

Abstract
Dysplastic nevi (DN) are benign lesions with atypical features intermediate between that of common melanocytic nevi (CMN) and malignant melanoma (MM). Debate remains over whether DN represent progressive lesions from CMN. Through gene expression profiling and analysis of molecular gene signatures, our study revealed progressive increases in immune activation and regulation, along with pathways implicated in melanomagenesis, from CMN to DN to MM. Using criteria of 1.5 fold change and false discovery rate ≤ 0.05, we found differential expression of 7,186 probes (6,370 unique genes) with the largest difference detected between DN and MM from the standpoint of genomic melanoma progression. Despite progressive increases in the T-helper type 1 (Th1) inducing gene (IL-12), RT-PCR indicated impaired Th1 or cytotoxic T-cell response (decreased IFN-γ) in MM. Concordantly, our results indicated progressive increases in molecular markers associated with regulatory T-cells, exhausted T-cells, and tolerogenic dendritic cells, including detection of increased expression of suppressor of cytokine signaling 3 (SOCS3) in dendritic cells associated with MM. All together, our findings suggest that the increased immunosuppressive microenvironment of melanoma may contribute to unhampered proliferation of neoplastic cells. In addition, the detection of increased markers associated with tolerogenic dendritic cells in MM suggest that targeting these suppressive immune cell types may represent an alternative avenue for future immunotherapy.
Keywords: Immune checkpoints, exhausted T-cell, immunoregulation, tumor immunology, regulatory dendritic cell
Graphical Abstract
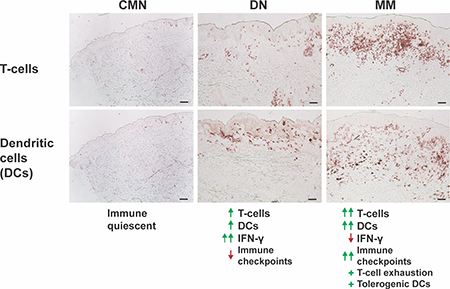
Gene expression profiling revealed progressive increases in immune system activity from common melanocytic nevi to dysplastic nevi to melanoma, with negative immunoregulation emerging strongly only in melanoma. The first identification of suppressor of cytokine signaling-3 (SOCS3) in dendritic cells of melanomas represents an alternative suppressor pathway for future targeted immunotherapy.
INTRODUCTION
Dysplastic nevi (DN) are intermediate melanocytic lesions that are important to distinguish from common melanocytic nevi (CMN) given their associated increased malignant melanoma (MM) risk[1]. Although no definitive clinical criteria for DN exists, they generally contain some of the following features: ≥5 mm, variable pigmentation, indistinct or irregular borders, and asymmetry[1, 2]. Histologically, DN are distinguished from CMN by architectural disorder and cytological atypia[3, 4]. DN can be further graded with mild, moderate, or severe atypia, which correlates with increased melanoma risk[5]. MM contains increased severity and uniformity of atypical cells and demonstrates dermal invasion[3, 4].
Uncertainty remains around the progressive nature and the relationship between DN and melanoma. Proposed in 1984, the Clark model of melanoma development described the malignant transformation of CMN as occurring through a linear, stepwise series of mutations, with intermediate precursors[6]. Current understandings of MM formation support the idea of multiple trajectories towards oncogenesis, with the majority of lesions never progressing to malignancy[7-9]. Rendering support for distinct evolutionary lineages, genetic sequencing of driver mutations has revealed that most CMN have BRAFV600E mutations, while only some DN contain BRAFV600E mutations and other DN contain alternative oncogenic mutations, including BRAF non-V600E and NRAS mutations[8].
Clinically, the presence of a single DN confers a two-fold increased risk of MM compared to no increased risk from a CMN[1]. Relative to CMN and MM, DN exhibit intermediate levels of cellular proliferation[10, 11] and point-mutational burden, upregulation of telomere maintenance mechanisms (TMMs), and heterozygous loss of CDKN2A function[8]. Telomerase reverse transcriptase promoter mutations (TPMs) are common somatic, noncoding mutations that occur in many cancers, including melanoma[12-14]. In a gene sequencing study of melanocytic lesions, 77% of DN and melanoma in situ contained TPMs[8], which contribute to genomic instability and allow for additional time for cellular replication and associated mutations and the development of functional TMMs to bypass replicative senescence and achieve cellular immortalization[12, 15]. However, an estimated equal percentage of 20–50% of melanomas arise adjacent to CMN or DN[16, 17], arguing against an increased risk of MM development from DN. Thus, there is still debate about the progressive nature and oncogenic potential of DN, particularly in comparison with CMN[7, 8, 16].
In this study, we investigated the relationship between CMN, DN, and MM using differential gene expression and pathway analyses. Our findings posit the DN as a lesion exhibiting intermediary molecular behaviors and activation of pathways implicated in melanoma development. Moreover, we further characterize the immune microenvironment of the three classes of melanocytic lesions and find that increased negative immunoregulatory molecules, immune checkpoints, and dysfunctional, exhausted immune cell types characterize the immune progression from CMN to DN to MM.
MATERIALS AND METHODS
Patient enrollment and tissue samples
This is an institutional review board-approved study with written informed consent obtained prior to patient enrollment. The study was performed in accordance with the Declaration of Helsinki Principals. Whole tissue samples were obtained via excisional biopsies at the Rockefeller University, Charite University, and Tel-Hashomer Hospital. The biopsies were bisected with one-half processed in formalin for routine hematoxylin and eosin staining and pathological diagnosis and the other half was frozen in OCT compound for RNA and immunohistochemistry analysis. Tissue was never destroyed en bloc in the event that the pathologist thought diagnostic information was needed from the frozen sections. As described in our previous study[11], DN were classified by an experienced dermatopathologist using cytological and architectural features[4]. Melanocytic nuclear atypia was noted if irregular, hyperchromatic, or with prominent nucleoli. Architectural disorder included elongation and bridging of rete ridges and the “shoulder” phenomenon[4]. MM were distinguished by increased severity and uniformity in atypical cells, pagetoid migration of melanocytes, and dermal invasion. Six normal skin (NRML), five CMN, seven DN, and sixteen MM samples were processed for microarray analysis. For RT-PCR, 11 different NRML samples and an additional seven CMN, seven DN, and three MM samples were included. Patient demographics can be found in Supplementary Table S1.
RNA extraction
Total RNA extraction was performed from frozen tissue sections using the RNeasy Micro Kit (QIAGEN, Valencia, CA) according to the manufacturer’s protocol.
cDNA microarray analysis
Target amplification and labeling to the Human Genome U133 2.0 plus arrays was performed according to the manufacturer’s instructions (Affymetrix, Santa Clara, CA). The data can be found at the Gene Expression Omnibus repository (GSE accession number GSE114445).
Quantitative RT-PCR
Manufacturer’s instructions (Applied Biosystems, Foster City, CA) were followed for preamplification and mRNA expression measurements. Data was normalized to RPLP0/hARP. The primers and probes used are listed in Supplementary Table S2.
Immunohistochemistry and immunofluorescence
Frozen skin sections were prepared and standard protocol was followed. Antibodies used are listed in Supplementary Table S2.
Statistical analyses
Microarray analyses were performed in R/Bioconductor packages. Quality control of microarray chips was carried out with standard R package and metrics. Images were scrutinized for spatial artifacts with Harshlight. Expression measures were obtained with the GCRMA algorithm. Batch effect, corresponding to the hybridization date, was detected by using principal component analysis and adjusted with the ComBat function from R’s sva package. Probe sets with at least 15 samples with expression values greater than 3 were kept for further analyses. Expression values were modeled using a mixed-effect model with lesional categories as fixed factors and random effect for each patient. Fold changes (FCH) for the comparison between lesions were estimated under the general framework for linear models in the R limma package. P-values from t-tests were adjusted for multiple hypotheses using Benjamini-Hochberg procedure. Differentially expressed genes (DEGs) were defined by FCH≥1.5 or ≤−1.5 and false discovery rate (FDR≤0.05).
For pathway analysis, log2FCH least squares means and standard error of the means were estimated for sets of genes considering pathway and lesional groups as fixed factors and genes as random factors in a mixed effect model with lme function from the R nmle package (list of genes in pathways are in Supplementary Table S3). Further pathway analysis was performed using the Ingenuity Pathway Analysis Tool (Ingenuity H Systems, Redwood City, CA). Gene-based enrichment analysis was conducted via eXploring Genomic Relations (XGR).
In comparisons of gene expression across samples from melanoma patients and normal subjects, upregulated expression was determined by evaluating the samples that were statistically located outside the 95% confidence interval for NRML. Fisher’s exact test for count data was performed, with a p<0.05 used to determine statistical significance.
RESULTS
Gene expression similarities and differences between CMN and MM
We first sought to evaluate the molecular similarities and differences between CMN and MM. Upon examining similarities in the gene expression profiles (GEPs) of CMN and MM relative to normal skin (NRML), among the most highly expressed genes in both lesions were involved in pigmentation, including TYR, TRPM1, GPR143, MLANA, and SLC45A2 (Table 1). A study from 2017 found 4,638 differentially expressed genes (DEGs) between CMN and MM using RNA-sequencing[18]. In our microarray analysis, there were 2,687 DEGs (fold change, FCH≥1.5 or ≤−1.5; false discovery rate, FDR≤0.10) between CMN and MM, with an overlap of 1,336 DEGs (49.7%) compared to the aforementioned study (Supplementary Fig. S1a). Gene-based enrichment analysis of these 1,336 DEGs indicated that the major biological processes enriched in both datasets included innate immune response, IFN-gamma signaling (T-helper type 1 response), cell adhesion, angiogenesis, and apoptosis. Of known cancer-associated antigens, both methods showed high expression levels of PRAME (preferentially expressed antigen in melanoma), but MAGE (melanoma-associated antigen) isoforms were detected only by microarray and also confirmed by RT-PCR (data presented later in Fig. 2). Analysis of the 1,351 DEGs uniquely present in our dataset revealed that the major biological processes enriched included stem cell division, vesicular trafficking, keratinocyte development, and epigenetic modifications (Supplementary Fig. S1b). Contrasting with the data from the Badal et al report[18], we did not detect enrichment for the biological processes of translation regulation, IL-4 activity (T-helper type 2 polarization), and cytokinesis that were detected in 3,302 unique genes from the RNA-sequencing analysis (Supplementary Fig. S1c). Given differences in immune activity detected in this study versus the Badal et. al report[18] and that neither RNA-sequencing or gene arrays have sufficient sensitivity for quantification of priming cytokines of polar T-cell subsets, we used more sensitive RT-PCR to measure T-cell subset activity in these melanocytic growths, as detailed later in results.
Table 1. Upregulated probesets in melanoma and common melanocytic nevi compared with normal skin with similar levels of expression (false discovery rate, FDR≤0.10).
Abbreviations: MM: malignant melanoma, CMN: common melanocytic nevi, FCH: fold change
| MM | CMN | |||||
|---|---|---|---|---|---|---|
| Probe | Symbol | FCH | FDR | FCH | FDR | Description |
| 1555505_a_at | TYR | 20.38 | 0.000433 | 15.26 | 0.038355 | tyrosinase |
| 237070_at | TRPM1 | 10.41 | 0.00296 | 40.05 | 0.00638 | transient receptor potential cation channel, subfamily M, member 1 |
| 206630_at | TYR | 8.78 | 0.00365 | 8.61 | 0.072692 | tyrosinase |
| 206696_at | GPR143 | 8.42 | 0.00041 | 6.38 | 0.044982 | G protein-coupled receptor 143 |
| 206427_s_at | MLANA | 8.05 | 0.00807 | 16.29 | 0.03035 | melan-A |
| 220245_at | SLC45A2 | 7.25 | 0.000744 | 5.73 | 0.055976 | solute carrier family 45, member 2 |
| 228375_at | IGSF11 | 6.91 | 0.00372 | 12.76 | 0.020684 | immunoglobulin superfamily, member 11 |
| 232504_at | LOC285628 | 6.73 | 0.000484 | 8.76 | 0.013231 | MIR146A host gene | microRNA 146a |
| 205376_at | INPP4B | 6.55 | 4.30E-05 | 4.4 | 0.028029 | inositol polyphosphate-4-phosphatase type II B |
| 228245_s_at | OVOS2 | 6.49 | 0.000141 | 4.13 | 0.052376 | ovostatin 2 | ovostatin | ovostatin homolog 2 |
| 204273_at | EDNRB | 6.46 | 0.00098 | 14.06 | 0.00638 | endothelin receptor type B |
| 207116_s_at | GAPDHS | 6.32 | 0.00439 | 8.88 | 0.037084 | glyceraldehyde-3-phosphate dehydrogenase, spermatogenic |
| 212190_at | SERPINE2 | 6.3 | 0.000649 | 8.07 | 0.016159 | serpin peptidase inhibitor, clade E (nexin, plasminogen activator inhibitor type 1), member 2 |
| 220615_s_at | FAR2 | 5.88 | 0.00238 | 6.7 | 0.040755 | fatty acyl CoA reductase 2 |
| 206376_at | SLC6A15 | 5.72 | 0.00286 | 8.56 | 0.025244 | solute carrier family 6 (neutral amino acid transporter), member 15 |
| 206426_at | MLANA | 5.64 | 0.0209 | 10.99 | 0.051151 | melan-A |
| 236972_at | TRIM63 | 5.61 | 0.00348 | 7.34 | 0.036115 | tripartite motif containing 63, E3 ubiquitin protein ligase |
| 215695_s_at | GYG2 | 5.54 | 0.000273 | 5.54 | 0.019558 | glycogenin 2 |
| 231666_at | PAX3 | 5.53 | 0.00856 | 10.58 | 0.02856 | paired box 3 |
| 221558_s_at | LEF1 | 5.49 | 7.36E-05 | 3.54 | 0.044409 | lymphoid enhancer-binding factor 1 |
Figure 2. Dysplastic nevi demonstrate intermediate molecular cell behaviors and pathways, with immune progression from common nevi to dysplastic nevi to melanoma.

Log2 fold change (Log2FCH) least squares mean of common melanocytic nevi (CMN), dysplastic nevi (DN), and malignant melanoma (MM) versus normal skin (NRML) for differentially expressed genes in various gene expression signatures of molecular pathways and behaviors implicated in cancer demonstrating progressive changes (a) and non-progressive changes (b) with statistically significant differences between groups indicated by a * located under the group being compared to another lesional category as specified by the corresponding star color. (c) Immunohistochemistry of CD3+ and CD11c+ infiltrates in the lesions. Scale bar = 100 μm. RT-PCR of cytokines from Th1, Th2, and Th17 immune axes (d-h) and melanoma-associated antigens (i-k) with fold changes +/− standard error of the mean (SEM) and * indicating statistically significant differences. Increasing number of stars indicates greater statistical significance, with * corresponding to p<0.05, ** corresponding to p<0.01, and *** corresponding to p<0.001.
Nevi and melanoma have distinct gene expression profiles versus normal skin
To our knowledge, previous studies have not incorporated the comparative molecular GEPs of DN into the progressive model of CMN and MM. To elucidate the relationship between the three lesions, we investigated the DEGs between NRML, CMN, DN, and MM. There were 7,186 differentially expressed probes (FCH≥1.5 or ≤−1.5, FDR≤0.05) between NRML (n=6), CMN (n=5), DN (n=7), and MM (n=16), as seen in Fig. 1a. Principal component analysis (PCA) demonstrated clear separation between NRML and melanocytic lesions along the PC-1 axis (Fig. 1b). The CMN and DN groups exhibited some overlap along the PC-1 and PC-2 axes, but were clustered separately from MM. The DN cluster was located between the CMN and MM clusters.
Figure 1. Benign nevi are molecularly distinct from malignant melanoma.

(a) Hierarchical clustered heatmap with 7,186 differentially expressed probes (fold change, FCH≥1.5 or ≤−1.5, false discovery rate, FDR≤0.05) between normal skin (NRML), common melanocytic nevi (CMN), dysplastic nevi (DN), and malignant melanoma (MM) samples. (b) Principal component analysis of the differentially expressed probes along the PC-1 (36%) and PC-2 (13%) axes. (c) Venn diagram depicting number of different and shared genes between the gene expression profiles (GEPs) of CMN, DN, and MM. (d) Venn diagram of the GEPs and differentially expressed genes (DEGs) between DN and MM. (e) Enriched biological processes by gene-ontology (GO) terms using the eXploring Genomic Relations (XGR) tool for the 70 genes contained within both GEPs of DN and MM (f) Top 20 pathways of the 860 DEGs between MM and DN that are contained within the GEP of MM via the Ingenuity Pathway Analysis (IPA) Tool.
Comparing the GEPs of CMN, DN, and MM, approximately 51.8%, 57.7%, and 85.9% of the DN GEP was shared among all three lesions, with CMN only, and with MM only, respectively (Fig. 1c). The 44 genes shared by all three lesions were enriched for biological processes including melanocyte differentiation and pigmentation, apoptosis regulation, and regulation of transcription from RNA polymerase II promoters (Supplementary Fig. S2a). CMN contained 380 unique DEGs, with enrichment for biological processes revolving around extracellular matrix organization, including endodermal cell differentiation, collagen catabolism, and cell adhesion (Supplementary Fig. S2b). Previous work demonstrated that DN can be molecularly distinguished from CMN, with DN being characterized by dysplasia of the epidermal-melanin unit and aberrant expression of hair follicle-related proteins[11]. Thus, we focused our analysis on molecular differences and similarities between DN and MM.
Differences in immune system activation distinguish the DN and MM transcriptomes
To better understand the relationship between DN and MM, we directly compared their GEPs (Fig. 1d). There were 70 genes contained within both GEPs that were not differentially expressed. In other words, these 70 genes account for the similar ways in which DN and MM differ from NRML. Upon examination of these genes, the enriched biological processes included melanocyte differentiation, negative regulation of apoptosis, cell proliferation, and positive DNA transcription (Fig. 1e).
Three genes (A2ML1, NR1D2, SYT8) were contained within both the DN and MM GEPs and also differentially expressed. There were 7 (4+3) genes within the DN GEP that were differentially expressed from MM (A2ML1, BCAN, HOXA1, LOC339260, MCOLN3, NR1D2, SYT8). Three of the genes (NR1D2, MCOLN3, SYT8) are involved in autophagy and endosome trafficking[19-21]. Within the MM GEP, there were 860 (857+3) genes that were differentially expressed from DN. Approximately 99.7% (857/860) of these DEGs were not contained within the DN GEP, suggesting that these genes are critical in the characterization and formation of MM. Pathway analysis of the 860 DEGs using Ingenuity Pathway Analysis (IPA) revealed a preponderance of immune system activation in melanoma, including T-helper type 1 (Th1) and Th2 activation, pathogenesis of multiple sclerosis, NFAT regulation of the immune system, and macrophage and monocyte phagocytosis (Fig. 1f). There were 305 DEGs between MM and DN that were not differentially expressed from NRML. The biological processes enriched included cell adhesion, T-cell receptor signaling in naïve CD4+ T cells, growth factor signaling pathways (IGF1, downstream hepatocyte growth factor receptor (c-MET) signaling, including PI3K and MAPK), and Fas signaling (Supplementary Fig. S3a).
In our analysis, there were several sets of genes that were not differentially expressed between MM and DN and also were not differentially expressed either between MM and NRML or DN and NRML. There were 8 genes within the DN GEP that were not differentially expressed between DN and MM or between MM and NRML (LOC284513, DBN1, TP73-AS1, SLC17A5, MEF2D, ENTPD1, POLR1E, WDR63), suggesting that these genes have intermediate expression levels in MM without meeting our criteria for DEGs. Of note, TP73-AS1 has been reported to have tumor suppressive properties in melanoma[22]. There were 3,755 genes within the MM GEP that were not differentially expressed between MM and DN or between DN and NRML, suggesting that DN may display intermediate expression levels of these genes without meeting our criteria for DEGs. Analysis of the 3,755 genes revealed enrichment for biological processes including neutrophil degranulation, regulation of Rho protein signal transduction, negative regulation of T cell receptor signaling, cell proliferation, and apoptosis (Supplementary Fig. S3b).
DNs display intermediate expression levels of molecular gene signatures implicated in melanoma
Given that DN clustered in an intermediary position in the PCA plot (Fig. 1b), we investigated the underlying molecular behavior and changes in pathway activation in the three melanocytic lesions. An unbiased analysis of molecular gene signatures implicated in oncogenesis and melanoma were curated from the literature and the Broad Institute, as listed in Supplementary Table S3[23-27]. Progressive increases in cell proliferation pathways from CMN to DN to MM, with statistically significant and prominent fold changes in MM (Fig. 2a), were observed. Similarly, progressive increases were found in other pathways implicated in oncogenesis, including epithelial to mesenchymal transition (EMT), metabolic reprogramming (upregulation in glycolysis), apoptosis, and immunoregulation. Pathways expressed to a similar degree in DN and MM included hypoxia and angiogenesis. Additionally, genes upregulated as a response to ultraviolet (UV) irradiation and in CD271+ cells, which are melanoma-initiating cells[28] associated with increased tissue invasion[23], were also statistically and progressively increased. To investigate for differences in the cell cycle checkpoints, genes involved in the progression through G1/S (E2F targets) and the G2/M checkpoint pathways were queried, with significant changes between MM and the other two lesions observed. Lastly, cell-signaling pathways implicated in MM development, including MAPK/KRAS/MEK, PI-3K/AKT/mTOR/PTEN, and Hedgehog were examined[29, 30]. Of these pathways, MAPK/KRAS/MEK, PI-3K/AKT/mTOR, and Hedgehog signaling revealed progressive increases in activation. Clear progressive changes in Notch, Myc, p53, and Wnt/β-catenin pathways were not appreciated (Fig. 2b).
Progressive increases in mitogenic cytokines and melanogenic growth factors from CMN to DN to MM
Given the prominent increase in the cell proliferation gene signature in MM relative to the benign lesions, an investigation into growth factors and receptors was conducted. By RT-PCR, an increasing trend in the chemokine CXCL1 and its receptor CXCR2 was found (Supplementary Fig. S4a-b). CXCL1 and CXCR2 are involved in an autocrine growth factor loop in melanoma cells and proliferating melanocytes[31]. Increases in IL-6 and IL-8, cytokines involved in melanoma cell growth[32-36], were also appreciated (Supplementary Fig. S4c-d). Similarly, OSM, an IL-6 family cytokine, was progressively increased from CMN to DN to MM (Supplementary Fig. S4e). NGF, GM-CSF, VEGFA, and PDGFB also displayed increased levels in MM (Supplementary Fig. S4f-i).
Immune progression from CMN to DN to MM
With the significant immune activity and immunoregulation observed from the differential gene expression profiles, pathway, and molecular gene signature analyses, we sought to further characterize the immunologic microenvironment of the three melanocytic lesions. Immunohistochemistry (IHC) demonstrated increased amounts of dendritic cell and lymphocytic infiltrate from CMN to DN to MM (Fig. 2c). Consistent with the aforementioned analyses, progressive increases in Th1 (IL-12), Th2 (IL-4), and Th17 (IL-23A, IL-17A) could be appreciated from CMN to DN to MM via RT-PCR (Fig. 2d-g). Despite the progressive increase in the Th1-inducing gene (IL-12), a decrease in Th1 output (IFN-γ) was observed from DN to MM, suggesting impaired Th1 or type-1 cytotoxic T-cell (Tc1) immune response in MM (Fig. 2h). With the immune progression seen from CMN to DN to MM, we hypothesized that there could be transformed neoplastic cells in DN eliciting an immune response. RT-PCR of the melanoma-associated antigens, MAGE-A3, MAGE-A12, and PRAME, was performed, which revealed progressive increases, though the expression was quantitatively much larger between DN and MM (Fig. 2i-k).
Progressive increases in dysfunctional, immunosuppressive immune cells from CMN to DN to MM
Given evidence of a decreased Th1 or Tc1 immune response in MM and progressive increases in melanoma-associated antigens, we examined if immunoregulatory and/or tolerogenic immune cells were similarly progressively increased. Recent literature has shown that the presence of increased lymphocytic infiltrates in MM alone may not be as significant as the functional activity of these cells in combating MM[37]. Thus, we queried immune cell-specific gene transcript signatures[38-41], which revealed progressive increases in exhausted T-cells, regulatory T-cells (Tregs), and tolerogenic dendritic cells (DCs) gene signatures (Fig. 3a-c). RT-PCR confirmed progressive increases in Treg activity (TGF-β, IL-10), and immunoregulatory markers (CTLA-4, PD-L1, PD-L2, IDO1) associated with exhausted T-cells (Fig. 3d-i).
Figure 3. Negative immunoregulation is progressively increased from common melanocytic nevi to dysplastic nevi to melanoma.

Heatmap of immune cell-type specific gene transcripts for regulatory T-cells (a), exhausted T-cells (b), and tolerogenic dendritic cells (c) between normal skin (NRML), common melanocytic nevi (CMN), dysplastic nevi (DN), and malignant melanoma (MM). RT-PCR for regulatory T-cell cytokines (d-e) and negative immunoregulatory molecules (f-i) with fold changes between tissue types and statistically significant results indicated by *. Increasing number of stars indicates greater statistical significance, with * corresponding to p<0.05, ** corresponding to p<0.01, and *** corresponding to p<0.001.
SOCS3 is upregulated in dendritic cells in MM and presents a potential alternative immunotherapeutic avenue
With increased expression of multiple molecules associated with tolerogenic DCs in MM, we performed IHC staining for SOCS3, a marker of tolerogenic DCs[42, 43]. Compared with NRML, which displayed rare SOCS3+ DCs, MM displayed prominent SOCS3 staining in a DC pattern (Fig. 4a). Double immunofluorescence confirmed increased CD11c+ DC expression of SOCS3 in MM, but only rare expression of SOCS3+ DCs in NRML (Fig. 4b). Comparing the expression levels between MM and NRML, we found similarities in the consistency of upregulated SOCS3 expression across samples compared to current immunotherapeutic targets, including CTLA4 and CD274 (no statistically significant differences in proportions), as shown in Fig. 4c. In contrast, compared to CTLA4, there was heterogeneity of expression levels in MM for MAGE-A3 (p=0.02) and MAGE-A6 (p=0.04). Given the immunosuppressive properties associated with SOCS3+ DCs and the consistency in its expression across melanoma samples, targeting of these cell types may represent a novel avenue in the reprogramming of the immune microenvironment to more effectively combat MM.
Figure 4. SOCS3+, a marker associated with tolerogenic dendritic cells, is increased in melanoma.

(a) Immunohistochemistry of SOCS3+ (normal, NRML; malignant melanoma, MM) and a negative control (MM). Black arrow labeled “M” indicates melanophages. (b) Double immunofluorescence staining of SOCS3+ (red) and CD11c+ (green) on representative NRML and MM samples. White arrow indicates a sparse double positive cell (yellow) in NRML. White line delineates the dermoepidermal junction. (c) Comparison of gene expression levels of melanoma-associated antigens, negative immunoregulatory molecules, and tolerogenic dendritic cell markers in NRML and MM samples. Boxed samples indicate upregulation in MM, with expression levels higher than the 95% confidence interval for normal tissue for the corresponding gene. Bar with p-value indicates statistically significant differences in the proportion of MM samples with upregulated expression levels compared to NRML. Scale bar = 100 μm.
DISCUSSION
In this study, we elucidated the molecular and cellular characteristics of DN, along with its relationship with respect to CMN and MM, using a rigorous statistical method that has previously been successfully applied to the study of many cutaneous diseases, including psoriasis[27, 44, 45], alopecia areata[46], atopic dermatitis[47-49], and skin cancers[50, 51]. Studies investigating the molecular profiles of melanocytic lesions that include DN are limited. In the only previous study that directly compared DN and MM, gene transcripts in the DN were found to differ from that of melanoma in the regulation of transcription and the mismatch repair system[52]. Similarly, in our study, we identified differences between MM and DN that encompassed differential regulation of transcription and cellular response to DNA damage. However, Scatolini et. al were only able to identify small subsets of DEGs between melanocytic lesions and additionally, did not find significant immune system differences detected between nevi and MM. The more robust results generated from our study might be accounted for by several factors: greater selection for and characterization of DN with moderate to severe architectural disorder and cellular atypia, increased gene microarray sensitivity in detecting gene expression changes, the use of extensive confirmatory RT-PCR with the ability to detect T-cell cytokines at low levels of expression not detected by microarray, and the application of rigorous statistical methodology to compare the melanocytic lesions. Our results were notable not only for increased immune activation along the Th1/Th2/Th17 axes in the progression from CMN to DN to MM, but also for a significant immunosuppressive signature that likely involves both T-cells and dendritic cells. Additionally, we found that DN display intermediary molecular characteristics and behavior skewing towards malignant transformation. Altogether, in this schema, one can hypothesize that some neoplastic cells of the DN might transition to malignant cells of melanoma, but then might be recognized by expression of cancer-associated neoantigens and thus, restrained or eliminated by immunosurveillance involving Th1 or Tc1 cells.
In this study, we found that large percentages of the DN GEP were shared with CMN and MM, respectively, suggesting that there are relatively few unique biological processes that define the DN. Indeed, our results demonstrated that DN display intermediate levels for molecular gene signatures and pathways implicated in oncogenesis, including the hallmarks of cancer, UV damage response, and cell-signaling pathway dysregulation, suggesting that DN exhibit cell behaviors skewing towards malignancy. The hallmarks of cancer describe characteristics common to malignancies, including sustained proliferative signaling, evading apoptosis and growth suppressors, bypassing replicative senescence, inducing angiogenesis, developing the capacity to invade and metastasize, reprogramming cellular metabolism, and escaping immune surveillance[53]. The hallmarks of cancer have been criticized as a framework for understanding oncogenesis given that invasion and metastasis is the only one of the original six hallmarks that is not also characteristic of benign tumors[54]. However, the increasing trend in gene signatures associated with tissue invasion and metastasis (EMT, CD271+ cells) support the model of DN as intermediate lesions with molecular behavior progressing towards malignancy.
One of the more prominent changes observed in our study was the progressive upregulation in immune and inflammatory pathways from CMN to DN to MM, with increased activation of Th1/Th2/Th17 immune axes. Indeed, a significantly increased ratio of DEGs in the pathogenesis of multiple sclerosis pathway was appreciated. Lending support to our findings, multiple sclerosis is an autoimmune demyelinating disease that is associated with alterations in Th1/Th2/Th17 cells[55-57]. From our results, we hypothesized that similar to the immune clearance that tempers the oncogenic transformation of pre-malignant, senescent hepatocytes[58], somatic mutations in neoplastic melanocytes can lead to the expression of antigens which stimulate immune system activation and clearance. It has been proposed that cellular proliferation in the DN is balanced by cellular attrition factors, including immunosurveillance[7]. MM would therefore represent a disruption in the balance between immunosurveillance and neoplastic cell transformation and proliferation. Indeed, we found that DN and MM microenvironments displayed progressively increased levels of negative immunoregulation.
In accordance with the increased immunosuppressive landscape in melanoma, our study identified increased DCs expressing suppressive markers that have previously been associated with tolerogenic, immunoregulatory function. Similar to SOCS2, which was previously identified as a protein expressed by dendritic cells that is critical for limiting anti-tumor immune response in MM[59], SOCS3 is also a member of the suppressor of cytokine signaling proteins (SOCS) family. SOCS3 expression by dendritic cells has not previously been described in human melanoma. Increased cytokine IL-10 in the immune microenvironment has been reported to increase SOCS3 expression and IL-10 secretion by DCs, which prolong Treg cell function[40]. Treg cells, in turn, contribute to T-cell exhaustion[60], which mount ineffective immune responses against cancer. SOCS3+ DCs also direct T-cell polarization towards a Th2 phenotype[42, 61]. Additionally, T-cell effector responses can be hampered by tolerogenic DCs, as antigen presentation in SOCS3+ DCs is disrupted in tumors[62], with lower expression of MHC class II and co-stimulatory molecules[42]. SOCS3 in tumor-associated DCs can also inhibit the activation of DCs from type I IFN signaling[63]. The increase in immunosuppressive and exhausted, dysfunctional immune cells, including tolerogenic DCs, in melanoma may contribute to unrestrained proliferation of neoplastic cells.
The results of this study have implications for immunotherapy of melanoma. Approaches to boost anti-melanoma immune responses via vaccination with or without DCs have been described[64, 65]. We found significant heterogeneity in expression of MAGE isoforms in primary melanomas. For example, MAGE-A3 was upregulated in only 10 out of 16 melanoma samples. Thus, one might need to individualize immunogenic antigens to a patient’s melanoma “expression” genotype. More recent approaches in melanoma treatment have involved checkpoint inhibitors, including CTLA4 and PD-L1 antibodies—an approach highly supported by our profiling data, which revealed greater consistency of upregulation among melanoma patients. Since negative immune regulators associated with tolerogenic DCs, such as SOCS3 and CD37, are similarly increased in MM and not exclusively in melanomas with PD-L1 upregulation, an additional avenue for future immunotherapy might be to antagonize some of these alternate suppressor pathways.
Supplementary Material
Comparison of differentially expressed genes between malignant melanoma and common melanocytic nevi from Badal et. al publication and our dataset. The Top 20 biological processes enriched using gene-ontology terms for unique and overlapping genes in the two datasets via eXploring Genomic Relations (XGR) are shown (a-c).
Overall comparison of the gene expression profiles of common melanocytic nevi, dysplastic nevi, and melanoma. Gene-based enrichment analysis via eXploring Genomic Relations (XGR) for biological processes based upon gene-ontology terms are shown for the 44 genes shared by all three melanocytic lesions (a) and for the unique gene sets in common melanocytic nevi (b).
Top 20 biological processes enriched based on gene-ontology terms for dysplastic nevi and malignant melanoma. (a) Top 20 biological process gene-ontology (GO) terms enriched in the 305 differentially expressed genes (DEGs) between malignant melanoma (MM) and dysplastic nevi (DN), but not contained within either gene expression profile (GEPs) (b) Top 20 biological process GO terms enriched in the 3,755 genes contained in the MM GEP with intermediate expression levels in DN. Gene-based enrichment analyses were performed using the eXploring Genomic Relations (XGR) tool.
Progressively increased growth factors and cytokines from common melanocytic nevi to dysplastic nevi to melanoma. RT-PCR of growth factors and cytokines (a-i) in normal skin (NRML), common nevi (CN), dysplastic nevi (DN), and malignant melanoma (MM) with fold changes and * indicating statistically significant changes. Increasing number of stars indicates greater statistical significance, with * corresponding to p<0.05, ** corresponding to p<0.01, and *** corresponding to p<0.001.
ACKNOWLEDGEMENTS
We acknowledge N. Scott McNutt for important early work on this project. This research was supported by the Milstein Foundation and in part by National Institutes of Health (NIH) grant UL1TR001866 (BYY, JGK) and KL2TR000151 (HM, JGK). NG was supported by NIH MSTP grant T32GM07739.
Footnotes
CONFLICTS OF INTEREST
The authors state no conflict of interest.
REFERENCES
- [1].Tucker MA; Halpern A; Holly EA; Hartge P; Elder DE; Sagebiel RW; Guerry D. t.; Clark WH Jr., Clinically recognized dysplastic nevi. A central risk factor for cutaneous melanoma. Jama 1997, 277, (18), 1439–44. [PubMed] [Google Scholar]
- [2].Bergman W; van Voorst Vader PC; Ruiter DJ, [Dysplastic nevi and the risk of melanoma: a guideline for patient care. Nederlandse Melanoom Werkgroep van de Vereniging voor Integrale Kankercentra]. Ned Tijdschr Geneeskd 1997, 141, (42), 2010–4. [PubMed] [Google Scholar]
- [3].Elder DE, Dysplastic naevi: an update. Histopathology 2010, 56, (1), 112–20. [DOI] [PubMed] [Google Scholar]
- [4].Elder DE; Elenitsas R Jr., B. L. J.; Murphy GF; Xu X, Lever’s Histopathology of the Skin. Tenth ed.; Lippincott Williams & Wilkins: 2009. [Google Scholar]
- [5].Arumi-Uria M; McNutt NS; Finnerty B, Grading of atypia in nevi: correlation with melanoma risk. Mod Pathol 2003, 16, (8), 764–71. [DOI] [PubMed] [Google Scholar]
- [6].Clark WH Jr.; Elder DE; Guerry D. t.; Epstein MN; Greene MH; Van Horn M, A study of tumor progression: the precursor lesions of superficial spreading and nodular melanoma. Hum Pathol 1984, 15, (12), 1147–65. [DOI] [PubMed] [Google Scholar]
- [7].Shain AH; Bastian BC, From melanocytes to melanomas. Nat Rev Cancer 2016, 16, (6), 345–58. [DOI] [PubMed] [Google Scholar]
- [8].Shain AH; Yeh I; Kovalyshyn I; Sriharan A; Talevich E; Gagnon A; Dummer R; North J; Pincus L; Ruben B; Rickaby W; D’Arrigo C; Robson A; Bastian BC, The Genetic Evolution of Melanoma from Precursor Lesions. N Engl J Med 2015, 373, (20), 1926–36. [DOI] [PubMed] [Google Scholar]
- [9].Elder DE, Melanoma progression. Pathology 2016, 48, (2), 147–54. [DOI] [PubMed] [Google Scholar]
- [10].Lebe B; Pabuccuoglu U; Ozer E, The significance of Ki-67 proliferative index and cyclin D1 expression of dysplastic nevi in the biologic spectrum of melanocytic lesions. Appl Immunohistochem Mol Morphol 2007, 15, (2), 160–4. [DOI] [PubMed] [Google Scholar]
- [11].Mitsui H; Kiecker F; Shemer A; Cannizzaro MV; Wang CQF; Gulati N; Ohmatsu H; Shah KR; Gilleaudeau P; Sullivan-Whalen M; Cueto I; McNutt NS; Suarez-Farinas M; Krueger JG, Discrimination of Dysplastic Nevi from Common Melanocytic Nevi by Cellular and Molecular Criteria. J Invest Dermatol 2016, 136, (10), 2030–2040. [DOI] [PubMed] [Google Scholar]
- [12].Chiba K; Lorbeer FK; Shain AH; McSwiggen DT; Schruf E; Oh A; Ryu J; Darzacq X; Bastian BC; Hockemeyer D, Mutations in the promoter of the telomerase gene TERT contribute to tumorigenesis by a two-step mechanism. Science 2017, 357, (6358), 1416–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Horn S; Figl A; Rachakonda PS; Fischer C; Sucker A; Gast A; Kadel S; Moll I; Nagore E; Hemminki K; Schadendorf D; Kumar R, TERT promoter mutations in familial and sporadic melanoma. Science 2013, 339, (6122), 959–61. [DOI] [PubMed] [Google Scholar]
- [14].Huang FW; Hodis E; Xu MJ; Kryukov GV; Chin L; Garraway LA, Highly recurrent TERT promoter mutations in human melanoma. Science 2013, 339, (6122), 957–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Shay JW, New insights into melanoma development. Science 2017, 357, (6358), 1358–1359. [DOI] [PubMed] [Google Scholar]
- [16].Colebatch AJ; Scolyer RA, Trajectories of premalignancy during the journey from melanocyte to melanoma. Pathology 2018, 50, (1), 16–23. [DOI] [PubMed] [Google Scholar]
- [17].Pampena R; Kyrgidis A; Lallas A; Moscarella E; Argenziano G; Longo C, A meta-analysis of nevus-associated melanoma: Prevalence and practical implications. J Am Acad Dermatol 2017, 77, (5), 938–945.e4. [DOI] [PubMed] [Google Scholar]
- [18].Badal B; Solovyov A; Di Cecilia S; Chan JM; Chang LW; Iqbal R; Aydin IT; Rajan GS; Chen C; Abbate F; Arora KS; Tanne A; Gruber SB; Johnson TM; Fullen DR; Raskin L; Phelps R; Bhardwaj N; Bernstein E; Ting DT; Brunner G; Schadt EE; Greenbaum BD; Celebi JT, Transcriptional dissection of melanoma identifies a high-risk subtype underlying TP53 family genes and epigenome deregulation. JCI Insight 2017, 2, (9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sulli G; Rommel A; Wang X; Kolar MJ; Puca F; Saghatelian A; Plikus MV; Verma IM; Panda S, Pharmacological activation of REV-ERBs is lethal in cancer and oncogene-induced senescence. Nature 2018, 553, (7688), 351–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Choi S; Kim HJ, The Ca2+ channel TRPML3 specifically interacts with the mammalian ATG8 homologue GATE16 to regulate autophagy. Biochem Biophys Res Commun 2014, 443, (1), 56–61. [DOI] [PubMed] [Google Scholar]
- [21].Kanda M; Shimizu D; Tanaka H; Tanaka C; Kobayashi D; Hayashi M; Iwata N; Niwa Y; Yamada S; Fujii T; Sugimoto H; Murotani K; Fujiwara M; Kodera Y, Significance of SYT8 For the Detection, Prediction, and Treatment of Peritoneal Metastasis From Gastric Cancer. Ann Surg 2018, 267, (3), 495–503. [DOI] [PubMed] [Google Scholar]
- [22].Hu H; Liu JM; Hu Z; Jiang X; Yang X; Li J; Zhang Y; Yu H; Khaitovich P, Recently evolved tumor suppressor transcript TP73-AS1 functions as sponge of human-specific miR-941. Mol Biol Evol 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Restivo G; Diener J; Cheng PF; Kiowski G; Bonalli M; Biedermann T; Reichmann E; Levesque MP; Dummer R; Sommer L, low neurotrophin receptor CD271 regulates phenotype switching in melanoma. Nat Commun 2017, 8, (1), 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Venet D; Dumont JE; Detours V, Most random gene expression signatures are significantly associated with breast cancer outcome. PLoS Comput Biol 2011, 7, (10), e1002240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liberzon A; Birger C; Thorvaldsdottir H; Ghandi M; Mesirov JP; Tamayo P, The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 2015, 1, (6), 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Liberzon A; Subramanian A; Pinchback R; Thorvaldsdottir H; Tamayo P; Mesirov JP, Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, (12), 1739–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Gulati N; Suarez-Farinas M; Correa da Rosa J; Krueger JG, Psoriasis is characterized by deficient negative immune regulation compared to transient delayed-type hypersensitivity reactions. F1000Res 2015, 4, 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Boiko AD; Razorenova OV; van de Rijn M; Swetter SM; Johnson DL; Ly DP; Butler PD; Yang GP; Joshua B; Kaplan MJ; Longaker MT; Weissman IL, Human melanoma-initiating cells express neural crest nerve growth factor receptor CD271. Nature 2010, 466, (7302), 133–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hayward NK; Wilmott JS; Waddell N; Johansson PA; Field MA; Nones K; Patch AM; Kakavand H; Alexandrov LB; Burke H; Jakrot V; Kazakoff S; Holmes O; Leonard C; Sabarinathan R; Mularoni L; Wood S; Xu Q; Waddell N; Tembe V; Pupo GM; De Paoli-Iseppi R; Vilain RE; Shang P; Lau LMS; Dagg RA; Schramm SJ; Pritchard A; Dutton-Regester K; Newell F; Fitzgerald A; Shang CA; Grimmond SM; Pickett HA; Yang JY; Stretch JR; Behren A; Kefford RF; Hersey P; Long GV; Cebon J; Shackleton M; Spillane AJ; Saw RPM; Lopez-Bigas N; Pearson JV; Thompson JF; Scolyer RA; Mann GJ, Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, (7653), 175–180. [DOI] [PubMed] [Google Scholar]
- [30].O’Reilly KE; de Miera EV; Segura MF; Friedman E; Poliseno L; Han SW; Zhong J; Zavadil J; Pavlick A; Hernando E; Osman I, Hedgehog pathway blockade inhibits melanoma cell growth in vitro and in vivo. Pharmaceuticals (Basel) 2013, 6, (11), 1429–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mockenhaupt M; Peters F; Schwenk-Davoine I; Herouy Y; Schraufstatter I; Elsner P; Norgauer J, Evidence of involvement of CXC-chemokines in proliferation of cultivated human melanocytes. Int J Mol Med 2003, 12, (4), 597–601. [PubMed] [Google Scholar]
- [32].Chen GL; Luo Y; Eriksson D; Meng X; Qian C; Bauerle T; Chen XX; Schett G; Bozec A, High fat diet increases melanoma cell growth in the bone marrow by inducing osteopontin and interleukin 6. Oncotarget 2016, 7, (18), 26653–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Schadendorf D; Moller A; Algermissen B; Worm M; Sticherling M; Czarnetzki BM, IL-8 produced by human malignant melanoma cells in vitro is an essential autocrine growth factor. J Immunol 1993, 151, (5), 2667–75. [PubMed] [Google Scholar]
- [34].Singh RK; Varney ML, IL-8 expression in malignant melanoma: implications in growth and metastasis. Histol Histopathol 2000, 15, (3), 843–9. [DOI] [PubMed] [Google Scholar]
- [35].Wang L; Yi T; Kortylewski M; Pardoll DM; Zeng D; Yu H, IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J Exp Med 2009, 206, (7), 1457–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lu C; Kerbel RS, Interleukin-6 undergoes transition from paracrine growth inhibitor to autocrine stimulator during human melanoma progression. J Cell Biol 1993, 120, (5), 1281–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Weiss SA; Han SW; Lui K; Tchack J; Shapiro R; Berman R; Zhong J; Krogsgaard M; Osman I; Darvishian F, Immunologic heterogeneity of tumor-infiltrating lymphocyte composition in primary melanoma. Hum Pathol 2016, 57, 116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Chung W; Eum HH; Lee HO; Lee KM; Lee HB; Kim KT; Ryu HS; Kim S; Lee JE; Park YH; Kan Z; Han W; Park WY, Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat Commun 2017, 8, 15081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Schinnerling K; Garcia-Gonzalez P; Aguillon JC, Gene Expression Profiling of Human Monocyte-derived Dendritic Cells - Searching for Molecular Regulators of Tolerogenicity. Front Immunol 2015, 6, 528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Devi KS; Anandasabapathy N, The origin of DCs and capacity for immunologic tolerance in central and peripheral tissues. Semin Immunopathol 2017, 39, (2), 137–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Manicassamy S; Pulendran B, Dendritic cell control of tolerogenic responses. Immunol Rev 2011, 241, (1), 206–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Li Y; Chu N; Rostami A; Zhang GX, Dendritic cells transduced with SOCS-3 exhibit a tolerogenic/DC2 phenotype that directs type 2 Th cell differentiation in vitro and in vivo. J Immunol 2006, 177, (3), 1679–88. [DOI] [PubMed] [Google Scholar]
- [43].Liu X; Qu X; Chen Y; Liao L; Cheng K; Shao C; Zenke M; Keating A; Zhao RC, Mesenchymal stem/stromal cells induce the generation of novel IL-10-dependent regulatory dendritic cells by SOCS3 activation. J Immunol 2012, 189, (3), 1182–92. [DOI] [PubMed] [Google Scholar]
- [44].Bissonnette R; Suarez-Farinas M; Li X; Bonifacio KM; Brodmerkel C; Fuentes-Duculan J; Krueger JG, Based on Molecular Profiling of Gene Expression, Palmoplantar Pustulosis and Palmoplantar Pustular Psoriasis Are Highly Related Diseases that Appear to Be Distinct from Psoriasis Vulgaris. PLoS One 2016, 11, (5), e0155215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Mitsui H; Suarez-Farinas M; Belkin DA; Levenkova N; Fuentes-Duculan J; Coats I; Fujita H; Krueger JG, Combined use of laser capture microdissection and cDNA microarray analysis identifies locally expressed disease-related genes in focal regions of psoriasis vulgaris skin lesions. J Invest Dermatol 2012, 132, (6), 1615–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Suarez-Farinas M; Ungar B; Noda S; Shroff A; Mansouri Y; Fuentes-Duculan J; Czernik A; Zheng X; Estrada YD; Xu H; Peng X; Shemer A; Krueger JG; Lebwohl MG; Guttman-Yassky E, Alopecia areata profiling shows TH1, TH2, and IL-23 cytokine activation without parallel TH17/TH22 skewing. J Allergy Clin Immunol 2015, 136, (5), 1277–87. [DOI] [PubMed] [Google Scholar]
- [47].Brunner PM; Israel A; Zhang N; Leonard A; Wen HC; Huynh T; Tran G; Lyon S; Rodriguez G; Immaneni S; Wagner A; Zheng X; Estrada YD; Xu H; Krueger JG; Paller AS; Guttman-Yassky E, Early-onset pediatric atopic dermatitis is characterized by TH2/TH17/TH22-centered inflammation and lipid alterations. J Allergy Clin Immunol 2018, 141, (6), 2094–2106. [DOI] [PubMed] [Google Scholar]
- [48].Guttman-Yassky E; Lowes MA; Fuentes-Duculan J; Zaba LC; Cardinale I; Nograles KE; Khatcherian A; Novitskaya I; Carucci JA; Bergman R; Krueger JG, Low expression of the IL-23/Th17 pathway in atopic dermatitis compared to psoriasis. J Immunol 2008, 181, (10), 7420–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Guttman-Yassky E; Lowes MA; Fuentes-Duculan J; Whynot J; Novitskaya I; Cardinale I; Haider A; Khatcherian A; Carucci JA; Bergman R; Krueger JG, Major differences in inflammatory dendritic cells and their products distinguish atopic dermatitis from psoriasis. J Allergy Clin Immunol 2007, 119, (5), 1210–7. [DOI] [PubMed] [Google Scholar]
- [50].Mitsui H; Suarez-Farinas M; Gulati N; Shah KR; Cannizzaro MV; Coats I; Felsen D; Krueger JG; Carucci JA, Gene expression profiling of the leading edge of cutaneous squamous cell carcinoma: IL-24-driven MMP-7. J Invest Dermatol 2014, 134, (5), 1418–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kaporis HG; Guttman-Yassky E; Lowes MA; Haider AS; Fuentes-Duculan J; Darabi K; Whynot-Ertelt J; Khatcherian A; Cardinale I; Novitskaya I; Krueger JG; Carucci JA, Human basal cell carcinoma is associated with Foxp3+ T cells in a Th2 dominant microenvironment. J Invest Dermatol 2007, 127, (10), 2391–8. [DOI] [PubMed] [Google Scholar]
- [52].Scatolini M; Grand MM; Grosso E; Venesio T; Pisacane A; Balsamo A; Sirovich R; Risio M; Chiorino G, Altered molecular pathways in melanocytic lesions. Int J Cancer 2010, 126, (8), 1869–1881. [DOI] [PubMed] [Google Scholar]
- [53].Hanahan D; Weinberg RA, Hallmarks of cancer: the next generation. Cell 2011, 144, (5), 646–74. [DOI] [PubMed] [Google Scholar]
- [54].Lazebnik Y, What are the hallmarks of cancer? Nat Rev Cancer 2010, 10, (4), 232–3. [DOI] [PubMed] [Google Scholar]
- [55].Oreja-Guevara C; Ramos-Cejudo J; Aroeira LS; Chamorro B; Diez-Tejedor E, TH1/TH2 Cytokine profile in relapsing-remitting multiple sclerosis patients treated with Glatiramer acetate or Natalizumab. BMC Neurol 2012, 12, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Hedegaard CJ; Krakauer M; Bendtzen K; Lund H; Sellebjerg F; Nielsen CH, T helper cell type 1 (Th1), Th2 and Th17 responses to myelin basic protein and disease activity in multiple sclerosis. Immunology 2008, 125, (2), 161–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Arellano G; Acuna E; Reyes LI; Ottum PA; De Sarno P; Villarroel L; Ciampi E; Uribe-San Martin R; Carcamo C; Naves R, Th1 and Th17 Cells and Associated Cytokines Discriminate among Clinically Isolated Syndrome and Multiple Sclerosis Phenotypes. Front Immunol 2017, 8, 753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Kang TW; Yevsa T; Woller N; Hoenicke L; Wuestefeld T; Dauch D; Hohmeyer A; Gereke M; Rudalska R; Potapova A; Iken M; Vucur M; Weiss S; Heikenwalder M; Khan S; Gil J; Bruder D; Manns M; Schirmacher P; Tacke F; Ott M; Luedde T; Longerich T; Kubicka S; Zender L, Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, (7374), 547–51. [DOI] [PubMed] [Google Scholar]
- [59].Nirschl CJ; Suarez-Farinas M; Izar B; Prakadan S; Dannenfelser R; Tirosh I; Liu Y; Zhu Q; Devi KSP; Carroll SL; Chau D; Rezaee M; Kim TG; Huang R; Fuentes-Duculan J; Song-Zhao GX; Gulati N; Lowes MA; King SL; Quintana FJ; Lee YS; Krueger JG; Sarin KY; Yoon CH; Garraway L; Regev A; Shalek AK; Troyanskaya O; Anandasabapathy N, IFNgamma-Dependent Tissue-Immune Homeostasis Is Co-opted in the Tumor Microenvironment. Cell 2017, 170, (1), 127–141.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Penaloza-MacMaster P; Kamphorst AO; Wieland A; Araki K; Iyer SS; West EE; O’Mara L; Yang S; Konieczny BT; Sharpe AH; Freeman GJ; Rudensky AY; Ahmed R, Interplay between regulatory T cells and PD-1 in modulating T cell exhaustion and viral control during chronic LCMV infection. J Exp Med 2014, 211, (9), 1905–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Dimitriou ID; Clemenza L; Scotter AJ; Chen G; Guerra FM; Rottapel R, Putting out the fire: coordinated suppression of the innate and adaptive immune systems by SOCS1 and SOCS3 proteins. Immunol Rev 2008, 224, 265–83. [DOI] [PubMed] [Google Scholar]
- [62].Zhang Z; Liu Q; Che Y; Yuan X; Dai L; Zeng B; Jiao G; Zhang Y; Wu X; Yu Y; Zhang Y; Yang R, Antigen presentation by dendritic cells in tumors is disrupted by altered metabolism that involves pyruvate kinase M2 and its interaction with SOCS3. Cancer Res 2010, 70, (1), 89–98. [DOI] [PubMed] [Google Scholar]
- [63].Zeng B; Li H; Liu Y; Zhang Z; Zhang Y; Yang R, Tumor-induced suppressor of cytokine signaling 3 inhibits toll-like receptor 3 signaling in dendritic cells via binding to tyrosine kinase 2. Cancer Res 2008, 68, (13), 5397–404. [DOI] [PubMed] [Google Scholar]
- [64].Alvarez-Dominguez C; Calderon-Gonzalez R; Teran-Navarro H; Salcines-Cuevas D; Garcia-Castano A; Freire J; Gomez-Roman J; Rivera F, Dendritic cell therapy in melanoma. Ann Transl Med 2017, 5, (19), 386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Grenier JM; Yeung ST; Khanna KM, Combination Immunotherapy: Taking Cancer Vaccines to the Next Level. Front Immunol 2018, 9, 610. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Comparison of differentially expressed genes between malignant melanoma and common melanocytic nevi from Badal et. al publication and our dataset. The Top 20 biological processes enriched using gene-ontology terms for unique and overlapping genes in the two datasets via eXploring Genomic Relations (XGR) are shown (a-c).
Overall comparison of the gene expression profiles of common melanocytic nevi, dysplastic nevi, and melanoma. Gene-based enrichment analysis via eXploring Genomic Relations (XGR) for biological processes based upon gene-ontology terms are shown for the 44 genes shared by all three melanocytic lesions (a) and for the unique gene sets in common melanocytic nevi (b).
Top 20 biological processes enriched based on gene-ontology terms for dysplastic nevi and malignant melanoma. (a) Top 20 biological process gene-ontology (GO) terms enriched in the 305 differentially expressed genes (DEGs) between malignant melanoma (MM) and dysplastic nevi (DN), but not contained within either gene expression profile (GEPs) (b) Top 20 biological process GO terms enriched in the 3,755 genes contained in the MM GEP with intermediate expression levels in DN. Gene-based enrichment analyses were performed using the eXploring Genomic Relations (XGR) tool.
Progressively increased growth factors and cytokines from common melanocytic nevi to dysplastic nevi to melanoma. RT-PCR of growth factors and cytokines (a-i) in normal skin (NRML), common nevi (CN), dysplastic nevi (DN), and malignant melanoma (MM) with fold changes and * indicating statistically significant changes. Increasing number of stars indicates greater statistical significance, with * corresponding to p<0.05, ** corresponding to p<0.01, and *** corresponding to p<0.001.