

Abstract
Fetal alcohol spectrum disorders (FASD) represent a wide array of defects that arise from ethanol exposure during development. However, the underlying molecular mechanisms are limited. In the current report, we aimed to further evaluate the cannabinoid receptor type 1 (CB1R)-mediated mechanisms in a postnatal ethanol-exposed animal model. We report that the exposure of postnatal day 7 (P7) mice to ethanol generates p25, a CDK5-activating peptide, in a time- and CB1R-dependent manner in the hippocampus and neocortex brain regions. Pharmacological inhibition of CDK5 activity before ethanol exposure prevented accumulation of cleaved caspase-3 (CC3) and hyperphosphorylated tau (PHF1) (a marker for neurodegeneration) in neonatal mice and reversed cAMP response element-binding protein (CREB) activation and activity-regulated cytoskeleton-associated protein (Arc) expression. We also found that postnatal ethanol exposure caused a loss of RhoGTPase-related, Rac1, gene expression in a CB1R and CDK5 activity-dependent manner, which persisted to adulthood. Our epigenetic analysis of the Rac1 gene promoter suggested that persistent suppression of Rac1 expression is mediated by enhanced histone H3 lysine 9 dimethylation (H3K9me2), a repressive chromatin state, via G9a recruitment. The inhibition of CDK5/p25 activity before postnatal ethanol exposure rescued CREB activation, Arc, chromatin remodeling and Rac1 expression, spatial memory, and long-term potentiation (LTP) abnormalities in adult mice. Together, these findings propose that the postnatal ethanol-induced CB1R-mediated activation of CDK5 suppresses Arc and Rac1 expression in the mouse brain and is responsible for persistent synaptic plasticity and learning and memory defects in adult mice. This CB1R-mediated activation of CDK5 signaling during active synaptic development may slow down the maturation of synaptic circuits and may cause neurobehavioral defects, as found in this FASD animal model.
Subject terms: Developmental biology, Learning and memory
Introduction
Alcohol consumption during pregnancy results in deleterious effects on multiple organ systems in the developing fetus, collectively called as fetal alcohol spectrum disorders (FASD) [1, 2]. FASD are the leading cause of mental retardation in Western countries. Currently, 2–5% of children in the United States are affected by FASD [3], with nearly 40,000 cases reported each year [4]. The brain is the most susceptible organ affected by prenatal alcohol exposure, which results in reduced academic performance; an increased frequency of learning disabilities; impaired executive functions; poor hand/eye coordination; tremors; gait and balance difficulties; and impairments in attention, reaction time, motor skills, memory, and social and adaptive functions [1, 5–7]. Our laboratory and others have previously demonstrated that exposure of mice to ethanol at postnatal day 7 (P7), which is corresponding to the third trimester of human pregnancy, affects synaptogenesis, induces neuronal cell death, and results in long-lasting neurobehavioral dysfunctions [8–13].
Cyclin-dependent kinase 5 (CDK5) is a proline-directed serine/threonine kinase that plays a critical role in the central nervous system. CDK5 is involved in the normal physiological activities of the brain, such as cytoskeletal organization, axonal guidance, neuron migration, neurite outgrowth and support, and synaptogenesis, and plays an important role in neurotransmission, synaptic plasticity, and cognitive functions [14–19]. However, overexpression or aberrant activity of CDK5 results in neuronal dysfunction, leading to a variety of neurodegenerative diseases [17, 20–22]. The activity of CDK5 depends on cell-specific activators. In neurons, CDK5 is activated through direct binding of the proteins p35/p39 and their cleavage products p25/p29 [23, 24]. The deregulation of CDK5 is mainly due to its association with p25, a C-terminal fragment of p35 [21, 25]. Under various conditions, p35 is cleaved to p25 and p10. P25 has a longer half-life compared to p35 and hence prolongs the activation of CDK5, leading to increased phosphorylation of its target proteins, which ultimately results in neurodegeneration [17, 22]. Additionally, inhibition of the CDK5/p25 hyperactivation state results in reduced neurodegeneration and improved cognitive function [17, 26]. Thus it is evident that aberrant CDK5 activity leads to neurodegeneration and regulation of its activity is of therapeutic importance. However, the role of CDK5 in alcohol-induced neuronal anomalies is poorly understood.
The third trimester of pregnancy is also important in humans, as synaptogenesis begins in this period, which is characterized by heightened neuronal signaling, neurite outgrowth, and synapse formation [27–29]. Rho family small GTPases (Rac1, RhoA, and Cdc42) are widely implicated in the regulation of neurite formation [30]. By regulating the cytoskeletal rearrangement, these proteins affect neurite extension, neurite retraction, neuronal polarization, and plasticity, which are important for cognitive function [30, 31]. Because ethanol exposure of P7 mice results in cognitive defects [8, 11–13, 32], it is important to understand CDK5 and Rho GTPase signaling during this period. Hence, we explored the functions of CDK5 and Rac1 in P7 ethanol-induced neurodegeneration and their impact on long-lasting learning and memory abnormalities.
Materials and methods
The experimental procedures used in this study are described in Supplementary Materials and Methods. This section provides precise details on ethanol, drug administration, western blotting assays, immuno microscopy procedures, quantitative PCR (qPCR) assays, the chromatin immunoprecipitation (ChIP) procedure, behavioral tests, long-term potentiation (LTP), and statistical analyses.
Results
P7 ethanol treatment enhances CDK5 activity through increased generation of p25
The exposure of P7 mice to low (1.0 g × 2) and high (2.5 g × 2) (subcutaneously at 0 h and again at 2 h) doses of ethanol resulted in blood ethanol levels (BELs) of 0.20 ± 0.025 and 0.35 ± 0.03 g/dl at 4 h that were gradually reduced to 0.09 ± 0.017 and 0.21 ± 0.06 g/dl, respectively, at 9 h after the first ethanol injection [12, 33]. This treatment has been shown to induce moderate (low dose of ethanol) to widespread (high dose of ethanol) activation of caspase-3 (neuroapoptosis) as measured using a cleaved caspase-3 (CC3) antibody [12, 34]. Next, we evaluated the activity of CDK5 by measuring the levels of p35 and p25 in P7 ethanol-treated mouse brain extracts by western blot analysis. Neither the low- nor the high-dose ethanol treatment in P7 mice had a significant (p > 0.05) effect on the levels of CDK5 in either the hippocampus (HP) or the neocortex (NC) (Fig. 1b, c). However, the levels of p35 were significantly (one-way analysis of variance (ANOVA) with Bonferroni’s post hoc tests) decreased by the low (4, 8, and 24 h) and high doses (8 and 24 h) of ethanol treatment both in HP (1 g: F1,46 = 25; 2.5 g: F1,46 = 35, p < 0.05) and NC (1 g: F1,46 = 22; 2.5 g: F1,46 = 38, p < 0.05) compared to the saline treatment. In contrast, the levels of p25 significantly increased both in HP (1 g: F1,46 = 28; 2.5 g: F1,46 = 32, p < 0.05) and NC (1 g: F1,46 = 25; 2.5 g: F1,46 = 37, p < 0.05) after low (4, 8, and 24 h) and high (8 and 24 h) doses of ethanol treatment. The ratio of p25/p35 significantly increased both in HP (1 g: F1,46 = 20; 2.5 g: F1,46 = 18, p < 0.05) and NC (1 g: F1,46 = 18; 2.5 g: F1,46 = 27, p < 0.05) after low (4, 8, and 24 h) and high (8 and 24 h) doses of ethanol treatment (Fig. 1b, c).
Fig. 1.

Postnatal ethanol exposure increases p25 protein levels in the P7 mouse brain HP and NC in a caspase-3- and CB1R-dependent manner. Schematic diagram to show CDK5 (inactive) association with the activator p35 (active and unstable) and the interaction with the proteolytic product p25 (highly active and stable) (a). Hippocampus (HP) and neocortex (NC) samples, obtained 4–24 h after the first saline or low- (b) and high-dose (c) ethanol exposure. CDK5 (i), p35 (ii) and p25 (iii) protein levels, as well as p25/p35 ratios (iv), were determined by western blot analysis. The protein samples were equally loaded, confirmed with Ponceau S staining, and normalized to β-actin. Increased p25/p35 (iv) ratios were rescued by preadministration of P7 mice with broad-spectrum caspase inhibitor (Q-VD-OPh) (d), CB1R antagonist (SR141617A) (e), or genetic ablation of CB1R (CB1R KO) (f). For the 0 h ethanol group, saline was administered. [*p < 0.05 vs. saline (0 h); #p < 0.05 vs. E]. Error bars, SEM (n = 12 pups/group)
Q-VD-OPh prevents p35 protein degradation and generation of p25 protein in P7 ethanol-treated mice
We have previously reported that injection of high-dose ethanol to P7 mice significantly increases the CC3 levels at 8 and 24 h postinjection [12, 34]. Activated caspase-3 can degrade many proteins in the cell [34–36]. Therefore, we next evaluated the role of caspase 3 activation in p35 protein degradation using a broad-spectrum caspase inhibitor (Q-VD-OPh). We have previously shown that preadministration of an optimum dose (1 mg/kg) of Q-VD-OPh before ethanol treatment in P7 mice effectively prevented the formation of CC3 without altering BELs [11, 34]. Thus, in the present study, a 1 mg/kg dose of Q-VD-OPh was used. Preadministration of Q-VD-OPh 30 min before ethanol injection prevented the degradation of the p35 protein to p25 in both the HP (p35: F1,20 = 25; p25: F1,20 = 20; p25/p35: F1,20 = 30, p < 0.05) and NC (p35: F1,20 = 32; p25: F1,20 = 30; p25/p35: F1,20 = 37, p < 0.05) (two-way ANOVA with Bonferroni’s post hoc test) brain regions (Fig. 1d) and significantly normalized the p25/p35 ratio to control levels.
CDK5 activity in P7 ethanol-treated mice is dependent on cannabinoid receptor type 1 (CB1R) activation
Pharmacological inhibition or genetic blockade of CB1R gives protection against ethanol-induced activation of caspase-3 [12, 37]. Thus, to further understand the function of CB1R on CDK5 activity, we used a specific CB1R antagonist (SR) and CB1R knockout (KO) mice in our study. Preadministration of an optimum dose of SR 30 min before ethanol injection was previously shown to effectively prevent caspase 3 activation without affecting ethanol metabolism [12, 37]. The SR significantly prevented the degradation of p35 to p25 in both the HP (p35: F1,20 = 32; p25: F1,20 = 30; p25/p35: F1,20 = 39, p < 0.05) and NC (p35: F1,20 = 34; p25: F1,20 = 37; p25/p35: F1,20 = 41, p < 0.05) (two-way ANOVA with Bonferroni’s post hoc test) brain regions (Fig. 1e). As seen with the SR treatment, p35 protein degradation was not found in ethanol-treated CB1R KO mice compared to WT mice in HP (p35: F1,20 = 38; p25: F1,20 = 33; p25/p35: F1,20 = 41, p < 0.05) and NC (p35: F1,20 = 30; p25: F1,20 = 35; p25/p35: F1,20 = 42, p < 0.05) (two-way ANOVA with Bonferroni’s post hoc test) (Fig. 1f) tissues.
Pharmacological inhibition of CDK5 prevents ethanol-induced caspase 3 activation in P7 mice
From the above studies, it is evident that ethanol increases the degradation of p35 to p25 through the CB1R pathway, which results in CDK5 activation. To further examine the role of CDK5 in postnatal ethanol effects, we used roscovitine (RV), a pharmacological inhibitor of CDK5. To understand the dose response, different concentrations of RV (0–5 mg/kg, s.c.) were preadministered to P7 mice 30 min before the first ethanol injection, and the levels of CC3 were evaluated (8 h) in NC extracts. RV prevented the activation of caspase 3 in a dose-dependent manner as evidenced by decreased CC3 protein levels in NC (F3,35 = 20, p < 0.05) (two-way ANOVA with Bonferroni’s post hoc test) (Fig. 2a). A significant reduction in CC3 levels was found at a dose as low as 1.25 mg/kg of RV, and the optimum dose was found to be 2.5 mg/kg. In our initial experiments, we assessed whether RV would alter ethanol metabolism. The administration of a higher dose of RV (5 mg/kg) (E+RV; BEL peaked at 4 h at 0.36 ± 0.20 g/dl and was steadily reduced to 0.23 ± 0.12 g/dl at 9 h) 30 min before the ethanol treatment (E+V; BEL peaked at 4 h at 0.33 ± 0.22 g/dl and was steadily reduced to 0.25 ± 0.11 g/dl at 9 h) did not alter the BELs significantly (p > 0.05). The optimum dose of RV (2.5 mg/kg) was used in all the proceeding experiments. Also, optimum dose of RV rescued CC3 levels as measured by immunostaining (HP: F3,28 = 45; retrosplenial cortex: F3,28 = 56, p < 0.05) (Fig. 2b), as well as by immunoblot in HP (HP: F3,20 = 23, p < 0.05) (two-way ANOVA with Bonferroni’s post hoc test) (Fig. 2c). The CDK5/p25 kinase activity hyperphosphorylates tau (major microtubule-associated protein), alters the cytoskeleton, and promotes neuronal death [38]. Therefore, we further evaluated PHF1 (phosphorylated tau) levels in the HP and NC brain regions. Injection of high dose of ethanol strongly increased PHF1, whereas RV preadministration significantly prevented enhanced PHF1 levels in both brain regions (HP: F1,20 = 28; NC: F1,20 = 30, p < 0.05) (two-way ANOVA with Bonferroni’s post hoc test) (Fig. 2d).
Fig. 2.

Pharmacological inhibition of CDK5/p25 activity with roscovitine (RV) dose-dependently prevents the accumulation of cleaved caspase-3 (CC3), phosphorylated tau (PHF1), and impaired pERK1/2 and pCREB caused by ethanol exposure in P7 mice. Mice were exposed to a high dose of ethanol after preadministration (30 min) with various doses of RV (0–5 mg/kg) or vehicle, and CC3 levels were determined in NC brain samples by a western blot analysis (a). Saline and 8 h ethanol-exposed mice were pretreated with 2.5 mg/kg RV for 30 min, the free-floating coronal brain sections (HP and retrosplenial cortex (RSC)) were subjected to IHC analysis with anti-rabbit-CC3 and CC3-positive cells were counted in the HP and RSC brain regions. The arrows indicate the CC3-positive neurons in the HP and RSC. Scale bars = 200 μm (b). The hippocampal region was enlarged to show the CC3-positive cells (asterisk (*)). CC3 levels were also determined in HP brain region by a western blot analysis (c). The HP and NC tissue fractions were subjected to western blot analysis with specific PHF1 (d), pERK1/2, ERK1/2 (e, i), and CREB and pCREB (e, ii) antibodies. The protein samples were equally loaded, confirmed with Ponceau S staining, and normalized to total proteins (ERK1/2 and CREB) followed by β-actin. (*p < 0.05 vs. S; #p < 0.05 vs. E). Error bars, SEM (n = 8 pups/group)
CDK5 activity regulates the phosphorylated extracellular signal–regulated kinase 1/2 (pERK1/2) and phosphorylated cAMP response element-binding protein (pCREB) pathways in P7 ethanol-treated mice
It was shown that CDK5 activity inhibits the phosphorylation of ERK1/2 through mitogen-activated extracellular signal-regulated kinase 1 (MEK1) phosphorylation [39]. Our previous studies have demonstrated that P7 ethanol treatment significantly reduces ERK1/2 and CREB phosphorylation and causes neurodegeneration in a CB1R-dependent manner [12, 37]. To further understand the possible link between increased CDK5 activity and decreased ERK1/2 and CREB phosphorylation in P7 ethanol-treated mice, we measured the levels of pERK1/2 and pCREB in RV-treated samples. Preadministration of 2.5 mg/kg RV 30 min prior to the first ethanol injection significantly rescued pERK1/2 and pCREB levels both in the HP (pERK1/2: F1,20 = 22; pCREB: F1,20 = 40, p < 0.05) and NC (pERK1/2: F1,20 = 40; pCREB: F1,20 = 45, p < 0.05) (two-way ANOVA with Bonferroni’s post hoc test) brain tissues (Fig. 2e). These findings suggest that ethanol-induced activation of CDK5 inhibits the pERK1/2 and pCREB pathways in P7 mice.
Downstream activities of CDK5 in P7 ethanol-treated mice is dependent on Rac1
To further understand the mechanism through which CDK5 exhibits neurodegeneration, first we studied the Rac1 levels in P7 ethanol-treated mice by western blot analysis using a specific antibody. Treatment of the P7 pups with low and high doses of ethanol reduced the levels of Rac1 in a time-dependent manner both in the HP (1 g: F1,46 = 35; 2.5 g: F1,46 = 40, p < 0.05) and NC (1 g: F1,46 = 32; 2.5 g: F1,46 = 42, p < 0.05) compared to saline treatment (one-way ANOVA with Bonferroni’s post hoc test). Consistent with the protein levels, Rac1 mRNA levels were also reduced in 8 h ethanol-injected HP and NC tissues compared to saline treatment (Fig. 3a, b) without significantly (p > 0.05) affecting the housekeeping gene (Hprt) expression.
Fig. 3.

Postnatal ethanol exposure reduces Rac1 expression in a CB1R- and CDK5-dependent manner in the P7 mouse brain. Hippocampus (HP) and neocortex (NC) samples, procured 4–24 h following the first saline or low- (a) and high-dose (b) ethanol exposure, were used in these experiments. Rac1 protein levels (i) were determined using western blot analysis. The protein samples were equally loaded, confirmed with Ponceau S staining, and normalized to β-actin. Rac1 mRNA levels (ii) were determined in samples exposed to ethanol for 8 h by qPCR analysis. Hprt mRNA was used as the internal control for normalization of Rac1 mRNA. For the 0 h ethanol group, saline was administered. Mice exposed to a high dose of ethanol for 8 h after preadministration (30 min) with vehicle or SR (1 mg/kg) (c) or genetically ablated CB1R mice (CB1R KO) (d) or RV (2.5 mg/kg) (e) were used to understand the role of CB1R on Rac1 expression. Rac1 mRNA levels (f) were determined in samples exposed to saline or ethanol for 8 h with or without RV (2.5 mg/kg) by qPCR analysis. Hprt mRNA was used as the internal control for normalization of Rac1 mRNA. (*p < 0.05 vs. S; #p < 0.05 vs. E). Error bars, SEM (n = 10 pups/group)
To further dissect whether CB1Rs regulate Rac1 in P7 ethanol-exposed mice, we used SR and CB1R KO mice. Preadministration of SR (1 mg/kg) 30 min prior to high-dose ethanol exposure significantly prevented the ethanol-induced reduction in Rac1 levels in both the brain regions measured (HP: F3,33 = 36, p < 0.05; NC: F3,33 = 30, p < 0.05) (two-way ANOVA with Bonferroni’s post hoc test) (Fig. 3c). Consistent with the SR results, Rac1 levels were not altered in the HP (F1,20 = 3, p > 0.05) and NC (F1,20 = 2, p > 0.05) tissues of the ethanol-treated CB1R KO mice compared to those of the WT mice (Fig. 3d). Further, preadministration of RV (2.5 mg/kg) 30 min prior to the high dose of ethanol injection significantly rescued the ethanol-induced loss of Rac1 levels in both the HP (F3,33 = 32, p < 0.05) and NC (F3,33 = 44, p < 0.05) (two-way ANOVA with Bonferroni’s post hoc test) brain regions (Fig. 3e). Consistent with the Rac1 protein results, Rac1 mRNA levels were also rescued by preadministration of RV in both the HP (F3,33 = 23, p < 0.05) and NC (F3,33 = 24, p < 0.05) (two-way ANOVA with Bonferroni’s post hoc test) brain regions (Fig. 3f) and suggest that Rac1 expression is regulated by transcriptional suppression of the Rac1 gene by ethanol in P7 mice. Together, these results suggest that the CB1R pathway regulates Rac1 expression via CDK5 to induce neurodegeneration in postnatal ethanol-exposed neonatal mice.
Postnatal ethanol exposure reduced Rac1 expression to adulthood via epigenetic mechanisms; postnatal ethanol-induced deficits in activity-regulated cytoskeleton-associated protein (Arc), pCREB, and Rac1 levels in adult mice were rescued by pretreatment with RV
We have previously demonstrated that P7 ethanol treatment causes persistent impairment of pCREB followed by Arc expression in adult animals [11, 37]. To understand whether RV treatment at P7 rescues these signaling deficits in the adulthood, we analyzed Arc and pCREB protein levels in HP tissues of adult mice treated with RV at P7. RV treatment 30 min prior to high-dose ethanol injection rescued both pCREB (F3,33 = 30, p < 0.05) and Arc (F3,33 = 23, p < 0.05) expression impairments in the adult mice (two-way ANOVA with Bonferroni’s post hoc test) (Fig. 4a). We have also evaluated the effect of P7 ethanol treatment on Rac1 levels in the adult mice. Similar to the P7 data, the Rac1 protein (F1,20 = 32, p < 0.05) and mRNA (F1,20 = 21, p < 0.05) levels in HP were persistently impaired in adult mice exposed to ethanol at P7 (one-way ANOVA with Bonferroni’s post hoc test) (Fig. 4b). RV treatment has no significant effect (p > 0.05) on housekeeping gene (Hprt) expression. We further extended our studies to analyze whether epigenetic suppression of gene expression regulates reduced transcription of the Rac1 gene. It is well documented that histone modification by the addition of methyl groups or by the removal of acetyl groups silences gene transcription [40]. Therefore, we performed ChIP experiments using Rac1 gene promoter-specific primers. The results demonstrated significantly increased H3K9me2 (F1,20 = 28, p < 0.05) and G9a (F1,20 = 39, p < 0.05) levels in the Rac1 gene promoter region (one-way ANOVA with Bonferroni’s post hoc test), while H3K14ac, H4K8ac, histone deacetylase 1 (HDAC1), and CREB-binding protein (CBP) levels were not significantly altered (p > 0.05) (Fig. 4c). Preadministration of mice with RV (Fig. 4d) or SR (Fig. 4e) before ethanol exposure at P7 normalized H3K9Me2 (RV: F3,33 = 21, p < 0.05; SR: F3,33 = 23, p < 0.05) enrichment and G9a (RV: F3,33 = 28, p < 0.05; SR: F3,33 = 21, p < 0.05) recruitment on the Rac1 gene promoter region of the adult mice. Further, pretreatment with RV at P7 significantly ameliorated the ethanol-induced loss of the Rac1 protein (F3,33 = 31, p < 0.05) and mRNA (F3,33 = 22, p < 0.05) (Fig. 4f) impairment in the adult HP tissues. Similarly, SR pretreatment at P7 rescued the loss of Rac1 protein in the adult HP tissues (F3,33 = 19, p < 0.05) (Fig. 4g). These results together imply that postnatal ethanol-activated CB1R or CDK5 persistently reduces Rac1 expression in the adult mice through increased suppressive chromatin at the Rac1 gene promoter.
Fig. 4.

Preadministration of RV rescues pCREB and Arc expression; enhanced H3K9me2 and G9a in the Rac1 gene promoter suppresses Rac1 expression and is rescued by preadministration of RV or SR in the adult mouse hippocampus caused by ethanol exposure in P7 mice. Mice were exposed to saline or high dose of ethanol for 8 h after administration (30 min) with or without RV (2.5 mg/kg), and pCREB (ai) and Arc (aii) protein levels were determined in HP brain samples by a western blot analysis. HP tissue extracts from adult mice exposed to saline or ethanol were subjected for western blot analysis to determine Rac1 expression (bi). The protein samples were equally loaded, confirmed with Ponceau S staining, and normalized with total proteins (CREB) followed by β-actin. Rac1 mRNA levels (bii) were determined in samples exposed to saline and ethanol (8 h) by qPCR analysis. Hprt mRNA was used as the internal control for normalization of Rac1 mRNA. Epigenetic analysis at the promoter region of the Rac1 gene (c–e). ChIP analysis of the Rac1 gene promoter in HP tissues from the saline and ethanol groups with anti-H3K9me2, anti-H3K14ac, anti-H4K8ac, anti-G9a, anti-HDAC1, or ant-CBP antibodies (c). HP tissues from saline and ethanol groups preadministered (30 min before) with or without RV (d) or SR (e) were immunoprecipitated with anti-H3K9me2 or anti-G9a antibodies. Levels of Rac1 gene promoter chromatin enrichment in the IPs were measured by RT-qPCR. IgG was used as negative control. Enrichment values were normalized to input values and represented as percentage of input. Mice were exposed to a high dose of ethanol for 8 h after preadministration (30 min) with vehicle or RV (2.5 mg/kg), and Rac1 protein levels were determined in HP tissue samples by a western blot analysis (fi). The protein samples were equally loaded, confirmed with Ponceau S staining, and normalized to β-actin. Rac1 mRNA levels (fii) were determined in samples exposed to saline and ethanol (8 h) with and without RV by qPCR analysis. Hprt mRNA was used as the internal control for normalization of Rac1 mRNA. Mice were exposed to a high dose of ethanol for 8 h after preadministration (30 min) with vehicle or SR (1 mg/kg), and Rac1 protein levels were determined in HP tissue samples by a western blot analysis (g). The protein samples were equally loaded, confirmed with Ponceau S staining, and normalized to β-actin (*p < 0.05 vs. S; #p < 0.05 vs. E). Error bars, SEM (n = 12 pups/group)
Preadministration of CDK5 inhibitor at P7 rescues P7 ethanol-induced memory loss in adult mice
We examined spatial recognition memory (SRM) using a Y-maze. A two-way ANOVA revealed that the S+V and S+RV-treated male and female mice entered more frequently into (Arm entry: male: F3,21 = 22; female: F3,21 = 19, p < 0.05) (Fig. 5a) and spent more time in (Dwell time: male: F3,21 = 34, p < 0.05; female: F3,21 = 37, p < 0.05) (Fig. 5b) the novel, previously unvisited arm of the maze. In contrast, the P7 E+V-treated animals showed a reduced preference toward the novel arm (p < 0.05) and spent less time (Dwell time) (p < 0.05) in the novel arm compared to the P7 S+V male and female mice at 24 h retention. RV preadministration prevented ethanol-induced SRM deficits (E+RV) with a preference toward exploration of the novel arm (p < 0.05) and time spent (p < 0.05) in the novel arm. While all S+V- and S+RV-treated animals (combined male and female due to lack of gender effects) preferred the novel arm as the first choice, E+V-treated animals showed a reduced preference for the novel arm, and this was prevented by RV preadministration (E+RV) (F3,45 = 40, p < 0.05) (Fig. 5c). These results indicate that CDK5 plays an important role in the P7 ethanol-induced SRM loss in adult mice.
Fig. 5.

Pretreatment of P7 mice with RV rescues behavioral and synaptic plasticity abnormalities found in adult mice exposed to postnatal ethanol. The spatial working memory was determined with the Y-maze in adult male and female mice treated with saline (S+V) or ethanol (E+V) with or without RV (S+RV and E+RV) at the P7 developmental stage. The discrimination ratio [preference for the novel arm over the familiar other arm (Novel/Novel+Other)] for arm entries (a) and dwell time (time spent in each arm) (b) of the S+V-, E+V-, S+RV-, and E+RV-administered mice 24 h after the first encounter with the partially opened maze. The percentage of mice choosing the novel arm as the first choice is shown for S+V-, E+V-, S+RV-, and E+RV-administered mice, 24 h after the first encounter with the partially opened maze (c). The spontaneous alternation memory of adult mice treated with S+V, E+V, S+RV, and E+RV at P7 was determined using the Y-maze. The percentage of spontaneous alternation performance by mice is shown for all the treatment groups (d). The percentage of social investigation is shown for all the treated mice (S+V, E+V, S+RV, and E+RV), 24 h after the first encounter with the same juvenile mouse. (*p < 0.05 vs. saline; #p < 0.05 vs. ethanol, n = 8 mice/group) (e). A schematic illustration demonstrates the position of the stimulating and recording electrode in the CA1 region of the hippocampus (f). The average fEPSP slope at various time points of P7 S+V-, E+V-, S+RV-, or E+RV-treated adult male mice is shown (g, h). Inset: Representative traces are shown for the saline group. For each slice, the fEPSP slopes were normalized against the average slope over the 10-min recording before LTP stimulation. Arrows show the time of TBS (4 pulses at 100 Hz, with bursts repeated at 5 Hz, and each tetanus including 3 different 10-burst trains separated by 15 s) (i, j). Inset: Representative traces before and after TBS are shown for all the treatment groups. Scale: 1 mV; 10 ms. Bar graph demonstrates the average of the fEPSP slopes at multiple time points after TBS for all the treated groups (k) (*p < 0.05 vs. S; #p < 0.05 vs. E). Error bars, SEM (n = 5 mice/group; 10 slices/group)
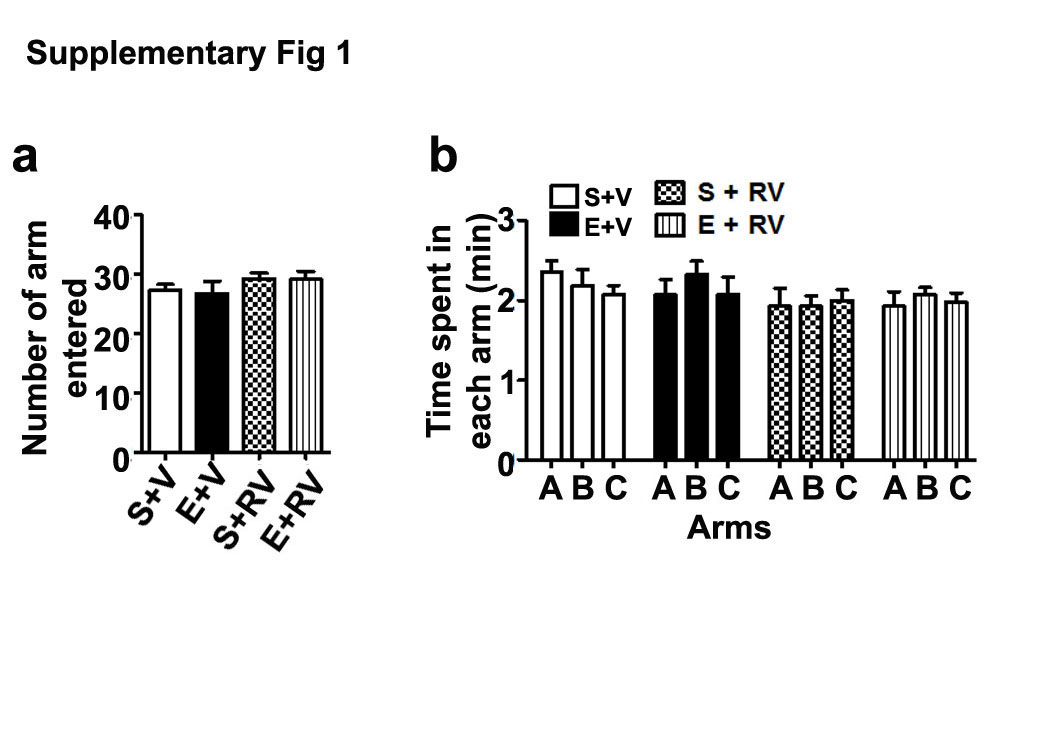
We measured spontaneous alternation as an additional measure of spatial memory test. Statistical analysis suggested that the S+V, S+E, S+RV, and E+RV treatment caused no significant effect (p > 0.05) on the adult (male and female) exploratory activities, as determined by the number of arm entries (Supl Fig. 1a) and time spent (Supl Fig. 1b) in each arm during the Y-maze task. Owing to a lack of sex effect, we have combined both male and female data for final statistical analysis. Two-way ANOVA with Bonferroni’s post hoc analysis indicated that the ethanol treatment (E+V group) showed significantly impaired spontaneous alternation compared with S+V group. RV administration prior to ethanol (E+RV group) prevented these impairments (F3,33 = 32, p < 0.05) (Fig. 5d). S+RV group mice exhibited normal spontaneous alternation (p > 0.05). Altogether, these results indicated that inhibition of CDK5 activity prevent the impact of ethanol exposure at P7 on spatial memory defects in adult mice.
The social investigation results revealed that E+V-treated male and female mice exhibited significantly reduced social recognition memory performance compared to the S+V-treated animals. A two-way ANOVA analysis (male and female combined) revealed that RV preadministration (E+RV) prevented the ethanol-induced social recognition memory deficits compared to the vehicle (E+V)-treated animals [F3,45 = 38, p < 0.05] (Fig. 5e). Further, S+RV alone had no substantial influence (p > 0.05), and these mice displayed normal social recognition memory behavior. Aggressive encounter was observed in <4% of the animals, which were excluded from the statistical analysis. These results propose that activated CDK5 contributes to P7 ethanol-induced social recognition memory loss in adult mice.
Preadministration of a CDK5 inhibitor before ethanol treatment in P7 mice prevents LTP deficits in adult mice
LTP experiments were performed using male mice due to the lack of gender effects in behavioral studies. Hippocampal slices prepared from the adult male animals treated with S+V, E+V, S+RV, and E+RV at P7 were subjected to the input/output (I/O) responses followed by LTP in the Schaffer collateral pathway (Fig. 5f). By performing the stimuli at a series of increasing intensities, we established an I/O relation between fiber volley (FV) and field excitatory postsynaptic potential (fEPSP) slope for all the treatment groups. The I/O curve was found to be similar between the treated and control groups (S+V, E+V, S+RV, and E+RV) (p > 0.05) (Fig. 5g, h). The baseline fEPSP was recorded at 60 s intervals with a stimulation intensity equivalent to ~35% of the maximum evoked response. After recording stable baseline responses for 10 min, we delivered tetanic stimulation, and then test pulses were resumed for an additional 110 min (Fig. 5i, j). In this situation, the tetanic stimulation evoked typical LTP in slices from adult mice treated on P7 with S+V, E+V, S+RV, and E+RV. These responses were stable over 110 min. However, the tetanic stimulation evoked a significantly reduced LTP in slices (n = 10 slices/5 mice/group) prepared from P7 E+V animals compared to S+V animals (p < 0.05) with a significant group interaction (two-way ANOVA) (F1,36 = 34, p < 0.05; post hoc test). S+V vs. E+V were significantly different at all post-tetanic stimulation time intervals (p < 0.05). The LTP in slices prepared from S+RV-treated mice were similar to those in S+V-treated mice (p > 0.05). RV preadministration completely prevented the P7 ethanol-induced LTP deficits (p < 0.05) (Fig. 5k). This experiment indicated that CDK5 plays an important role in ethanol-induced delays in synaptic maturation during development.
Discussion
In this study, the use of a postnatal ethanol-exposed mouse model that induces pleiotropic effects including caspase-3 activation and enhances the generation of the CDK5-activating peptide, p25, allowed us to delineate both the neurodegeneration mechanisms in neonatal mice and neurobehavioral consequences in adult mice. Our results show that p25 functions as a CB1R downstream mediator of ethanol-induced neurodegeneration and that blockade of CDK5 protects neonatal mice from neurodegeneration. Additionally, we provide multiple lines of evidence demonstrating that p25 generation caused by postnatal ethanol induces persistent impaired synaptic plasticity, as well as reduced cognitive capacity, and thereby contributes to the development of FASD-like neurobehavioral abnormalities. CDK5 has been implicated in various neurodegenerative disorders. P25, the cleavage product of p35, results in hyperactivity of CDK5. CDK5/p35 activity is required for physiological processes; however, increased CDK5/p25 kinase activity has been implicated in several neuropathological conditions. Our study demonstrated that postnatal ethanol treatment increases the p35 cleavage product, p25, in the cortex and HP region of the brain. Several lines of evidence obtained using a caspase-3 inhibitor, a CB1R antagonist and CB1R KO suggest that the increase in p25 levels is mediated by postnatal ethanol-activated CB1R, and therefore, CDK5 activation is the downstream target of CB1R. In our previous studies, postnatal ethanol caused neurodegeneration via upregulation of endocannabinoids (anandamide), epigenetically upregulated CB1R expression, and impairment of neuronal survival pathways, such as pERK1/2/pCREB regulated Arc expression [11, 12, 37]. In the current study, inhibition of CDK5/p25 kinase activity before ethanol exposure prevented the loss of pERK1/2/pCREB-regulated Arc expression and neurodegeneration (caspase-3 activation and tau hyperphosphorylation), suggesting that CDK5 activity is part of the CB1R-mediated pERK1/2/pCREB/Arc pathway that is responsible for postnatal ethanol-induced neurodegeneration in neonatal mice. Although the role of ethanol-activated CDK5 during postnatal development is not known, it has been shown that enhanced CDK5 activity is associated with activation of caspase-3 [41] and inhibition of ERK1/2 phosphorylation via inhibition of MEK1 catalytic activity [39]. Further, inhibition of CDK5 rescues neurite retraction and neurodegeneration (tau hyperphosphorylation) [42]. Additionally, CDK5 silencing increased CREB and ERK expression and prevented neuronal loss [43]. In contrast, conditional ablation of CDK5 in the HP exhibited loss of the cAMP pathway and impaired CREB phosphorylation [44]. Interestingly, ethanol exposure throughout the gestational period was shown to reduce p35 and CDK5 levels in the adolescent medial frontal cortical tissues [45]. These observations together suggest that postnatal ethanol-activated CB1R facilitates the generation of the CDK5 activator, p25 (CDK5 activation), and impairs the downstream signaling leading to neurodegeneration in neonatal mice. It also appears that upregulation of CDK5/p25 activity has an influence on additional activation of caspase-3 in this model and studies are underway to further investigate this pathway.
Rac1, a member of the RhoGTPase family proteins, is critical for neurodevelopment, structural plasticity of dendrites, and dendritic spines [46]; is involved in the response to environmental insult; and plays a significant role in the balance between neuronal survival and death [31, 47]. Thus we speculated that Rac1 may be involved in ethanol-induced neurodegeneration in neonatal mice. To our surprise, we found that the expression and total protein content of Rac1 were significantly decreased in the NC and HP regions of postnatal ethanol-exposed neonatal mouse brains, which are persistent to adulthood. We have previously reported that the CB1R pathway plays a key regulatory function in controlling ethanol-induced structural and behavioral changes in a mouse model of FASD [11, 12, 37, 48]. Therefore, experiments in mice exposed to a CB1R antagonist or in CB1R KO mice prevented the ethanol-induced loss of Rac1 protein expression in HP and NC tissues. These results suggest that the loss of Rac1 protein expression is downstream of CB1R activity. In addition, inhibition of CDK5 activity also prevented the loss of Rac1 protein expression. Although the mechanism by which CB1R or CDK5 activation impairs Rac1 expression is not known, several studies suggest that CDK5 regulates Rac1 function in a variety of conditions, including the neurodegeneration environment [49–51]. The Rac1 GTPase-deficient mouse lens exhibits reduced cell survival [52]. Genetic ablation or pharmacological inhibition of CDK5 provides neuronal protection in a Rac1-dependent manner [53, 54]. Further, it is well established that Rac1 activation is involved in actin polymerization while inhibition results in actin depolymerization [30] and the inhibition of spine formation [55]. Actin depolymerization has been shown to function in the process leading to apoptosis [56–58]. Our data are consistent with previous findings in developmental ethanol studies in which ethanol has been shown to inhibit Rac1 activation [59], cause impaired neurite formation, and induce apoptosis in cerebellar granule neurons [28]. Therefore, it is possible that, by inhibiting Rac1 expression in the cell, postnatal ethanol-activated CB1R/CDK5 may be involved in destabilizing actin dynamics, which ultimately causes neurodegeneration in neonatal mice.
Several investigators have demonstrated neurobehavioral defects in adult mice exposed to postnatal ethanol [8–13, 37, 60]. We have recently shown that persistent impairments in pCREB signaling followed by impaired expression of the Arc gene in a CB1R-dependent manner [11, 12, 37] were responsible for postnatal ethanol-induced neurobehavioral defects in adult mice. We have also demonstrated that persistent deficits in Arc expression in adult mice are mediated by enhanced H3K9me2 and reduced H3K14ac in the Arc gene promoter region [11, 37]. These studies indicate that postnatal ethanol-induced long-lasting signaling and epigenetic defects play an important and unprecedented role in neuronal abnormalities in adult animals. Inhibition of CDK5 before ethanol exposure in P7 mice prevented persistent pCREB and Arc levels in adult mice as observed with a CB1R antagonist in our previous studies [37]. In addition, the postnatal ethanol-induced loss of Rac1 expression was long-lasting to adulthood. Because decreased Rac1 expression is regulated via a reduced Rac1 gene transcription, we have evaluated the epigenetic regulation of the Rac1 gene, and we found that postnatal ethanol increased H3K9me2 and G9a without affecting H4K8ac, H3K14ac, HDAC1, and CBP levels in the Rac1 gene promoter region. It was shown that enhanced H3K9me2, a mark of heterochromatin, transcriptionally inhibits gene expression [61], and this process is catalyzed by H3K9-specific methyltransferases, specifically G9a. We [10, 33, 61, 62] and others [63] have shown that developmental ethanol exposure activates G9a, increases H3K9me2 levels, and inhibits gene expression. Our current findings further suggested that both H3K9me2 enrichment and recruitment of G9a on Rac1 gene promoter are downstream of CB1R and CDK5 signaling. Although no studies have demonstrated this mechanism in any of the developmental ethanol studies, regulation of Rac1 expression via epigenetic processes has been demonstrated under many conditions. A similar repressive chromatin state in the Rac1 gene promoter was found in the human postmortem nucleus accumbens of depressed subjects, which was linked to reduced Rac1 gene transcription [64]. Enhanced H3K27me2 levels on the Rac1 gene promoter region, and the subsequent decrease in Rac1 gene expression, was observed in a chronic social defeat stress animal model [65]. Together, our findings suggest that Rac1 is transcriptionally downregulated in the HP through an epigenetic mechanism in an animal model of FASD.
While the molecular mechanisms are not clear, developmental ethanol exposure causes persistent neurobehavioral abnormalities in adult mice [8–13, 37, 60, 66–68]. In our previous studies, we have demonstrated that postnatal ethanol-induced long-lasting neurobehavioral abnormalities could be rescued by blocking either CB1R or G9a before ethanol exposure, suggesting that CB1R-mediated signaling and G9a-regulated epigenetic modifications play an important role in the development of neurobehavioral abnormalities found in many animal models of FASD [5, 61]. In the current study, administration of RV before ethanol exposure prevented pCREB, Arc, and Rac1 expression deficits in the HP brain region. These findings suggest that CB1R/CDK5 activation causes persistent impairments in signaling and gene expression that are vital for synaptic plasticity and learning and memory behavior. In line with these findings, both spatial memory and social recognition memory deficits in adult mice were rescued by preadministration of RV before ethanol exposure in P7 mice. Consistent with our previous findings [11], spatial memory was not different in male and female mice, suggesting that postnatal ethanol or RV effects are not dependent on sex. Postnatal ethanol-induced LTP defects [8, 9, 11–13, 60] were rescued by preadministration of RV, suggesting that inhibition of CDK5/p25 activity not only prevented neurodegeneration in neonatal mice but also facilitated the proper maturation of synaptic circuits that was reflected in both synaptic plasticity and behavioral outcomes in adult mice. In other neurodegenerative disorders, administration of RV significantly attenuated the neurodegeneration, rescued the CA1 synaptic dysfunction, and improved the spatial and novel object recognition memory [69]. Deficits in cognition are a common phenomenon observed in most neurodegenerative and developmental disorders, which highlights the importance of learning and memory [70]. CDK5 kinase activity has been widely implicated in regulating synaptic plasticity and learning and memory formation [71]. In addition, CDK5 epigenetically regulates the transcription of genes involved in synaptic plasticity [72]. Therefore, further understanding of the CDK5/Rac1 signaling mechanism and its influence on the transcription of additional genes is of great importance in developing effective treatment strategies for FASD. We have shown previously that postnatal ethanol exposure impairs signaling events required for proper development of activity-dependent synaptic plasticity in adult mice including enhanced phosphorylation of methyl-CpG-binding protein 2 (pMeCP2) [11]. Our findings are consistent with previous findings that CDK5-mediated phosphorylation of MeCP2 and transcriptional regulation have been shown to control activity-dependent dendrite development [73]. It was shown that conditional knockdown of Rac1 in excitatory neurons in the forebrain in vivo not only affects spine structure but also impairs synaptic plasticity, as well as defects in HP-dependent spatial learning [74]. Additionally, Rac1 mutants exhibit impairments in working/episodic-like memory tasks, suggesting that Rac1 plays an important regulatory role in encoding novel spatial information in vivo [74]. Furthermore, inactivation of Rac1 is associated with impaired long-term plasticity [75]. These observations along with current findings strongly suggest that CB1R/CDK5-mediated signaling events, as well as epigenetic regulation of gene transcription such as Arc and Rac1, play an important role in the development of neurobehavioral abnormalities found in FASD, and further characterization of CB1R-mediated signaling events would aid in the development of potential targets to treat FASD.
In summary, the current findings suggest that postnatal ethanol exposure generates p25, a CDK5-activating peptide, and causes suppression of Rac1 expression in a CB1R-dependent manner. Inhibition of CDK5 activity prevents the ethanol-induced loss of Rac1 expression in neonatal mice. Rac1 expression is persistently impaired to adulthood, and its expression is regulated by the presence of H3K9me2 and G9a, a suppressive chromatin, in the Rac1 gene promoter region. Administration of RV in P7 mice also rescued neurodegeneration in neonatal mice and protected the pERK1/2, pCREB, and Arc signaling defects and loss of Rac1 gene expression, synaptic plasticity, and behavioral abnormalities in adult mice exposed to P7 ethanol. These findings indicate that CB1R-mediated [12, 33] activation of CDK5/p25 activity followed by the persistent loss of pERK and pCREB and epigenetic suppression of Arc [11] and Rac1 expression is responsible for the long-lasting neurobehavioral defects in postnatal ethanol-exposed adult mice. Further understanding of the CB1R/CDK5/Rac1 signaling mechanism may present a novel therapeutic target to treat neurobehavioral abnormalities found in FASD [76–84].
Electronic supplementary material
{kind=link}
Acknowledgements
We thank Neha Balapal for editing the final version of the manuscript. This research was generously funded by NIH grants R01 AA019443 (BSB).
Author contributions
BSB conceived and designed the experiments. All authors performed the experiments and contributed to data analysis. BSB and VJ wrote the manuscript. BSB contributed to study supervision.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary Information accompanies this paper at (10.1038/s41386-018-0181-y).
References
- 1.Guerri C, Bazinet A, Riley EP. Foetal alcohol spectrum disorders and alterations in brain and behaviour. Alcohol Alcohol. 2009;44:108–14. doi: 10.1093/alcalc/agn105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sokol RJ, Delaney-Black V, Nordstrom B. Fetal alcohol spectrum disorder. JAMA. 2003;290:2996–9. doi: 10.1001/jama.290.22.2996. [DOI] [PubMed] [Google Scholar]
- 3.Lunde ER, Washburn SE, Golding MC, Bake S, Miranda RC, Ramadoss J. Alcohol-induced developmental origins of adult-onset diseases. Alcohol Clin Exp Res. 2016;40:1403–14. doi: 10.1111/acer.13114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Medina AE. Fetal alcohol spectrum disorders and abnormal neuronal plasticity. Neuroscientist. 2011;17:274–87. doi: 10.1177/1073858410383336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Basavarajappa BS. Fetal alcohol spectrum disorder: potential role of endocannabinoids signaling. Brain Sci. 2015;5:456–93. doi: 10.3390/brainsci5040456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kodituwakku PW. Neurocognitive profile in children with fetal alcohol spectrum disorders. Dev Disabil Res Rev. 2009;15:218–24. doi: 10.1002/ddrr.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mattson SN, Crocker N, Nguyen TT. Fetal alcohol spectrum disorders: neuropsychological and behavioral features. Neuropsychol Rev. 2011;21:81–101. doi: 10.1007/s11065-011-9167-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Noel M, Norris EH, Strickland S. Tissue plasminogen activator is required for the development of fetal alcohol syndrome in mice. Proc Natl Acad Sci USA. 2011;108:5069–74. doi: 10.1073/pnas.1017608108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sadrian B, Subbanna S, Wilson DA, Basavarajappa BS, Saito M. Lithium prevents long-term neural and behavioral pathology induced by early alcohol exposure. Neuroscience. 2012;206:122–35. doi: 10.1016/j.neuroscience.2011.12.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Subbanna S, Basavarajappa BS. Pre-administration of G9a/GLP inhibitor during synaptogenesis prevents postnatal ethanol-induced LTP deficits and neurobehavioral abnormalities in adult mice. Exp Neurol. 2014;261:34–43. doi: 10.1016/j.expneurol.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Subbanna S, Nagre NN, Shivakumar M, Joshi V, Psychoyos D, Kutlar A, et al. CB1R-mediated activation of caspase-3 causes epigenetic and neurobehavioral abnormalities in postnatal ethanol-exposed mice. Front Mol Neurosci. 2018;11:45. doi: 10.3389/fnmol.2018.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Subbanna S, Shivakumar M, Psychoyos D, Xie S, Basavarajappa BS. Anandamide-CB1 receptor signaling contributes to postnatal ethanol-induced neonatal neurodegeneration, adult synaptic and memory deficits. J Neuoscience. 2013;33:6350–66. doi: 10.1523/JNEUROSCI.3786-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilson DA, Peterson J, Basavaraj BS, Saito M. Local and regional network function in behaviorally relevant cortical circuits of adult mice following postnatal alcohol exposure. Alcohol Clin Exp Res. 2011;35:1974–84. doi: 10.1111/j.1530-0277.2011.01549.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dhavan R, Tsai LH. A decade of CDK5. Nat Rev Mol Cell Biol. 2001;2:749–59. doi: 10.1038/35096019. [DOI] [PubMed] [Google Scholar]
- 15.Fischer A, Sananbenesi F, Pang PT, Lu B, Tsai LH. Opposing roles of transient and prolonged expression of p25 in synaptic plasticity and hippocampus-dependent memory. Neuron. 2005;48:825–38. doi: 10.1016/j.neuron.2005.10.033. [DOI] [PubMed] [Google Scholar]
- 16.Mita N, He X, Sasamoto K, Mishiba T, Ohshima T. Cyclin-dependent kinase 5 regulates dendritic spine formation and maintenance of cortical neuron in the mouse brain. Cereb Cortex. 2016;26:967–76. doi: 10.1093/cercor/bhu264. [DOI] [PubMed] [Google Scholar]
- 17.Shupp A, Casimiro MC, Pestell RG. Biological functions of CDK5 and potential CDK5 targeted clinical treatments. Oncotarget. 2017;8:17373–82. doi: 10.18632/oncotarget.14538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheung ZH, Fu AK, Ip NY. Synaptic roles of Cdk5: implications in higher cognitive functions and neurodegenerative diseases. Neuron. 2006;50:13–8. doi: 10.1016/j.neuron.2006.02.024. [DOI] [PubMed] [Google Scholar]
- 19.Tan TC, Valova VA, Malladi CS, Graham ME, Berven LA, Jupp OJ, et al. Cdk5 is essential for synaptic vesicle endocytosis. Nat Cell Biol. 2003;5:701–10. doi: 10.1038/ncb1020. [DOI] [PubMed] [Google Scholar]
- 20.Nguyen PV, Woo NH. Regulation of hippocampal synaptic plasticity by cyclic AMP-dependent protein kinases. Prog Neurobiol. 2003;71:401–37. doi: 10.1016/j.pneurobio.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 21.Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402:615–22. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- 22.Cruz JC, Tseng HC, Goldman JA, Shih H, Tsai LH. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron. 2003;40:471–83. doi: 10.1016/s0896-6273(03)00627-5. [DOI] [PubMed] [Google Scholar]
- 23.Lew J, Huang QQ, Qi Z, Winkfein RJ, Aebersold R, Hunt T, et al. A brain-specific activator of cyclin-dependent kinase 5. Nature. 1994;371:423–6. doi: 10.1038/371423a0. [DOI] [PubMed] [Google Scholar]
- 24.Tsai LH, Delalle I, Caviness VS, Jr., Chae T, Harlow E. p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature. 1994;371:419–23. doi: 10.1038/371419a0. [DOI] [PubMed] [Google Scholar]
- 25.Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature. 2000;405:360–4. doi: 10.1038/35012636. [DOI] [PubMed] [Google Scholar]
- 26.Sundaram JR, Poore CP, Sulaimee NH, Pareek T, Asad AB, Rajkumar R, et al. Specific inhibition of p25/Cdk5 activity by the Cdk5 inhibitory peptide reduces neurodegeneration in vivo. J Neurosci. 2013;33:334–43. doi: 10.1523/JNEUROSCI.3593-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dobbing J, Sands J. Comparative aspects of the brain growth spurt. Early Hum Dev. 1979;3:79–83. doi: 10.1016/0378-3782(79)90022-7. [DOI] [PubMed] [Google Scholar]
- 28.Joshi S, Guleria RS, Pan J, Bayless KJ, Davis GE, Dipette D, et al. Ethanol impairs Rho GTPase signaling and differentiation of cerebellar granule neurons in a rodent model of fetal alcohol syndrome. Cell Mol Life Sci. 2006;63:2859–70. doi: 10.1007/s00018-006-6333-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olney JW, Young C, Wozniak DF, Jevtovic-Todorovic V, Ikonomidou C. Do pediatric drugs cause developing neurons to commit suicide? Trends Pharmacol Sci. 2004;25:135–9. doi: 10.1016/j.tips.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 30.Govek EE, Newey SE, Van Aelst L. The role of the Rho GTPases in neuronal development. Genes Dev. 2005;19:1–49. doi: 10.1101/gad.1256405. [DOI] [PubMed] [Google Scholar]
- 31.Linseman DA, Loucks FA. Diverse roles of Rho family GTPases in neuronal development, survival, and death. Front Biosci. 2008;13:657–76. doi: 10.2741/2710. [DOI] [PubMed] [Google Scholar]
- 32.Subbanna S, Joshi V, Basavarajappa BS. Activity-dependent signaling and epigenetic abnormalities in mice exposed to postnatal ethanol. Neuroscience. 2018. 10.1016/j.neuroscience.2018.07.011. [DOI] [PMC free article] [PubMed]
- 33.Subbanna S, Nagre NN, Shivakumar M, Umapathy NS, Psychoyos D, Basavarajappa BS. Ethanol induced acetylation of histone at G9a exon1 and G9a-mediated histone H3 dimethylation leads to neurodegeneration in neonatal mice. Neuroscience. 2014;258:422–32. doi: 10.1016/j.neuroscience.2013.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nagre NN, Subbanna S, Shivakumar M, Psychoyos D, Basavarajappa BS. CB1-receptor knockout neonatal mice are protected against ethanol-induced impairments of DNMT1, DNMT3A, and DNA methylation. J Neurochem. 2015;132:429–42. doi: 10.1111/jnc.13006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fischer U, Janicke RU, Schulze-Osthoff K. Many cuts to ruin: a comprehensive update of caspase substrates. Cell Death Differ. 2003;10:76–100. doi: 10.1038/sj.cdd.4401160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kamada S, Kikkawa U, Tsujimoto Y, Hunter T. Nuclear translocation of caspase-3 is dependent on its proteolytic activation and recognition of a substrate-like protein(s) J Biol Chem. 2005;280:857–60. doi: 10.1074/jbc.C400538200. [DOI] [PubMed] [Google Scholar]
- 37.Subbanna S, Nagre NN, Umapathy NS, Pace BS, Basavarajappa BS. Ethanol exposure induces neonatal neurodegeneration by enhancing CB1R Exon1 histone H4K8 acetylation and up-regulating CB1R function causing neurobehavioral abnormalities in adult mice. Int J Neuropsychopharmacol. 2015;18:1–15. doi: 10.1093/ijnp/pyu028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seo J, Kritskiy O, Watson LA, Barker SJ, Dey D, Raja WK, et al. Inhibition of p25/Cdk5 attenuates tauopathy in mouse and iPSC models of frontotemporal dementia. J Neurosci. 2017;37:9917–24. doi: 10.1523/JNEUROSCI.0621-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sharma P, Veeranna, Sharma M, Amin ND, Sihag RK, Grant P, et al. Phosphorylation of MEK1 by cdk5/p35 down-regulates the mitogen-activated protein kinase pathway. J Biol Chem. 2002;277:528–34. doi: 10.1074/jbc.M109324200. [DOI] [PubMed] [Google Scholar]
- 40.Graff J, Kim D, Dobbin MM, Tsai LH. Epigenetic regulation of gene expression in physiological and pathological brain processes. Physiol Rev. 2011;91:603–49. doi: 10.1152/physrev.00012.2010. [DOI] [PubMed] [Google Scholar]
- 41.Wang WY, Luo Y, Jia LJ, Hu SF, Lou XK, Shen SL, et al. Inhibition of aberrant cyclin-dependent kinase 5 activity attenuates isoflurane neurotoxicity in the developing brain. Neuropharmacology. 2014;77:90–9. doi: 10.1016/j.neuropharm.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 42.Maldonado H, Ramirez E, Utreras E, Pando ME, Kettlun AM, Chiong M, et al. Inhibition of cyclin-dependent kinase 5 but not of glycogen synthase kinase 3-beta prevents neurite retraction and tau hyperphosphorylation caused by secretable products of human T-cell leukemia virus type I-infected lymphocytes. J Neurosci Res. 2011;89:1489–98. doi: 10.1002/jnr.22678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gutierrez-Vargas JA, Munera A, Cardona-Gomez GP. CDK5 knockdown prevents hippocampal degeneration and cognitive dysfunction produced by cerebral ischemia. J Cereb Blood Flow Metab. 2015;35:1937–49. doi: 10.1038/jcbfm.2015.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guan JS, Su SC, Gao J, Joseph N, Xie Z, Zhou Y, et al. Cdk5 is required for memory function and hippocampal plasticity via the cAMP signaling pathway. PLoS ONE. 2011;6:e25735. doi: 10.1371/journal.pone.0025735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goggin SL, Caldwell KK, Cunningham LA, Allan AM. Prenatal alcohol exposure altersp35, CDK5 and GSK3beta in the medial frontal cortex and hippocampus of adolescent mice. Toxicol Rep. 2014;1:544–53. doi: 10.1016/j.toxrep.2014.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li Z, Aizenman CD, Cline HT. Regulation of rho GTPases by crosstalk and neuronal activity in vivo. Neuron. 2002;33:741–50. doi: 10.1016/s0896-6273(02)00621-9. [DOI] [PubMed] [Google Scholar]
- 47.Ma DK, Jang MH, Guo JU, Kitabatake Y, Chang ML, Pow-Anpongkul N, et al. Neuronal activity-induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science. 2009;323:1074–7. doi: 10.1126/science.1166859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Subbanna S, Psychoyos D, Xie S, Basavarajappa BS. Postnatal ethanol exposure alters levels of 2-arachidonylglycerol-metabolizing enzymes and pharmacological inhibition of monoacylglycerol lipase does not cause neurodegeneration in neonatal mice. J Neurochem. 2015;134:276–87. doi: 10.1111/jnc.13120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kesavapany S, Amin N, Zheng YL, Nijhara R, Jaffe H, Sihag R, et al. p35/cyclin-dependent kinase 5 phosphorylation of ras guanine nucleotide releasing factor 2 (RasGRF2) mediates Rac-dependent extracellular signal-regulated kinase 1/2 activity, altering RasGRF2 and microtubule-associated protein 1b distribution in neurons. J Neurosci. 2004;24:4421–31. doi: 10.1523/JNEUROSCI.0690-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liebl J, Weitensteiner SB, Vereb G, Takacs L, Furst R, Vollmar AM, et al. Cyclin-dependent kinase 5 regulates endothelial cell migration and angiogenesis. J Biol Chem. 2010;285:35932–43. doi: 10.1074/jbc.M110.126177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nikolic M, Chou MM, Lu W, Mayer BJ, Tsai LH. The p35/Cdk5 kinase is a neuron-specific Rac effector that inhibits Pak1 activity. Nature. 1998;395:194–8. doi: 10.1038/26034. [DOI] [PubMed] [Google Scholar]
- 52.Maddala R, Chauhan BK, Walker C, Zheng Y, Robinson ML, Lang RA, et al. Rac1 GTPase-deficient mouse lens exhibits defects in shape, suture formation, fiber cell migration and survival. Dev Biol. 2011;360:30–43. doi: 10.1016/j.ydbio.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Posada-Duque RA, Lopez-Tobon A, Piedrahita D, Gonzalez-Billault C, Cardona-Gomez GP. p35 and Rac1 underlie the neuroprotection and cognitive improvement induced by CDK5 silencing. J Neurochem. 2015;134:354–70. doi: 10.1111/jnc.13127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Posada-Duque RA, Palacio-Castaneda V, Cardona-Gomez GP. CDK5 knockdown in astrocytes provide neuroprotection as a trophic source via Rac1. Mol Cell Neurosci. 2015;68:151–66. doi: 10.1016/j.mcn.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 55.Tashiro A, Minden A, Yuste R. Regulation of dendritic spine morphology by the rho family of small GTPases: antagonistic roles of Rac and Rho. Cereb Cortex. 2000;10:927–38. doi: 10.1093/cercor/10.10.927. [DOI] [PubMed] [Google Scholar]
- 56.Franklin-Tong VE, Gourlay CW. A role for actin in regulating apoptosis/programmed cell death: evidence spanning yeast, plants and animals. Biochem J. 2008;413:389–404. doi: 10.1042/BJ20080320. [DOI] [PubMed] [Google Scholar]
- 57.Lemkuil BP, Head BP, Pearn ML, Patel HH, Drummond JC, Patel PM. Isoflurane neurotoxicity is mediated by p75NTR-RhoA activation and actin depolymerization. Anesthesiology. 2011;114:49–57. doi: 10.1097/ALN.0b013e318201dcb3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Levee MG, Dabrowska MI, Lelli JL, Jr., Hinshaw DB. Actin polymerization and depolymerization during apoptosis in HL-60 cells. Am J Physiol. 1996;271:C1981–92. doi: 10.1152/ajpcell.1996.271.6.C1981. [DOI] [PubMed] [Google Scholar]
- 59.Lindsley TA, Shah SN, Ruggiero EA. Ethanol alters BDNF-induced Rho GTPase activation in axonal growth cones. Alcohol Clin Exp Res. 2011;35:1321–30. doi: 10.1111/j.1530-0277.2011.01468.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sadrian B, Lopez-Guzman M, Wilson DA, Saito M. Distinct neurobehavioral dysfunction based on the timing of developmental binge-like alcohol exposure. Neuroscience. 2014;280:204–19. doi: 10.1016/j.neuroscience.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Basavarajappa BS, Subbanna S. Epigenetic mechanisms in developmental alcohol-induced neurobehavioral deficits. Brain Sci. 2016;6:12. doi: 10.3390/brainsci6020012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Subbanna S, Shivakumar M, Umapathy NS, Saito M, Mohan PS, Kumar A, et al. G9a-mediated histone methylation regulates ethanol-induced neurodegeneration in the neonatal mouse brain. Neurobiol Dis. 2013;54:475–85. doi: 10.1016/j.nbd.2013.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bekdash RA, Zhang C, Sarkar DK. Gestational choline supplementation normalized fetal alcohol-induced alterations in histone modifications, DNA methylation, and proopiomelanocortin (POMC) gene expression in beta-endorphin-producing POMC neurons of the hypothalamus. Alcohol Clin Exp Res. 2013;37:1133–42. doi: 10.1111/acer.12082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Golden SA, Christoffel DJ, Heshmati M, Hodes GE, Magida J, Davis K, et al. Epigenetic regulation of RAC1 induces synaptic remodeling in stress disorders and depression. Nat Med. 2013;19:337–44. doi: 10.1038/nm.3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mahgoub M, Monteggia LM. Epigenetics and psychiatry. Neurotherapeutics. 2013;10:734–41. doi: 10.1007/s13311-013-0213-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sanchez Vega MC, Chong S, Burne TH. Early gestational exposure to moderate concentrations of ethanol alters adult behaviour in C57BL/6J mice. Behav Brain Res. 2013;252:326–33. doi: 10.1016/j.bbr.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 67.Vaglenova J, Pandiella N, Wijayawardhane N, Vaithianathan T, Birru S, Breese C, et al. Aniracetam reversed learning and memory deficits following prenatal ethanol exposure by modulating functions of synaptic AMPA receptors. Neuropsychopharmacology. 2008;33:1071–83. doi: 10.1038/sj.npp.1301496. [DOI] [PubMed] [Google Scholar]
- 68.Cui ZJ, Zhao KB, Zhao HJ, Yu DM, Niu YL, Zhang JS, et al. Prenatal alcohol exposure induces long-term changes in dendritic spines and synapses in the mouse visual cortex. Alcohol Alcohol. 2010;45:312–9. doi: 10.1093/alcalc/agq036. [DOI] [PubMed] [Google Scholar]
- 69.Li L, Zhang C, Zi X, Tu Q, Guo K. Epigenetic modulation of Cdk5 contributes to memory deficiency induced by amyloid fibrils. Exp Brain Res. 2015;233:165–73. doi: 10.1007/s00221-014-4100-0. [DOI] [PubMed] [Google Scholar]
- 70.Bibb JA, Mayford MR, Tsien JZ, Alberini CM. Cognition enhancement strategies. J Neurosci. 2010;30:14987–92. doi: 10.1523/JNEUROSCI.4419-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hawasli AH, Benavides DR, Nguyen C, Kansy JW, Hayashi K, Chambon P, et al. Cyclin-dependent kinase 5 governs learning and synaptic plasticity via control of NMDAR degradation. Nat Neurosci. 2007;10:880–6. doi: 10.1038/nn1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim D, Frank CL, Dobbin MM, Tsunemoto RK, Tu W, Peng PL, et al. Deregulation of HDAC1 by p25/Cdk5 in neurotoxicity. Neuron. 2008;60:803–17. doi: 10.1016/j.neuron.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liang Z, Ye T, Zhou X, Lai KO, Fu AK, Ip NY. Cdk5 regulates activity-dependent gene expression and dendrite development. J Neurosci. 2015;35:15127–34. doi: 10.1523/JNEUROSCI.1443-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Haditsch U, Leone DP, Farinelli M, Chrostek-Grashoff A, Brakebusch C, Mansuy IM, et al. A central role for the small GTPase Rac1 in hippocampal plasticity and spatial learning and memory. Mol Cell Neurosci. 2009;41:409–19. doi: 10.1016/j.mcn.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Martinez LA, Tejada-Simon MV. Pharmacological inactivation of the small GTPase Rac1 impairs long-term plasticity in the mouse hippocampus. Neuropharmacology. 2011;61:305–12. doi: 10.1016/j.neuropharm.2011.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Basavarajappa BS, Nagre NN, Xie S, Subbanna S. Elevation of endogenous anandamide impairs LTP, learning, and memory through CB1 receptor signaling in mice. Hippocampus. 2014;24:808–18. doi: 10.1002/hipo.22272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hendrickx A, Pierrot N, Tasiaux B, Schakman O, Kienlen-Campard P, De Smet C, et al. Epigenetic regulations of immediate early genes expression involved in memory formation by the amyloid precursor protein of Alzheimer disease. PLoS ONE. 2014;9:e99467. doi: 10.1371/journal.pone.0099467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ikonomidou C, Bittigau P, Ishimaru MJ, Wozniak DF, Koch C, Genz K, et al. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science. 2000;287:1056–60. doi: 10.1126/science.287.5455.1056. [DOI] [PubMed] [Google Scholar]
- 79.Basavarajappa BS, Subbanna S. CB1 receptor-mediated signaling underlies the hippocampal synaptic, learning and memory deficits following treatment with JWH-081, a new component of spice/K2 preparations. Hippocampus. 2014;24:178–88. doi: 10.1002/hipo.22213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 81.Lundquist F. The determination of ethyl alcohol in blood and tissue. In: Glick D, editor. Methods in biochemical analysis. Interscience Publishers, Inc. New York 1959. p. 217–51.
- 82.Rao MV, McBrayer MK, Campbell J, Kumar A, Hashim A, Sershen H, et al. Specific calpain inhibition by calpastatin prevents tauopathy and neurodegeneration and restores normal lifespan in tau P301L mice. J Neurosci. 2014;34:9222–34. doi: 10.1523/JNEUROSCI.1132-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sarnyai Z, Sibille EL, Pavlides C, Fenster RJ, McEwen BS, Toth M. Impaired hippocampal-dependent learning and functional abnormalities in the hippocampus in mice lacking serotonin(1A) receptors. Proc Natl Acad Sci USA. 2000;97:14731–6. doi: 10.1073/pnas.97.26.14731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Thor DH, Wainwright KL, Holloway WR. Persistence of attention to a novel conspecific: some developmental variables in laboratory rats. Dev Psychobiol. 1982;15:1–8. doi: 10.1002/dev.420150102. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.