

Abstract
High-content screening data derived from physiologically-relevant in vitro models promise to improve confidence in data-integrative groupings for read-across in human health safety assessments. The biological data-based read-across concept is especially applicable to bioactive chemicals with defined mechanisms of toxicity; however, the challenge of data-derived groupings for chemicals that are associated with little or no bioactivity has not been explored. In this study, we apply a suite of organotypic and population-based in vitro models for comprehensive bioactivity profiling of twenty E-Series and P-Series glycol ethers, solvents with a broad variation in toxicity ranging from relatively non-toxic to reproductive and hematopoetic system toxicants. Both E-Series and P-Series glycol ethers elicited cytotoxicity only at high concentrations (mM range) in induced pluripotent stem cell-derived hepatocytes and cardiomyocytes. Population-variability assessment comprised a study of cytotoxicity in 94 human lymphoblast cell lines from 9 populations and revealed differences in inter-individual variability across glycol ethers, but did not indicate population-specific effects. Data derived from various phenotypic and transcriptomic assays revealed consistent bioactivity trends between both cardiomyocytes and hepatocytes, indicating a more universal, rather than cell-type specific mode-of-action for the tested glycol ethers in vitro. In vitro bioactivity-based similarity assessment using Toxicological Priority Index (ToxPi) showed that glycol ethers group according to their alcohol chain length, longer chains were associated with increased bioactivity. While overall in vitro bioactivity profiles did not correlate with in vivo toxicity data on glycol ethers, in vitro bioactivity of E-series glycol ethers were indicative of and correlated with in vivo irritation scores.
Keywords: new assessment methodologies, glycol ethers, in vitro, ToxPi, read-across, safety assessment
1. Introduction
Most commodity chemicals that are registered under the European Union REACH (Registration, Evaluation, Authorization and Restriction of Chemicals) regulatory framework have data gaps, primarily the information from experimental toxicity studies in animals (Ball et al., 2014). Read-across has become the go-to method for filling data gaps for substances with deficiencies in testing data based on the available information of another substance(s) (Berggren et al., 2015; European Chemicals Agency, 2015). Both analogue and category read-across approaches are used in an attempt to reduce or replace the need for additional testing (Patlewicz et al., 2014; Patlewicz et al., 2015). Grouping of substances into a category may be achieved based on the similarity in structure, physico-chemical, toxicological, or eco-toxicological properties that are likely to be analogous or follow a regular pattern (European Chemicals Agency, 2015).
The REACH regulatory guidance stipulates that “for a read-across adaptation to be assessed and potentially accepted by the Agency, registrants have to show with clear reasoning and supporting data, that the substances involved in the read-across are structurally similar” (European Chemicals Agency, 2015). However, it is well established that structural and physicochemical property similarity is insufficient to justify read-across, because even minor structural changes to a molecule may introduce significant changes in bioavailability, metabolism, and resulting bioactivity (Aasmoe et al., 1998; Boatman, 2005; Grimm et al., 2015a; Gu et al., 2009; Staples et al., 1998). Consequently, each similarity argument in read-across needs to be supported through additional data streams, preferably in the context of chemical- or category-specific biological activity (Ball et al., 2016; National Academies of Sciences Engineering and Medicine, 2017; Rusyn et al., 2012; Zhu et al., 2016). The availability of in vitro data through large-scale toxicity testing programs (Thomas et al., 2018; Tice et al., 2013) creates opportunities to use this information as “bioactivity” that may support grouping of substances in read across. Indeed, several examples of how these data can underpin chemical and complex substance groupings have been published (Grimm et al., 2016; Low et al., 2013).
While a strategy to use bioactivity from in vitro assays to support grouping for read-across is predicated on chemicals of interest exhibiting effects that can be used for comparisons, a particular challenge in this respect are chemicals that are either inert or are missing clear indications for mechanism-based toxicity screening (Berggren et al., 2015). Glycol ethers are one representative case example. These are a class of high-production volume solvents that consist of an ether and an alcohol group and are based on either ethylene (E-series glycol ethers) or propylene (P-series glycol ethers) (Garlantezec et al., 2012; Poet et al., 2016). While glycol ethers are generally considered to be relatively non-hazardous substances, toxicity is observed after exposure to high doses (above 500 mg/kg bw/day) in animal models and can vary considerably among the individual glycol ethers (see Supplemental Tables). In humans, systemic toxicity from acute accidental and intentional ingestion of these compounds at high doses has been also reported (Cogliano et al., 2004). A key difference between E-series and P-series glycol ethers is the propensity of primary alcohols in E-series solvents to serve as substrates for alcohol dehydrogenase (ADH) thereby enabling bioactivation of the parent compound into a toxic acid metabolite (Aasmoe et al., 1998; Ghanayem et al., 1987; Miller et al., 1984b). P-series glycol ethers on the other hand possess a secondary alcohol group that cannot be metabolized by ADH which prevents the formation of toxic intermediates (Miller et al., 1984a); however, both primary and secondary alcohols can undergo a variety of conjugation reactions (Poet et al., 2016). While there is little information about the bioactivities associated with specific conjugated metabolites, these are considered non-toxic and readily excreted.
In this study, we evaluated several strategies to group 20 commercially relevant glycol ethers (Figure 1, Supplemental Table 1) into categories. We used in vivo rodent toxicity and other information that was submitted as part of REACH registration dossiers, as well as conducted additional in vitro studies to test for bioactivity in three different human cell types, hepatocytes, cardiomyocytes and lymphoblasts. Both high-content imaging (Grimm et al., 2015b) and gene expression data were collected, and inter-individual variability in responses was queried in vitro using lymphoblasts from ~100 individuals (Abdo et al., 2015b; Chiu et al., 2017). Multi-dimensional data streams were integrated and visualized using the Toxicological Prioritization Index (ToxPi) approach (Reif et al., 2013).
Figure 1.
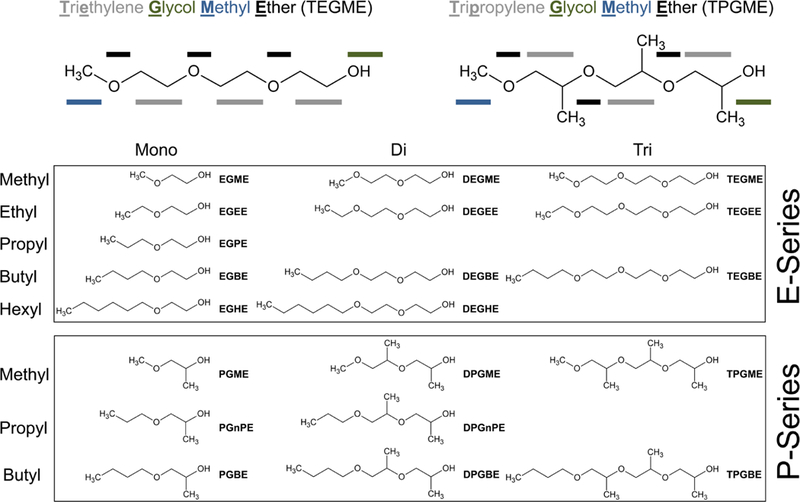
Chemical structures and nomenclature of P-series and E-series glycol ethers. Certain glycol ether preparations may consist of multiple isomers.
2. Materials & Methods
2.1. Data Acquisition and Transformation:
Data from a variety of toxicological studies on the compounds of interest (Figure 1, Supplemental Table 1) were collected from the REACH registration dossiers (http://echa.europa.eu/web/guest/information-on-chemicals/registered-substances) and assembled into a single spreadsheet where each row was assigned to a specific chemical, and the individual columns represent distinct endpoints for which a single value can be input. For acute toxicity endpoints, the Globally Harmonized System of Classification and Labelling of Chemicals (GHS) hazard categories were used to convert raw dose values to categories (United Nations, 2015). For repeated dose toxicity, developmental and reproductive toxicity, skin/eye irritation and skin sensitizing potential, Supplemental Table 2 shows how the in vivo study data were transformed into categories and normalized to be bounded by the interval [0, 1] to be compatible with the ToxPi input (Marvel et al., 2018). The data matrix resulting from this conversion is included as Supplemental Table 3. In addition to these endpoints, hematopoietic toxicity, hemolysis and testicular toxicity were recorded and scored separately with either a 1 or 0 when present or absent, respectively. These endpoints address specific target organs for several of the glycol ethers (E-series). The binary ‘presence/absence’ categorization system for reproductive and developmental toxicity endpoints was based on whether the substances would or would not be classified in accordance with GHS for these endpoints. If classification was deemed appropriate, a score of 1 was assigned.
2.2. Chemicals and Biologicals:
All P-series and E-series glycol ether samples (Figure 1, Supplemental Table 1) were provided by The Dow Chemical Company (Midland, MI) and were directly dissolved in cell media. iCell cardiomyocytes (cat. no.: CMC-100-010-001) and hepatocytes (cat. no.: PHC-100-020-001), including their respective plating and maintenance media were purchased from Cellular Dynamics International (Madison, WI). 94 human lymphoblastoid cell lines (Supplemental Table 4) were obtained from the Coriell Institute (Camden, NJ). EarlyTox Cardiotoxicity kits (cat. no.: R8211), containing reference chemicals isoproterenol (CAS 7683-59-2), sotalol (CAS 3930-20-9), and propranolol (CAS 525-66-6), were obtained from Molecular Devices (San Jose, CA). CellTiter-Glo Luminescent Cell Viability Assay kits (cat. no. G7573) were purchased from Promega (Madison, WI). RPMI 1640 medium (cat. no.: 11875093), B-27 medium supplement (cat. no.: 17504044), penicillin/ streptomycin solution (cat. no.: 10378016), gentamicin (50 mg/ml, cat. no.: 15750060), Hoechst 33342 (cat. no.: H3570), MitoTracker Orange CMTMRos reagent (cat. no.: M7510), CellROX Deep Red reagent (cat. no.: C10422), and HCS LipidTOX (cat. no.: H34477) were purchased from Life Technologies (Grand Island, NY). Fetal bovine serum (cat. no.: F2442), saponin (CAS 8047-15-2, cat. no.: 47036), cisapride monohydrate (CAS 260779-88-2, cat. no.: C4740), tetraoctyl ammonium bromide (CAS 14866-33-2, cat. no.: 294136), menadione (CAS 58-27-5, cat. no.: M5625), aflatoxin b1 (CAS 1162-65-8, cat. no.: A6636), amiodarone hydrochloride (CAS 19774-82-4, cat. no.: A8423), dimethyl sulfoxide (DMSO, CAS 67-68-5, cat.no. D2438), and formaldehyde (CAS 50-00-0, cat. no.: F8775) were purchased from SigmaAldrich (St. Louis, MO). Dexamethazone, hydrogen peroxide (3%), and recombinant oncostatin M (cat. no. 295OM010) were obtained from Fisher Scientific (Waltham, MA).
2.3. Cell Culture:
iCell cardiomyocytes were cultured in accordance with the manufacturer’s guidelines and as published previously (Grimm et al., 2015b). Briefly, cells were seeded in tissue-culture treated 384 well plates that were gelatinized for two hours at 37ºC and 5 % CO2 following the addition 25 µl of a 0.1 % (w/v) gelatin solution per well. iCell cardiomyocytes were thawed at 37°C in a waterbath for 4 minutes, before the contents of each vial were resuspended in 10 ml provided plating medium. The medium was equilibrated at room temperature and containing 1:500 (v/v) penicillin/streptomycin solution. The number of live cells in the suspension was determined by the trypan blue stain exclusion method using disposable hemocytometers. The suspension was then further diluted to a final cell density of 2*105 viable cells/ml and 25 µl (5000 cells), were then transferred per well after the gelatin solution had been discarded. Following a 30 minute resting period at room temperature, iCell cardiomyocytes were transferred to an incubator and maintained at 37ºC and 5 % CO2. The plating medium was completely exchanged with 30 µl of fresh maintenance medium (likewise containing 1:500 (v/v) penicillin/ streptomycin solution) 48 hours after initial cell plating. Cell media were changed every 48 hours for 12 days. On the experiment day, medium was completely exchanged with either 25 µl (for Ca2+ flux assay) or 40 µl (for high-content imaging) of fresh medium.
iCell hepatocytes 2.0 culture conditions were adapted from previous reports and reflect the manufacturer’s recommendations (Grimm et al., 2015b). Individual vials of iCell hepatocytes 2.0 were thawed for 3 minutes in a water bath at 37ºC. Cells were then resuspended in plating medium (RPMI 1640 containing 0.1µM dexamethasone, 2% (v/v) B27 supplement, 25 µg/ml Gentamicin, 20 ng/ml Oncostatin-M and 2% (v/v) iCell hepatocyte medium supplement) to a concentration of 7.2 × 105cells/ml based on viable cell count validation using the trypan blue exclusion method. 25 µl of cell suspension, i.e. 1.8×104 viable cells, were plated per well on Collagen I-coated 384-well plates. Following a 30 minute incubation at room temperature cells were transferred to an incubator at 37ºC and 5 % CO2. The plating medium was fully replaced with an equal volume of plating medium after 4 hours. Plating medium was subsequently exchanged daily for an additional 4 days. On day 5 post-plating, the plating medium was replaced by 25 µl/well maintenance medium (RPMI, 0.1 µM dexamethasone, 2% (v/v) B27 supplement, 25 µg/ml Gentamicin and 2% (v/v) iCell hepatocyte medium supplement). Maintenance medium was replaced every 48 hours. Prior to chemical treatments, medium was replaced with 20 µl of fresh maintenance medium.
Lymphoblastoid cell lines (n=46 female donors, n=48 male donors) comprising nine different populations, Utah residents with European Ancestry (CEU, n=13), Han Chinese from Beijing, China (CHB, n=14), Colombians from Medellin, Colombia (CLB, n=11), British from England and Scotland (GBR, n=9), Japanese from Tokyo, Japan (JPT, n=10), Luhya from Webuye, Kenya (LWK, n=8), Los Angeles residents with Mexican Ancestry (MXL, n=10), Tuscans from Italy (TSI, n=12), and Yoruba from Ibadan, Nigeria (YRI, n=11) were randomly assigned into four experimental batches and cultured as described previously (Abdo et al., 2015a; Abdo et al., 2015b). Cell aliquots were thawed and transferred to 15 ml sterile centrifuge tubes containing 6 ml cell medium (RPMI 1640 media containing 15% fetal bovine serum and 100 U/mL penicillin/100 mg/mL streptomycin). Cells were washed by centrifugation at 200xg for 10 min followed by suspending the cell pellet in 10 ml medium and filtration of the cell suspension using a 40 µm cell strainer. Cell density was determined through automated cell counting using the trypan blue exclusion method and the medium volume was adjusted to a final cell density of 2×105 cells/ml. Cells were transferred to a tissue culture flask (final volume 20 ml) and incubated for 5 days at 37ºC and 5% CO2. On day 6 post-plating, cell suspension was diluted to 4×105 cells/ml and 12.5 µl aliquoted into each well on a 384 well plate (final density of 5000 cells/well).
2.4. Chemical Preparation and Cell Treatments:
Chemical master plates were prepared as 5x concentrated stocks for iCell cardiomyocytes and 2x concentrated stocks for iCell hepatocytes and lymphoblasts cell lines. Glycol ethers, ethylene glycol, and propylene glycol were soluble in the specific cell media and thus did not require the addition of a solvent. Chemical master plates included duplicate samples of all test chemicals in concentration-response: 0.01, 0.1, 1, and 10 mM for iCell Cardiomyocytes, 0.0001, 0.001, 0.01, 0.1, 1, and 10 mM for iCell hepatocytes, and 0.1, 1, 3, and 10 mM for lymphoblasts. Concentration ranges were selected to span several orders of magnitude as recommended for in vitro toxicity screening (Parham et al., 2009). The top concentration was selected to be near the highest reported human blood concentration, around 5 mM, detected after intentional ingestion of around 500 mL of 2-butoxyethanol-containing consumer product (Gualtieri et al., 2003). Stock solutions of positive assay controls tetraoctyl ammonium bromide (all cell types), isoproterenol, propranolol, cisapride, and sotalol (iCell Cardiomyocytes), and menadione, aflatoxin b1, and amiodarone (iCell hepatocytes) were prepared in DMSO. The final DMSO concentration in medium was 1% (v/v). Appropriate vehicle controls were assigned to the plate design to exclude vehicle effects for reference controls. All wells on specific 384 well plates were treated simultaneously with chemicals (10 µl of 5x chemicals for iCell cardiomyocytes, 20 µl of 2x chemicals for iCell hepatocytes, and 12.5 µl of 2x chemicals for lymphoblasts) using the automated liquid handler in the FLIPR tetra (Molecular Devices, Sunnyvale, CA).
2.5. Ca2+ Flux Measurements:
Intracellular Ca2+ flux in iCell cardiomyocytes was measured as described in more detail elsewhere (Sirenko et al., 2017). iCell cardiomyocytes (25 µl media volume) were incubated after addition of 25 µl of pre-warmed calcium dye reagent for 2 hrs at 37ºC. Following this equilibrating period, 12.5 µl of 5x concentrated test chemicals and controls were added using the internal liquid handler of the FLIPR® tetra cellular screening system (Molecular Devices). Treated iCell cardiomyocytes were then incubated for 90 min. After 90 min incubation at 37°C and 5% CO2, the intracellular Ca2+ flux was measured at 515–575 nm (λexc=470–495 nm) for 100 seconds using a frequency of 8 Hz, i.e. 0.125 s/read. Instrument settings were adjusted as follows: exposure time per read −0.05s, gain – 2000, excitation intensity −30%, internal temperature −37ºC. Ca2+ flux traces were processed using Screenworks 4.0 (Molecular Devices) and quantitative descriptors Peak Frequency, Peak Amplitude, Peak Width, Peak Width at 10% Amplitude, Peak Baseline, Peak Spacing, Peak Rise Time, and peak Decay Time were extracted.
2.6. High-content cellular imaging:
For quantitative evaluation of cytotoxicity and mitochondrial integrity, iPSC cardiomyocytes were treated with test chemicals as specified above. After 24 hours of treatment, 50 µl pre-equilibrated 2x concentrated staining solution (iCell cardiomyocyte maintenance medium supplemented with 4 µg/ml Hoechst 33342 and 0.4 µM MitoTracker Orange) was added to each sample well and cells were incubated for 15 min at 37ºC and 5% CO2. Next, the cell medium was discarded and cells were washed with maintenance medium prior to the addition of 25 µl 3.7% formaldehyde solution. After 15 min of incubation at 37ºC and 5% CO2, cells were washed three times with 25 µl PBS. Images were acquired on the ImageXpress Micro XL (Molecular Devices), using the DAPI (Hoechst 33342) and Cy3 (MitoTracker Orange) filters.
High-content imaging in iPSC hepatocytes was carried out following exposure of cells to test chemicals for 48 hours (total media volume 40 µl). Cells were incubated for 15 min at 37°C and 5% CO2 after addition of an equal volume of 2x concentrated staining solution containing 4 µg/ml Hoechst 33342, 0.4 µM MitoTracker® Orange, and 10 µM CellROX® Deep Red before they were washed with media and fixed for 15 min in 25 µl 3.7% formaldehyde. Cells were then washed three times with 25 µl PBS and image acquisition was completed on the ImageXpress Micro XL using the DAPI (for Hoechst 33342) and Cy3 (for MitoTracker® Orange), and Cy5 (for CellROX®) filters.
A second set of iPSC hepatocyte samples was washed with PBS followed by 3.7% formaldehyde fixation for 15 min at 37°C and 5% CO2. Cells were then washed with PBS and incubated for 1 hour with 25 µl of cell permeabilization solution (0.02% saponin plus 2% (w/v) FBS in PBS), followed by staining with 165 nM AlexaFluor® 488 Phalloidin in PBS for 120 min at room temperature. Cells were then washed twice with PBS after which cells were stained for 20 min at room temperature with 25 µl LipidTOX™ Deep Red solution (1000x diluted in PBS). Images were recorded on the ImageXpress Micro XL using the FITC (for AlexaFluor® 488 Phalloidin) and Cy5 (for LipidTOX™ Deep Red) filters. Acquired images were processed using the instrument-specific MetaXpress software (Molecular Devices). Quantification of imaging-based parameters for concentration-response assessment was achieved using the multiwavelength cell scoring application module.
2.7. Cell Viability Screening in Human Lymphoblasts:
Cytotoxicity in human lymphoblasts was evaluated using by quantification of ATP levels using the Cell-Titer Glo® Luminescent Cell Viability Assay (Promega, Madison, WI). After initial cell plating, samples were treated with an equal volume (12.5 µl) of 2x concentrated chemical solutions, followed by incubation for 48 hours at 37ºC and 5% CO2. Microplates were then equilibrated at room temperature for 30 min before 25 µl Cell-Titer Glo® Luminescent Cell Viability Assay solution (Promega) were added per well. Contents were mixed at 300 rpm using an orbital shaker to induce cell lysis. Following additional incubation for 10 min at room temperature, the luminescent signal was recorded on the FLIPR® tetra and Screenworks 4.0 (Molecular Devices).
2.8. TempO-seq Gene Expression Profiling:
Differential gene expression was analyzed by targeted RNA sequencing using TempO-Seq™ (BioSpyder Technologies, Carlsbad, CA) as detailed in (Grimm et al., 2016; House et al., 2017). iCell hepatocytes were treated with glycol ethers in concentration-response as described above. After 48 hr cell medium was aspirated and cells were lysed by the additional of 10 µl TempO-Seq lysis buffer. Following 15 min of mixing at 300 rpm on a bench-top plate micro plate shaker, lysates were stored at −80°C until further processing.
The sequencing library was prepared according to the manufacturer’s protocol. Briefly, 2 µl of each cell lysate were hybridized with an equal volume of Tox Panel detector oligo mix, targeted at 2,983 pre-selected mRNAs (10 min at 70°C followed by a temperature gradient to 45°C over 40 min). Following a nuclease digestion step (90 min at 37°C), the hybridization products were ligated (60 min at 37°C followed by 30 min enzyme denaturation at 80°C) to provide a pool of amplification templates (final volume 52 µl). Ten µl of each ligation product were then transferred to their respective positions on a new 96-well plate. Each well contained specific, “barcoded” primer pairs that ensure unambiguous matching of sequencing reads for each sample. Product amplification was carried out in a Roche Light Cycler 96 (Roche Diagnostics, Indianapolis, IN) using the following settings: 10 min at 37°C, 2 min at 95°C, followed by 25 cycles of 30 sec at 95°C, 30 sec at 58°C and 60 sec at 72°C. After completion of the last cycle an additional 60 sec at 72°C was added before the reaction mix was cooled to 25°C. 5 µl of each amplicon were then combined into a sequencing library. Libraries were cleaned up using a PCR clean-up kit (Clontech, Mountain View, CA) prior to sequencing using both lanes on a rapid flow cell on a HiSeq 2500 Ultra-High-Throughput Sequencer (Illumina, San Diego, CA).
2.9. Transcriptomics Data Analysis:
Differential gene expression in glycol ether-treated hepatocytes was evaluated using the DESeq2 R package as detailed elsewhere (Grimm et al., 2016). Briefly, read counts for each gene and sample were normalized under consideration of compositional bias i.e. library size and sample-specific differences in read-counts. A correction factor was based on the median ratio of a specific transcript count over the geometric average of all counts for that specific gene within a dataset. Adjusted counts were further processed by Benjamini-Hochberg false discovery rate (FDR) correction to reveal significant changes in differential gene expression based on a q-value of <0.1. Cluster analysis of global differential gene expression changes was conducted using the hclust package in R. For each chemical and 1541 mapped genes, the DESeq2 calculated log2 fold change (10 mM dose vs. vehicle) and associated q-values were loaded into Ingenuity®. Genes and pathways were clustered and a heatmap (average linkage) was generated.
2.10. Data Integration in ToxPi:
For global ranking and qualitative comparison of bioactivity of the chemicals tested in this study, POD values were integrated and visualized using the Toxicological Priority Index Graphical User Interface (Reif et al., 2013). POD values for 20 bioactivity phenotypes (see Supplemental Table 5) from hepatocytes, cardiomyocytes, and lymphoblasts were each normalized and scaled from 0 to 1 using logPOD which more appropriately represents changes across several orders of magnitude. For in vivo data (Supplemental Table 3), 21 endpoints were used in ToxPi analysis (Supplemental Table 5). The values of 0 reflect the highest observed POD or the highest tested concentration for nonbioactive chemicals and the value of 1 reflects the lowest observed POD value, i.e., the most bioactive chemical with lowest POD, for each individual phenotype. The scaled POD values were then transferred into a ToxPi compatible data matrix and used to visualize chemicalspecific bioactivity profiles. In the resulting pie charts, each slice is reflective of the bioactivity in each cell type and bioactivity, and the area of the slice is proportional to the relative bioactivity within this data set. Slices were scaled based on the number of phenotypes included. A ToxPi score is the sum of all slice areas.
2.11. Assay Quality Controls:
A number of assay-specific performance criteria were assessed. Assay robustness of in vitro screening in iPSC-derived cardiomyocytes and hepatocytes was shown by determination of coefficients of variation (%CV) (Supplemental Table 6). Intra-plate variability was assessed where appropriate by regression (Pearson) and rank-based (Spearman) correlation (Supplemental Table 6). Inter-plate replicability was not assessed, as experiments were conducted on a single plate. Assay-specific phenotypic quality controls were included to evaluate each phenotype. For cardiomyocytes, cell performance was evaluated with known cardio-active drugs isoproterenol (positive inotrope), propranolol (negative inotrope), and cisapride (potassium channel inhibitor). Tetraoctylammonium bromide was used as a cytotoxic control. Positive control chemicals for hepatocytes included menadione (ROS formation, cytotoxicity), aflatoxin b1 (cytotoxicity), and amiodarone (steatotic agent). Intra-plate (Supplemental Figure 1) and batch-to-batch reproducibility (Supplemental Figure 2) of lymphoblast screening experiments was assessed using regression (Pearson) and rank-based (Spearman) correlation analysis. High-throughput transcriptomics quality criteria included blank controls (lysis buffer) for exclusion of potential well contamination, and lysate samples from untreated and vehicle-treated cells. Positive controls for assay performance was the universal human reference RNAs.
3. Results
3.1. High-Content Screening in iPSC Hepatocytes and Cardiomyocytes:
The utility of in vitro bioactivity profiling to facilitate decision-making through grouping of chemicals has been recently demonstrated (Chiu et al., 2018; Grimm et al., 2016). Therefore, we evaluated bioactivity of glycol ethers, including ethylene glycol and propylene glycol (Figure 1), in concentration-response (0.001 to 10 mM) for five endpoints in hepatocytes and ten in cardiomyocytes. Figure 3 shows concentration-response profiles for several representative phenotypes in each cell type. Threshold values of the effects (points of departure, POD) were calculated for each compound, phenotype and cell type and are listed in Supplemental Table 7. We found that all substances exerted no cytotoxicity up to 1 mM concentration. Hexyl etherderivative DEGHE was cytotoxic at 10 mM in both hepatocytes (POD=1.18 mM) and cardiomyocytes (POD=1.43 mM), whereas EGHE (POD=6.16 mM) and TGBE (POD=4.58 mM) had an effect only in hepatocytes.
Figure 3.
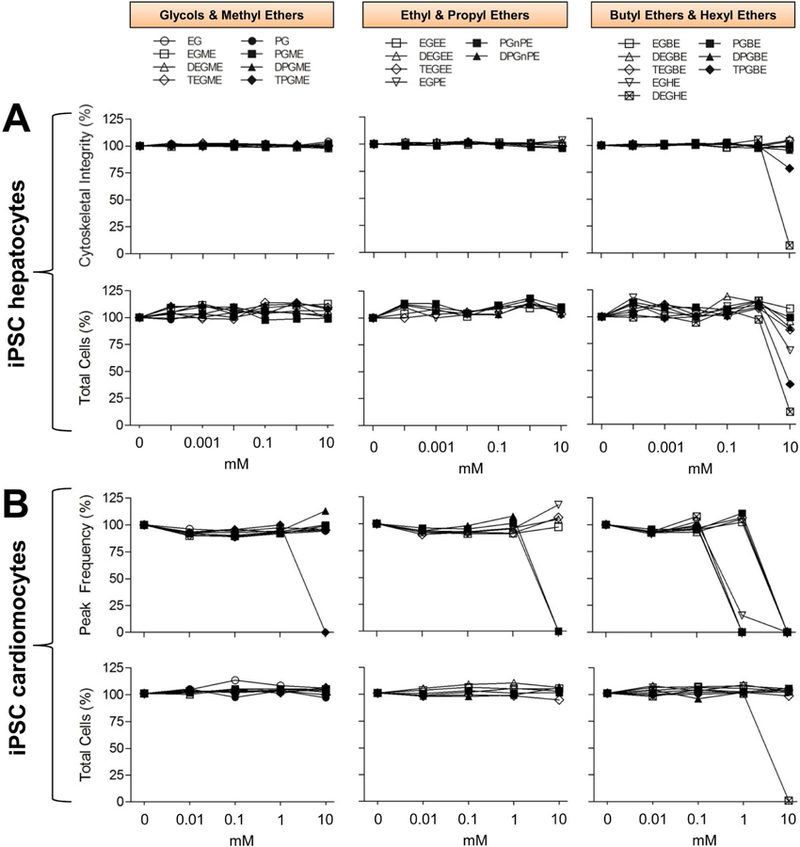
Concentration-response plots for representative phenotypes of iPSC hepatocytes (A) and cardiomyocytes (B). Individual plots indicate averages of duplicate measurements of cardiomyocyte peak frequency (beats per min, BPM), live cell count (both cardiomyocytes and hepatocytes) and cytoskeletal integrity (hepatocytes) for simple glycols and methyl ethers (left column), ethyl & propyl ethers (middle column) and butyl and hexyl ethers (right column).
With respect to the effects on the functional endpoints in cardiomyocytes, several substances showed effects at sub-cytotoxic high concentrations. At 10 mM concentration, TPGME, PGnPE, DPGnPE, and all butyl- and hexyl ether derivatives showed an effect on beating rate and other functional parameters. Both hexyl ethers (EGHE and DEGHE) and di- and tri-propylene glycol butyl ethers (DPGBE and TPGBE) exhibited effects at 1 mM concentration (POD range 0.12–0.35 mM). Interestingly, these compounds were bioactive in cardiomyocytes at approximately one order of magnitude lower concentrations than ethylene glycol-based butyl ethers (EGBE, DEGBE, and TEGBE) and PGBE (POD range 2.5–3.5 mM). Overall, concentration-response assessment of bioactivity in these cell types and endpoints showed chemical group-specific trends indicating that the alcohol chain length is a likely determinant for in vitro effects.
3.2. Population Variability Screening in Human Lymphoblasts:
We used a human in vitro population-based model (Abdo et al., 2015b; Eduati et al., 2015) to evaluate inter-individual variability in cytotoxicity. We used a total of 94 human lymphoblast cell lines derived from genetically distinct individuals (Supplemental Table 4), a number selected based on the previous analysis demonstrating that a ~100 lymphoblast cell line sample size is sufficient to accurately estimate both the mean and variability in cytotoxic effects of chemicals in a population (Chiu et al., 2017). For quality control, we evaluated both intra-plate, as well as inter-plate and batch-tobatch variability (Supplemental Figures 1 and 2). There was good correlation between sample replicates (Pearson’s r range 0.67 to 0.98, and Spearman’s ρ range 0.66 to 0.96). Batch-to-batch correlation revealed consistently good linearity (Pearson’s r range 0.74 to 0.95, Spearman’s ρ range 0.36 to 0.81).
Chemical-specific differences in the potency and inter-individual variability in cytotoxic effects of glycol ethers was observed (Figure 4, Supplemental Figure 3). Example concentration-response profiles across 94 lymphoblast cell lines for two tested substances are shown in Figure 4A. A wide range in effects on the individual cell lines was found for both compounds, with no significant difference among the sub-populations of origin. However, while P-series glycol ethers exhibited largely consistent effects with population means around 1 mM, E-series glycol ethers showed a wider range in the population mean activity among the compounds in this class (Figure 4B).
Figure 4.
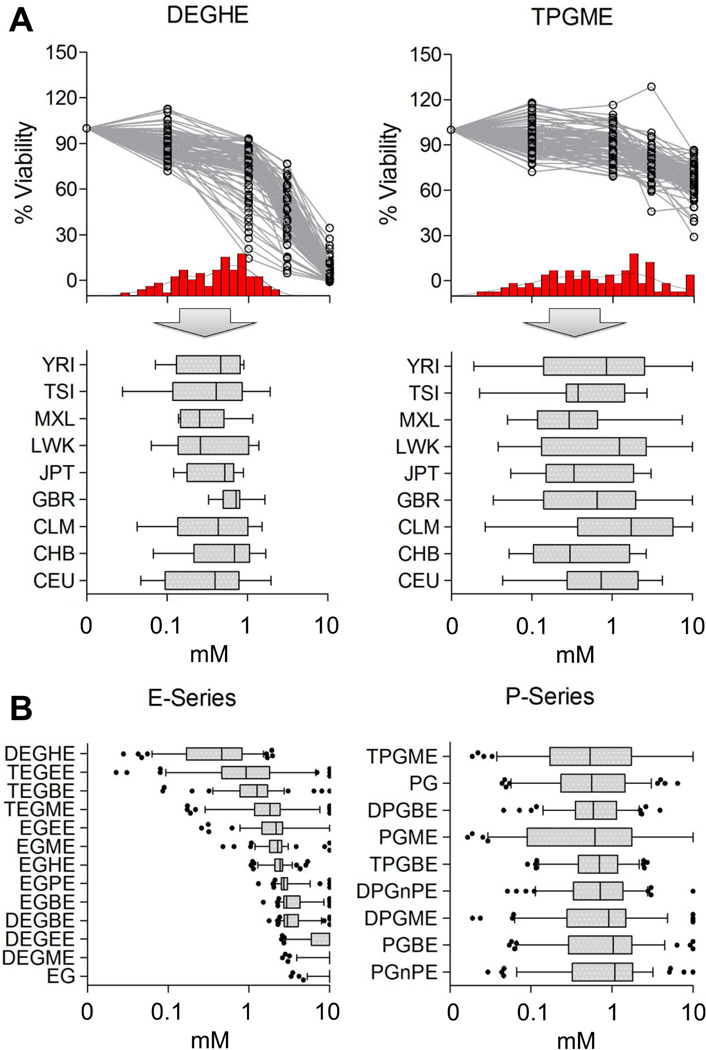
Population variability assessment of E-Series and P-Series glycol ethers in human lymphoblast cell lines. Panel A provides an overview of the distribution of calculated Point-of-Departure (POD) values for each one selected E-Series (DEGHE) and P-Series (TPGME) glycol ether across all tested cell lines (n=94). Individual concentration response plots and relative frequency distribution of calculated point-of-departure (POD) values (red inserts) are shown in the top part, the POD frequency distribution for all nine tested donor populations is shown on the bottom. Panel B highlights the distribution of POD values for both E-Series and P-Series glycol ethers across all populations. Individual box and whisker plots indicate the median and span across the 5–95 percentile. Statistical outliers are depicted as individual dots. Abbreviations: CEU=Utah residents with European Ancestry (n=13); CHB=Han Chinese from Beijing, China (n=14); CLB=Colombians from Medellin, Colombia (n=11); GBR=British from England and Scotland (n=9); JPT=Japanese from Tokyo, Japan (n=10); LWK=Luhya from Webuye, Kenya (n=8); MXL=Los Angeles residents with Mexican Ancestry (n=10); TSI=Tuscans from Italy (n=12); YRI=Yoruba from Ibadan, Nigeria (n=11).
3.3. Transcriptomic Profiling in iPSC Hepatocytes:
While apical in vitro bioactivity phenotypes have proven useful in grouping substances for decision-making (Chiu et al., 2018; Grimm et al., 2016), molecular signatures (e.g., gene expression patterns) are also a valuable data stream for evaluating similarities in chemical effects (De Abrew et al., 2016; De Abrew et al., 2015). Here, the effect of glycol ethers on gene expression in hepatocytes was explored. We evaluated gene expression differences between untreated cells, or cells treated with 10 mM concentration of the tested compounds, in replicate analyses and observed excellent reproducibility (Supplemental Figure 5).
Differential gene expression was examined across all P- and E-series glycol ethers (Figure 5A-C). Few transcripts were altered by more than 2-fold (Figure 5A) with most effects observed in cells treated with di- (DEGBE, DPGME, DPGnPE, DPGBE) and tri-ether (TEGBE) compounds. Transcriptional effects, if present, were highly concordant across compounds (Figure 5B). The similarities in gene expression across all substances, as well as among the highest correlated ones (DEGBE, DPGnPE, TEGBE and DPGBE), is also apparent when the data are presented as a heat map (Figure 5C). Furthermore, we evaluated the biological processes that were affected by treatment with glycol ethers (Figure 5D). Most pronounced effects were observed on the biological processes involved in lipid and energy metabolism (beta-oxidation fatty acids, metabolism and movement of lipids, steroids, sterols, and cholesterol), effects that are concomitant with the detergent properties of glycol ethers and their likely primary effect on cell membranes.
Figure 5.

TempO-seq Gene Expression. P- and E-series glycol ethers were assessed via TempO-seq for expression of 1616 genes. Counts were normalized with DESeq2, and expression ratios computed (10mM treated counts / vehicle treated counts). The data were then visualized in the following ways: (A) Boxplots of absolute log2(ratio), (B) correlation matrix of the log2(ratio); the magnitude of these values are visualized in the lower triangular region using circle size/color combinations, and (C) heatmap of log2(ratio). Functional Pathway Analysis (D) using expression log2(ratio) were analyzed for dose-response association. A clustered heatmap (average linkage) is shown for the top 15 disease and biological functions. Negative scores indicate pathway downregulation, positive scores are associated with functional pathway induction.
3.4. Grouping of P- and E-Series Glycol Ethers with in vitro Data.
High-content screening (Figures 3-4) and transcriptomic (Figure 5) data were integrated into the overall bioactivity profiles (Figure 6) using the ToxPi approach (Reif et al., 2013). There were 20 in vitro-data derived phenotypes that were used for ToxPi analysis shown in this figure (see Supplemental Table 8 for the complete data matrix and description of each phenotype). First, the substances were ranked according to their ToxPi score (Figure 6A). No clear distinction between P- and Eseries glycol ethers was evident in their overall ToxPi score; however, clear trends in the potency based on the alcohol chain length were observed when ToxPi’s for individual substances were arrayed based on this chemical property (Figure 6B). The longer alcohol chain length (i.e. hexyl & butyl ethers) substances were more bioactive than propyl-, ethyl- and methyl-containing glycol ethers. Similarly, in many instances increases in bioactivity were concordant with an increase in the length of the ether, i.e. mono-ethers were less bioactive than di-ethers and triethers. Hierarchical clustering analysis (Figure 6C) confirmed no clear separation between E- and P-series glycol ethers, but showed that certain compounds with similar secondary chemical characteristics, e.g. butyl and hexyl-containing glycol ethers (EGBE and EGHE, or DEGHE, DPGBE, and DEGBE) were concordant with their in vitro bioactivity. Principal components analysis (Figure 6D) provided additional support for the observation that the alcohol chain length is the primary determinant for observable in vitro bioactivity, showing spatial separation between shorter (methyl, ethyl, propyl) and longer alcohol-containing (butyl, hexyl) ethers, while accounting for 85% of the observable variation.
Figure 6.
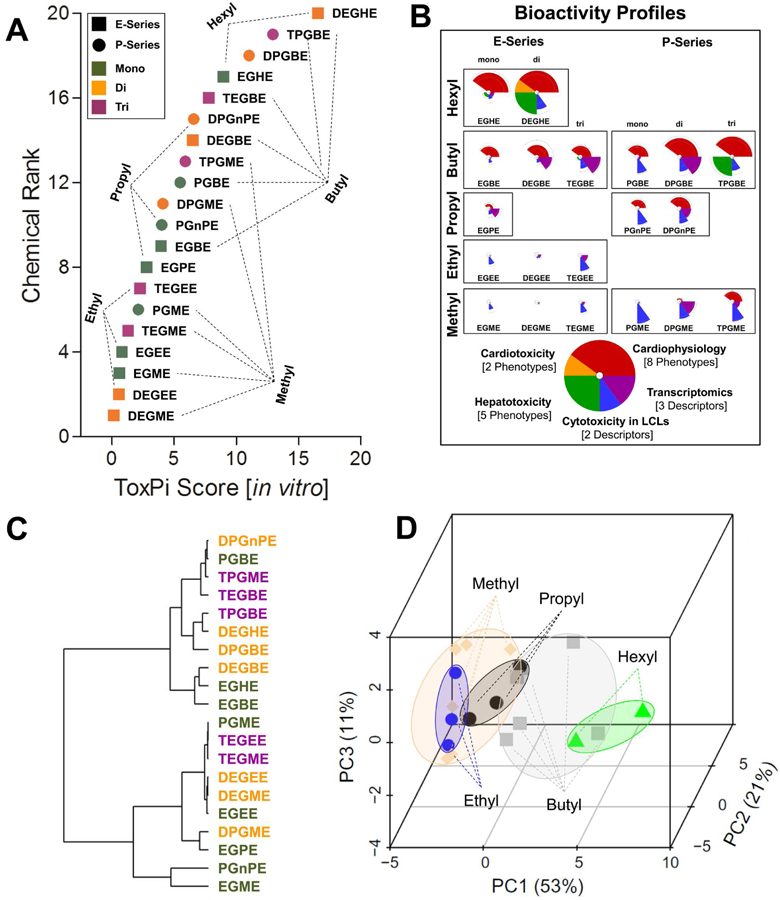
Biological data-integrative grouping of P-series and E-series glycol ethers. Unbiased global ranking of chemicals according to their cumulative ToxPi score was achieved using 20 in vitro derived phenotypic descriptors (Supplemental Table 8). (A). Individual bioactivity profiles are shown in panel (B). A correlation dendrogram based on Spearman’s ρ correlation coefficients and principal components analysis across all phenotypic descriptors are shown in panels C and D.
3.5. Evaluation of the similarities between in vitro and in vivo effects of glycol ethers.
Last, we compared in vitro bioactivity of glycol ethers to their effects in guideline in vivo toxicity studies. In vivo toxicity data for each endpoint were rescaled using GHS-like transformation criteria (Supplemental Table 2) to enable ToxPi analysis. Scaled in vivo phenotypes (Supplemental Table 3) were integrated into in vivo effect profiles using 5 toxicity categories: acute toxicity (5 phenotypes), irritation (3 phenotypes), repeat-dose toxicity (6 phenotypes), developmental toxicity (3 phenotypes), and reproductive toxicity (4 phenotypes) and rank-ordered according to their overall ToxPi score (Figure 7A, Supplemental Table 9 for the complete data matrix and description of each phenotype). ToxPi-based ranking showed that E-series monoesters (EGBE, EGPE, EGEE, EGHE, and EGME) had most effects in vivo; however, no other patterns of in vivo effects were evident among E- and P-series glycol ethers. Next, we plotted correlation between ToxPi scores from in vitro and in vivo data and found that overall effects of these substances in vivo and in vitro are not concordant, a finding that is in agreement with known limitations of relying solely on in vitro data to predict hazard (Becker et al., 2017). To narrow comparisons of concordance to more closely related endpoints, we examined correlations between individual mammalian toxicity categories and in vitro ToxPi scores. This analysis revealed that for E-series glycol ethers, but not P-series, in vitro bioactivity was significantly correlated with in vivo irritation (Figure 7B). Importantly, the rank order for compounds in both dataset was reflective of the alcohol chain length-associated increase in irritation potential.
Figure 7.
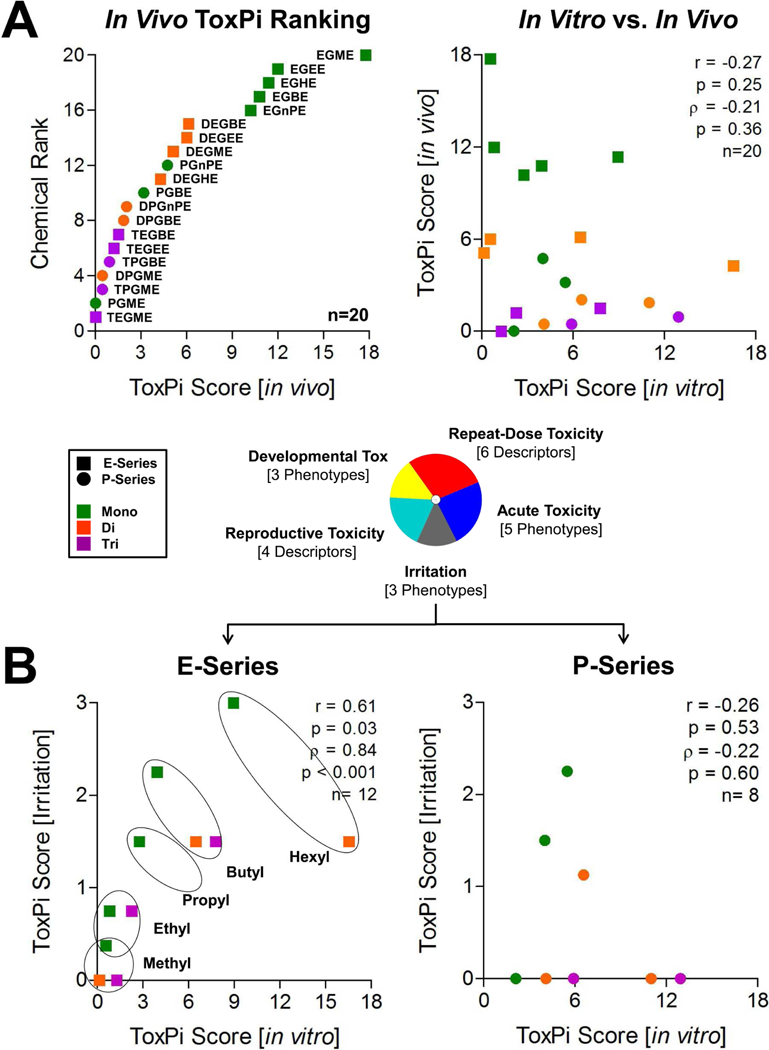
Implementation of multidimensional in vitro screenings for biological data-integrative read-across applications. GHS-like transformation criteria (Supplemental Table 1) for 21 in vivo phenotypes (Supplemental Table 9) from five major toxicity categories provided the basis for comprehensive in vivo toxicity profiling and ranking of 20 glycol ethers (A). In vivo data were accessible through official REACH submissions of The Dow Chemical Company. Correlation analysis between in vitro and in vivo, as well as in vitro and in vivo irritation ToxPi scores for E- and P-Series glycol ethers are shown in panels A and B. Pearson’s r and Spearman’s ρ correlation coefficients including significance levels are indicated.
4. Discussion
Integration of novel in vitro and computational data streams into practice of regulatory decision-making is both an opportunity and a challenge in moving the toxicology into the 21st century. There is great anticipation that novel technologies will help redefine chemical safety assessments, thereby exerting a considerable impact on improving public health decisions (National Academies of Sciences Engineering and Medicine, 2017; National Research Council, 2007). Moreover, integration of in vitro models in combination with state-of-the-art screening approaches, including high-content imaging and “omics” technologies, into hazard and risk assessments is a practical next step in fulfilling the vision for the 21st century toxicology (Thomas et al., 2018). Despite significant progress, regulatory acceptance of these data has been slow, as the challenge lies in developing a framework that can guide the interpretation of the data streams generated using in vitro methods such that it is clear where they can or cannot fit into the existing workflows for hazard and risk assessments (Chiu et al., 2018; Guyton et al., 2018a; Rusyn and Greene, 2018; Thomas et al., 2018). This challenge is compounded by the constant evolution in the testing methods and data integration approaches, innovations that range from complex tissue-chip systems (Marx et al., 2016) to adverse outcome pathways (Kleinstreuer et al., 2016).
While it is still debated whether in vitro data have utility for hazard identification (Becker et al., 2017; Goodman et al., 2018; Guyton et al., 2018b; Thomas et al., 2012), there are a number of examples where bioactivity, alone or in combination with structural and physicochemical properties, is informative for increasing confidence in hazard classifications (Chiu et al., 2018; Guyton et al., 2018a), or groupings that underpin category read-across (Ball et al., 2016; Grimm et al., 2016; Low et al., 2013; Patlewicz et al., 2014; Zhu et al., 2016). Indeed, even though current regulatory guidelines largely confine read-across to the apical toxicity data from rodent and other traditional experimental models, data from alternative methods can be used as supporting evidence (European Chemicals Agency, 2015). However, only few studies explored whether in vitro data can result in groupings that are similar to those based on the more traditional data streams and thus increase confidence in their use for category formation.
This study attempted to examine the utility of in vitro bioactivity, data that combined a wide range of endpoints in several cell types, for grouping substances that comprise several closely related chemical classes. Glycol ethers are a relevant group of substances identified as a suitable case study for assessing the use of novel testing methods to strengthen the evidence base for decision making though read-across (Berggren et al., 2015). Specifically, propylene glycol ethers are considered as substances with no or very low toxicity. In contrast, the ethylene glycol ethers have been associated with both hematopoietic and reproductive toxicity (Wess, 1992), effects that are thought to be mediated by alkoxyacid metabolites (Lockley et al., 2005). Indeed, including both propylene and ethylene glycols into a case study was meant to facilitate understanding of the difference in bioactivity in vitro and to assist in gaining confidence in readout result from the assumed non-toxic substances (Berggren et al., 2015).
In this case study, we used several diverse, organotypic and population-based in vitro models, human iPSC-derived hepatocytes and cardiomyocytes, and lymphoblasts. These three cell types were chosen to include a metabolically-competent cell type (i.e., iCell hepatocytes (Grimm et al., 2015b; Sirenko et al., 2014)), a cell type that replicates organ-specific function yet is not a metabolically-active cell type (i.e., iCell cardiomyocytes (Rana et al., 2012; Sirenko et al., 2013; Sirenko et al., 2017)), and a blood-derived cell type that is also a population-based in vitro model (i.e., lymphoblasts (Abdo et al., 2015a; Abdo et al., 2015b)). This set of cell types represented a reasonable number of in vitro models without bias to the potential organ-specific effects from in vivo studies. In addition, successful grouping of complex substances based on bioactivity data from two of these cell types was recently demonstrated (Grimm et al., 2016). While this approach relies on pre-selected biological targets and cells, thereby potentially limiting the interpretability from a hazard assessment perspective, this experimental design is sensible for using bioactivity in data-informed similarity evaluations. The goal was to evaluate the similarity among a group of chemicals using multiple, diverse biological data streams to improve confidence in assigned groupings, a “weight of evidence” approach that is consistent with recommended application of alternative test methods in legislative frameworks, including REACH (European Chemicals Agency, 2015) and the Lautenberg Chemical Safety Act (Krimsky, 2017).
Overall, we observed low cytotoxicity potential of both E- and P-series glycol ethers as the effects were observed only at the highest concentration tested (10 mM); albeit the functional endpoints, i.e. iPSC-derived cardiomyocyte beating-derived parameters, were more sensitive compared to cytotoxicity. The variation in the chemical structure of substances tested herein was primarily determined by two factors, the alcohol chain length and presence of either ethylene or propylene moiety. We found that in vitro bioactivity increased with increasing alcohol chain lengths, whereas presence of either ethylene or propylene had no impact. This observation is consistent with solvent properties being the primary determinant of observable cytotoxic effects of glycol ethers as a broader class. Transcriptomic analysis in glycol ether-treated hepatocytes revealed that most substances tested had little to no effects on gene expression, in accord with no effect on viability. Concomitantly, a group of glycol ethers with similar alcohol length (DEGBE, DPGnPE, TEGBE and DPGBE) affected gene expression and exhibited cytotoxicity. Interestingly, we found that while there was no sub-population that was particularly sensitive to any of the substances tested, inter-individual variability did vary considerably among tested substances in the ethylene glycol category.
By combining the wide range of bioactivity data for each substance using ToxPi, clustering and principal components analysis we sought to present a transparent and unbiased relative ranking/grouping of the tested substances to examine if patterns emerged that can assist in evaluating bioactivity-based similarity. These analyses revealed several trends further indicating that longer alcohol chain lengths is associated with higher bioactivity to a greater extent than the number of E and P units. These results indicate that solvent effects of the substances most likely impact the integrity of the cellular membranes. To further contextualize findings from the in vitro bioactivity profiling we performed correlation analysis with mammalian toxicity profiles. This exercise clearly indicated no correlation between the totality of the in vitro bioactivity data and a combination of mammalian toxicity data, or sub-categories such as acute, repeat-dose, reproductive, and developmental toxicity. However, consistent with the hypothesis on solvent properties of glycol ethers as the primary driver of adverse effects, in vitro data for E-series glycol ethers showed positive correlation with in vivo irritation data.
A related consideration for data integration across in vivo and in vitro studies is a challenge with dose comparison (Wetmore, 2015). In this study, we chose to discretize the animal data into GHS categories, or in the case of reproductive and developmental studies into binary hazard classes. This may be perceived as leading to a loss of dose-response information on the effects in the animal studies and thus may be related to the apparent lack of correlations observed in this study. While we agree that quantitative comparisons and in vitro to in vivo extrapolation using reverse toxicokinetic modeling are a desired strategy, in the case of glycol ethers this type of correlation is challenging as the animal studies database is rather sparse and divergent with respect to the doses used. In addition, interpretation of some of the effects as reproductive or developmental may depend on the regulatory context and we reasoned that for this set of compounds, the most sensible approach was to use GHS categories.
Overall, these studies support the utility of novel data streams for groupings of chemicals for subsequent read-across. Both ethylene and propylene-based glycol ethers grouped primarily according to their alcohol chain length, which indicates the concordance between chemical and biological similarity. This also provides a plausible explanation for observed cytotoxic effects, a finding that is also consistent with in vivo irritation effects that can be related to the same underlying mechanism of toxicity, i.e. cell lysis due to solvent effects. However, considering the ongoing debate on implementation of emerging technologies into regulatory decision making, this study clearly indicates the need for care in the interpretation of in vitro data, and warn against over-generalization as they still represent a fit-for-purpose approach that is limited in its ability to integrate specific mechanisms of action, i.e. those that require metabolic activation and/or non-standard cell systems. This is best reflected by the lack of correlation of in vitro bioactivity scores of glycol ethers and their mammalian toxicity counterparts for acute, repeatdose, reproductive, and developmental toxicity endpoints, which involve metabolic activation as a key event for progression from initial exposure to measurable adverse outcome (Ghanayem et al., 1987; Miller et al., 1984a; Miller et al., 1984b; Poet et al., 2016).
Supplementary Material
Figure 2.
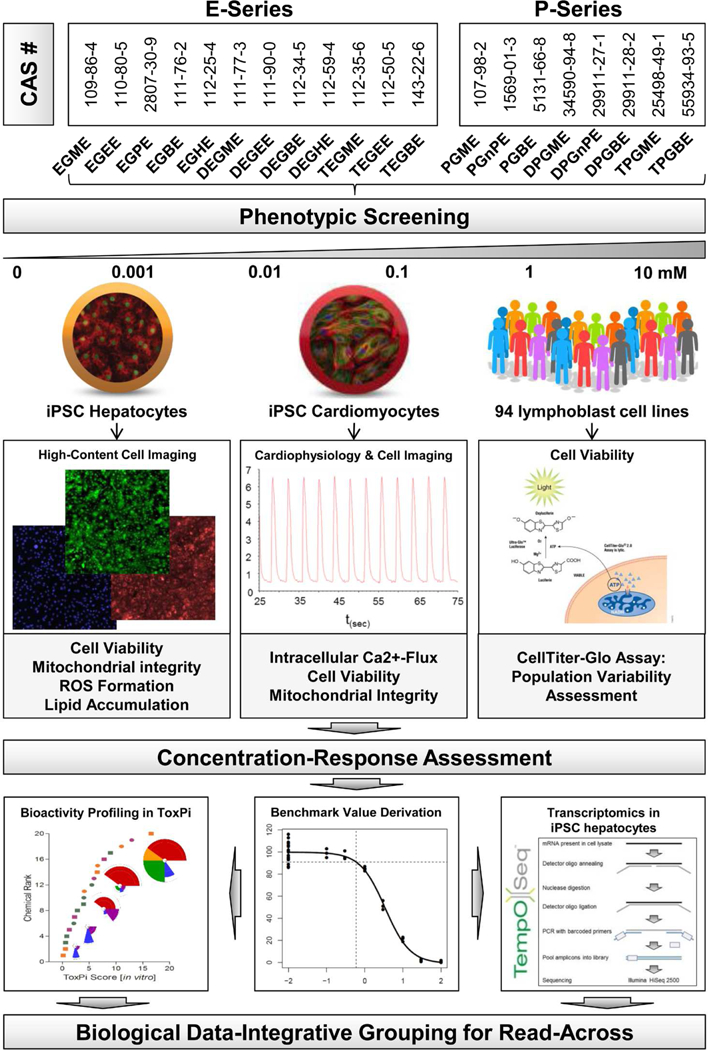
Overall study design. 12 E-Series and 8 P-Series glycol ethers (plus ethylene glycol and propylene glycol) were screened in a phenotypic and transcriptomic in vitro screening approach using human iPSC-derived cardiomyocytes and hepatocytes, as well as a population-based human lymphoblast cell model. Functional and high-content imaging data was analyzed for concentration-response and resulting Point-of-Departure values were integrated into bioactivity profiles and served as a basis for sample selection for targeted transcriptomics analysis using the TempO-seq assay. Both phenotypic screening and transcriptomics data were used for comprehensive biological data-integrative grouping of the glycol ethers.
Highlights.
New Approach Methodologies [NAMs] are now part of assessing and managing chemicals under TSCA
This study tested whether in vitro bioactivity may be applied to grouping of chemicals
Twenty E-Series and P-Series glycol ethers were tested in a suite of in vitro models
In vitro data showed that glycol ethers grouped according to their alcohol chain length
Overall in vitro bioactivity profiles did not correlate with in vivo toxicity data except for irritation scores
Acknowledgements
The authors acknowledge technical support from Ms. Qianwen Ouyang and useful discussions with Dr. J. Craig Rowlands (The Dow Chemical Company). This work was supported, in part, by funding from the US EPA (STAR RD83516602 and RD83580202), NIH (P42 ES027704) and the institutional support from Texas A&M University. The views expressed are those of the authors and do not necessarily reflect the statements, opinions, views, conclusions, or policies of the U.S. EPA, NIH, or the United States government. F.A. Grimm was the recipient of the Society of Toxicology’s 2015 Colgate-Palmolive Postdoctoral Fellowship and 2017 Syngenta Fellowship. Research materials tested in these studies (glycol ethers) were kindly provided by The Dow Chemical Company; no other funding for these studies was received from Dow.
Abbreviations
- CEU
Utah residents with European Ancestry
- CHB
Han Chinese from Beijing, China
- CLB
Colombians from Medellin, Colombia
- cm.amp
Average peak amplitude in iCell cardiomyocytes
- cm.bpm
Beat frequency in iCell cardiomyocytes (bpm = beats per min)
- cm.mito
Mitochondrial integrity in iCell cardiomyocytes
- cm.pbl
Average peak baseline in iCell cardiomyocytes
- cm.pdt
Average peak decay time in iCell cardiomyocytes
- cm.prt
Average peak rise time in iCell cardiomyocytes
- cm.ps
Average peak spacing in iCell cardiomyocytes
- cm.tc
Nuclei based cell count (tc=total cells) of iCell cardiomyocytes
- cm.w10a
Average peak width at 10% amplitude in iCell cardiomyocytes
- cm.width
Average peak width in iCell cardiomyocytes
- CO2
Carbon dioxide
- DEGBE
Diethylene glycol monobutyl ether
- DEGEE
Diethylene glycol monoethyl ether
- DEGHE
Diethylene glycol monohexyl ether
- DEGME
Diethylene glycol monomethyl ether
- DMSO
Dimethyl sulfoxide
- DPGBE
Dipropylene glycol n-butylether
- DPGnPE
Dipropylene glycol n-propyl ether
- DPGME
Dipropylene glycol monomethyl ether
- EG
Ethanediol (Ethylene glycol)
- EGBE
Ethylene glycol monobutyl ether
- EGEE
Ethylene glycol monoethyl ether
- EGHE
Ethylene glycol monohexyl ether
- EGME
Ethylene glycol monomethyl ether
- EGPE
Ethylene glycol monopropyl ether
- FBS
Fetal bovine serum
- GBR
British from England and Scotland
- hep.cyto
Cytoskeletal integrity in iCell hepatocytes
- hep.lipid
Lipid accumulation in iCell hepatocytes
- hep.mito
Mitochondrial integrity in iCell hepatocytes
- hep.ros
Reactive oxygen formation in iCell hepatocytes
- hep.tc
Nuclei based cell count (tc=total cells) of iCell hepatocytes iPSC Induced pluripotent stem cell
- JPT
Japanese from Tokyo, Japan
- LWK
Luhya from Webuye, Kenya
- MXL
Los Angeles residents with Mexican ancestry
- PBS
Phosphate-Buffered Saline
- PG
Propane-1,2-diol (propylene glycol)
- PGBE
1-butoxy-2-propanol (propylene glycol butyl ether)
- PGME
1-methoxy-2-propanol (propylene glycol methyl ether)
- PGnPE
Propylene glycol n-propyl ether
- TEGBE
Triethylene glycol monobutyl ether
- TEGEE
Triethylene glycol monoethyl ether
- TEGME
Triethylene glycol monomethyl ether
- TPGBE
Tripropylene glycol monobutyl ether
- TPGME
Tripropylene glycol methyl ether
- TSI
Tuscans from Italy
- YRI
Yoruba from Ibadan, Nigeria
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement
Nicholas Ball is an employee of The Dow Chemical Company, the manufacturer of the glycol ethers. Other co-authors have no relevant conflicts of interest to declare.
References
- Aasmoe L et al. , 1998. The role of liver alcohol dehydrogenase isoenzymes in the oxidation of glycolethers in male and female rats. Toxicol Appl Pharmacol 150, 86–90. [DOI] [PubMed] [Google Scholar]
- Abdo N et al. , 2015a. In vitro screening for population variability in toxicity of pesticide-containing mixtures. Environ Int 85, 147–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdo N, et al. , 2015b. Population-based in vitro hazard and concentration-response assessment of chemicals: the 1000 genomes high-throughput screening study. Environ Health Perspect 123, 458–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball N, et al. , 2014. The challenge of using read-across within the EU REACH regulatory framework; how much uncertainty is too much? Dipropylene glycol methyl ether acetate, an exemplary case study. Regul Toxicol Pharmacol 68, 212–21. [DOI] [PubMed] [Google Scholar]
- Ball N, et al. , 2016. Toward Good Read-Across Practice (GRAP) guidance. ALTEX 33, 149–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker RA, et al. , 2017. How well can carcinogenicity be predicted by high throughput “characteristics of carcinogens” mechanistic data? Regul Toxicol Pharmacol 90, 185–196. [DOI] [PubMed] [Google Scholar]
- Berggren E, et al. , 2015. Chemical Safety Assessment Using Read-Across: Assessing the Use of Novel Testing Methods to Strengthen the Evidence Base for Decision Making. Environ Health Perspect 123, 1232–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boatman RJ, 2005. International industry initiatives to improve the glycol ether health effects knowledge base. Toxicol Lett 156, 39–50. [DOI] [PubMed] [Google Scholar]
- Chiu WA, et al. , 2018. Use of high-throughput in vitro toxicity screening data in cancer hazard evaluations by IARC Monograph Working Groups. ALTEX 35, 51–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu WA, et al. , 2017. A tiered, Bayesian approach to estimating of population variability for regulatory decision-making. ALTEX 34, 377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogliano V, et al. , 2004. Advice on formaldehyde and glycol ethers. Lancet Oncol 5, 528. [DOI] [PubMed] [Google Scholar]
- De Abrew KN, et al. , 2016. Grouping 34 Chemicals Based on Mode of Action Using Connectivity Mapping. Toxicol Sci 151, 447–61. [DOI] [PubMed] [Google Scholar]
- De Abrew KN, et al. , 2015. A novel transcriptomics based in vitro method to compare and predict hepatotoxicity based on mode of action. Toxicology 328, 29–39. [DOI] [PubMed] [Google Scholar]
- Eduati F, et al. , 2015. Prediction of human population responses to toxic compounds by a collaborative competition. Nat Biotechnol 33, 933–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- European Chemicals Agency, Read-Across Assessment Framework (RAAF) European Chemicals Agency, Helsinki, Finland, 2015. [Google Scholar]
- Garlantezec R, et al. , 2012. Urinary biomarkers of exposure to glycol ethers and chlorinated solvents during pregnancy: determinants of exposure and comparison with indirect methods of exposure assessment. Occup Environ Med 69, 62–70. [DOI] [PubMed] [Google Scholar]
- Ghanayem BI, et al. , 1987. Metabolism and disposition of ethylene glycol monobutyl ether (2butoxyethanol) in rats. Drug Metab Dispos 15, 478–84. [PubMed] [Google Scholar]
- Goodman JE, et al. , 2018. Letter to the editor re: Guyton et al. (2018), ‘Application of the key characteristics of carcinogens in cancer hazard identification’. Carcinogenesis 39, 1089–1090. [DOI] [PubMed] [Google Scholar]
- Grimm FA, et al. , 2015a. Metabolism and metabolites of polychlorinated biphenyls. Crit Rev Toxicol 45, 245–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm FA, et al. , 2015b. High-Content Assay Multiplexing for Toxicity Screening in Induced Pluripotent Stem Cell-Derived Cardiomyocytes and Hepatocytes. Assay Drug Dev Technol 13, 529–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm FA, et al. , 2016. A chemical-biological similarity-based grouping of complex substances as a prototype approach for evaluating chemical alternatives. Green Chem 18, 4407–4419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu CG, et al. , 2009. DFT study on the bromination pattern dependence of electronic properties and their validity in quantitative structure-activity relationships of polybrominated diphenyl ethers. SAR QSAR Environ Res 20, 287–307. [DOI] [PubMed] [Google Scholar]
- Gualtieri JF, et al. , 2003. Repeated ingestion of 2-butoxyethanol: case report and literature review. J Toxicol Clin Toxicol 41, 57–62. [DOI] [PubMed] [Google Scholar]
- Guyton KZ, et al. , 2018a. Application of the key characteristics of carcinogens in cancer hazard identification. Carcinogenesis 39, 614–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyton KZ, et al. , 2018b. Re: ‘Application of the key characteristics of carcinogens in cancer hazard evaluation’: response to Goodman, Lynch and Rhomberg. Carcinogenesis 39, 1091–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- House JS, et al. , 2017. A pipeline for high throughput concentration response modeling of gene expression for toxicogenomics. Front Genet 8, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinstreuer NC, et al. , 2016. Adverse outcome pathways: From research to regulation scientific workshop report. Regul Toxicol Pharmacol 76, 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krimsky S, 2017. The unsteady state and inertia of chemical regulation under the US Toxic Substances Control Act. PLoS Biol 15, e2002404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockley DJ, et al. , 2005. Cutaneous metabolism of glycol ethers. Arch Toxicol 79, 160–8. [DOI] [PubMed] [Google Scholar]
- Low Y, et al. , 2013. Integrative chemical-biological read-across approach for chemical hazard classification. Chem Res Toxicol 26, 1199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvel SW, et al. , 2018. ToxPi Graphical User Interface 2.0: Dynamic exploration, visualization, and sharing of integrated data models. BMC Bioinformatics 19, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx U, et al. , 2016. Biology-inspired microphysiological system approaches to solve the prediction dilemma of substance testing. ALTEX 33, 272–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RR, et al. , 1984a. Propylene glycol monomethyl ether acetate (PGMEA) metabolism, disposition, and short-term vapor inhalation toxicity studies. Toxicol Appl Pharmacol 75, 521–30. [DOI] [PubMed] [Google Scholar]
- Miller RR, et al. , 1984b. Ethylene glycol monomethyl ether and propylene glycol monomethyl ether: metabolism, disposition, and subchronic inhalation toxicity studies. Environ Health Perspect 57, 233–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Academies of Sciences Engineering and Medicine, Using 21st Century Science to Improve Risk-Related Evaluations Washington, DC, 2017. [PubMed] [Google Scholar]
- National Research Council, 2007. Toxicity testing in the 21st century: A vision and a strategy The National Academies Press, Washington, DC. [Google Scholar]
- Parham F, et al. , 2009. Dose-response modeling of high-throughput screening data. J Biomol.Screen 14, 1216–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patlewicz G, et al. , 2014. Read-across approaches--misconceptions, promises and challenges ahead. ALTEX 31, 387–96. [DOI] [PubMed] [Google Scholar]
- Patlewicz G, et al. , 2015. Building scientific confidence in the development and evaluation of read-across. Regul Toxicol Pharmacol 72, 117–33. [DOI] [PubMed] [Google Scholar]
- Poet T, et al. , 2016. Deriving Biomonitoring Equivalents for selected E- and P-series glycol ethers for public health risk assessment. Int J Hyg Environ Health 219, 88–100. [DOI] [PubMed] [Google Scholar]
- Rana P, et al. , 2012. Characterization of human-induced pluripotent stem cell-derived cardiomyocytes: bioenergetics and utilization in safety screening. Toxicol Sci 130, 117–31. [DOI] [PubMed] [Google Scholar]
- Reif DM, et al. , 2013. ToxPi GUI: an interactive visualization tool for transparent integration of data from diverse sources of evidence. Bioinformatics 29, 402–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusyn I, Greene N, 2018. The Impact of Novel Assessment Methodologies in Toxicology on Green Chemistry and Chemical Alternatives. Toxicol Sci 161, 276–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusyn I, et al. , 2012. Predictive modeling of chemical hazard by integrating numerical descriptors of chemical structures and short-term toxicity assay data. Toxicol Sci 127, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirenko O, et al. , 2013. Multiparameter in vitro assessment of compound effects on cardiomyocyte physiology using iPSC cells. J Biomol Screen 18, 39–53. [DOI] [PubMed] [Google Scholar]
- Sirenko O, et al. , 2017. In vitro cardiotoxicity assessment of environmental chemicals using an organotypic human induced pluripotent stem cell-derived model. Toxicol Appl Pharmacol 322, 60–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirenko O, et al. , 2014. High-content assays for hepatotoxicity using induced pluripotent stem cell-derived cells. Assay Drug Dev Technol 12, 43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staples CA, et al. , 1998. Ethylene glycol ethers: an environmental risk assessment. Chemosphere 36, 1585–613. [DOI] [PubMed] [Google Scholar]
- Thomas RS, et al. , 2012. A comprehensive statistical analysis of predicting in vivo hazard using high-throughput in vitro screening. Toxicol Sci 128, 398–417. [DOI] [PubMed] [Google Scholar]
- Thomas RS, et al. , 2018. The US Federal Tox21 Program: A strategic and operational plan for continued leadership. ALTEX 35, 163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tice RR, et al. , 2013. Improving the human hazard characterization of chemicals: a Tox21 update. Environ Health Perspect 121, 756–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- United Nations, Globally Harmonized System of Classification and labelling of Chemicals (GHS) Vol. Sixth revised edition, Geneva, Switzerland, 2015. [Google Scholar]
- Wess JA, 1992. Reproductive toxicity of ethylene glycol monomethyl ether, ethylene glycol monoethyl ether and their acetates. Scand J Work Environ Health 18 Suppl 2, 43–5. [PubMed] [Google Scholar]
- Wetmore BA, 2015. Quantitative in vitro-to-in vivo extrapolation in a high-throughput environment. Toxicology 332, 94–101. [DOI] [PubMed] [Google Scholar]
- Zhu H, et al. , 2016. Supporting read-across using biological data. ALTEX 33, 167–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.