

Abstract
The inflammasome is a multiprotein complex necessary for the onset of inflammation. The adapter protein ASC assembles inflammasome components by acting as a molecular glue between danger-signal sensors and procaspase-1. The assembly is mediated by ASC self-association and protein interactions via its two Death Domains, PYD and CARD. Truncated versions of ASC have been shown to form filaments, but information on the filaments formed by full-length ASC is needed to construct a meaningful model of inflammasome assembly. To gain insights into this system, we used a combination of transmission EM, NMR, and computational analysis to investigate intact ASC structures. We show that ASC forms ∼6–7-nm-wide filaments that stack laterally to form bundles. The structural characteristics and dimensions of the bundles indicate that both PYD and CARD are integral parts of the filament. A truncated version of ASC with only the CARD domain (ASCCARD) forms different filaments (∼3–4-nm width), providing further evidence that both domains work in concert in filament assembly. Ring-shaped protein particles bound to pre-existing filaments match the size of ASC dimer structures generated by NMR-based protein docking, suggesting that the ASC dimer could be a basic building block for filament formation. Solution NMR binding studies identified the protein surfaces involved in the ASCCARD–ASCCARD interaction. These data provide new insights into the structural underpinnings of the inflammasome and should inform future efforts to interrogate this important biological system.
Keywords: inflammasome, apoptosis, Death Domain, inflammation, protein assembly, ASC, CARD, NMR, PYD, transmission electron microscopy
Introduction
Members of the Death Domain protein superfamily are involved in apoptotic and inflammatory processes (1, 2) by self-assembling into large signaling complexes through homotypic interactions (1–4). Death Domains typically fold into a six-helical bundle arranged in a Greek-key topology known as the Death fold (1, 4). Structural differences between subfamilies within this fold are likely responsible for the observed homotypic binding specificity (5). These Death Domain interactions have been classified in three different types (type I, II, and III (6)) based on high-resolution studies of Death Domain complexes and are key in the formation of large molecular platforms (7, 8). The binding interfaces resulting from the interactions do not overlap, thus making it possible for a single Death Domain to establish multiple interactions with other domains (2, 8).
ASC (apoptosis-associated specklike protein containing a CARD) is a member of the Death Domain superfamily that self-oligomerizes to form large molecular assemblies such as the inflammasome and the pyroptosome (9, 10). The latter is described as a supramolecular complex composed of oligomerized ASC dimers that induces inflammatory cell death (pyroptosis) via the activation of procaspase-1 (10, 11). The inflammasome is composed of sensor proteins (NLRs,2 also members of the Death Domain superfamily), ASC, and procaspase-1. ASC bears two Death Domains, PYD and CARD, and functions as an adapter for PYD-containing sensors by bridging them with the CARD of procaspase-1 via homotypic interactions. ASC is a regulator of inflammation by recruiting procaspase-1 (12).
The oligomerization process involved in inflammasome assembly is critical for procaspase-1 autoactivation, which in turn matures interleukin-1β and interleukin-18, thus triggering the inflammatory response (13). Recently, we contributed to increase our understanding of the role of Death Domains in inflammasome assembly by determining the 3D solution structure of NLRP3PYD (PDB code 2NAQ (14)) and reporting a detailed study of ASCPYD self-association and its interaction with NLRP3PYD by solution NMR and analytical ultracentrifugation (14). We found that the ASCPYD–ASCPYD and ASCPYD–NLRP3PYD complexes share similar binding interfaces and that the heteromolecular interaction shows higher affinity. Based on these data, we built models of the supramolecular complexes formed by ASCPYD self-association, and of the intermolecular ASCPYD–NLRP3PYD oligomerization, providing insight into potential first stages of inflammasome assembly.
EM studies have revealed critical structural characteristics of inflammasomes formed by sensors with CARDs (15), where the NLRs oligomerize into nanometer-size discs. The molecular bases of the activation of disc-type inflammasomes have also been unveiled by cryo-EM. Specifically, it has been found that the NAIP-NLRC4 inflammasome is a nucleated oligomerization (16, 17) and that the activation of NLRC4 self-association by the NLR protein NAIP is triggered by the relative rotation of two domains in both NAIP and NLRC4, resulting in the partial opening of the overall structure, thus exposing several binding surfaces that prompt self-assembly (18). However, other types of inflammasomes, particularly those formed by NLRP3, without a CARD, appear as large micrometer-size filamentous rings in the cytosol, that concentrically bind to analogous rings formed by the adapter ASC, leaving oligomerized caspase in the center of this cytosolic micrometer-size structure (19).
It has also been shown by cryo-EM that a truncated version of human ASC, consisting only of its PYD, forms a hollow right-handed helix with outer and inner diameters of ∼9 and 2 nm, respectively (8). More recently, cryo-EM combined with solid-state NMR data has been used to determine the structure of the mouse version of the truncated protein (ASCPYD) (20), resulting in a helical tube with close dimensions and similar overall characteristics to those originally reported. In this study, solid-state NMR data on filaments formed by full-length mouse ASC indicate that the CARD is unfolded. An unfolded CARD is a surprising result, taking into account that the NMR solution structure of full-length human ASC shows a properly folded CARD adopting the expected six-helix bundle motif (21).
The cryo-EM ultrastructure of the PYD of human ASC (8) and the solution NMR structure of the full-length protein (21) have been used jointly to propose a model for the assembly of ASC within the nanometer-ring inflammasome. According to this model, solely the PYDs of ASC oligomerize to form the helical ultrastructure, whereas the folded CARDs protrude from the helix connected by the semiflexible linker, ready to interact with the CARDs of procaspase-1 molecules (8). The model for inflammasome assembly based on the structure of truncated PYD-only ASC is depicted in Fig. 1.
Figure 1.
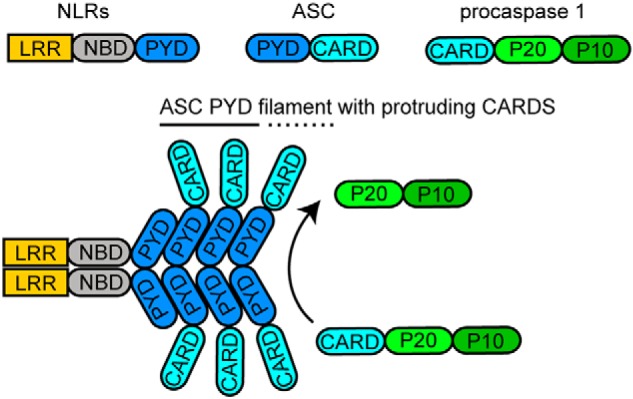
Schematic representation of inflammasome assembly based on ASCPYD filament resulting in procaspase 1 activation. The main components of the ASC inflammasome are shown, including the sensor-signal NLR proteins, the adapter ASC, and the interleukin processing enzyme, procaspase 1.
Compelling evidence indicates that PYD and CARD domains self-associate and tend to form filamentous structures and filament bundles upon oligomerization (8, 20–25). These structures could be very relevant to understand the inflammasome assembly of micrometer-size filamentous rings that are >100 nm in width (19). However, there is currently no structural information on the filaments formed by full-length ASC. Thus, the role of the ASCCARD in filament formation is not well understood. As a result, there are currently large gaps in our understanding of how these structures are formed and scarce information on the structural characteristics and dimensions of the different filaments.
Here, we use TEM to show that full-length ASC tends to form filament bundles of dimensions that can only be explained if both CARD and PYD form integral parts of the filament. Thus, our data deviate from the current model for inflammasome assembly in which the CARDs protrude from the polymerized PYDs forming the filament core. Moreover, our TEM images show ring-shaped structures of ∼7-nm diameter that are attached to pre-existing filaments, suggesting a mode of lateral growth for filament stacking. This information could have important implications to explain the assembly of micrometer-size rings formed by ASC in the cytosol of active macrophages (19).
The size of the ring-shaped structures observed in TEM images agrees with the size of ASC dimers calculated with protein docking based on experimental NMR data, which suggests that this oligomer could be the building block in filament assembly. This result is in agreement with previously reported data indicating that the ASC dimer is the minimal oligomer unit in pyroptosome assembly (10). In addition, we show that filament and filament stacking can be controlled by pH, pointing to a potential role of pH in inflammasome assembly.
Because of the existing controversy in relation to the role played by ASCCARD in filament formation, we have studied by TEM the oligomerization properties of this Death Domain in the absence of the PYD. We found that ASCCARD assembles into filaments with two different ultrastructures, which in turn tend to associate to form highly dense, directional bundles. More importantly, these filaments have significantly different structural characteristics from those of full-length ASC. Finally, we show by solution NMR that ASCCARD, while folded as a six-helix bundle, is able to self-associate, and we identify the binding interfaces responsible for this interaction and estimate the apparent dissociation constant.
Results
Influence of pH in the formation of human ASC filaments
The influence of pH on the aggregation properties of full-length human ASC in solution has been reported in a previous NMR structural study (21). ASC cannot be detected by SDS-PAGE in the supernatant of a solution of the protein at pH 7, indicating very low solubility at neutral pH. However, solubility increases greatly at acidic pH (pH ∼4); thus, the 3D solution structure of human full-length ASC at atomic resolution was determined at this pH (12) (PDB code 2KN6). Both PYD and CARD of ASC are properly folded into the six-helix bundle motif typical of Death Domains and are connected by a semiflexible 23-amino acid-long linker.
To improve our understanding on the effect of pH on the oligomerization capabilities of ASC, we have used TEM to analyze the potential formation of filaments under different conditions. Starting with a protein solution at pH 3, human full-length ASC mainly forms amorphous aggregates as well as few rough-edged filaments by rapid pH increase to less acidic values. However, a slow pH increase by dialysis and constant stirring favors filament formation, as observed by better-defined filament and filament bundles and a significantly higher population of these structures (Fig. 2). The filaments formed by human ASC are typically several hundreds of nm long (∼600–800 nm), although filaments longer than 1 μm have also been observed.
Figure 2.

Filament formation by full-length human ASC can be controlled by pH. Shown are TEM images of full-length human ASC filaments formed by slow pH increase using dialysis from pH value 3 to 4.1 (A), 6.5 (B), and 7.5 (C). Once assembled, the population of filaments is significantly reduced when the pH value is decreased back to 4.1, although some still persist (D). The number of filaments observed in 10 images at similar magnification (scale bars of 100–200 nm) under each condition of pH is as follows: pH 4.1, 0 filaments; pH 5.1 (image not shown), 22 filaments; pH 6.5, 76 filaments; pH decrease to 4.1 (after 6.5), 19 filaments. Arrows point to potentially forming or already formed filaments.
To study the influence of pH on ASC filament formation, we obtained TEM images at pH values 4.1, 5.1, 6.5, and 7.5. At the more acidic pH, some filaments seem to be already starting to form (Fig. 2A). At pH 5.1, filaments are already visible (data not shown), and at pH 6.5 and 7.5, the number of filaments significantly increases (Fig. 2, B and C). Filaments are observed within a 3-h period after slow pH change by dialysis, and no increase in filament population is apparent after 24 h when checked by TEM after 3 and 4 days of dialysis.
Acidification of the filament environment results in partial disassembly of human ASC filaments, as some filaments were still observed after pH decrease to 4.1 (Fig. 2D). Only by lowering the pH to 3.5 are all filaments disassembled, probably due to protein unfolding under these acid conditions. To provide a qualitative evaluation of filament formation, we have counted the number of filaments observed in 10 images obtained under different pH conditions. Once filaments were observed at low magnification, typically 10 images at high magnification were obtained. Images of protein material were obtained in the case where no fully formed filaments were observed (pH 4.1). The number of filaments at the remaining pH conditions was as follows: pH 5.1, 22 filaments; pH 6.5, 76 filaments; pH decrease to 4.1 (after 6.5), 19 filaments. Inspection of additional images supports the conclusion that the great majority of filaments are formed at neutral or close to neutral pH.
Human ASC filaments tend to stack laterally, forming bundles with potential implications in inflammasome assembly
ASC filaments show a high tendency to stack laterally, forming bundles, and appear in isolation less frequently (Figs. 2 (B and C) and 3 (A and B)). Bundles are typically composed of 2–7 filaments, the three- and four-filament arrangements being more abundant. We used scanning EM (SEM) to analyze larger filament bundles formed by full-length ASC. Our SEM data show higher-order structural organizations seemingly held together through cross-linking, with bundles >100 nm in width (Fig. 3B).
Figure 3.
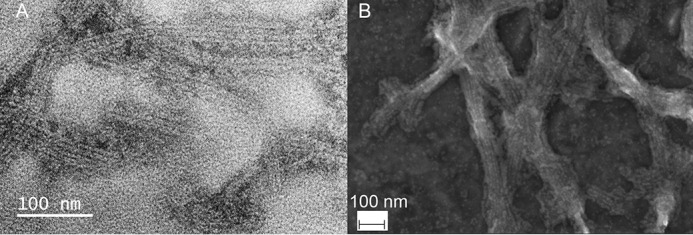
Human ASC filaments stack laterally, forming bundles with potential implications in micrometer-size inflammasomes. A, TEM image of full-length human ASC filament bundles. B, SEM image of ASC filament bundles of a width >100 nm, with potential implication in inflammasome assembly. Filaments were formed by slow pH increase from 3 to 8.2.
To further characterize full-length ASC ultrastructures, we show in Table 1 measurements of the dimensions of bundles composed of different numbers of laterally stacked filaments. Individual filaments, whether in isolation or within a bundle, are ∼6–7 nm wide. Surprisingly, the total width of the different bundles is multiple of the width of one filament, and the multiplication factor coincides with the number of filaments in the bundle (Table 1). This is a striking result in light of the current model for ASC filament formation.
Table 1.
Full-length human ASC filament and bundle dimensions
| Filament number in bundle | Average bundle widtha | Expected bundle widthb | Average filament widthc |
|---|---|---|---|
| nm | nm | nm | |
| 1 | 6.4 ± 0.8 | 6–7 | 6.4 ± 0.8 |
| 2 | 14.0 ± 1.1 | 16–18 | 7.0 ± 0.6 |
| 3 | 22.5 ± 1.3 | 26–29 | 7.5 ± 0.4 |
| 4 | 29.9 ± 1.8 | 36–40 | 7.5 ± 0.4 |
| 5 | 35.9 ± 1.2 | 46–51 | 7.2 ± 0.2 |
aMeasured average and S.D. from a population of 208 (1 filament/bundle), 34 (2 filaments/bundle), 42 (3 filaments/bundle), 16 (4 filaments/bundle), and 6 (5 filaments/bundle).
b Expected value according to the model derived from the ASCPYD helical tube and assuming filament width values of 6–7 nm.
c Calculated average filament width from bundle width divided by number of filaments.
The cryo-EM structure of human ASCPYD indicates that this domain oligomerizes into a helical tube with an outer diameter of 9 nm (Fig. 4, A and B), a similar result to the width of the filament formed by full-length ASC (Table 1). In principle, a larger diameter for an individual filament of the full-length protein could be expected because of the presence of the CARD (Fig. 4, C and D). However, if the CARD is mobile within the filament as a result of the flexible linker connecting the PYD and CARD in ASC, the TEM images would probably reflect only the PYD tube, and thus ASCPYD and full-length ASC filaments would be similar in size. Nonetheless, the width of the stacked filaments should be larger than the sum of the width of the individual filaments, as the filaments are assumed to bind by CARD–CARD interactions (Fig. 4E). Therefore, the bundle width should reflect the presence of the CARD and linker.
Figure 4.

Stacked filaments of human ASC show dimensions that deviate significantly from the model based on the cryo-EM structure of the truncated protein. Shown are top (A) and lateral (B) views of the human ASCPYD helical tube (cryo-EM, PDB code 3J63) with the dimensions specified in the original published work (8) and containing the 15 ASCPYD domains of the PDB entry. Each group of five ASCPYD domains is colored yellow, red, and blue to show the helical arrangement (A and B). Shown are top (C) and side (D) views of the ASCPYD tube with protruding ASCCARD domains and linker to show CARD availability for potential interactions. The PYDs of 15 full-length NMR structures of ASC molecules (21) (PDB code 2KN6) are superimposed onto the 15 PYD domains of the cryo-EM structure. After superposition, the NMR PYDs were deleted, and only the cryo-EM PYDs and the NMR CARDs are shown. One side of the helical tube is colored red, and the corresponding CARDs are shown in orange to facilitate understanding of directionality. The dimension shown was calculated with MOLMOL (41) (C and D). Shown is a modeled bundle of an arrangement of four ASCPYD helical filaments with protruding CARDs and linker from D. The red side of the PYD tube and corresponding CARDs in orange (from C and D) are shown to follow directionality. CARD domains in filament interfaces are assumed to stack vertically (overlapping as a worst-case scenario), thus occupying approximately the dimension of the ASCCARD (∼3.5 nm). The dimensions shown are as follows: 1) the minimum width of the four-filament bundle, which is estimated by assuming a filament width of 9 nm from the cryo-EM structure and 2) the estimated width range according to the model shown in D for the four-filament bundle (E), which is calculated assuming a filament width of 6–7 nm from our TEM studies.
Considering that ASCCARD has a diameter of ∼3–4 nm (according to the NMR structure), a three-filament bundle that arranges laterally by CARD–CARD interactions, as proposed by the current model, should show a thickness of ∼26–29 nm with a filament width of 6–7 nm (Table 1). Analogously, four-filament (Fig. 4E) and five-filament arrangements should show a total width of ∼36–40 nm and ∼46–51 nm, respectively (Table 1). These expected dimensions, according to the proposed model, are significantly larger than the observed values in our TEM images, and the difference increases with the number of filaments in the bundle (Table 1). The estimated bundle widths are minimum values because we have assumed the CARDs to stack vertically upon filament interaction, occupying only the size of one CARD (Fig. 4E).
The discrepancy between the observed and expected values (Table 1) suggests that there is no room for the CARDs in the structural model for the ASC filament proposed on the basis of the cryo-EM structure of the truncated protein. Therefore, it raises the question of whether the ultrastructure of the filament formed by full-length ASC is different from the reported cryo-EM structure of ASCPYD filaments.
Particles with the size of ASC dimers bind to pre-existing filaments, suggesting a mode for filament lateral growth
In many TEM images of ASC filaments, we observe bulges of protein material protruding from the individual filaments (Fig. 5). This effect is observed both in preparations where the pH is increased, albeit slightly acidic (Fig. 5A), and at neutral or basic pH (Fig. 5B for pH 8.2). The size of these bulges is close to the filament width (∼7 nm). It could be argued that the lateral protein material corresponds to CARD domains protruding from the filament core, as previously suggested on the basis of the cryo-EM structure of the ASCPYD helical tube. However, this is not an adequate explanation, because 1) the size of the bulges does not match that CARD size (∼3–4 nm); 2) the CARDs should populate the whole filament length (Fig. 4, C and D) instead of appearing only at certain positions along the pre-existing filament; and 3) the possibility of observing the CARD by TEM would imply that it is structurally fixed relative to the PYD, although the current model suggests it is flexible.
Figure 5.

Protein particles ∼7 nm in diameter bind to pre-existing filaments, suggesting a mode for filament lateral growth. Shown are TEM images of full-length human ASC formed at pH 6.5 (A) and pH 8.2 (B). The arrows point to protruding protein material.
TEM images of human ASC filaments using a staining protocol that results in higher contrast are shown in Fig. 6. ASC filament bundles predominate over isolated filaments, as observed previously. Filament width (7.0 ± 0.8 nm) is also similar to the values shown in Table 1. Filaments and bundles are slightly larger in this image, albeit still within the error expected from the use of a different staining protocol.
Figure 6.

Filaments of human full-length ASC are formed by the stacking of ring-shaped particles of ∼7 nm in diameter. A, TEM images of ASC filaments formed using method 2 as described under “Experimental procedures.” Rectangles show enlarged areas of A that compose B (bottom rectangle) C (top rectangle). The arrows point to several ring-shaped structures laterally attached to pre-existing filaments. An additional enlargement of the top center region of C forms D. The circle shows stacked rings forming the filaments. Bar length is indicated for each panel.
Nonetheless, bundle width values from Fig. 6 are also close to the sum of the width of the individual filaments composing the bundle (Table 2). Measurements for seven-filament arrangements are significantly lower (∼53 nm) than the expected values according to the PYD-only filament model (73–80 nm), again pointing to a different structural architecture of the filament formed by the full-length protein relative to the truncated PYD-only construct.
Table 2.
Full-length human ASC filament and bundle dimensions (Fig. 6)
| Filament number in bundle | Average bundle widtha | Average filament width |
|---|---|---|
| nm | nm | |
| 1 | 7.0 ± 0.8 | 7.0 ± 0.8 |
| 2 | 15.1 ± 1.2 | 7.6 ± 0.6 |
| 3 | 23.7 ± 0.4 | 8.0 ± 0.1 |
| 4 | 30.5 ± 0.7 | 7.6 ± 0.2 |
| 7 | 52.9 ± 1.7 (73–80)b | 7.6 ± 0.2 |
aAverage and S.D. from a population of 35 (1 filament/bundle), 4 (2 filaments/bundle), 6 (3 filaments/bundle), 2 (4 filaments/bundle), and 3 (7 filaments/bundle).
b Shown in parentheses is the bundle width expected for a 7-filament arrangement according to a model derived from the ASCPYD helical tube and assuming filament width values of 7–8 nm.
The higher-contrast image allows us to discern additional structural characteristics in some bundles that are well-separated (Fig. 6, B and C). Importantly, the protein material protruding from pre-existing filaments is clearly ring-shaped with a diameter of 7.7 ± 0.4 nm (of a population of 19), similar to the filament width (Fig. 6, B and C). These rings appear to form linear stacks that constitute the filaments (Fig. 6D) and also appear bound to pre-existing filaments (Fig. 6, B–D), thus suggesting a mode for lateral growth. Additional micrographs representative of ASC filament bundles and ring-shaped particles attached to preexisting filaments are shown in Fig. S1.
To determine whether the ring-shaped particles are repeating units constituting the filaments, we have analyzed the profile intensities along the long axis of 22 filaments in Fig. 6 to identify maximum and minimum intensity values (Fig. 7). The lowest intensities could correspond to the spacing between monomers and also to the spacing between the holes of the rings (Fig. 6), whereas the maximum intensities will be associated with the presence of protein material, the actual ring. Thus, the profile intensities were averaged to obtain a measurement of the repeating distance between maxima corresponding to 5.0 ± 0.6 nm, which should reflect the size of the repeating unit (Fig. 7). This value is slightly smaller than the measurement of the rings attached to preexisting filaments, which could reflect some compression happening upon filament formation. The bottom of Fig. 6B illustrates this idea, as it shows a ring that appears to be detached from the end of the filament (highlighted with an arrow), which is larger than the other protomers within the filament. In addition, the different ways of measuring particle size, intensity profiles versus distances, can contribute to the deviation of these values.
Figure 7.
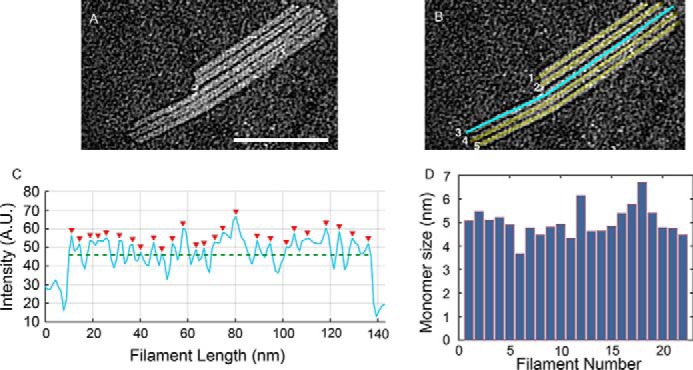
Protomer spacing in bundled human ASC filaments. A, subsection of TEM micrograph of ASC filaments shown in Fig. 6A. Shown are line cuts of 3 pixels, corresponding to 3 nm, using ImageJ along the long axis of each filament in the region of interest. B, as an example, a line cut along filament number 3 is shown in cyan. C, resulting average of intensity profiles over this width. Red arrowheads indicate locations of pixel intensity values above the mean intensity along the filament (green dashed line). A.U., arbitrary units. D, mean values for the separation of maximum peaks from averaged intensity profiles for each of the filaments/line cuts across a total number of 22 filaments. The mean protomer separation is 5.0 nm ± 0.6 nm. Scale bar (A), 100 nm.
The size of the ring-shaped structures agrees with the size of an ASC dimer, according to the NMR solution structure of full-length ASC (21). To obtain an experimental, structural model for the full-length ASC dimer, we have used protein docking protocols based on NMR data (HADDOCK server (26)). The docking procedure uses the solution structure of ASC and, as the main driving force for the interaction, the amino acids identified from NMR titration experiments to participate in the ASCPYD–ASCPYD oligomerization (14) as well as in the ASCCARD–ASCCARD self-association (details of these experiments are described below).
The docking was performed using two ASC structures of the 20 in the NMR ensemble, including the one with the lowest energy. A semiflexible segment was defined during protein docking to allow interdomain mobility from amino acid 90 to 112, which corresponds to the linker region. The 15 dimer structures with lowest energy and largest buried surface area resulting from the docking protocol show homotypic interactions between the CARDs and PYDs of both ASC molecules. The dimensions of the dimer (∼6 nm) calculated from the lowest-energy structures (Fig. 8) agree with the overall shape and size of the rings observed in the TEM images and are also close to the value obtained by profile intensity averaging of filament micrographs (Fig. 7).
Figure 8.
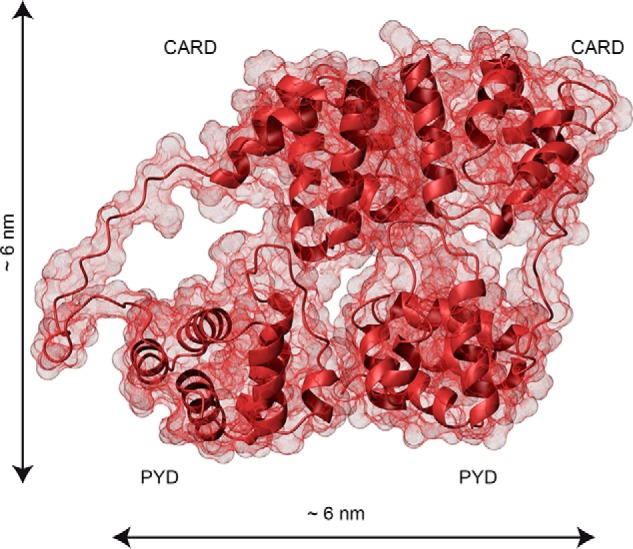
The dimensions of the ASC dimer agree with the size of the ring-shaped TEM particles. Shown is the contact surface of the ASC dimer structure derived from NMR-based protein docking with HADDOCK (26). The PYD and CARD domains are labeled. Dimensions were calculated with MOLMOL (41).
The ASC dimer was proposed earlier as a building block for ASC self-assembly based on cross-linking studies on the pyroptosome (10). These studies agree with our TEM data and the NMR-based docking structure, altogether suggesting a potential role of the ASC dimer in filament formation. All in all, our results indicate that both PYD and CARD in ASC form an integral part of the filament. Therefore, full-length human ASC seems to form filaments and bundles with structural features that significantly deviate from those of the ASCPYD helical tube and ASCCARD bundles.
The different structural arrangement of ASC filaments could have important implications in inflammasome assembly, particularly in relation to the binding of ASC to NLR proteins and procaspase-1. Specifically, ASC forms hollow specks or filamentous rings of micrometer size in active macrophages (19). These rings are bound in a concentric fashion to NLRP3 rings, leaving caspase molecules in the center. Because of the high propensity of ASC filaments to stack laterally and the tendency to cross-link, we speculate whether ASC rings observed in the cytosol of active macrophages could be assembled in a similar way.
Human ASCCARD and ASCPYD form filaments with different structural characteristics
To further understand the role played by the CARD in ASC filament formation, we studied its self-association capabilities by TEM. A slow pH increase from 3 to 7 of a solution of ASCCARD by dialysis results in the formation of highly organized bundles of fibrils or filaments (Fig. 9). In contrast to full-length ASC (Figs. 3 and 6), ASCCARD self-assembles into almost unidirectional bundles that are ∼150 nm wide and ∼0.5–1 μm long, from which narrower bundles branch out (Fig. 9, A and B). A closer look at these bundles shows that ASCCARD assembles into thin fibrils (labeled as Type A in Fig. 9) of ∼3.4 ± 0.5 nm (average from a population of 116 fibrils), which appear to self-organize into thicker structures (labeled as Type B in Fig. 9) of 10.0 ± 0.8 nm in width (average from a population of 84 filaments) (Fig. 9, C–E). The transition from the thin fibril to the thicker one is formed by protein material that appears to be unorganized. The size of the thicker fibrils, which includes some interfibril space, suggests that they could be composed of two thin fibrils.
Figure 9.

ASCCARD self-associates into filaments structurally different from full-length ASC filaments. ASCCARD forms ∼0.5–1-μm-long bundles with preferred directionality (A and B). Narrower bundles branch out from the thicker bundles at certain positions. ASCCARD forms thin filaments or fibrils of ∼4-nm width (labeled as Type A) and wider filaments probably composed of two narrower filaments (labeled as Type B) (C–E).
The different characteristics of the ultrastructures formed by ASCCARD compared with those of full-length ASC show that the individual Death Domains in ASC could oligomerize into different architectures. Thus, it is not surprising that according to our TEM data, ASC filaments are structurally different than those formed by the truncated PYD-only construct used to derive the nanometer-size inflammasome model. In fact, the significant decrease in the branching capacity of ASCCARD compared with ASC also suggests that both domains are necessary for additional growth in different directions; thus, the PYD may also participate in filament ramification.
It is interesting to compare the different characteristics of known CARD ultrastructures with our TEM data for ASCCARD. For example, the cryo-EM structure of the CARD of caspase-1 is a helical tube of approximately four protomers per turn and an outer diameter of 8 nm (22). The cryo-EM study of the CARD of MAVS protein shows a helical tube with an outer diameter of ∼9–10 nm (24). The width of ASCCARD type A fibrils is significantly smaller than the width of these helical filaments. The discrepancy is larger than the expected experimental error and suggests that CARDs of different proteins could assemble into ultrastructures with significantly different features.
ASCCARD filaments have been reported in a previous study using a SUMO protein construct to decrease filament aggregation (25). The dimensions of these filaments are not mentioned in this study. Our protein construct does not include additional proteins and/or domains attached to ASCCARD and thus would better represent the oligomerization process.
We have also studied by TEM the characteristics of filaments formed by ASCPYD under our experimental conditions. We found that ASCPYD tends to form thick filament bundles of varying widths (Fig. S2). Individual filaments composing these bundles are hard to distinguish. This result already suggests a very different polymerization mechanism as compared with full-length ASC, for which the individual filaments within the bundle are clearly distinguishable (Figs. 2, 3, 5, and 6). In addition, ASCPYD filaments do not show additional ultrastructure, as ring-shaped protein material attached to them or composing the filament itself, that is observed for the full-length protein. This seamless appearance of ASCPYD might be a result of the continuity of the helical arrangement of protomers that does not exist in full-length ASC. A reasonably large population of individual filaments for ASCPYD was observed (21 individual filaments) to obtain an average width of 8.0 ± 1.0 nm. This value is very similar to the data reported by the cryo-EM study (9 nm for the outer diameter of the helical tube) (8). Under similar conditions for filament formation, it is clear that ASCCARD, ASCPYD, and full-length ASC polymerize into macrostructures and large assemblies with significantly different characteristics.
Human ASCCARD self-associates predominantly through surfaces involving helices 2, 3, 5, and 6
Our TEM data indicate that CARD–CARD interactions in ASC are involved in filament formation. We have used solution NMR to further study this self-association process at the atomic/amino acid level. ASCCARD has a high tendency to oligomerize and precipitate, forming large bundles of fibrils (Fig. 9) at neutral pH and protein concentrations routinely used in NMR studies (sub-millimolar to millimolar range). Thus, our NMR experiments were performed at a 10 μm concentration of 13C,15N-uniformly labeled protein and pH 3.8. To overcome the low signal/noise ratio resulting from the very low protein concentration, we applied fast acquisition techniques (27) in a high magnetic field (800 MHz). The low pH value contributes to shift the oligomerization equilibrium toward the monomeric species, most likely as a result of the protonation of acidic residues known to be involved in electrostatic interactions characteristic of Death Domain self-association.
A series of 1H-15N SOFAST-HMQC experiments (27) of the uniformly labeled protein were recorded at increasing concentrations of unlabeled ASCCARD during the titration. The presence of a single set of signals during the titration indicates that ASCCARD self-association is fast on the NMR chemical shift time scale (Fig. 10). Thus, the observed chemical shift values represent the chemical shifts of the different species present in solution, averaged by their respective populations.
Figure 10.
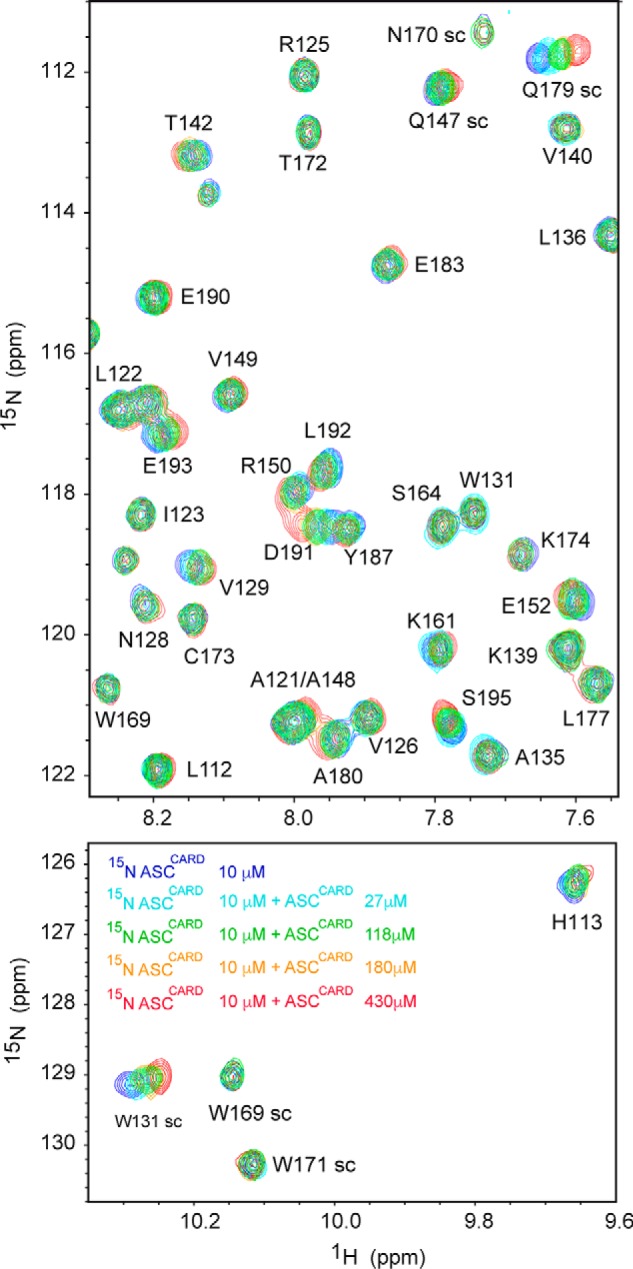
ASCCARD amino acids involved in the CARD–CARD interaction are mainly located in helices 2, 3, 5, and 6. Shown are superimposed SOFAST-HMQC spectra (27) resulting from the titration of 15N,13C-labeled ASCCARD (constant concentration) with unlabeled ASCCARD (increasing concentrations color-coded in the bottom spectrum). Some signals (e.g. NH side chain of Trp-169, amide NH of Arg-125, Thr-142, and Ser-164) remain unmodified, whereas others show clear changes in δ upon binding (e.g. NH side chain of Trp-131 and Gln-179, amide NH of Thr-142, Arg-150, and Asp-191).
The changes in chemical shift upon titration are relatively small (Fig. 10) compared with other Death Domain interactions monitored by NMR techniques, such as the PYD–PYD interaction of ASC (14). This probably indicates that most protein molecules have already oligomerized at the initial stage of the titration, resulting in small perturbations upon the interaction.
Amino acids with chemical shift changes that are equal to or larger than 0.02 ppm and solvent-exposed are mapped in the structure of ASCCARD (Fig. 11). The amino acids involved in ASCCARD self-association are located in the turn preceding helix 1 and mainly in helices 2, 3, 5, and 6. These helices participate in the three interaction types I, II, and III. Three contact regions in the domain surface of ASCCARD can be described from the NMR data: one formed by the N and C terminus of H1 and H6, respectively; a second one composed of the C terminus of H5 and the N terminus of H6; and the third region, which includes helices H2 and H3.
Figure 11.
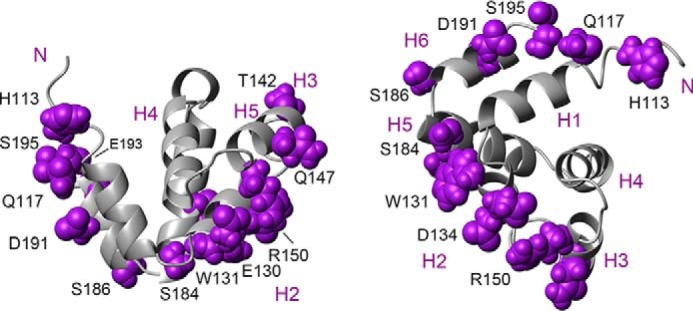
Interacting surfaces in ASCCARD self-association. ASCCARD structure (21) is shown as a ribbon diagram in two different views. The amino acids that are exposed to the solvent and with the largest chemical shift perturbations are depicted in purple and labeled in black. Helices are numbered in purple.
It is possible to question the physiological relevance of the interactions identified by the NMR experiments at low pH. Both hydrophobic and electrostatic interactions play an important role in Death Domain self-association. In particular, we have reported in previous studies that ASCPYD shows a stronger tendency to oligomerize in the presence of NaCl (14), as determined by NMR and analytical ultracentrifugation, indicating that hydrophobic interactions have an important function in Death Domain oligomerization. Acidic pH values are not expected to have an influence in hydrophobic interactions. For example, residues Trp-131 and Thr-142, which show some of the largest chemical shift perturbations in our titration experiments, are probably involved in ASCCARD oligomerization at both low pH and physiological pH. In contrast, acidic pH will result in the protonation of different populations of the side chains of acidic residues, such as Glu and Asp. Specifically, ∼50% of the Asp side chains (pKa = 3.8), and a smaller population of Glu side chains (pKa = 4.2) will be protonated at pH 3.8 (conditions used in the NMR titration of ASCCARD). Thus, still relatively large populations of Asp and Glu side chains will be able to participate in electrostatic interactions at pH 3.8, and these populations can be detected by NMR. Furthermore, Fig. 2A shows that ASC is capable of forming nascent filaments at pH 4.1. This result most likely indicates that the interactions driving filament formation at low pH are also present at physiological pH. With all of these considerations, we suggest that the interactions identified by our NMR titration experiments of ASCCARD self-association at pH 3.8 are relevant at neutral pH.
Interestingly, a recent study by X-ray crystallography of the complex between a double-mutant of ASCCARD (N128A/E130R) and an inhibitor of its self-association points to helix 6, and therefore the type II interaction, as relevant for complex formation (25). In addition, the crystallization of the complex could only be achieved for the monomeric ASCCARD double mutant, which includes a mutation in Glu-130, a residue with one of the largest chemical shift perturbations according to our NMR data. This result indicates that Glu-130 is a critical residue for ASCCARD self-association. Furthermore, an additional mutation, Y187A (located in helix 6), is shown to be involved in the interaction between ASCCARD and the CARD of procaspase-1 (25).
Because there are three main regions involved in the interaction, the chemical shift perturbation data of all of the amino acids shown in Fig. 11 were simultaneously incorporated as experimental restraints in the docking protocol HADDOCK (26) to avoid any bias when creating a model for ASC dimerization. The 15 structures of the ASC dimers with the largest buried surface area that resulted from the docking experiments show homotypic interactions between both Death Domains. However, the interacting regions are not the same in all of those structures. This result can be explained by considering the influence of the different interacting regions in ASC. Nonetheless, the dimensions of the different dimer structures (Fig. 8) are similar and close to the size of the ring-shaped particle structures observed in the TEM images (Fig. 6) as well as close to the size of the repeating unit in the filaments from the profile intensity analysis (Fig. 7).
The self-association of ASCCARD shows the expected hyperbolic dependence, when plotting the changes in chemical shift versus the concentration of the unlabeled protein up to values of 400 μm (Fig. 12). At higher concentrations, there is an additional increase in chemical shift variation, which reveals a more complex process in ASCCARD polymerization. It is possible that higher-order oligomeric species continue growing as more unlabeled protein is added. Excluding the chemical shift perturbation at the highest concentration, the data fit nicely to a rectangular hyperbola (Fig. 12). From this fitting, we have obtained apparent values for the dissociation constant (KD) that range from 6 to 94 μm, with an average value of 50 μm. The self-association of ASCPYD shows similar values (KD(av) = 65 μm), and NMR perturbation data indicate a major involvement of helices 1, 4 and helices 2, 3, pointing to a predominance of the type I interaction (14).
Figure 12.
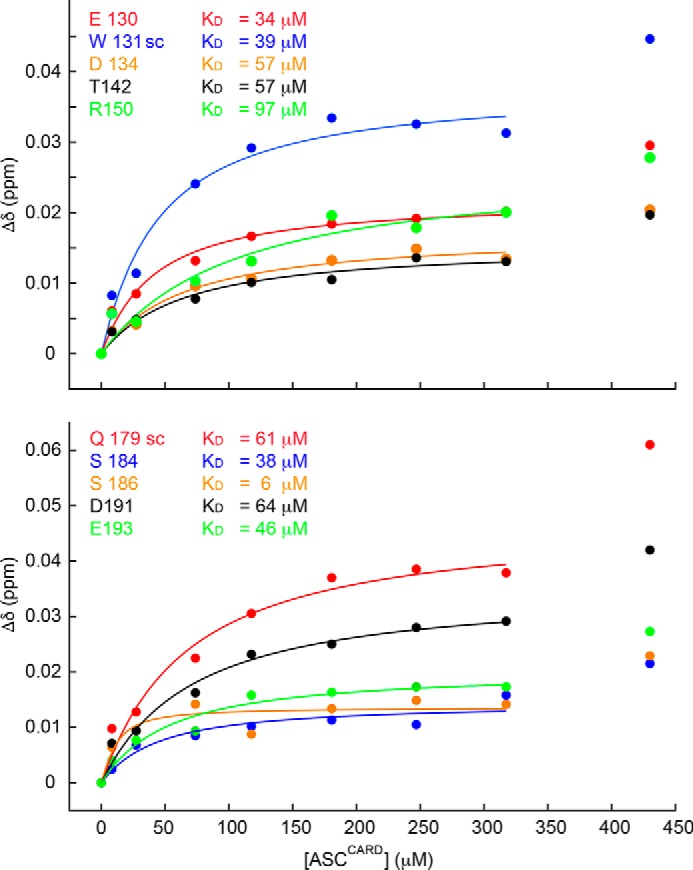
ASCCARD self-associates with KD ∼50 μm. Shown are chemical shift perturbations of 13C,15N-uniformly labeled ASCCARD versus unlabeled ASCCARD concentration. Representative residues exposed to the solvent with the largest perturbations are shown in both the top and bottom panels, together with the corresponding KD values.
To test the validity of the NMR results in relation to the specific amino acids involved in the interaction (Fig. 11), we have designed three ASCCARD mutants and studied their oligomerizing capabilities by TEM. The mutations were selected on the basis of the NMR chemical shift perturbation data (Fig. 12). ASCCARD mutant 1 contains a total of seven mutations to Ala of the 10 residues shown in Fig. 12 (E130A, W131A, D134A, S184A, S186A, D191A, and D193A). This mutant could not be expressed in Escherichia coli, most likely due to protein instability caused by the large number of mutations that could have perturbed the protein structure. For this reason, the second mutant was designed with only three mutations. We specifically selected residue Glu-130 because its mutation to Arg, in addition to the mutation N128A, apparently precluded CARD oligomerization in a previously reported study on the structure of ASCCARD (25). We chose the more conservative mutation E130A and also mutated nearby residues with large chemical shift perturbations. Thus, ASCCARD mutant 2 contains mutations E130A, W131A, and D134A. This mutant was expressed and purified. ASCCARD mutant 2 is capable of forming filaments with very different structural characteristics and arrangements (Fig. S3) as compared with WT ASCCARD (Fig. 9). Micrographs of ASCCARD mutant 2 show short and thin filaments with no apparent superstructural organization, and no specific bundles were observed. This result indicates that the mutations significantly affect the oligomerizing capabilities of ASCCARD, although not to the extent of completely precluding self-association. The fact that ASCCARD mutant 2 oligomerizes, whereas the E130R mutant studied by Schmidt et al. (25) does not, can be explained by the nonconservative mutation of the latter. We designed a third mutant to include nonconservative mutations on negatively charged residues with the largest perturbation data, ASCCARD mutant 3 (E130R, W131A, D134R, D191R, and E193R). Mutant 3 was expressed and purified, and when it was subjected to the conditions for filament formation, we could observe a precipitate as in all other cases. This precipitated material was deposited on grids for extensive TEM analysis. ASCCARD mutant 3 is not capable of forming filaments, and the precipitated protein appears as amorphous aggregates lacking any regular structure (data not shown). The results on the oligomerization capabilities of ASCCARD mutants 2 and 3 support the NMR findings on the importance of specific amino acids in ASCCARD self-association and filament formation.
Discussion
Altogether, our results provide new insight into the structural characteristics and factors involved in ASC-dependent inflammasome assembly. Specifically, we found that ASC filament formation is favored at neutral pH, whereas smaller oligomers are predominant at low pH. It is well-known that inflammatory loci are commonly found in acidic environments (28) and that macrophages create acidic extracellular compartments to help in protein digestion (29) and need acidification for autophagy (30). It is tempting to speculate whether the pH dependence of ASC self-association could play a role in inflammasome assembly and disassembly mediated by pH. Work in this direction that will help to test this hypothesis is still necessary.
In addition, our results indicate that ASC filaments show a high tendency to stack laterally, forming ∼100-nm wide bundles. This association could be a possible mechanism by which micrometer-size inflammasomes assemble in the cytosol of activated macrophages. More importantly, our TEM data show that ASC polymerization into filaments is mediated by both the CARD and the PYD. This result deviates from the current model for ASC filament formation based on the cryo-EM structure of the truncated protein containing only the PYD, which proposes that only ASCPYD participates in the filament core, whereas the CARDs protrude away from it (Figs. 1 and 4 (C and D)).
The concomitant participation of both PYD and CARD in ASC filament formation implies that the filament contains two sides, each occupied with PYDs and CARDs, respectively, and each capable of interacting with the PYDs of oligomerized NLRP3 and the CARDs of procaspase-1. The structural arrangement of this new model, shown in Fig. 13, suggests that the oligomerization of ASC with NLRP3 and procaspase-1 might be more directionally controlled than expected based on the model with protruding CARDs. This additional control in directionality from the filament structural arrangement is in better agreement with the presence of concentric rings of NLRP3 bound to ASC and of oligomerized procaspase-1 in the center, as observed in the cytosol of activated macrophages (19). However, it is important to mention that our experimental conditions for ASC filament and filament bundle formation differ from the physiological conditions necessary for inflammasome activation, as it happens in most biochemical and biophysical studies, because the cell context is absent. Thus, our results obtained in vitro indicate structural propensities of ASC and ASCCARD that might show variations in vivo.
Figure 13.
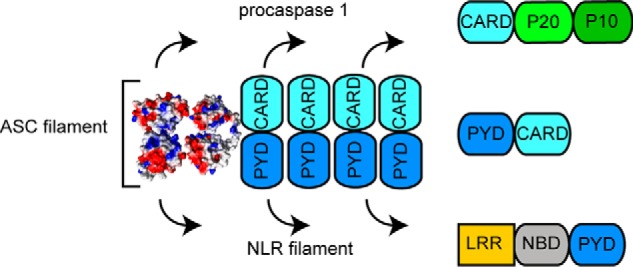
Model for ASC-dependent inflammasome assembly based on TEM data. ASC dimer is shown as the minimal building block. The ASC filament shows two interacting sides, one for recruiting procaspase-1 CARD and the other for interaction with the PYD of NLR sensor proteins.
Our TEM data suggest that the ASC dimer could be the structural building block of the filament and could participate in bundle formation by self-attaching to pre-existing filaments. Dimer formation and simultaneous participation of the CARD and PYD of ASC in filament assembly will result in a decrease of mobility of both Death Domains relative to one another. Interestingly, recent work using FRET to study conformational changes associated with ASC polymerization have found that the PYD and CARD domains of ASC come close to one another upon oligomerization (31). The ASC rings observed in our TEM data agree with a reduced flexibility of the linker, whereas a PYD filament core with protruding CARD implies flexibility in the CARD relative to the PYD facilitated by linker mobility. Moreover, chemical cross-linking experiments of the purified pyroptosome, a caspase-1–activating platform formed by oligomerized ASC, show that the ASC dimer is the minimal oligomerization unit (10).
Additionally, our data on ASCCARD fibril and bundle dimensions provide insight into its oligomerization capabilities, which were previously modeled based on the NMR structure to form ring assemblies (32). Altogether, this information points to a role of ASCCARD in the integral assembly of the ASC filament in addition to its binding to procaspase-1 via recruitment by CARD–CARD interaction. These results also indicate that full-length ASC and truncated versions of ASC (PYD-only and CARD-only) self-assemble into different ultrastructures.
Finally, another indication that Death Domains operate differently at the atomic and molecular levels despite sharing a common fold is the participation of three interacting regions in ASCCARD self-association, compared with a prominent role of the type I interaction for ASCPYD oligomerization (14). Information on the subtle differences in Death Domain interactions is critical to understand inflammasome inhibition and regulation using decoy proteins, such as the Pyrin- or CARD-only proteins POPs and COPs (33, 34), that could be used as pharmacological targets.
Experimental procedures
Protein production and purification
Human ASC (residues 1–195), ASCPYD (residues 1–90), and ASCCARD (residues 111–195) with UniProt accession number Q9ULZ3 were cloned into pET15b expression vectors. The three ASCCARD mutants (mutant 1 (E130A, W131A, D134A, S184A, S186A, D191A, and D193A); mutant 2 (E130A, W131A, and D134A); and mutant 3 (E130R, W131A, D134R, D191R, and E193R)) were also cloned in pET15b vectors. The correct DNA sequences of all constructs were verified by standard DNA-sequencing techniques. Proteins were expressed in BL21(DE3) E. coli strain (Invitrogen). For the unlabeled proteins, cells were grown in LB medium, whereas isotopic labeling was achieved in M9 minimal medium supplemented with 15NH4Cl and [13C]glucose as the only sources of nitrogen and carbon, respectively. Bacteria were grown overnight at 37 °C and diluted the following day to reach an A600 nm of ∼0.8. After 30 min of equilibration at 37 °C, overexpression of the recombinant proteins was induced with 1 mm isopropyl β-d-1-thiogalactopyranoside for 4 h. Pellets for ASC and ASCCARD were dissolved in 20 mm Tris-HCl, 500 mm NaCl, 5 mm imidazole, 6 m guanidine HCl (pH 7.9) (binding buffer). Equivalent buffer, but replacing guanidine HCl with 8 m urea (pH ∼8.0), was used for ASCPYD and ASCCARD mutants. All constructs carried an N-terminal six-histidine tag and were purified by Ni2+-affinity chromatography. The proteins were attached to the resin using the binding buffer and washed in the presence of 20 mm imidazole. Elution was achieved by HPLC gradient in the presence of 500 mm imidazole. Immediately after elution, the proteins were subjected to several dialysis steps against H2O (pH ∼4.0) to slow protein self-association processes. All proteins, except ASCPYD and some ASC samples, were subsequently purified by reverse-phase chromatography using a C4 column (Higgings Analytical) and water/acetonitrile mixtures in the presence of 0.1% TFA. Proteins were lyophilized. After purification, all proteins were >95% pure, as monitored by SDS-PAGE, and the correct molecular weight was obtained from electrospray MS data. Correct protein folding for ASC and ASCCARD was verified by 2D 1H-15N HSQC and other standard NMR experiments.
Protein filament and filament bundle preparation
Protein filaments and bundles for ASC were prepared by three different methods with no significant differences in the observed structural characteristics and dimensions of the resulting ultrastructures. Amorphous protein aggregates in addition to filaments and bundles were also observed using the three methods. In method 1, the lyophilized ASC was dissolved at different concentrations in the micromolar range (3 to ∼50 μm) in HPLC grade water at pH 3.0 and dialyzed against Milli-Q water at different pH values from 4.0 to 8.2 or against buffer containing 150 mm NaCl and 20 mm Tris, pH 8.2. In method 2, the lyophilized ASC was dissolved in 6 m guanidine HCl, 20 mm Tris, pH 8.2, 100 mm NaCl and dialyzed against 20 mm Tris, pH 8.2, and 100 mm NaCl. In method 3, protein eluted from the Ni2+-affinity chromatography was dialyzed against 20 mm Tris, pH 8.2, and 100 mm NaCl. Filaments appeared in the precipitated protein material and in solution for ASC and ASCCARD. Protein filament formation for ASCCARD mutants 2 and 3 and ASCPYD followed methods 1 and 3, respectively.
TEM and TEM sample preparation
A volume of 4 μl of precipitated protein and solution, both containing filaments, was deposited on a carbon-coated copper grid for 10 min. The grid was washed three times in 40-μl droplets of HPLC grade water and stained in 40-μl droplets of 1–2% phosphotungstic acid adjusted at pH 7–7.5. Staining proceeded for 5 min, and the excess staining solution was wiped out with filter paper. Images of the ultrastructures were obtained with a JEOL JEM 2010 transmission electron microscope equipped with LaB6 filament at 200 kV. A Gatan camera of 1350 × 1040 pixels was used. Images were inspected with Digital Micrograph, and filament and bundle dimensions were determined with ImageJ.
For Fig. 6, lacey carbon-coated copper mesh grids were used. The grids were glow-discharged briefly, immediately before adsorbing 10 μl of the protein sample solution for 1 min. Excess solution was wiped out, and the sample in the grid was washed by adding 10 μl of water on the surface. Excess solution was wiped out, and 10 μl of 3% (w/v) uranyl acetate staining solution was added on the surface. Excess staining solution was wiped out, and the grid was left to air-dry.
SEM and SEM sample preparation
Protein samples for SEM studies were prepared as described under “Protein filament and filament bundle preparation,” stained, and deposited on TEM grids as explained under “TEM and TEM sample preparation.” SEM images were acquired with a field emission gun scanning electron microscope (Zeiss Gemini 500) with an electron high tension of 3 kV and a working distance of 3.5 mm.
Filament counting and measurement from TEM images
TEM micrographs were used for filament counting, and measurements were acquired at magnifications ranging from ×400,000 to ×250,000, which were perfectly adequate to ensure differentiation between the filaments and bundles. The size of the analyzed areas ranged from ∼400 × 300 nm to 600 × 500 nm. Each filament was counted only once and measured only once. The number of filaments counted for the measurements is shown at the bottom of Tables 1 and 2. The tables include the average and S.D. of the measurement. Bundles with more than three filaments were less abundant, and thus there is a smaller number of them.
Single filament structure analysis and monomer spacing measurement
TEM micrographs were used to determine the average spacing between adjacent ASC monomers. Raw images of ASC filaments were analyzed in Fiji (ImageJ) to define the regions of interest, spanning different lengths, depending on the filament. Each filament identified in a bundle was averaged over using a 3-pixel-wide line mask. Average intensity profiles along each filament of various lengths (n = 22 filaments) were extracted and imported into Matlab. The “findpeaks” function in Matlab was used with appropriate constraints to determine the average spacing of intensity peaks along each filament profile, which provides a measurement of the average spacing of the protomers within the filaments.
NMR sample preparation
NMR samples used to check correct fold and to obtain resonance assignments were prepared at a concentration of 200 μm by weight of the lyophilized 15N,13C-labeled protein and dissolved in a buffer containing 20 mm glycine-d5, 1 mm tris(2-carboxyethyl)phosphine, and 2.5% D2O at pH 3.8. Samples for titration contained 10 μm 15N,13C-labeled ASCCARD in the same buffer, and the unlabeled ligand protein in lyophilized form was added directly to the protein solution followed by pH adjustment.
NMR spectroscopy and titration experiments
NMR experiments were acquired on a Bruker Avance III HD 800-MHz spectrometer equipped with a triple-resonance cryogenic probe at 298 K. Fast 2D 1H-15N correlation spectra (SOFAST-HMQC) (27) were acquired to monitor chemical shift (δ) changes upon protein binding. Amide 1H-15N chemical shift values for ASCCARD were obtained from previous assignment of full-length ASC (35) and confirmed by 3D 15N-1H NOESY-HSQC (110-ms mixing time), CBCACONH, and HNCACB (36). NMR spectra were processed with NMRPipe (37) and analyzed with SPARKY (38). Chemical shift perturbations (Δδav) were obtained as follows,
| (Eq. 1) |
where subscripts “b” and “f” indicate bound and free forms, respectively. To obtain apparent dissociation constants (KD), Δδav versus ligand protein concentration plots were fit to the equation,
| (Eq. 2) |
where P0 is protein concentration (10 μm) and L is total ligand concentration (39). The ligand concentrations used in the titration experiments were 0, 8, 27, 74, 118, 180, 247, 317, and 430 μm. Data fitting was done with Matlab. Only amino acids with Δδav values larger than 0.02 upon self-association and with a total solvent-exposed area ≥30% as calculated by GETAREA (40) were used to estimate apparent dissociation constants.
Molecular docking
Molecular docking was performed with the program HADDOCK (26) using the full-length ASC solution structure (PDB code 2KN6) (21). Those residues showing Δδav values higher than 0.02 upon ligand binding and with a total solvent exposure index larger than 30% as calculated by GETAREA (40) were defined as active for the interaction. These residues create a list of ambiguous interaction restraints used in the docking calculation. Specifically, the interfaces (active residues) used for the docking calculations were those formed by the α-helices H1 and H4 and the N terminus of H5 (residues Gly-2, Arg-3, Asp-6, Asp-10, Thr-16, Leu-50, Asp-54, Glu-62, Thr-63, Glu-67, and His-90) and by helices H2 and H3 and the C terminus of H5 (residues Glu-19, Lys-22, Lys-24, Leu-25, Ser-29, Val-30, Leu-45, and Asp-75) from ASCPYD (14) and residues His-113, Gln-117, Glu-130, Trp-131, Asp-134, Thr-142, Gln-147 side chain, Arg-150, Gln-179 side chain, Ser-184, Ser-186, Asp-191, Glu-193, and Ser-195 from ASCCARD. Passive residues were selected by the docking protocol around those defined as active. We used the HADDOCK protocol defined as expert interface that allows the backbone and side chain of semiflexible segments to move during refinement. The semiflexible segment was defined from residue 90 to 112 (ASC interdomain linker). The N and C termini were considered charged. The number of starting structures is 1000, out of which 200 are considered for semiflexible refinement. Solvated docking was used with water as solvent for the iterations. The top five clusters were analyzed and inspected with MOLMOL (41). The 15 best dimers showed the experimentally observed type I interaction for the PYD–PYD intermolecular association and the homotypic interaction between the CARDs.
Author contributions
R. J. T. N., S. I. S., and E. d. A. data curation; R. J. T. N., S. I. S., D. A. Q., and E. d. A. formal analysis; R. J. T. N., S. I. S., and E. d. A. visualization; R. J. T. N., S. I. S., D. M. S., and E. d. A. methodology; R. J. T. N., S. I. S., and E. d. A. writing-review and editing; E. d. A. conceptualization; E. d. A. resources; E. d. A. supervision; E. d. A. validation; E. d. A. investigation; E. d. A. writing-original draft; E. d. A. project administration.
Supplementary Material
Acknowledgments
We thank the University of California Merced Imaging Facility and manager, Michael Dunlap, for assistance in TEM image acquisition. We are grateful to Robert Tycko (NIDDK, National Institutes of Health), and Eva Nogales and Patricia Grob (University of California, Berkeley) for advice on TEM sample preparation and preliminary image acquisition and analysis. We thank Mourad Sadqi (University of California, Merced) for MS data of proteins used in this work and Victor Muñoz (University of California, Merced) for helpful discussions. We thank the NMR Facility at the University of California, Santa Cruz, and its Director Jack Lee (National Institutes of Health Grant 1S10OD018455 for funding the University of California, Santa Cruz, 800-MHz NMR spectrometer). We are grateful for the support of the NSF-CREST Center for Cellular and Biomolecular Machines (to D. A. Q. and Mourad Sadqi). We acknowledge University of California, Merced, undergraduate students Andrea Gould and Itati Campos for assisting in protein expression and purification.
This work was supported by start-up funds from the University of California (Merced, CA). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S3.
- NLR
- Nod-like receptor
- 3D
- three-dimensional
- 2D
- two-dimensional
- PDB
- Protein Data Bank
- TEM
- transmission EM
- SEM
- scanning EM
- SUMO
- small ubiquitin-like modifier
- HSQC
- heteronuclear single-quantum coherence
- HMQC
- heteronuclear multiple-quantum coherence.
References
- 1. Park H. H., Logette E., Raunser S., Cuenin S., Walz T., Tschopp J., and Wu H. (2007) Death domain assembly mechanism revealed by crystal structure of the oligomeric PIDDosome core complex. Cell 128, 533–546 10.1016/j.cell.2007.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Park H. H., Lo Y. C., Lin S. C., Wang L., Yang J. K., and Wu H. (2007) The death domain superfamily in intracellular signaling of apoptosis and inflammation. Annu. Rev. Immunol. 25, 561–586 10.1146/annurev.immunol.25.022106.141656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schroder K., and Tschopp J. (2010) The inflammasomes. Cell 140, 821–832 10.1016/j.cell.2010.01.040 [DOI] [PubMed] [Google Scholar]
- 4. Kersse K., Verspurten J., Vanden Berghe T., and Vandenabeele P. (2011) The death-fold superfamily of homotypic interaction motifs. Trends Biochem. Sci. 36, 541–552 10.1016/j.tibs.2011.06.006 [DOI] [PubMed] [Google Scholar]
- 5. Chu L. H., Gangopadhyay A., Dorfleutner A., and Stehlik C. (2015) An updated view on the structure and function of PYRIN domains. Apoptosis 20, 157–173 10.1007/s10495-014-1065-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kwon D., Yoon J. H., Shin S.-Y., Jang T.-H., Kim H.-G., So I., Jeon J.-H., and Park H. H. (2012) A comprehensive manually curated protein–protein interaction database for the Death Domain superfamily. Nucleic Acids Res. 40, D331–D336 10.1093/nar/gkr1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Weber C. H., and Vincenz C. (2001) The death domain superfamily: a tale of two interfaces? Trends Biochem. Sci. 26, 475–481 10.1016/S0968-0004(01)01905-3 [DOI] [PubMed] [Google Scholar]
- 8. Lu A., Magupalli V. G., Ruan J., Yin Q., Atianand M. K., Vos M. R., Schröder G. F., Fitzgerald K. A., Wu H., and Egelman E. H. (2014) Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 156, 1193–1206 10.1016/j.cell.2014.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lu A., and Wu H. (2015) Structural mechanisms of inflammasome assembly. FEBS J. 282, 435–444 10.1111/febs.13133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fernandes-Alnemri T., Wu J., Yu J. W., Datta P., Miller B., Jankowski W., Rosenberg S., Zhang J., and Alnemri E. S. (2007) The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 14, 1590–1604 10.1038/sj.cdd.4402194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wright J. A., and Bryant C. E. (2016) The killer protein Gasdermin D. Cell Death Differ. 23, 1897–1898 10.1038/cdd.2016.100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stehlik C., Lee S. H., Dorfleutner A., Stassinopoulos A., Sagara J., and Reed J. C. (2003) Apoptosis-associated speck-like protein containing a caspase recruitment domain is a regulator of procaspase-1 activation. J. Immunol. 171, 6154–6163 10.4049/jimmunol.171.11.6154 [DOI] [PubMed] [Google Scholar]
- 13. Rathinam V. A., Vanaja S. K., and Fitzgerald K. A. (2012) Regulation of inflammasome signaling. Nat. Immunol. 13, 333–342 10.1038/ni.2237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Oroz J., Barrera-Vilarmau S., Alfonso C., Rivas G., and de Alba E. (2016) ASC pyrin domain self-associates and binds NLRP3 protein using equivalent binding interfaces. J. Biol. Chem. 291, 19487–19501 10.1074/jbc.M116.741082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Faustin B., Lartigue L., Bruey J.-M., Luciano F., Sergienko E., Bailly-Maitre B., Volkmann N., Hanein D., Rouiller I., and Reed J. C. (2007) Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol. Cell 25, 713–724 10.1016/j.molcel.2007.01.032 [DOI] [PubMed] [Google Scholar]
- 16. Halff E. F., Diebolder C. A., Versteeg M., Schouten A., Brondijk T. H., and Huizinga E. G. (2012) Formation and structure of a NAIP5-NLRC4 inflammasome induced by direct interactions with conserved N- and C-terminal regions of flagellin. J. Biol. Chem. 287, 38460–38472 10.1074/jbc.M112.393512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang L., Chen S., Ruan J., Wu J., Tong A. B., Yin Q., Li Y., David L., Lu A., Wang W. L., Marks C., Ouyang Q., Zhang X., Mao Y., and Wu H. (2015) Cryo-EM structure of the activated NAIP2-NLRC4 inflammasome reveals nucleated polymerization. Science 350, 404–409 10.1126/science.aac5789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tenthorey J. L., Haloupek N., López-Blanco J. R., Grob P., Adamson E., Hartenian E., Lind N. A., Bourgeois N. M., Chacón P., Nogales E., and Vance R. E. (2017) The structural basis of flagellin detection by NAIP5: a strategy to limit pathogen immune evasion. Science 358, 888–893 10.1126/science.aao1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Man S. M., Hopkins L. J., Nugent E., Cox S., Glück I. M., Tourlomousis P., and John A., Wright J. A., Cicuta P., Monie T. P., Bryant C. E. (2014). Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc. Natl. Acad. Sci. U.S.A. 111, 7403–7408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sborgi L., Ravotti F., Dandey V. P., Dick M. S., Mazur A., Reckel S., Chami M., Scherer S., Huber M., Böckmann A., Egelman E. H., Stahlberg H., Broz P., Meier B. H., and Hiller S. (2015) Structure and assembly of the mouse ASC inflammasome by combined NMR spectroscopy and cryoelectron microscopy. Proc. Natl. Acad. Sci. U.S.A. 112, 13237–13242 10.1073/pnas.1507579112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. de Alba E. (2009) Structure and interdomain dynamics of apoptosis-associated speck-like protein containing a CARD (ASC). J. Biol. Chem. 284, 32932–32941 10.1074/jbc.M109.024273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lu A., Li Y., Schmidt F. I., Yin Q., Chen S., Fu T.-M., Tong A. B., Ploegh H. L., Mao Y., and Wu H. (2016) Molecular basis of caspase-1 polymerization and its inhibition by a new capping mechanism. Nat. Struct. Mol. Biol. 23, 416–425 10.1038/nsmb.3199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Humke E. W., Shriver S. K., Starovasnik M. A., Wayne J., Fairbrother W. J., and Dixit V. M. (2000) ICEBERG: A novel inhibitor of interleukin-1β generation. Cell 103, 99–111 10.1016/S0092-8674(00)00108-2 [DOI] [PubMed] [Google Scholar]
- 24. Wu B., Peisley A., Tetrault D., Li Z., Egelman E. H., Magor K. E., Walz T., Penczek P. A., and Hur S. (2014) Molecular imprinting as a signal-activation mechanism of the viral RNA sensor RIG-I. Mol. Cell 55, 511–523 10.1016/j.molcel.2014.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schmidt F. I., Lu A., Chen J. W., Ruan J., Tang C., Wu H., and Ploegh H. L. (2016) A single domain antibody fragment that recognizes the adaptor ASC defines the role of ASC domains in inflammasome assembly. J. Exp. Med. 213, 771–790 10.1084/jem.20151790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. van Zundert G. C. P., Rodrigues J. P. G. L. M., Trellet M., Schmitz C., Kastritis P. L., Karaca E., Melquiond A. S. J., van Dijk M., de Vries S. J., Bonvin A. M. J. J. (2016) The HADDOCK2.2 webserver: user-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 428, 720–725 10.1016/j.jmb.2015.09.014 [DOI] [PubMed] [Google Scholar]
- 27. Schanda P., and Brutscher B. (2005) Very fast two-dimensional NMR spectroscopy for real-time investigation of dynamic events in proteins on the time scale of seconds. J. Am. Chem. Soc. 127, 8014–8015 10.1021/ja051306e [DOI] [PubMed] [Google Scholar]
- 28. Lardner A. (2001) The effects of extracellular pH on immune function. J. Leukoc. Biol. 69, 522–530 [PubMed] [Google Scholar]
- 29. Haka A. S., Grosheva I., Chiang E., Buxbaum A. R., Baird B. A., Pierini L. M., and Maxfield F. R. (2009) Macrophages create an acidic extracellular hydrolytic compartment to digest aggregated lipoproteins. Mol. Biol. Cell 20, 4932–4940 10.1091/mbc.e09-07-0559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shi B., Huang Q. Q., Birkett R., Doyle R., Dorfleutner A., Stehlik C., He C., and Pope R. M. (2017) SNAPIN is critical for lysosomal acidification and autophagosome maturation in macrophages. Autophagy 13, 285–301 10.1080/15548627.2016.1261238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gambin Y., Giles N., O'Carroll A., Polinkovsky M. E., Johnston W., Hunter D. J., Alexandrov K., Schroder K., and Sierecki E. (2018) Single-molecule fluorescence reveals the oligomerization and folding steps driving the prion-like behavior of ASC. J. Mol. Biol. 430, 491–508 10.1016/j.jmb.2017.12.013 [DOI] [PubMed] [Google Scholar]
- 32. Santiveri C. M., Oroz J., and de Alba E. (2014) A ring-like model for ASC self-association via the CARD domain. Inflammasome 1, 44–54 [Google Scholar]
- 33. Dorfleutner A., Chu L., and Stehlik C. (2015) Inhibiting the inflammasome: one domain at a time. Immunol. Rev. 265, 205–2016 10.1111/imr.12290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ratsimandresy R. A., Chu L. H., Khare S., de Almeida L., Gangopadhyay A., Indramohan M., Misharin A. V., Greaves D. R., Perlman H., Dorfleutner A., and Stehlik C. (2017) The PYRIN domain-only protein POP2 inhibits inflammasome priming and activation. Nat. Commun. 8, 15556 10.1038/ncomms15556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. de Alba E. (2007) 1H, 15N and 13C backbone and side chain chemical shifts of human ASC (apoptosis-associated speck-like protein containing a CARD domain). Biomol. NMR Assign. 1, 135–137 10.1007/s12104-007-9036-x [DOI] [PubMed] [Google Scholar]
- 36. Bax A., and Grzesiek S. (1993) Methodological advances in NMR. Acc. Chem. Res. 26, 131–138 10.1021/ar00028a001 [DOI] [Google Scholar]
- 37. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 38. Goddard T. D., and Kneller D. G.. SPARKY 3, University of California, San Francisco [Google Scholar]
- 39. Fielding L. (2007) NMR methods for the determination of protein-ligand dissociation constants. Prog. Nucl. Magnet. Reson. Spec. 51, 219–242 10.1016/j.pnmrs.2007.04.001 [DOI] [PubMed] [Google Scholar]
- 40. Fraczkiewicz R., and Braun W. (1998) Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J. Comp. Chem. 19, 319–333 10.1002/(SICI)1096-987X(199802)19:3%3C319::AID-JCC6%3E3.0.CO%3B2-W [DOI] [Google Scholar]
- 41. Koradi R., Billeter M., and Wüthrich K. (1996) MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 14, 51–55, 29–32 10.1016/0263-7855(96)00009-4 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.