Next, we examined the molecular mechanisms governing cell cycle regulation by ADAR1 in HSPC. Since CDKN1A is the central hub (Figures 1L-M and S1H-1I), we analyzed the A-to-I RNA editing in CDKN1A transcript using the ADAR1 WT transduced normal HSPC RNA-seq dataset, but we did not find any direct A-to-I editing events. Thus, we hypothesized that ADAR1 may control CDKN1A expression by regulating the function of specific miRNAs (Jiang et al., 2017). Although the role of ADAR1 in miRNAs biogenesis has been studied in human cell lines and leukemia stem cells (LSC) (Mallela and Nishikura, 2012; Nishikura, 2010, 2016; Zipeto et al., 2016), a complete profile of the edited miRNome and implications in normal hematopoietic stem and progenitor cell function has not been elucidated. To investigate the role of ADAR1 in global miRNA regulation, we performed miRNome miScript PCR array analysis of 1008 miRNAs in cord blood CD34+ HSPCs overexpressing ADAR1 WT or ADAR1E912A. Using Diana miRNA target base (Chou et al., 2016), “cell cycle” was identified as the top cellular pathway significantly targeted by miRNAs regulated by ADAR1 WT but not ADAR1E912A (Figures 2A). Overall, 112 and 32 miRNAs were significantly differentially expressed following ADAR1 WT or ADAR1E912A expression, respectively (Figures 2B-D, Table S2). These data suggest that ADAR1 may regulate cell cycle transit through modulation of miRNA biogenesis (Figure 2A). Other than let-7 miRNAs, which were previously identified as ADAR1 editing targets (Zipeto et al., 2016), ADAR1 WT inhibited the expression of miR-2278, a tumor suppressor that targets STAT5 and restores tyrosine kinase inhibitor sensitivity in CML (Kaymaz et al., 2015), and miR-411, which induces cell proliferation in several human tumor types (Xia et al., 2015; Zhang et al., 2016b; Zhao et al., 2016) (Figure 2C). Notably, ADAR1-mediated A-to-I editing activity inhibited expression of miR-26a-5p, a tumor suppressor miRNA that is transcriptionally repressed by MYC (Salvatori et al., 2011; Sander et al., 2008) and frequently downregulated in hematological malignancies (Chen et al., 2016; Fatica and Fazi, 2013). Because overexpression of miR-26a is known to impair cell cycle progression, attenuate cell proliferation (Sander et al., 2008) and disrupt the let-7/LIN28B axis by directly targeting Lin28B (Fu et al., 2014), we hypothesized that inhibition of miR-26a expression by ADAR1 could accelerate cell cycle transit and increase self-renewal of HSPCs.
Figure 2. Regulation of miRNome by ADAR1 in normal cord blood CD34+ HSPC.
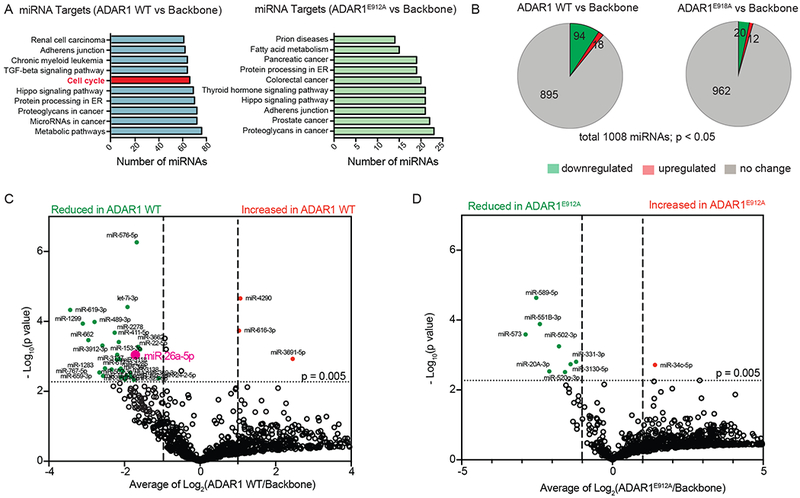
(A) Top ten significantly affected pathways by differentially expressed miRNAs targeted by ADAR1 WT or ADAR1E912A compared with lentiviral backbone. (B) Pie chart of differentially expressed miRNAs in cord blood CD34+ HSPC overexpression ADAR1 WT or ADAR1E912A compared with backbone vector control (n=3-4) derived from miRNome array of 1008 miRNAs. (C and D) Volcano plot analysis derived from miRNome showing significantly differentially expressed miRNAs (p<0.005, Student’s t-test, Log2Fold change >2) between cord blood CD34+ cells transduced with backbone vector control and those with lenti-ADAR1 WT (C) or between cord blood CD34+ cells transduced with backbone vector control and those with lenti-ADAR1E912A (D) (n=3-4). See also Table S2.
To test this hypothesis, we first examined the impact of miR-26a expression on normal HSPC survival and self-renewal using in vitro cord blood colony forming and replating assay system. Lentivirally enforced miR-26a expression reduced total colony number, replating capacity, and LIN28B expression, indicative of inhibited self-renewal ability (Zipeto et al., 2016) (Figures 3A-E and S2A). Moreover, lentiviral miR-26a overexpression was associated with increased CDKN1A mRNA expression and blocked G0 to G1 transition in normal cord blood HSPC (Figures 3F-H). This was further validated by a positive correlation between pri-miR-26a transcript level and CDKN1A expression, and increased CDKN1A protein level in HEK293T cells transduced with miR-26a overexpressing vector (Figure S2B-C). Interestingly, ADAR1 WT significantly enhanced the expression of enhancer of zeste homolog 2 (EZH2), a known target of miR-26a (Lu et al., 2011; Salvatori et al., 2011; Sander et al., 2008) (Figure 3I). As a polycomb protein that mediates global gene expression by histone 3 lysine 27 trimethylation (H3K27me3), EZH2 suppresses CDKN1A expression by altering H3K27me3 at the CDKN1A promoter region and transcriptional start site (TSS) (Figure S2D-E) (Beguelin et al., 2016; Fan et al., 2011; Kim and Roberts, 2016; Lund et al., 2014; Pawlyn et al., 2017; Xie et al., 2014). Both knockdown of ADAR1 with a shRNA and overexpression of miR-26a inhibited EZH2 expression in cord blood CD34+ HSPCs (Figures 3J-K). Taken together, these data suggest that ADAR1 editase regulates cell cycle transit, at least in part, through inhibition of miR-26a biogenesis, which indirectly suppresses CDKN1A via EZH2.
Figure 3. Important role of miR-26a in self-renewal capacity of normal hematopoietic progenitors.
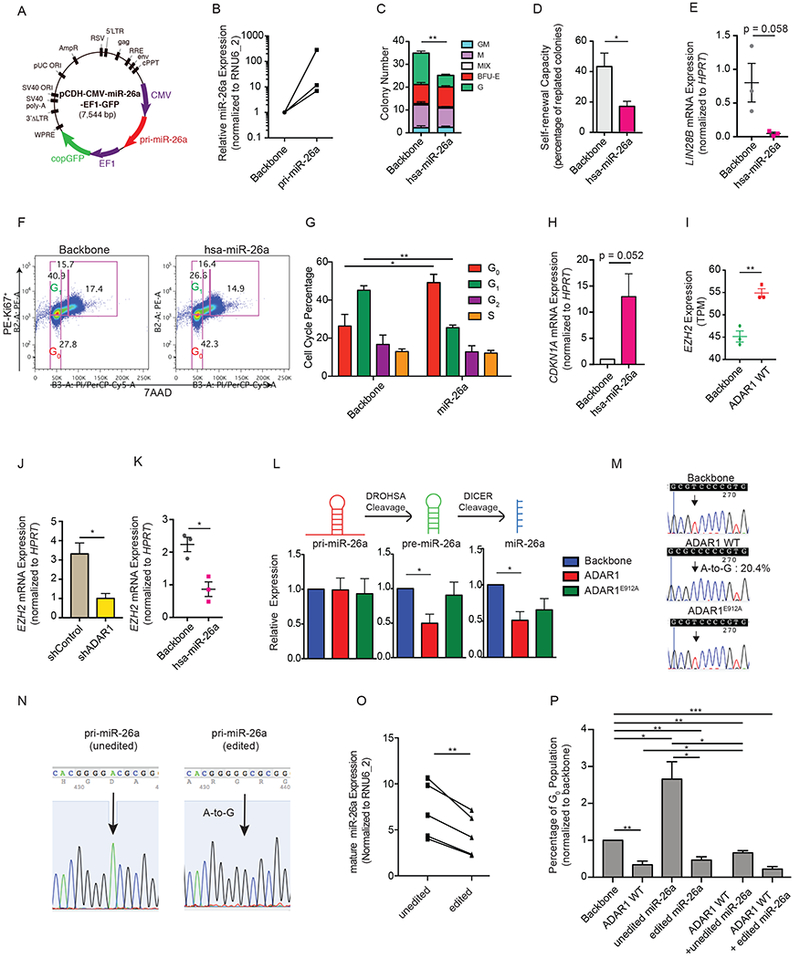
(A) Lentiviral construct for human primary (pri-) miR-26a expression. (B) The expression of mature miR-26a was determined by qRT-PCR in cord blood CD34+ HSPC transduced with pri-miR-26a lentivirus (n=3). (C and D) The total colony number (C) and the percentage of replated colonies in cord blood CD34+ cells overexpressing hsa-miR-26a or the backbone control (n=3). (E) Expression of LIN28B in cord blood CD34+ HSPC overexpressing hsa-miR-26a or the backbone control (n=3). (F) Representative cell cycle flow analysis of CB CD34+ HSPC (n=3) transduced with either backbone or hsa-miR-26a overexpression lentivirus. (G) Flow cytometry analysis of cell cycle transit in cord blood CD34+ HSPC overexpressing hsa-miR-26a or the backbone control (n=3). (H) Expression of CDKN1A mRNA level was determined by qRT-PCR in cord blood CD34+ HSPC overexpressing hsa-miR-26a or the backbone control (n=3). (I) EZH2 expression evaluated by RNA-seq in cord blood CD34+ cells transduced with backbone control of ADAR1 WT (n=3). (J and K). Examination of EZH2 expression in cord blood CD34+ cells with either ADAR1 knockdown by shRNA (J) or overexpression of hsa-miR-26a (K) (n=3). (L) Expression of primary (pri-), precursor (pre-) and mature miR-26a transcripts was measured by qRT-PCR in cord blood CD34+ HSPCs transduced with backbone, ADAR1 WT, or ADAR1E912A (n=3). (M) The A-to-I RNA editing efficiency was determined by TOPO sequencing of blood CD34+ cells overexpressing backbone, ADAR1 WT, or ADAR1E912A (n=3). 20.4% of colonies examined contain A-to-G change at the DROSHA cleavage site. The arrow pointed to the A-to-G mutation site, reverse sequenced as T-to-C change. (N) Confirmation of lentiviral constructs of “unedited” and “edited” pri-miR-26a. The arrow points to the A-to-I (G) mutation site. (O) HEK293T cells were transfected with “unedited” or “edited” pri-miR-26a lentivirus, and the mature miR-26a expression was determined by qRT-PCR (n=5 experiments). (P) Cell cycle flow analysis of G0 population in HEK293T cells transduced with ADAR1 WT in combination with “unedited” or “edited” miR-26a (n=3 experimental triplicate). All graphs show mean with SEM and statistical analysis was calculated using the Student’s t-test. *p<0.05, **p<0.005, ***p<0.0005. See also Figure S2.
ADAR1 impairs pri-miR-26a biogenesis by preventing DROSHA cleavage
As previously shown for miR-142 and let-7 family miRNAs, A-to-I editing of DROSHA or DICER cleavage sites can impair miRNA maturation (Yang et al., 2006; Zipeto et al., 2016). The editing dependent reduction of precursor (pre-) and mature miR-26a compared to unaltered primary (pri-) miRNA expression suggested that A-to-I editing may occur in the DROSHA cleavage site of primary pri-miR-26a (Figure 3L). Indeed, TOPO cloning of pri-miR-26a transcripts identified 20.4% A-to-I(G) RNA editing at the DROSHA cleavage site in ADAR1 WT expressing cells whereas the expression of ADAR1E912A did not induce any A-to-I(G) changes (Figure 3M). Cross-linking RNA Immunoprecipitation (CLIP) in the K562 leukemic cell line stably expressing ADAR1 WT or ADAR1E912A revealed that both ADAR1 WT and ADAR1E912A directly interact with pri-miR-26a transcripts (Figures S2F-G). Next, we performed site-directed mutagenesis at the DROSHA cleavage site in pri-miR-26a (Figures 3N and S2H). Compared to WT pri-miR-26a, editing of pri-miR-26a at the DROSHA cleavage site resulted in a significant reduction in mature miR-26a production (Figure 3O). In a manner similar to enforced ADAR1 WT expression, overexpression of edited miR-26a reduced the G0 population. A combination of ADAR1 WT with unedited miR-26a or edited miR-26a exhibited the same phenotype (Figure 3P). These data suggest that A-to-I RNA hyper-editing of miR-26a accelerated cell cycle progression in cord blood HSPCs.
Enforced miR-26a expression prevents CML progenitor self-renewal
Advances in functional miRNA studies in hematological malignancies have provided an array of potential biomarkers and therapeutic targets for leukemia patients (Lechman et al., 2016; Wojtowicz et al., 2016). The transition from pre-malignant progenitor to therapy resistant CSC is often accompanied by aberrant ADAR1 activation (Chen et al., 2013; Han et al., 2015; Jiang et al., 2013). Thus, we hypothesized that ADAR1-mediated impairment of miRNA biogenesis, including miR-26a, contributed to progression from CP to BC CML. A pilot study revealed significantly reduced expression of miR-26a during CP to BC transformation (Figure 4A). This was validated in CP CD34+ cells transduced with ADAR1 WT (Figures 4B-C). To assess the effect of miR-26a on BC LSC maintenance, miR-26a was introduced into BC CD34+ cells followed by colony forming and replating assays as well as transplantation into Rag2−/−γc−/− immunocompromised mice (Figure 4D-F). MiR-26a expression reduced total colony number and replating capacity of BC CD34+ cells (Figures 4D-E). Although no significant change was observed in total human CD45+ cells (Figures S3A-C), FACS analysis revealed that miR-26a expression significantly reduced engraftment of granulocyte-macrophage progenitors (GMP, CD45+Lin−CD34+CD38+CD123+CD45RA+) that harbored LSC self-renewal capacity (Abrahamsson et al., 2009; Jamieson et al., 2004) (Figure 4G and S3D). These data confirmed the important role of miR-26a as a tumor suppressor.
Figure 4. Reduced miR-26a enhances self-renewal capacity of CML Progenitors.
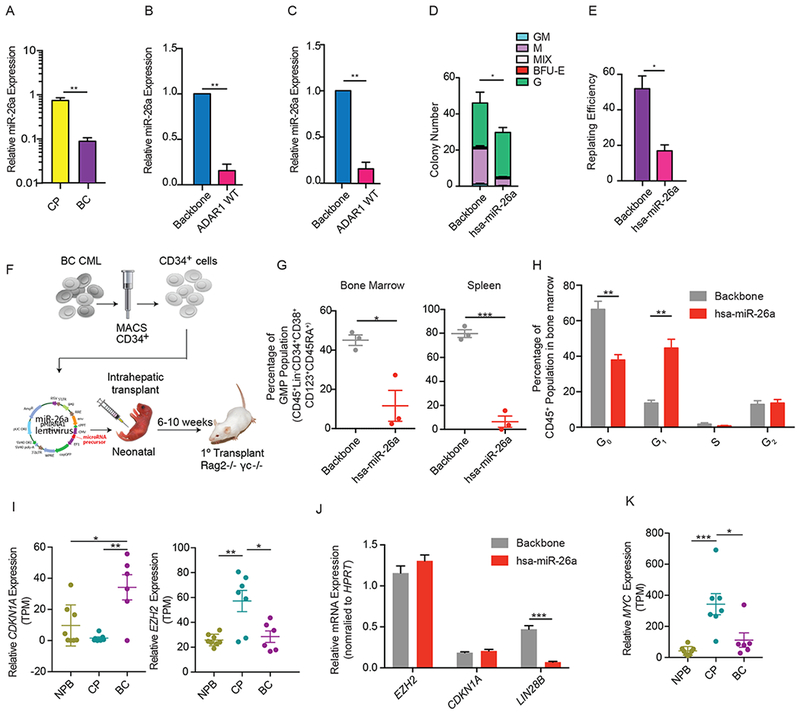
(A) Expression of mature miR-26a in CML CP (n=3) and CML BC (n=3) CD34+ cells measured by qRT-PCR (n=3). (B) miR-26a expression in CML CP CD34+ cells transduced with backbone control or ADAR1 WT examined by miRNA PCR array (n=3). (C) Validation of miR-26a expression in CP CML CD34+ cells transduced with backbone control or ADAR1 WT lentivirus by qRT-PCR (n=3). (D and E) Number of colonies formed in primary colony-formation assay (D) and percentage of secondary colonies formed after replating primary colonies (E) in BC CML CD34+ cells transduced with backbone control or hsa-miR-26a (n=3). (F) Experimental design of in vivo xenograft mouse studies. (G) Percentage of granulocyte macrophage progenitors (GMP) engraftment in the bone marrow or spleen of Rag2−/−γc−/− mice transplanted with BC CD34+ cells overexpressing control or hsa-miR-26a (n=3 mice per group). (H) Cell cycle flow analysis of backbone or lenti-miR-26a transduced CD34+ cells isolated from engrafted BC bone marrow (n=3 mice per group). (I) RNA-seq analysis of CDKN1A and EZH2 expression in CP (n=7) and BC CML (n=6). (J) Expression of EZH2, CDKN1A, and LIN28B in BC CD34+ cells transduced with backbone control or hsa-miR-26a (n=3). (K) RNA-seq analysis of the expression of MYC in CP (n=7) and BC CML (n=6). All graphs show mean with SEM and statistical analysis was calculated using the Student’s t-test. *p<0.05, **p<0.005, ***p<0.0005. See also Figure S3.
In human BC leukemia cells, miR-26a overexpression enhanced the G0 to G1 phase transition in the bone marrow niche and reduced LSC dormancy (Figure 4H and S3E). This was further validated using a single-stranded modified RNA specifically targeting miR-26a in K562 cells (Figure S3F). Moreover, knockdown of miR-26a resulted in a reduction in the G0 to G1 phase transition. Divergent effects of miRNA have been reported in normal HSPC and LSC (Lechman et al., 2016). To understand the differential effects of ADAR1-editing of pri-miR-26a on normal versus malignant hematopoiesis, we examined the differentially expressed miR-26a targets by RNA-seq analysis of normal HSPC overexpressing ADAR1 WT compared with CP and BC progenitors. RNA-seq analysis showed increased expression of miR-26a target transcripts in cord blood CD34+ cells transduced with ADAR1 WT compared to lentiviral backbone (Figure S3G). Remarkably, the miR-26a target mRNA profile revealed a different set of targets in BC CML compared to cord blood HSPC (Figures S3H). Several upregulated miR-26a target transcripts were unique to BC CML progenitors including SMAD1 and TP53INP1, which encode important transcription factors that activate CDKN1A. These data suggest that miR-26a may target different pathways in normal versus malignant progenitors and thus has divergent roles in cell cycle regulation.
Despite the activation of ADAR1 (Jiang et al., 2013; Zipeto et al., 2016) and miR-26a downregulation during BC transformation of CML, CDKN1A was upregulated by approximately 30-fold in BC progenitors (Figures 4I). While EZH2 expression was upregulated in CP compared to normal aged progenitors (Xie et al., 2016), it returned to low expression levels in BC progenitors (Figure 4I). To confirm this, we overexpressed miR-26a in CML CD34+ cells. Of note, miR-26a was able to drive LIN28B downregulation (Figure 4J). Taken together, these results suggest that accumulation of additional oncogenic events in CML BC progenitors may contribute to EZH2 downregulation.
MYC, which is frequently deregulated during tumor progression, has been reported to stimulate EZH2 expression through miR-26a inhibition (Sander et al., 2008) or by direct binding to regulatory elements to activate EZH2 transcription (Neri et al., 2012). We therefore analyzed the expression of MYC in CML progenitor RNA-seq dataset to determine if MYC is responsible for EZH2 downregulation in BC progenitors. Indeed, the expression of MYC followed the same trend as EZH2; it was highly upregulated in CP progenitors compared to normal aged controls and returned to a lower level during CP progenitor transformation into dormant BC progenitor LSC (Figure 4K). However, MYC expression was not altered by either ADAR1 overexpression or knockdown, suggesting that MYC expression changes in CML progression were unrelated to ADAR1 activation (Figures S3I-J). Thus, RNA editing independent MYC protein deregulation in BC progenitors likely results in inhibition of EZH2 transcription and contributes to CDKN1A upregulation despite reduced expression of miR-26a in BC progenitors.
ADAR1 editing of MDM2 3’ UTR prevents miRNA binding and inhibits TP53 transcription
Other than directly modifying miRNA sequences, A-to-I RNA editing has also been shown to alter the miRNA targeting sequences within 3’ UTR. ADAR1 can also directly compete with the RNA transport regulator STAU1 for 3’ UTR occupancy resulting in incomplete transcript suppression or translation (Jiang et al., 2017; Yang et al., 2017; Zhang et al., 2016a). However, the link between ADAR1 and disruption of 3’ UTR targeting by miRNA has not been well established in LSCs. We therefore investigated the location of A-to-I RNA editing within cell cycle transcripts. Compared to the lentiviral backbone, differential A-to-I editing in 3’ UTR was observed in CHEK1, TP53, and TFDP2 transcripts in cord blood CD34+ cells overexpressing ADAR1 WT (Figure 5A). A comparison between BC and CP progenitors revealed an increase of 3’ UTR editing during BC transformation commensurate with increased ADAR1 expression. Strikingly, most 3’UTR editing events occurred within MDM2 transcripts (Figures 5B). As an E3 ubiquitin ligase that binds to the N-terminal transactivation domain of p53, MDM2 inhibits transcriptional activation of p53. Thus, upregulation of MDM2 and the corresponding downregulation of p53 were associated with accelerated phase (AP) and BC CML (Trotta et al., 2003). Using the miRcode, whole transcriptome human miRNA target prediction tool (Jeggari et al., 2012), we discovered that several miRNA targeting sites overlapped with the RNA-editing regions. In CML, we observed a cluster of targeting sites for miR-200b/c, one site for miR-204/204b/211, and one site for miR-155 (Figure 5C and Table S3). These sites were located within a ~600 nt region, suggesting “hyper” RNA editing in the MDM2 3’UTR occurred specifically in BC progenitors thereby underscoring the cell type and context specific effects of ADAR1 editing. In comparison, there was only one A-to-I RNA editing site (#68843263) in MDM2 transcripts in cord blood CD34+ cells overexpressing ADAR1 WT compared to backbone control although it did not occur at a known miRNA targeting site (Table S3).
Figure 5. Differential A-to-I RNA editing in 3’UTR regions of normal HSPCs and BC LSC.
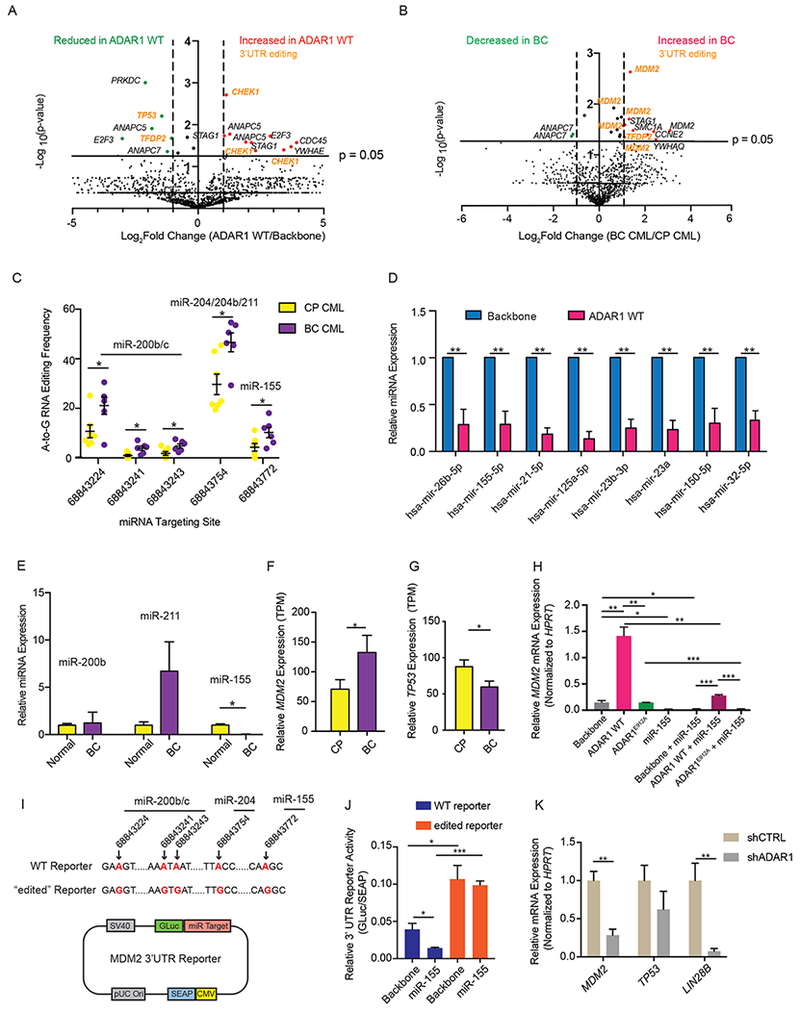
(A and B) Volcano plot showing the A-to-I(G) editome of cell cycle genes in ADAR1 WT-transduced cord blood CD34+ cells compared with lentiviral vector controls (n=3) (A) and in CP progenitors (n=7) compared with BC counterparts (n=6) (B). (C) A-to-I RNA editing of MDM2 3’UTR in individual CP (n=7) and BC (n=6) samples. The predicted miRNA binding sites within MDM2 3’UTR using miRcode transcriptome-wide miRNA target prediction tool were shown (Jeggari et al., 2012). (D) Relative miRNA expression determined by miRNA qPCR array of 84 miRNAs in CML CP CD34+ cells transduced with backbone or ADAR1 WT (n=3). (E) Relative miRNA expression in normal aged-matched (>55 year old) CD34+ cells (n=4) and BC CML CD34+ cells (n=3). (F and G) The expression of MDM2/p53 pathway transcripts, MDM2 (F) and TP53 (G) in progenitor population of normal peripheral blood (NPB), CML CP (n=7), and CML BC (n=6) determined by RNA-seq. (H) MDM2 expression in HEK293T cells transduced with ADAR1 WT, ADAR1E912A alone or in combination with miR-155 (n=3 experiments). (I) Structure of “wt” or “edited” MDM2 3’UTR reporter construct with A-to-(I)G changes introduced at miRNA targeting sites (highlighted in red). The genomic loci of A-to-(I)G changes were also indicated with arrows. (J) The MDM2 3’UTR reporters were transfected into HEK293T cells and then challenged with miR-155 overexpressing lentivirus (n=3 experimental triplicate). The miRNA targeting efficiency was measured as the relative luciferase activity (GLuc/SEAP ratio). (K) MDM2 and LIN28B expression in BC CML CD34+ cells with shControl or shADAR1 (n=3). All graphs show mean with SEM and statistical analysis was calculated using the Student’s t-test. *p<0.05, **p<0.005, ***p<0.0005. See also Figure S4 and Table S3.
Interestingly, miR-155, a miRNA that normally targets the SPI1 transcripts that encodes PU.1, was consistently suppressed in cord blood HSPC and CP progenitors transduced with ADAR1 WT and during CML BC transformation thereby explaining the previously observed increase in SPI1 in ADAR1 overexpressing progenitors (Figures 5D-E and S4A-B) (Jiang et al., 2013). Several other important miRNAs, including miR-125 and miR-150, also showed a reduced expression upon ADAR1 WT activation in CP CD34+ cells (Figure 5D). Interestingly, the inhibition of miR-155 biogenesis does not depend entirely on ADAR1’s A-to-I RNA editing since ADAR1E912A also reduced miR-155 expression compared to ADAR1 WT (Figures S4C-E). Therefore, it is likely that ADAR1 affects MDM2 expression by both 3’UTR editing thereby evading miRNA targeting and inhibition of miRNAs that directly target MDM2, such as miR-155. In keeping with the hypothesis that ADAR1-mediated 3’UTR hyper-editing and suppression of miRNA biogenesis allow transcripts to evade miRNA targeting, we observed a significant increase in MDM2 expression and a decrease in the transcript level of TP53 in BC that harbored high levels of ADAR1 compared to CP progenitors (Figures 5F-G). Expression of downstream targets in the MDM2/p53 pathway, such as p16INK4a and p14ARF, were also increased during BC transformation (Figures S4F-G).
To determine if increased MDM2 transcript abundance was due to ADAR1 activation in BC CML, we expressed ADAR1 WT or ADAR1E912A in combination with miR-155 and quantified MDM2 transcripts by qRT-PCR. Indeed, A-to-I editing activity of ADAR1 was required for upregulation of MDM2, and this increase was abolished by lentiviral transduction with miR-155 (Figure 5H). To directly show that the A-to-I RNA editing of MDM2 3’UTR prevented miR-155 targeting, we utilized a luciferase reporter with either wild-type (“unedited”) or mutant (“edited”) miRNA targeting sites (Figure 5I). The relative luciferase activity increased in the “edited” reporter compared to wild-type reporter, likely due to the endogenous miRNA fails to target the “edited” reporter. We challenged both wild-type and “edited” reporter with miR-155 expressing lentivirus. Only the “edited” reporter was insensitive to miR-155 (Figure 5J). These data suggest that 3’UTR RNA editing enables MDM2 to evade targeting by miR-155. Moreover, shRNA knockdown of ADAR1 in BC CML CD34+ cells reduced MDM2 expression as well as increased transcript levels of TP53 (Figure 5K). Lastly, ADAR1 overexpression in CML CP CD34+ cells led to increased protein level of MDM2 (Figures S4H-I). Together, these data reveal dual mechanism of ADAR1-dependent LSC generation involving 1) impaired biogenesis of cell cycle regulatory miRNAs, and 2) 3’UTR editing resulting in disruption of miRNA binding (Figures 6).
Figure 6.
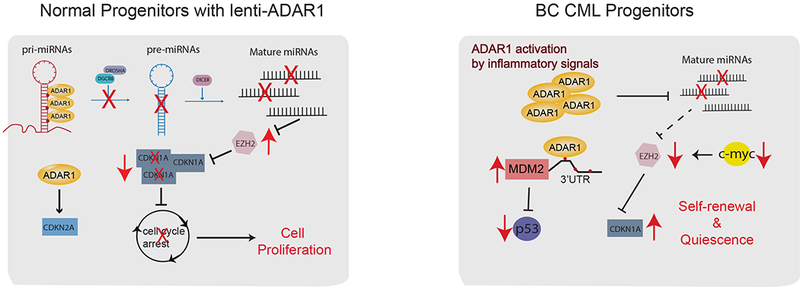
Summary of A-to-I RNA editing function in normal HSPCs and BC LSC.
DISCUSSION
Seminal murine studies underscore the importance of ADAR1 in murine hematopoiesis. Functional deletion of ADAR1 in embryonic stem cells induces embryonic lethality as a result of loss of erythropoiesis while conditional deletion in hematopoietic stem cells impairs multi-lineage reconstitution potential (Hartner et al., 2009; Wang et al., 2000). As a result of advances in RNA sequencing technology, RNA editing has emerged as a dynamic regulator of mammalian transcriptomic diversity (Ramaswami and Li, 2014, 2016; Tan et al., 2017). Striking differences in A-to-I editing between humans and mice are related, at least in part, to the propensity of ADAR1 to edit within double-stranded RNA (dsRNA) loops, which are frequently formed by inverted Alu repetitive elements that represent 11% of the human genome (Tan et al., 2017) but do not exist in mice. In addition to protecting (stem cells from retroviral integration, a vital physiological role of ADAR1 is to edit endogenous dsRNA to prevent sensing of endogenous dsRNA as non-self by MDA5 (Liddicoat et al., 2015). Recently, A-to-I RNA editing by ADAR1 was shown to play a key role in translational control and proteomic diversity (Chung et al., 2018; Peng et al., 2018). In addition, A-to-I editing events are dynamically regulated in a tissue specific manner. However, the functional role of ADAR1 in human benign and malignant hematopoietic stem and progenitor cell maintenance has not been clearly elucidated. Malignant deregulation of ADAR1-mediated RNA editing has been linked to progression and therapeutic resistance of at least twenty types of human cancer (Han et al., 2015; Jiang et al., 2013; Qi et al., 2014; Qin et al., 2014; Shah et al., 2009; Zipeto et al., 2016). Because the majority of A-to-I RNA editing events occur within dsRNA loops created by Alu repeat sequences (Deininger, 2011; Jiang et al., 2017; Tan et al., 2017), the functional role of ADAR1 in cancer progression is best studied mechanistically in humanized systems.
In this study, we observed that ADAR1 activation is sufficient to induce normal HSPC cell expansion by inducing differential expression of cell cycle transcripts. Tightly controlled expression of cell cycle regulatory genes is achieved by A-to-I RNA editing of pri-miRNAs and 3’UTR of transcripts in cell cycle pathways. Cytoscape analysis of RNA-seq revealed that CDKN1A represents a central hub in ADAR1 regulated cell cycle transit in normal HSPC. CDKN1A maintains HSC in a quiescent state after induction of DNA repair pathways and ADARI-regulated depletion of CDKN1A results in accelerated cell cycle transit. Moreover, the decreased expression of miR-26a and its role in cell cycle regulation supports our hypothesis that ADAR1-regulated miRNA biogenesis is essential for maintenance of HSC proliferation. miR-26a inhibits LIN28B expression in both normal HSPC and BC CML cells, suggesting that ADAR1-mediated miR-26a reduction is a parallel pathway of LIN28B/let-7 axis regulation (Zipeto et al., 2016). In pre-malignant progenitors with mutations that promote survival, such as BCR-ABL, deregulated ADAR1 contributes to the malignant reprogramming of progenitors into dormant LSCs. In this setting, ADAR1 mediated A-to-I hyper-editing prevents binding of miRNA to the 3’UTR of MDM2 mRNA, which results in increased MDM2 expression and repression of the p53 tumor suppressor. Thus, ADAR1 inhibition may represent a potent method for eradicating LSC.
A recent study of dynamic RNA editing in mammals showed that ADAR1 is the primary editor of repetitive sites and ADAR2 is the editor of non-repetitive coding region (Tan et al., 2017). Indeed, ADAR1 editing sites of cell cycle transcripts in both normal HSPC and CML progenitors occurs in noncoding regions such as Alu-rich intronic sequences and 3’ UTRs. However, ADAR1 clearly possesses disease-specific preferential targeting of certain editing sites, such as the MDM2 3’ UTR in BC progenitors. It is possible that this preferential A-to-I targeting is caused by disease- or cell type-specific expression of ADAR1 activity regulators as recently reported (Tan et al., 2017). The dichotomous role of A-to-I RNA editing in HSPC and LSC suggest that future studies of malignant CSC reprogramming should incorporate disease-, cell type-, and tissue-specific mechanisms.
These results also highlight a link between ADAR1 activation and EZH2 expression. EZH2 is the core subunit of the polycomb repressive complex 2 (PRC2) with histone methyltransferase activity that introduces H3K27me3 at target gene promoters thereby suppressing gene expression. EZH2 expression is tightly associated with cell proliferation (Margueron et al., 2008) and is upregulated in ADAR1-overexpressing HSPC through inhibition of miR-26a that directly targets EZH2 mRNA. However, EZH2 upregulation by ADAR1 is disrupted in LSC due to activation of oncogenes, such as MYC. This raises the possibility that post-transcriptional A-to-I RNA editing may influence normal HSPC maintenance and that disruption of this regulation by cancer-specific oncogenic pathways may lead to malignant progenitor propagation. Since deregulated RNA editing activity is associated with many types of cancer, further work is needed to elucidate ADARI’s role in epitranscriptomic disruption in other cancer types, as well as identification of the corresponding coding and non-coding RNA editing target transcripts. Understanding the cell type and context specific effects of A-to-I editing has become even more pressing since recent studies showed that a catalytically inactive Cas13 can be used to direct ADAR-mediated RNA editing to specific transcripts. The RNA Editing for Programmable A to I Replacement (REPAIR) system holds promise for treating intractable genetic diseases, particularly in post-mitotic cells (Cox et al., 2017).
In conclusion, we have uncovered a dichotomous role for ADAR1 in normal and malignant progenitor cell cycle regulation and maintenance that is resulted from suppression of miRNA biogenesis and 3’UTR hyper-editing of miRNA binding sites. This dual mechanism provides an efficient way to regulate gene expression through A-to-I RNA editing of noncoding sequences. Dormant BC CML LSCs in the bone marrow protective niche often escape therapies that target dividing cells thereby contributing to therapeutic resistance and disease relapse (Goff et al., 2013). Therefore, ADAR1 inhibition may represent an effective modality for eliminating dormant LSCs that evade tyrosine kinase inhibitor in CML but also in other advanced malignancies that co-opt ADAR1.
STAR METHODS
CONTACT for REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Catriona Jamieson (cjamieson@ucsd.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal Experiments
All mouse studies were conducted under protocols approved by the Institutional Animal Care and Use Committee (IACUC) of the University of California, San Diego and were in compliance with federal regulations regarding the care and use of laboratory animals: Public Law 99-158, the Health Research Extension Act, and Public Law 99-198, the Animal Welfare Act which is regulated by USDA, APHIS, CFR, Title 9, Parts 1, 2, and 3. Immunocompromised Rag2−/−γc−/− mice were bred and maintained in the Sanford Consortium for Regenerative Medicine vivarium according to IACUC approved protocols of the University of California, San Diego. Neonatal mice of both sexes were used in the study. BC CML CD34+ cells (1-2×105) were injected intrahepatically into neonatal Rag2−/−γc−/− mice. Leukemic engraftment was quantified by FACS analysis-based peripheral blood screening of human CD45+ population starting from week 6 for every 2 weeks until the engraftment exceeded 1%. Mice were then humanely sacrificed and cells were collected from hematological organs (bone marrow, spleen and liver) for FACS analysis.
Human Subjects
Primary adult non-leukemic blood and bone marrow as well as patient chronic myeloid leukemia (CML) samples were obtained from consenting patients at the University of California, San Diego in accordance with an approved human research protections program Institutional Review Board approved protocol (#131550) that meets the requirements as stated in 45 CFR 46.404 and 21 CFR 50.51. De-identified (IRB exempt) human cord blood and normal aged-match samples were purchased as purified CD34+ cells from AllCells Inc or StemCell Techologies Inc. Detailed patient information can be found in Table S4.
Cell culture
All human cell lines (HEK293T and K562) were cultured in 37°C in DMEM supplemented with 10% FBS and 2 mM L-glutamine and maintained according to ATCC protocols. All cell lines were confirmed to be mycoplasma-free with repeated testing and authenticated by short-tandem repeat (STR) profiling.
METHOD DETAILS
Patient sample preparation.
Peripheral blood mononuclear cells (PBMC) were extracted by Ficoll density centrifugation and were CD34-selected (MACS, Miltenyi), followed by FACS sorted with human-specific antibody according to published methods (Abrahamsson et al., 2009; Goff et al., 2013; Jiang et al., 2013; Zipeto et al., 2016) and analyzed and purified with the FACSAria and FlowJo software.
Cross-linking immunoprecipitation (CLIP)
K562 and HEK293T cell lines were purchased and cultured according to the manufacturer’s protocol (ATCC). All cells were tested negative for mycoplasma. For generating K562 and HEK293T cell lines stably expressing pCDH backbone control, ADAR1 WT, or ADAR1E912A cells were transduced at 5×104 cells in a 24 well plate with a multiplicity of infection (MOI) of 100 for 48-72 hr. Cells were then cultured in 6 well plate till the total cell number reached 1×106 and FACS-sorted based on GFP+ signal. CLIP in K562 were performed with an anti-ADAR1 antibody (ab168809, Abcam) using previously published protocol(Zipeto et al., 2016). The expression of ADAR1 was confirmed in HEK293T and K562 cells every 3-5 passages by qRT-PCR.
MDM2 3’UTR reporter assay
MDM2 3’UTR reporter construct was purchased from manufacturer (GeneCopoeia, HmiT065341-MT05) and A-to-I editing site changes were introduced with A-to-G site-directed mutagenesis. HEK293T cells were transduced with pCDH backbone or miR-155 overexpressing lentivirus at 50% confluence with a MOI of 50. After 48 hr, transduced HEK293T cells were collected and plated into 24-well plate and transfected with either “wild-type” or “edited” MDM2 3’UTR reporter at a MOI of 50. The media is collected 24 hr after transfection and the relative luciferase activity (Gluc/SEAP ratio) was determined according to the manufacturer’s protocol (GeneCopoeia, LF033).
Lentiviral construct and overexpression.
Lentiviral human miR-26a (pCDH-CMV-hsa-miR-26a-EF1-copGFP) was purchased (System Biosciences), and wild-type and mutant ADAR1E912A (pCDH-EF1-T2A-copGFP) were produced according to published protocol (Zipeto et al., 2016). All lentivirus was tested by transduction of HEK293T cells and efficiency was assessed by FACS analysis of GFP signal and qRT-PCR. Lentiviral transduction of primary patient samples was performed at a MOI of 100-200. The cells were cultured for 3-4 days in 96-well plate (2×105-5×105 cells per well) containing StemPro (Life Technologies) media supplemented with human IL-6, stem cell factor (SCF), Thrombopoietin (Tpo) and FLT-3 (all from R&D Systems) (Abrahamsson et al., 2009; Goff et al., 2013; Jiang et al., 2013; Zipeto et al., 2016). The transduced cells were collected for either in vitro flow analysis or in vivo transplant experiment. For in vitro flow analysis, the cells were stained with CD34-APC, CD38-PECY7, and a lineage cocktail-PECY5.5 (CD8, CD56, CD4, CD3, CD19, CD2, and CD14) and the percentage of stem cells (CD34+CD38−Lin−) and progenitors (CD34+CD3+Lin−) were evaluated.
RNA and microRNA extraction and quantitative real-time polymerase chain reaction.
Total RNA was isolated from 2×105-5×105 FACS-sorted or CD34+ selected cells from normal cord blood, CP CML, and BC CML, and complementary DNA was synthesized according to published methods (Abrahamsson et al., 2009; Goff et al., 2013; Jiang et al., 2013; Zipeto et al., 2016). qRT-PCR was performed in duplicate or triplicate on an iCycler with the use of SYBR GreenER qPCR SuperMix (Invitrogen), 5 ng of template mRNA and 0.2 μM of each forward and reverse primer (Table S5). Human specific HPRT primers were used as housekeeping control. The primers were listed in Table S5. MicroRNA extraction was performed using the RNeasy Micro Kit (Qiagen) according to the manufacturer’s instructions. Then 30 ng of cDNA was prepared in a reverse-transcription reaction using miScript II RT kit (Qiagen, 218161) and served as a template for the quantification of the expression of mature miRNA of interest. qRT-PCR was performed using mature miRNA human-specific primers (Qiagen) and miScript SYBR Green PCR Kit (Qiagen, 218076). MiScript primers, RNU6_2 (Qiagen), were used as housekeeping control.
Cell cycle qRT-PCR array
The isolated RNA (20 ng) were converted into cDNA using RT2 First Strand Kit (Qiagen, #330401) and pre-amplified for cell cycle pathway with RT2 PreAMP cDNA Synthesis Kit (Qiagen, #330451, PBH-020Z). The PCR array profiling of 84 cell cycle genes and 5 housekeeping genes were performed using the RT2 SYBR Green Fluor qPCR mastermix according to manufacturer’s protocol (Qiagen, #330512 and PAHS-020Z).
miRNome qPCR array, DIANA miRNA target pathway analysis, and miRNA binding site prediction
miRNome profiling was performed by using miScript miRNome PCR arrays (Qiagen, MIHS-216Z). 10 ng of miRNA were reverse transcribed by using miScript II RT-PCR according to the manufacturer’s instructions. The reverse transcribed cDNA functioned as a template for the pre-amplification. Pre-amplification of mature miRNA was performed by using miScript PreAmp PCR kit (Qiagen, 331451). 10 μl of cDNA were diluted into 40 μl of H2O. 5 μl of diluted cDNA were used as a template for the pre-amplification reactions. Three different pre-amplification reactions were set up for each sample, each one using a different set of primer mix to cover the entire miRNome (Qiagen, MBHS-3216Z). Following pre-amplification, pre-amplified miRNA was pulled in one tube and used for miRNome qPCR assay. The miRNA expression was normalized to RNU6_2 housekeeping gene and the fold change to pCDH lentiviral vector control was calculated. Significantly differentiated miRNAs were analyzed for mRNA targets using DIANA mirPath software (Vlachos et al., 2015) (http://diana.imis.athena-innovation.gr/DianaTools/index.php). The predicted miRNA binding sites were determined using miRcode transcriptome-wide miRNA target prediction tool (Jeggari et al., 2012) (http://mircode.org/index.php).
DiR staining and measurement by FACS
Cord blood CD34+ cells (1×105) were isolated and stained with 4 mg/mL DiR (Invitrogen) in PBS according to the manufacturer’s specifications as described previously (Goff et al., 2013). DiR stained cells were then washed and transduced with GFP+ lentiviral vectors. After 3 days, cells were collected and analyzed by FACS for GFP+ and DiR+ cells.
Flow cytometry cell cycle analysis
FACS cell cycle analysis was performed with 7-AAD and Ki-67 as previously described (Goff et al., 2013). Single cell suspensions of bone marrow cells of engrafted mice with either lentiviral backbone or miR-26a conditions were immunostained with Alexa405-conjugated anti-human CD45 (Invitrogen), Alexa647-anti-human CD38 (Ab Serotec) and biotin-anti-human CD34 (Invitrogen) plus Alexa488-strepavidin (Invitrogen) in 2% fetal bovine serum/ PBS- followed by live cell staining using the LIV&OEAD® Fixable Near-IR Dead Cell Stain Kit (Invitrogen). Surface stained cells were then fixed in 70% ethanol overnight and were immunostained with PE-conjugated anti-Ki-67 (BD) in 0.15% saponin/ 2% fetal bovine serum/ PBS, washed and incubated with 7-AAD (Invitrogen, 10 μg/mL in 0.1 M sodium citrate/ 5 mM EDTA pH8.0/ 0.15 M NaCl/ 0.5% BSA/ 0.02% saponin). For HEK293T cells, cells were transduced with lentiviral backbone or miR-26a for 3 days and then stained with the LIVE/DEAD® Fixable Near-IR Dead Cell Stain Kit (Invitrogen). Cells were then fixed in 70% ethanol for 4 hr at 4°C and immunostained with PE-conjugated anti-Ki-67 (BD) and 7-AAD as described. Stained samples were analyzed using a FACSAria and FlowJo.
Western blots
HEK293T cell lysate (10 mg) was mobilized onto nitrocellulose member after electrophoresis on a 4-20% gradient acrylamide gel. The member was blocked in 5% BSA/20 mM Tris-HCl for 1 hr. The blot was incubated with primary CDKN1A antibody (Abcam, ab18209) in 5% BSA/20 mM Tris-HCl/0.1% Tween-20 overnight at 4°C, followed by secondary HP R-linked Rabbit IgG antibody (Cell Signaling, #70745) for 2-4 hr at room temperature. The member was incubated in SuperSignal West Femto Substrate (ThermoFisher, #34096) for chemiluminescent reading on ChemiDoc System (Bio-Rad).
Immunofluorescent staining
Cord blood CD34+ cells (2×105 cells, 200 μL) was cytospun onto slides at 500 rpm for 5 min, fixed for 10 min in 4% PFA at room temperature, rinsed with PBS, and incubated with 5% normal donkey serum/0.2% Triton X-100 followed by incubation with primary antibodies overnight at 4°C. Primary antibodies used were anti-human PE-conjugated Ki-67 (BD) and anti-CDKN1A [CP74] Biotin (Abcam, ab79467). Stained slides were then incubated with secondary Alexa Fluor® 488 Goat Anti-Mouse IgG (H+L) Antibody (Life Technologies, A11029) and mounted using Prolong® Gold antifade with DAPI. Images were acquired using confocal fluorescence microscopy (Olympus Fluoview FV10i) and Adobe Photoshop CS5.
Hematopoietic colony formation assay.
After lentiviral transduction, normal cord blood or CML patient CD34+ cells were plated into Methocult Medium (50-100 cells per well, 12 well plate). After two weeks, colonies were scored and individual colonies were replated into fresh MethoCult media as previously described (Jiang et al., 2013). Secondary colonies were scored after an additional two weeks in culture.
Human progenitor xenotransplantation
BC CML CD34+ cells were transduced with lentiviral backbone or miR-26a with a MOI of 200 for 3 days. Neonatal mice were transplanted intrahepatically with 1×105-2×105 transduced BC CML CD34+ cells according to our published methods (Abrahamsson et al., 2009; Goff et al., 2013; Jiang et al., 2013; Zipeto et al., 2016). Transplanted mice were FACS screened for human engraftment in peripheral blood at 6-10 weeks. Once human engraftment was confirmed (>1% human CD45+ cells in peripheral blood), mice were euthanized and single cell suspensions of hematopoietic tissues were analyzed by FACS for human CD45+ engraftment and cell cycle analysis (Goff et al., 2013).
RNA-sequencing read preprocessing
Primary normal and CML samples were obtained and RNA-sequencing analysis were performed according to published methods (Abrahamsson et al., 2009; Goff et al., 2013; Jiang et al., 2013; Zipeto et al., 2016). For 50 bp paired end reads from previously aligned data, the reads were converted from bam to fastq using samtools bam2fq (Li et al., 2009). For 100 bp paired end reads, the reads were entered into pre-processing as is. Reads were cleaned using cutadapt to remove Illumina universal adapters (Martin, 2011).
H3K27me3 CHIP-sequencing
ChIP was performed with 2×105 CD34+ cord blood cells (per condition). CD34+ cord blood cells were crosslinked in 1% formaldehyde after transduction with pCDH backbone, ADAR1 WT or ADAR1E912A for 48 hr. The crosslinking reaction was quenched in 0.2 M glycine and lysed in Cell Lysate Buffer (50 mM Tris pH8.0, 10 nM EDTA, 1% SDS). Lysates were subjected to chromatin shearing according to the manufacturer’s protocol (truCHIP™ Chromatin Shearing Reagent Kit, Covaris). Sample libraries were prepared using ACCEL-NGS 2S Plus DNA library kits (Swift Biosciences) and sequenced on Illumina HiSeq 2500.
H3K27me3 CHIP-qPCR
ChIP-qPCR were performed according to the manufacturer’s instructions (LowCell#ChIP kit from Diagenode, cat#C01010072). In summary, cross-linking of HEK293T cells overexpressed ADAR1 WT and pCDH backbone was performed using 1% formaldehyde. Chromatin shearing was performed in Cell Lysate Buffer (50 mM Tris pH8.0, 10 nM EDTA, 1% SDS, 1× protease inhibitor) and then sonicated for 10 min in 0.5 min pulse intervals to obtain small fragments of DNA (between 300 and 500 kb). Antibodies (Rabbit IgG cat# C15410206 Diagenode; Histone H3K27me3 cat# 39155 Active Motif) were bound to protein A-coated magnetic beads and incubated for 2 hr in 4°C. The fragmented chromatin was added to antibody-coated beads and incubated on a rotating wheel overnight at 4°C. The samples were washed three times with Washing Buffer (50 mM HEPES pH7.5, 0.5 M LiCl, 1 mM EDTA, 1% NP-40) then two times with TE buffer. The sheared chromatin was eluted using the diagenode Elution Module (cat# mc-magme-002). The samples were subject to qRT-PCR using primers listed in Table S5.
QUANTIFICATION AND STATISTICAL ANALYSIS
Read alignment and gene counts
Reads were aligned using STAR’s two-pass alignment method, using the GRCh38.84 reference genome and corresponding Ensembl GTF (Aken et al., 2016; Guo et al., 2017). STAR was used to output a sorted genome-coordinate based BAM file, as well as a transcriptome-coordinate based BAM file (Dobin et al., 2013) (https://github.com/alexdobin/STAR). STAR also was used to output the number of reads aligned to each gene similar to hi-seq count. STAR settings were based on those used for the ENCODE STAR-RSEM pipeline. The infer_experiment.py script from the RSeQC package was used to confirm the strandedness option corresponding to the correct read counts (Li and Dewey, 2011; Wang et al., 2012) (http://rseqc.sourceforge.net/), and also to confirm the forward strand probability for input to RSEM. The total reads per million (TPM) (Mortazavi et al., 2008) over the total collapsed exonic regions represent the ‘gene’ expression level.
RNA editing analysis
Coordinates from the DARNED and RADAR databases were combined and converted to GRCh38 using Crossmap (Kiran and Baranov, 2010; Ramaswami and Li, 2014; Zhao et al., 2014). The resulting coordinates were used as input to the REDItoolKnown.py script from the REDItools package to determine the number of A, C, G, and T base calls at each coordinate (Picardi and Pesole, 2013) (http://srv00.recas.ba.infn.it/reditools/). Only coordinates with coverage greater than or equal to 5 in all samples for a given comparison were reported. The percentage of bases called as G at bases with reference A was reported. Coordinates with a percentage G of 0 in all samples for a given sample were not reported. Using percentage G at a coordinate as an input metric, the mean percentage G in each group, the log2 fold change of percentage G of one group versus another, the p values, and minus log10 p values by both the Wilcox and student t-tests were recorded for each coordinate similar to published methods (Jiang et al., 2013). Coordinates were annotated with the name of the closest gene using bedtools closest and bedtools intersect (Quinlan and Hall, 2010) (http://bedtools.readthedocs.io/en/latest/). The coordinates annotated with the names of genes in the KEGG cell cycle gene set were recorded.
Transcript and gene quantification and differential expression
The transcriptome-coordinate based BAM from the read alignment step was input to RSEM, using settings based on the ENCODE STAR-RSEM pipeline (Li and Dewey, 2011). RSEM was provided the GRCh38.84 reference genome and corresponding Ensembl GTF for its transcriptome reference. RSEM was used to provide TPM and expected counts for genes and transcripts. For genes, the gene count data generated by STAR in the alignment step was used as input to EdgeR (Dobin et al., 2013; Robinson et al., 2010) (http://bioconductor.org/packages/release/bioc/html/edgeR.html). For transcripts, the expected counts data from RSEM was used as input to EdgeR. Only features with a minimum CPM of 0.5 (in at least half the samples in the comparison) as measured by EdgeR were submitted to EdgeR’s differential expression, to yield log2 fold change, p value, and FDR for each feature for the comparison. The threshold for significant genes and transcripts was set at a p value less than 0.05 and an FDR less than 0.10. An additional threshold based on the absolute value of the log2 fold change was also used to filter features for inclusion in a heatmap. Heatmaps visualize the log2(TPM+1) transformed TPM quantity from RSEM for each feature, and were generated using GENE-E with default settings for a row and column clustered heatmap and dendrogram.
H3K27me3 CHIP-sequencing
ChIP-seq data is analyzed by following steps. Single-end ChIP-seq reads are mapped to the human reference genome hg19 using BWA (http://bio-bwa.sourceforge.net/) with default setting. Non-uniquely mapped reads (MAPQ < 30) are filtered and the remaining reads are sorted by genomic coordinates using samtools sort. PCR duplicates was removed using samtools rmdup (-s). Bigwig file was generated using deeptools (https://deeptools.readthedocs.io/en/develop/) and the data is visualized using IGV (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3346182/).
Other Statistical Analysis and Reproducibility
All experiments were performed with at least three biological or experimental replicates, with specific number of replicates stated in the figure legends. Unless otherwise stated, the statistical analyses were performed using GraphPad Prism (v7.0) and statistical significance were determined at p value < 0.05, with specific statistical test stated in the figure legends.
DATA AND SOFTWARE AVAILABILITY
The RNA-sequencing dataset used in this study were available from previous studies. The BC and CP progenitors RNA-sequencing dataset is available from (Jiang et al., 2013) (BioProject: PRJNA214016) and cord blood transduced with backbone or ADAR1 WT dataset is available from (Zipeto et al., 2016) (BioProject: PRJNA319866).
Supplementary Material
Table S1, Related to Figure 1. Cell Cycle qPCR array.
Table S2, Related to Figure 2. A-to-I edited miRNome by ADAR1 in cord blood HSPC.
Table S3, Related to Figure 5. A-to-I RNA editome of KEGG cell cycle transcripts in cord blood HSPC, CML CP and CML BC progenitors by RNA-seq.
Significance.
Cumulative reports demonstrate that ADAR1-mediated A-to-I RNA editing contributes to proteomic diversity, therapeutic resistance and progression of a broad array of malignancies. However, the precise molecular mechanistic underpinnings of ADAR1-mediated cancer resistance and progression had not been elucidated. Here we show that ADAR1 promotes malignant progenitor cell cycle deregulation through hyper-editing of cell cycle regulatory and tumor suppressor coding and non-coding transcripts. By defining the combined cell cycle and tumor suppressor regulatory mechanisms governed by A-to-I RNA editing in malignant compared with normal progenitors, prognostic RNA editing based biomarkers may be developed together with ADAR1 inhibitory strategies that provide a reasonable therapeutic index to treat various types of cancers.
Highlights.
A-to-I RNA editing alters cell cycle transit by impairing pri-miR-26a maturation
Enforced miR-26a expression reduces BC CML progenitor propagation in vivo
pri-miR-155 downregulation by ADAR1 stabilizes MDM2 in CML progenitors
Hyper-editing of 3’UTR of MDM2 prevents miR-155 binding in CML progenitors
Jiang et al. show that ADAR1 promotes HSPC proliferation by indirectly reducing CDKN1A expression whereas it enhances the propagation of blast crisis chronic myeloid leukemia progenitors, which express high levels of CDKN1A, by editing the MDM2 regulatory miRNA and the miRNA binding site to stabilize MDM2 mRNA.
ACKNOWLEDGEMENTS
This work was supported by NIH/NCI R01CA205944; NIH/NIDDK R01DK114468-01; NIH NCI R21CA189705; NIH NIGMS 5K12GM068524; NIH NCI 2P30CA023100-28; The Sanford Stem Cell Clinical Center; the Koman Family Foundation; the Strauss Family Foundation; and the Moores Family Foundation. N.D.S was supported by NIH grant T15LM011271. The authors wish to thank Dr. Ida Deichaite for scientific advice, and the Scripps and Canada’s Michael Smith Genome Sciences Centre Library Construction, Sequencing and Bioinformatics teams.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
C. Jamieson was a co-founder of Impact Biomedicines Inc, which was acquired by Celgene, and holds stock in Forty Seven Inc.
REFERENCES
- Abrahamsson AE, Geron I, Gotlib J, Dao KH, Barroga CF, Newton IG, Giles FJ, Durocher J, Creusot RS, Karimi M, et al. (2009). Glycogen synthase kinase 3beta missplicing contributes to leukemia stem cell generation. Proceedings of the National Academy of Sciences of the United States of America 106, 3925–3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aken BL, Ayling S, Barrell D, Clarke L, Curwen V, Fairley S, Fernandez Banet J, Billis K, Garcia Giron C, Hourlier T, et al. (2016). The Ensembl gene annotation system. Database (Oxford) 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batzer MA, and Deininger PL (2002). Alu repeats and human genomic diversity. Nat Rev Genet 3, 370–379. [DOI] [PubMed] [Google Scholar]
- Beguelin W, Teater M, Gearhart MD, Calvo Fernandez MT, Goldstein RL, Cardenas MG, Hatzi K, Rosen M, Shen H, Corcoran CM, et al. (2016). EZH2 and BCL6 Cooperate to Assemble CBX8-BCOR Complex to Repress Bivalent Promoters, Mediate Germinal Center Formation and Lymphomagenesis. Cancer Cell 30, 197–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Zhang K, Xu Y, Gao Y, Li C, Wang R, and Chen L (2016). The role of microRNA-26a in human cancer progression and clinical application. Tumour Biol 37, 7095–7108. [DOI] [PubMed] [Google Scholar]
- Chen L, Li Y, Lin CH, Chan TH, Chow RK, Song Y, Liu M, Yuan YF, Fu L, Kong KL, et al. (2013). Recoding RNA editing of AZIN1 predisposes to hepatocellular carcinoma. Nature medicine 19, 209–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou CH, Chang NW, Shrestha S, Hsu SD, Lin YL, Lee WH, Yang CD, Hong HC, Wei TY, Tu SJ, et al. (2016). miRTarBase 2016: updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res 44, D239–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung H, Calis JJA, Wu X, Sun T, Yu Y, Sarbanes SL, Dao Thi VL, Shilvock AR, Hoffmann HH, Rosenberg BR, et al. (2018). Human ADAR1 Prevents Endogenous RNA from Triggering Translational Shutdown. Cell 172, 811–824 e814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox DBT, Gootenberg JS, Abudayyeh OO, Franklin B, Kellner MJ, Joung J, and Zhang F (2017). RNA editing with CRISPR-Cas13. Science. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews LA, Balaian L, Delos Santos NP, Leu HS, Court AC, Lazzari E, Sadarangani A, Zipeto MA, La Clair JJ, Villa R, et al. (2016). RNA Splicing Modulation Selectively Impairs Leukemia Stem Cell Maintenance in Secondary Human AML. Cell Stem Cell [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deininger P (2011). Alu elements: know the SINEs. Genome Biol 12, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan T, Jiang S, Chung N, Alikhan A, Ni C, Lee CC, and Hornyak TJ (2011). EZH2-dependent suppression of a cellular senescence phenotype in melanoma cells by inhibition of p21/CDKN1A expression. Mol Cancer Res 9, 418–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatica A, and Fazi F (2013). MicroRNA-regulated pathways in hematological malignancies: how to avoid cells playing out of tune. Int J Mol Sci 14, 20930–20953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X, Meng Z, Liang W, Tian Y, Wang X, Han W, Lou G, Wang X, Lou F, Yen Y, et al. (2014). miR-26a enhances miRNA biogenesis by targeting Lin28B and Zcchc11 to suppress tumor growth and metastasis. Oncogene 33, 4296–4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff DJ, Recart AC, Sadarangani A, Chun HJ, Barrett CL, Krajewska M, Leu H, Low-Marchelli J, Ma W, Shih AY, et al. (2013). A Pan-BCL2 inhibitor renders bone-marrow-resident human leukemia stem cells sensitive to tyrosine kinase inhibition. Cell Stem Cell 12, 316–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Dai Y, Yu H, Zhao S, Samuels DC, and Shyr Y (2017). Improvements and impacts of GRCh38 human reference on high throughput sequencing data analysis. Genomics 109, 83–90. [DOI] [PubMed] [Google Scholar]
- Han L, Diao L, Yu S, Xu X, Li J, Zhang R, Yang Y, Werner HM, Eterovic AK, Yuan Y, et al. (2015). The Genomic Landscape and Clinical Relevance of A-to-I RNA Editing in Human Cancers. Cancer Cell 28, 515–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartner JC, Walkley CR, Lu J, and Orkin SH (2009). ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat Immunol 10, 109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamieson CH, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL, Gotlib J, Li K, Manz MG, Keating A, et al. (2004). Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med 351, 657–667. [DOI] [PubMed] [Google Scholar]
- Jeggari A, Marks DS, and Larsson E (2012). miRcode: a map of putative microRNA target sites in the long non-coding transcriptome. Bioinformatics 28, 2062–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Crews LA, Holm F, and Jamieson CHM (2017). RNA editing-dependent epitranscriptome diversity in cancer stem cells. Nat Rev Cancer 17, 381–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Crews LA, and Jamieson CH (2013). ADAR1 promotes malignant progenitor reprogramming in chronic myeloid leukemia. Proceedings of the National Academy of Sciences of the United States of America 110, 1041–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J, and Smith T (1988). A fundamental division in the Alu family of repeated sequences. Proceedings of the National Academy of Sciences of the United States of America 85, 4775–4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaymaz BT, Gunel NS, Ceyhan M, Cetintas VB, Ozel B, Yandim MK, Kipcak S, Aktan C, Gokbulut AA, Baran Y, et al. (2015). Revealing genome-wide mRNA and microRNA expression patterns in leukemic cells highlighted “hsa-miR-2278” as a tumor suppressor for regain of chemotherapeutic imatinib response due to targeting STAT5A. Tumour Biol 36, 7915–7927. [DOI] [PubMed] [Google Scholar]
- Kim KH, and Roberts CW (2016). Targeting EZH2 in cancer. Nature medicine 22, 128–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiran A, and Baranov PV (2010). DARNED: a DAtabase of RNa EDiting in humans. Bioinformatics 26, 1772–1776. [DOI] [PubMed] [Google Scholar]
- Lechman ER, Gentner B, Ng SW, Schoof EM, van Galen P, Kennedy JA, Nucera S, Ciceri F, Kaufmann KB, Takayama N, et al. (2016). miR-126 Regulates Distinct Self-Renewal Outcomes in Normal and Malignant Hematopoietic Stem Cells. Cancer Cell 29, 602–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, and Walkley CR (2015). RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349, 1115–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, He ML, Wang L, Chen Y, Liu X, Dong Q, Chen YC, Peng Y, Yao KT, Kung HF, et al. (2011). MiR-26a inhibits cell growth and tumorigenesis of nasopharyngeal carcinoma through repression of EZH2. Cancer Res 71, 225–233. [DOI] [PubMed] [Google Scholar]
- Lund K, Adams PD, and Copland M (2014). EZH2 in normal and malignant hematopoiesis. Leukemia 28, 44–49. [DOI] [PubMed] [Google Scholar]
- Mallela A, and Nishikura K (2012). A-to-I editing of protein coding and noncoding RNAs. Crit Rev Biochem Mol Biol 47, 493–501. [DOI] [PubMed] [Google Scholar]
- Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, Nellaker C, Vesely C, Ponting CP, McLaughlin PJ, et al. (2014). The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep 9, 1482–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcu-Malina V, Goldberg S, Vax E, Amariglio N, Goldstein I, and Rechavi G (2016). ADAR1 is vital for B cell lineage development in the mouse bone marrow. Oncotarget. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Li G, Sarma K, Blais A, Zavadil J, Woodcock CL, Dynlacht BD, and Reinberg D (2008). Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol Cell 32, 503–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt Removes Adapter Sequences From High-Throughput Sequencing Reads. EMBnetjournal 17. [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, and Wold B (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5, 621–628. [DOI] [PubMed] [Google Scholar]
- Neri F, Zippo A, Krepelova A, Cherubini A, Rocchigiani M, and Oliviero S (2012). Myc regulates the transcription of the PRC2 gene to control the expression of developmental genes in embryonic stem cells. Mol Cell Biol 32, 840–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikura K (2010). Functions and regulation of RNA editing by ADAR deaminases. Annu Rev Biochem 79, 321–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikura K (2016). A-to-I editing of coding and non-coding RNAs by ADARs. Nat Rev Mol Cell Biol 17, 83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlyn C, Bright MD, Buros AF, Stein CK, Walters Z, Aronson LI, Mirabella F, Jones JR, Kaiser MF, Walker BA, et al. (2017). Overexpression of EZH2 in multiple myeloma is associated with poor prognosis and dysregulation of cell cycle control. Blood Cancer J 7, e549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X, Xu X, Wang Y, Hawke DH, Yu S, Han L, Zhou Z, Mojumdar K, Jeong KJ, Labrie M, et al. (2018). A-to-I RNA Editing Contributes to Proteomic Diversity in Cancer. Cancer Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picardi E, and Pesole G (2013). REDItools: high-throughput RNA editing detection made easy. Bioinformatics 29, 1813–1814. [DOI] [PubMed] [Google Scholar]
- Qi L, Chan TH, Tenen DG, and Chen L (2014). RNA editome imbalance in hepatocellular carcinoma. Cancer Res 74, 1301–1306. [DOI] [PubMed] [Google Scholar]
- Qin YR, Qiao JJ, Chan TH, Zhu YH, Li FF, Liu H, Fei J, Li Y, Guan XY, and Chen L (2014). Adenosine-to-inosine RNA editing mediated by ADARs in esophageal squamous cell carcinoma. Cancer Res 74, 840–851. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami G, and Li JB (2014). RADAR: a rigorously annotated database of A-to-I RNA editing. Nucleic Acids Res 42, D109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami G, and Li JB (2016). Identification of human RNA editing sites: A historical perspective. Methods. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvatori B, Iosue I, Djodji Damas N, Mangiavacchi A, Chiaretti S, Messina M, Padula F, Guarini A, Bozzoni I, Fazi F, et al. (2011). Critical Role of c-Myc in Acute Myeloid Leukemia Involving Direct Regulation of miR-26a and Histone Methyltransferase EZH2. Genes Cancer 2, 585–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander S, Bullinger L, Klapproth K, Fiedler K, Kestler HA, Barth TF, Moller P, Stilgenbauer S, Pollack JR, and Wirth T (2008). MYC stimulates EZH2 expression by repression of its negative regulator miR-26a. Blood 112, 4202–4212. [DOI] [PubMed] [Google Scholar]
- Shah SP, Morin RD, Khattra J, Prentice L, Pugh T, Burleigh A, Delaney A, Gelmon K, Guliany R, Senz J, et al. (2009). Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature 461, 809–813. [DOI] [PubMed] [Google Scholar]
- Tan MH, Li Q, Shanmugam R, Piskol R, Kohler J, Young AN, Liu KI, Zhang R, Ramaswami G, Ariyoshi K, et al. (2017). Dynamic landscape and regulation of RNA editing in mammals. Nature 550, 249–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotta R, Vignudelli T, Candini O, Intine RV, Pecorari L, Guerzoni C, Santilli G, Byrom MW, Goldoni S, Ford LP, et al. (2003). BCR/ABL activates mdm2 mRNA translation via the La antigen. Cancer Cell 3, 145–160. [DOI] [PubMed] [Google Scholar]
- Vlachos IS, Zagganas K, Paraskevopoulou MD, Georgakilas G, Karagkouni D, Vergoulis T, Dalamagas T, and Hatzigeorgiou AG (2015). DIANA-miRPath v3.0: deciphering microRNA function with experimental support. Nucleic Acids Research 43, W460–W466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wang S, and Li W (2012). RSeQC: quality control of RNA-seq experiments. Bioinformatics 28, 2184–2185. [DOI] [PubMed] [Google Scholar]
- Wang Q, Khillan J, Gadue P, and Nishikura K (2000). Requirement of the RNA editing deaminase ADAR1 gene for embryonic erythropoiesis. Science 290, 1765–1768. [DOI] [PubMed] [Google Scholar]
- Wojtowicz EE, Lechman ER, Hermans KG, Schoof EM, Wienholds E, Isserlin R, van Veelen PA, Broekhuis MJ, Janssen GM, Trotman-Grant A, et al. (2016). Ectopic miR-125a Expression Induces Long-Term Repopulating Stem Cell Capacity in Mouse and Human Hematopoietic Progenitors. Cell Stem Cell 19, 383–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia K, Zhang Y, Cao S, Wu Y, Guo W, Yuan W, and Zhang S (2015). miR-411 regulated ITCH expression and promoted cell proliferation in human hepatocellular carcinoma cells. Biomed Pharmacother 70, 158–163. [DOI] [PubMed] [Google Scholar]
- Xie H, Peng C, Huang J, Li BE, Kim W, Smith EC, Fujiwara Y, Qi J, Cheloni G, Das PP, et al. (2016). Chronic Myelogenous Leukemia-Initiating Cells Require Polycomb Group Protein EZH2. Cancer Discov 6, 1237–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H, Xu J, Hsu JH, Nguyen M, Fujiwara Y, Peng C, and Orkin SH (2014). Polycomb repressive complex 2 regulates normal hematopoietic stem cell function in a developmental-stage-specific manner. Cell Stem Cell 14, 68–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CC, Chen YT, Chang YF, Liu H, Kuo YP, Shih CT, Liao WC, Chen HW, Tsai WS, and Tan BC (2017). ADAR1-mediated 3’ UTR editing and expression control of antiapoptosis genes fine-tunes cellular apoptosis response. Cell Death Dis 8, e2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Chendrimada TP, Wang Q, Higuchi M, Seeburg PH, Shiekhattar R, and Nishikura K (2006). Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat Struct Mol Biol 13, 13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Yang CS, Varelas X, and Monti S (2016a). Altered RNA editing in 3’ UTR perturbs microRNA-mediated regulation of oncogenes and tumor-suppressors. Sci Rep 6, 23226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Xu G, Liu G, Ye Y, Zhang C, Fan C, Wang H, Cai H, Xiao R, Huang Z, et al. (2016b). miR-411-5p inhibits proliferation and metastasis of breast cancer cell via targeting GRB2. Biochem Biophys Res Commun 476, 607–613. [DOI] [PubMed] [Google Scholar]
- Zhao H, Sun Z, Wang J, Huang H, Kocher JP, and Wang L (2014). CrossMap: a versatile tool for coordinate conversion between genome assemblies. Bioinformatics 30, 1006–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Qin L, and Li S (2016). miR-411 contributes the cell proliferation of lung cancer by targeting FOXO1. Tumour Biol 37, 5551–5560. [DOI] [PubMed] [Google Scholar]
- Zipeto MA, Court AC, Sadarangani A, Delos Santos NP, Balaian L, Chun HJ, Pineda G, Morris SR, Mason CN, Geron I, et al. (2016). ADAR1 Activation Drives Leukemia Stem Cell Self-Renewal by Impairing Let-7 Biogenesis. Cell Stem Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipeto MA, Jiang Q, Melese E, and Jamieson CH (2015). RNA rewriting, recoding, and rewiring in human disease. Trends Mol Med 21, 549–559. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1, Related to Figure 1. Cell Cycle qPCR array.
Table S2, Related to Figure 2. A-to-I edited miRNome by ADAR1 in cord blood HSPC.
Table S3, Related to Figure 5. A-to-I RNA editome of KEGG cell cycle transcripts in cord blood HSPC, CML CP and CML BC progenitors by RNA-seq.
Data Availability Statement
The RNA-sequencing dataset used in this study were available from previous studies. The BC and CP progenitors RNA-sequencing dataset is available from (Jiang et al., 2013) (BioProject: PRJNA214016) and cord blood transduced with backbone or ADAR1 WT dataset is available from (Zipeto et al., 2016) (BioProject: PRJNA319866).