

Abstract
Acute pancreatitis is characterized by premature intracellular protease activation and infiltration of inflammatory cells, mainly neutrophil granulocytes and macrophages, into the organ. The lysosomal proteases cathepsin B, D, and L have been identified as regulators of early zymogen activation and thus modulators of the severity of pancreatitis. Cathepsin C (CTSC, syn. dipeptidly-peptidase I) is a widely expressed, exo-cystein-protease involved in the proteolytic processing of various other lysosomal enzymes. We have studied its role in pancreatitis. We used CTSC-deleted mice and their WT littermates in two experimental models of pancreatitis. The mild model involved eight hourly caerulein injections and the severe model partial duct ligation. Isolated pancreatic acini and spleen-derived leukocytes were used for ex vivo experiments. CTSC is expressed in the pancreas and in inflammatory cells. CTSC deletion reduced the severity of pancreatitis (more prominently in the milder model) without directly affecting intra-acinar cell trypsin activation in vitro. The absence of CTSC reduced infiltration of neutrophil granulocytes impaired their capacity for cleaving E-cadherin in adherens junctions between acinar cells and reduced the activity of neutrophil serine proteases polymorphonuclear (neutrophil) elastase, cathepsin G, and proteinase 3, but not neutrophil motility. Macrophage invasion was not dependent on the presence of CTSC. CTSC is a regulator and activator of various lysosomal enzymes such as cathepsin B, D, and L. Its loss mitigates the severity of pancreatitis not by reducing intra-acinar cell zymogen activation but by reducing infiltration of neutrophil granulocytes into the pancreas. In this context one of its key roles is that of an activator of neutrophil elastase.
Keywords: pancreas, cell biology, cadherin-1 (CDH1) (epithelial cadherin) (E-cadherin), neutrophil, leukocyte, acute pancreatitis, cathepsin C (CTSC), dipeptidyl-peptidase I, lysosomal protease, neutrophil elastase
Introduction
Acute pancreatitis is a complex disorder that involves both premature protease activation (1, 2) and recruitment of inflammatory cells into the organ (3). Intracellular activation of the zymogen protease trypsinogen by limited proteolysis is one of the earliest events in acute pancreatitis and mediated by the lysosomal protease cathepsin B (CTSB)3 (4, 5). Other lysosomal proteases, specifically CTSL and CTSD, either enhance or counteract the action of CTSB and thus regulate the onset and severity of pancreatitis in a parallel manner (6, 7). Cathepsin C (CTSC, syn. dipeptidyl-peptidase I, EC 3.4.14.1) is a lysosomal protease that has been detected in a variety of tissues including inflammatory cells (8). It belongs to the papain superfamily, and its molecular structure is that of a tetramer consisting of four identical subunits with approximate molecular masses of 200 kDa (9). Under acidic conditions CTSC acts as a dipeptidase, cleaving at the N terminus of a propeptide (10), whereas at higher pH it adopts other functions such as that of a transferase, and it was even reported to reverse such a reaction (11, 12). CTSC is capable of activating several serine proteases that are constituents of immune cells, among them lymphocytes, mast cells, and neutrophil granulocytes (9, 13, 14). There appears to be a complex interaction of CTSC with other lysosomal hydrolases: autoactivation of CTSC depends on endopeptidases such as cathepsin L and to a lesser extent cathepsin S, whereas cathepsin D degrades both inactive and mature CTSC (15). Similarly, the combination of CTSC and active CTSB appears to process the N terminus of CTSB enzyme, yielding the correct N terminus of mature CTSB (16).
In view of the different interactions of CTSC with other proteases, we have investigated its role in acute pancreatitis using a mouse model with a complete CTSC knockout (CTSC−/−). Our results indicate that CTSC affects the severity of acute experimental pancreatitis, and its action mainly affects inflammatory cells, specifically neutrophil granulocytes, in which it acts as an activator of neutrophil elastase.
Results
CTSC is expressed in the pancreas and does not directly affect zymogen activation in acinar cells
Immunofluorescence stainings and immunoblots showed expression of CTSC in the pancreas. As expected, CTSC−/− pancreata were completely devoid of CTSC. Measurement of CTSC activity using the substrate Gly-Arg-AMC showed activity in WT pancreas homogenates but none in knockout tissue (Fig. 1A). Neither trypsinogen nor amylase content in the pancreas were affected by the deletion of CTSC (Fig. 1B). Stimulation of isolated acini with supramaximal cholecystokinin (CCK) concentrations led to an increase in intracellular CTSC activity (Fig. 1C). To test whether CTSC is a secretory product of acinar cells, we stimulated them with different concentrations of CCK. Although amylase, stored in zymogen granules, showed a peak secretion at a 100 pm CCK, there were no detectable levels of CTSC activity in the supernatant (Fig. 1D). Absence of CTSC from the secretory compartment was confirmed by co-immunolabelings of pancreatic tissue sections with trypsin, a zymogen marker, and barely any co-localization was detected. In contrast, CTSC highly superimposed upon CTSD, suggesting the mainly intralysosomal localization of CTSC (Fig. 1E). Because we observed that total trypsinogen levels were almost identical between CTSC−/− mice and wildtypes, we investigated whether direct activation or degradation of trypsinogen is CTSC-dependent. Co-incubation in vitro of trypsinogen with active CTSC enzyme, even at relatively high concentrations, did not lead to trypsinogen cleavage, whereas enterokinase, a known trypsinogen activator, rapidly resulted in a cleavage product (Fig. 1F). In addition, supramaximally CCK-stimulated acinar cells showed identical activation of the two secretory enzymes trypsin and chymotrypsin in CTSC−/− and control mice (Fig. 1G). Moreover, the extent of cellular necrosis, measured by propidium iodide exclusion, was not altered by CTSC deletion (Fig. 1H). These results indicate that zymogen activation within acinar cells is independent of the presence of CTSC and that acinar cell damage is not affected by CTSC, even though CTSC is expressed in acinar cell lysosomes and undergoes activation after supramaximal stimulation.
Figure 1.

Cathepsin C is expressed in the pancreas, activated in pancreatitis, but not involved in intra-acinar cell protease activation. A, cathepsin C is expressed in pancreatic exocrine tissue but is absent in CTSC−/− mice, as shown by antibody labeling and immunofluorescence staining, Western blotting from pancreas homogenates, and enzyme activity in acinar cells. B, trypsinogen content and amylase content are identical in CTSC−/− mice and controls. C, CTSC is intracellularly activated in isolated acinar cells upon supramaximal CCK stimulation. D, CTSC, unlike amylase, is not secreted from acinar cells. E, CTSC only marginally co-localizes with the zymogen marker trypsin but mainly co-localizes with the lysosomal protein CTSD in pancreatic acinar cells shown by immunofluorescence. F, CTSC, unlike enterokinase, does not directly activate trypsinogen in vitro. G, ex vivo activation of chymotrypsinogen and trypsinogen in CTSC−/− acinar cells upon supramaximal CCK concentrations does not differ from WT acini. H, cell death, measured as propidium iodide exclusion, was not different in CTSC−/− or WT acini. At least five animals were used for each experiment, and all experiments were performed in triplicate. All experiments were performed independently three or more times. The values represent means ± S.E. *, p < 0.05. Scale bar, 50 μm. con, control; DAPI, 4′,6′-diamino-2-phenylindole; RFU, relative fluorescence units; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Absence of CTSC ameliorates severity in a mild experimental model for acute pancreatitis and reduces neutrophil infiltration
To examine whether CTSC affects the course of acute pancreatitis, we first used the caerulein-induced acute pancreatitis model, which mimicks a mild disease course usually followed by complete restitution. At an early time point (1 h) only a slight increase in serum lipase was observed, which was independent of CTSC (Fig. 2A). Local pancreatic injury was mild at 1 h, showing no difference in CTSC−/− mice (Fig. 2B). At 8 h, we found a difference in severity with CTSC-deficient mice having less serum lipase activity and less histological damage. Intrapancreatic trypsinogen activation measured as active, trypsin was increased at 1 and 8 h and in parallel with the reduced pancreatic damage in CTSC−/− mice (Fig. 2C). Chymotrypsin, which is characteristically activated at an early time point of the disease, showed a prominent increase in activity at 1 h followed by a rapid decrease at 8 h. Both time points were independent of CTSC expression (Fig. 2D).
Figure 2.

Reduced severity of mild acute pancreatitis in CTSC−/− mice. A, after 8 h of pancreatitis, a decrease in serum lipase activity was observed in CTSC−/− mice. B, also at 8 h, histologic damage was reduced in CTSC knockout animals. C, trypsin activity was not altered in the initial phase of pancreatitis but was markedly reduced at 8 h in CTSC−/− mice. D, chymotrypsin activity shows a maximum of activity at 1 h and only a little activity at 8 h. No differences between CTSC−/− and wildtypes were observed. E and F, myeloperoxidase activity in lung and pancreatic tissue homogenates was reduced in CTSC−/− mice. G, in parallel with the reduced infiltration of neutrophil granulocytes (Ly6g labeling). H, the number of macrophages in the pancreas, stained by CD68 and F4/80, was not different. Nuclear staining was performed using 4′,6-diamidin-2-phenylindol (DAPI). For each experiment at least seven animals were used, and all experiments were performed in triplicate. The values are means ± S.E. *, p < 0.05. Scale bar, 50 μm.
Along with a reduction in the severity of pancreatitis in CTSC−/− mice at 8 h, we observed a reduced invasion of neutrophil granulocytes into the pancreas and the lungs. Myeloperoxidase (MPO) activity, a specific marker for neutrophils, decreased significantly in the lungs (Fig. 2E) and the pancreas (Fig. 2F) of CTSC knockout animals. In parallel, there was a reduced number of Ly6g-positive cells in the pancreatic tissue, confirming a decreased neutrophil granulocyte infiltration (Fig. 2G). At an earlier time point (1 h) no significant differences between WT and CTSC−/− mice were detected. Macrophages also infiltrated the pancreas at 8 h, shown by CD68 and F4/80 antigen staining, but this infiltration was not affected by the deletion of CTSC (Fig. 2H). Both neutrophils and macrophages are cell populations that have been proposed to play a major role in acute pancreatitis. Apparently CTSC affects the infiltration of different species of inflammatory cells into the pancreas in different ways. Although CTSC deletion decreased neutrophil infiltration in the pancreas, its effect on macrophages remained unaffected. This may explain the considerable remaining pancreatic damage in CTSC−/− mice.
Neutrophil granulocyte infiltration is reduced in a severe experimental model for acute pancreatitis in CTSC−/− mice
To investigate whether depletion of CTSC also affects disease severity in a severe model for acute pancreatitis, we used a partial pancreatic duct ligation. CTSC-deficient mice displayed less trypsin activity (Fig. 3A) in pancreatic homogenates, indicating a reduced zymogen activation. In parallel CTSC−/− mice showed less MPO activity (Fig. 3B) in pancreatic homogenates, and fewer Ly6g-positive neutrophils were recruited into the pancreas (Fig. 3C). The degree of pancreatic injury as measured by serum lipase activity (Fig. 3D) and histological damage (Fig. 3E) was significantly increased after 24 h in both WT and CTSC−/− mice with extensive necrotic areas in the pancreas. However, the severity did not differ between the groups. As for the caerulein model, we could not observe differences in macrophage infiltration into the pancreas, as visualized by F4/80 staining (Fig. 3F). From these results we conclude that the beneficial effect of CTSC deletion is more pronounced in the mild model of pancreatitis and factors unregulated by CTSC drive severity in more severe disease.
Figure 3.

Deletion of CTSC in severe acute pancreatitis. A–C, reduced trypsin activity was found in CTSC−/− mice at 24 h after pancreatic duct ligation (A), in parallel with a reduced pancreatic MPO activity (B), and a lower number of infiltrating neutrophil granulocytes (C) into the pancreas (Ly6g immunostaining). D and E, conversely, serum lipase (D) and total histological damage (E) were not different between CTSC−/− mice and WT controls. F, macrophage infiltration into the pancreas was independent of CTSC. At least seven animals were used for each experiment, and all experiments were performed in triplicate. The values are means ± S.E. *, p < 0.05. Scale bar, 50 μm.
CTSC regulates neutrophil elastase activation and E-cadherin cleavage in acute pancreatitis
The above data suggest that neutrophil granulocyte, but not macrophage infiltration, is altered in the absence of CTSC. Therefore CTSC−/− neutrophils were investigated in more detail. We could confirm a strong CTSC expression in neutrophil granulocytes by co-labeling with the neutrophil marker Ly6g (Fig. 4A) and a high enzymatic activity in homogenates of isolated neutrophils (Fig. 4B), which was absent in the CTSC−/− animals. Moreover, activity of the neutrophil serine protease neutrophil elastase (NE) was markedly reduced by around 50% in the knockout granulocytes (Fig. 4C). The other neutrophil serine proteases cathepsin G (CTSG) and proteinase 3 (PR3) were strongly reduced as well. When we added purified active CTSC to homogenates of isolated CTSC−/− neutrophils, we observed an increase in NE activity that did not occur when other proteolytic enzymes such as CTSB or trypsin were added, confirming CTSC to be a specific activator of NE (Fig. 4D). Because in both models infiltration of CTSC−/− neutrophils into the pancreas was significantly impaired, we studied whether neutrophil migration and motility measured in a modified Boyden chamber assay differed between CTSC+/+ and CTSC−/− neutrophils. Irrespective of the medium conditions that were used for the assay (Fig. 4E), we found no significant difference between the groups. On the other hand, in both models cleavage of the cell–cell contact molecule E-cadherin (120 kDa) was impaired in the absence of CTSC as shown by densitometry of the cleaved E-cadherin band at 105 kDa in pancreatic homogenates (Fig. 4F). These data suggest that the impaired tissue infiltration of CTSC−/− neutrophils is caused by reduced E-cadherin cleavage at pancreatic adherens junctions rather than by a reduced motility of neutrophils. NE expression was confirmed in neutrophils invading the pancreas, whereas tissue-infiltrating macrophages contained hardly any NE (Fig. 4G). The conclusion from these data is that maturation of neutrophil serine proteases, among them NE, is impaired in absence of CTSC. Loss of CTSC has a beneficial effect on severity of acute pancreatitis because of a reduced infiltration of inflammatory cells.
Figure 4.

NE activity and E-cadherin cleavage are dependent on CTSC. A and B, CTSC is expressed in pancreatic neutrophils shown by immunofluorescence of tissue sections and enzyme activity in isolated neutrophils. Specificity of CTSC signal was confirmed by using CTSC knockouts. Neutrophils were identified using the neutrophil marker Ly6g. C, in isolated CTSC−/− neutrophils activity of the azurophilic granule serine proteases neutrophil elastase, cathepsin G, and proteinase 3 were markedly reduced. Because NE also cleaves PR3 substrate, the samples were pretreated with NE inhibitor (elastase inhibitor II, 50 μg). Relative enzyme activity was determined in 105 isolated neutrophils. D, addition of active CTSC enzyme (1.5 units/ml) to isolated neutrophils from CTSC−/− mice increases neutrophil elastase activity (results shown as fold increase) indicating cross-activation. E, chemotaxis of isolated neutrophil granulocytes was similar in CTSC−/− neutrophils and WT cells. F, impaired cleavage of E-cadherin in CTSC−/− mice during acute pancreatitis (caerulein and ligation model) shown by immunoblotting. Equal protein loading was confirmed by reprobing with GAPDH antibody. Protein load was 50 μg. G, NE was expressed in neutrophils (Ly6g) but not in macrophages (CD68). At least five animals were used for each experiment, and all experiments were performed in triplicate. All experiments were done independently three or more times. The values are means ± S.E. *, p < 0.05. Scale bar, 20 μm. Con, control; DAPI, 4′,6′-diamino-2-phenylindole.
Neutrophil elastase cleaves E-cadherin and increases the biovolume of acini
The previous data from two experimental mouse models for acute pancreatitis suggest that cleavage of E-cadherin is mediated by neutrophil elastase and absence of CTSC reduced NE activity and decreased E-cadherin cleavage. Direct effects of NE on E-cadherin cleavage could be shown ex vivo in isolated acinar cells and acini. The addition of recombinant NE to acinar cells led to a cleavage that was comparable with the effects of supramaximal CCK stimulation, and such an effect was not found for cathepsin G and proteinase 3, two other main components of azurophilic granules. Band intensity of cleaved E-cadherin was quantified by densitometry (Fig. 5A). Measurement of the biovolume of living acini was used to assess the dissociation of cell contacts. Neutrophil elastase increased the amount of single acinar cells and simultaneously decreased the number of multicell acini as comparable with cholecystokinin. The effects of CTSG and PR3 on biovolume were less distinct (Fig. 5B).
Figure 5.
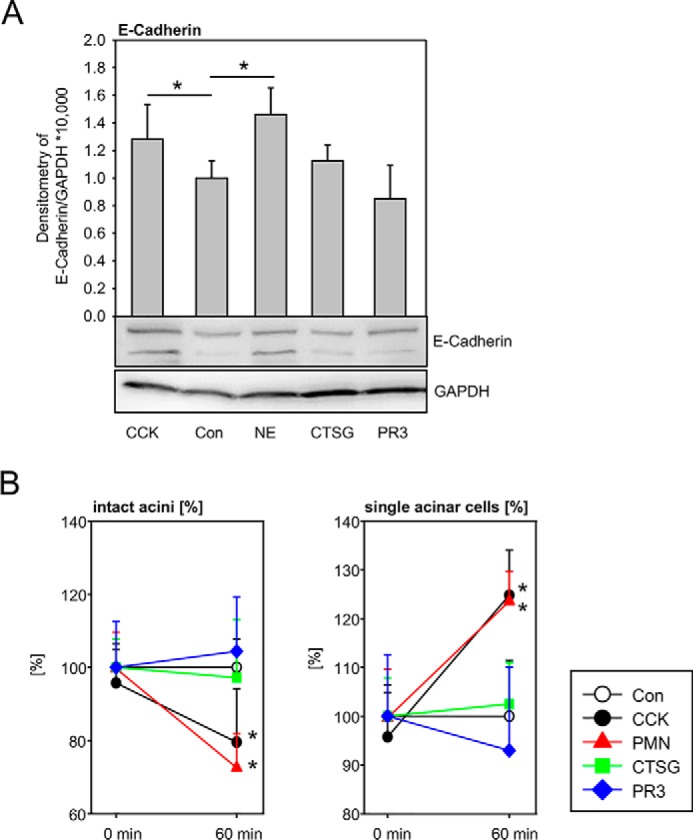
E-cadherin cleavage and acinar cell dissociation are dependent on neutrophil serine proteinases. A, addition of NE, CTSG, and PR-3 to isolated acinar cells induced cleavage of E-cadherin, which was most prominent when using NE. Treatment with cholecystokinin served as a positive control. B, neutrophil elastase decreased the number of intact acini, which was comparable with the effects of CCK. The effects were absent with CTSG and PR-3. Simultaneously the number of isolated acinar cells was increased by number by NE. CTSG and PR3 had no such effect. Average single cell size was calculated between the lower (11 mm) and upper (23 mm) diameter cutoffs. Particles with diameters below 11 mm are cell debris; particles above 23 mm are cell aggregates of three or more cells (acini). Con, control.
Discussion
Acute pancreatitis has primarily been regarded as an autodigestive disease, triggered by activation of acinar cell digestive proteases (17, 18). The clinical course is variable, ranging from a mild form with complete restitution up to a severe variety with many complications and a high mortality. Presently no reliable markers exist to predict the disease outcome at the time of the patients hospital admission (19, 20).
Inflammatory cells have also been identified to be essential for the pathogenesis of acute pancreatitis. When the capability of inflammatory cells to transmigrate into the pancreas is reduced, i.e. by depletion of CD18, a subunit of the MAC-1 complex, a reduction in the severity of experimental pancreatitis and a decrease of intrapancreatic protease activation occur (21). In acute pancreatitis neutrophil granulocytes and macrophages are the main leukocyte subpopulations undergoing transmigration into the pancreas. Depletion of neutrophils resulted in an attenuation of zymogen activation and pancreatic damage. This effect seems to be regulated by release of reactive oxygen species by NADPH oxidase activity in neutrophils (22) and drives a tumor necrosis factor-α–dependent activation of digestive proteases in acinar cells (21). Recently the importance of macrophages for the initiation and perpetuation of pancreatitis was highlighted and is based on two independent mechanisms. First, macrophages directly stimulate acini via tumor necrosis factor-α, not unlike neutrophils. They also intracellularly activate trypsinogen after endocytosis of zymogen-containing vesicles resulting in activation of macrophages and the release of cytokines (23). Here we investigated the effect of a lysosomal aminopeptidase, CTSC, that is highly expressed in inflammatory cells, e.g. neutrophil granulocytes, macrophages, and cytotoxic lymphocytes (8, 24, 25). We could demonstrate that CTSC is also expressed in pancreatic acinar cells, and this expression was predominantly found in their lysosomal compartment. This was shown by co-immunolabeling with cathepsin D, a well-characterized lysosomal protease, and did not involve the secretory compartment. In addition, intracellular CTSC activity rapidly increased after supramaximal CCK stimulation of acini in a manner that was similar to that of the lysosomal protease CTSB or digestive zymogens in early experimental pancreatitis (2). These observations led us to assume that CTSC, like CTSB (4), CTSL (6), and CTSD (7) may also affect intracellular zymogen activation and acinar cell injury. Surprisingly, no differences in intracellular digestive protease activation or cellular necrosis were found when we compared isolated acini from CTSC−/− animals with those of WT controls, indicating that CTSC is not involved in intra-acinar cell processes during pancreatitis. These observations are in line with results from the caerulein pancreatitis model, in which no significant differences in pancreatic injury were detected at an early time point (1 h). However, differences in disease severity became apparent at later time points (8 h), and CTSC−/− mice showed not only a milder course of pancreatitis but also less neutrophil granulocyte infiltration into the pancreas and the lungs. In contrast, macrophage infiltration into the pancreas was not altered and therefore appears independent of the presence of CTSC. This differential infiltration of neutrophil granulocytes and macrophages in the absence of CTSC was confirmed in a second, more severe model for acute pancreatitis. In this model pancreatic necrosis was prominent after 24 h, but similar in CTSC knockout animals and controls, despite a reduced neutrophil infiltration and trypsin activation. This suggests that the severe damage in this model is independent of CTSC, and other factors overwhelm the beneficial effect of impaired neutrophil infiltration or trypsinogen activation. The action of macrophages, which remain unaffected by the deletion of CTSC, has been reported to play a predominant role in this model (23, 26).
The differences in neutrophil invasion prompted a closer characterization of neutrophil granulocytes. Neutrophil elastase (NE) is a major component of azurophilic granules of neutrophil granulocytes, and its activity was dramatically decreased in CTSC−/− neutrophils. Conversely, addition of purified CTSC increased NE activity in neutrophil homogenates, indicating a rapid conversion from pro-NE to active NE. This neutrophil serine protease is known to modulate the severity in several other inflammatory diseases such as glomerulonephritis or arthritis. In addition, an attenuated influx of inflammatory cells into these inflamed organs upon depletion of CTSC was demonstrated (27–29). Activities of CTSG and PR3 were also strongly reduced in CTSC−/− neutrophils, indicating that CTSC is involved in activation of these neutrophil serine proteinases as well. NE and other leukocyte serine proteases such as proteinase 3 and cathepsin G share a similar molecular structure, and their activation is based on identical steps that include removal of a prodipeptide between the proenzyme and the mature enzyme (28, 30, 31).
Although we could not detect a reduced influx of macrophages in our experimental models, others have reported a CTSC-related effect on macrophages. One potential reason for this difference is that in the glomeronephritis model, inflammatory cytokine production, in particular IL-1β, was concomitantly decreased, causing less attraction of macrophages (27). Measurements of proinflammatory cytokines in our pancreatitis models showed similar levels between WT and CTSC knockout animals. Second, the release of cytokines from stimulated CTSC−/− neutrophils in ex vivo experiments was not altered (data not shown), which may permit a sustained macrophage stimulation and thus leave their infiltration into the pancreas unimpaired. Moreover, CTSC expression is much lower in macrophages than in neutrophils, which suggests that this lysosomal enzyme has no great impact on the biology of macrophages. Because macrophage infiltration was largely responsible for pancreatic damage during acute pancreatitis in a recent study (23), the presence of macrophages can explain why pancreatic damage still occurs in CTSC−/− mice during mild and severe experimental pancreatitis.
Depletion of CTSC does not result in an impairment of neutrophil chemotaxis shown by ex vivo experiments even under different conditions. Adkison et al. (28) have also described normal ex vivo migration of CTSC−/− neutrophils and an unaffected neutrophil penetration into the peritoneal cavity following nonspecific inflammation. In the pancreas the conditions are different. To transmigrate from blood vessels into the pancreas, granulocytes need to dissociate adherens junction (32), and they do so by secreting active NE to degrade E-cadherin bridges between exocrine cells (33). We could show that this is the mechanism operative in pancreatitis, that it depends on NE activation by CTSC, and thus that it is the milder disease course in the absence of CTSC. Other neutrophil proteases like cathepsin G and proteinase 3 may also cleave E-cadherin, so that the impaired transmigration of neutrophils into the pancreas of CTSC−/− mice is most likely a combined effect of several serine proteinases, although we could not demonstrate such an effect of CTSG and PR3 in the pancreas. Moreover, both NE and CTSG are known to cleave vascular endothelium cadherin, which further promotes neutrophil transmigration (34). Macrophages, which do not produce significant amounts of elastase, use alternative mechanisms for pancreatitis tissue infiltration and are therefore not affected by CTSC deletion.
In summary, we identified CTSC as a lysosomal enzyme that is expressed in the pancreas, affects the severity of pancreatitis mainly by altering the infiltration of neutrophil granulocytes (but not macrophages), but plays no direct role in the intra-acinar activation of digestive zymogens. Absence of CTSC does not directly alter the motility of neutrophils but is essential for activation of NE and other neutrophil proteases, which, in turn, cleave E-cadherin, a critical component of adherens junctions between epithelial cells and a defensive barrier against infiltrating granulocytes.
Experimental procedures
Reagents, substrates, and antibodies
Collagenase from Clostridium histolyticum (EC.3.4.24.3) was purchased from SERVA (lot no. 14007, Heidelberg, Germany) and used for acinar cell isolation. Trypsinogen from bovine pancreas, enterokinase from porcine intestine, cathepsin B enzyme from bovine spleen, and MPO enzyme from human polymorphonuclear leukocytes, proteinase 3, caerulein, and CCK were obtained from Sigma. Recombinant enzymes for neutrophil elastase, and cathepsin G were purchased from Merck. Ketamine and xylazine were from Selectavet (Weyarn-Holzolling, Germany). Amylase and lipase were quantified using a kit from Roche–Hitachi (Grenzach-Wyhlen, Germany). The following antibodies were used: anti-Ly6g (ab25377) from Abcam (Cambridge, UK)l anti-CD68 (ABIN181836) from Antibody-Online (Aachen, Germany)l anti-F4/80 (MCA497R) from AbD Serotec (Raleigh, NC)l anti-cathepsin C (sc-74590), anti-cathepsin D (sc-6486), and neutrophil elastase (sc-55548) from Santa Cruz Biotechnology (Dallas, TX); anti-glyceraldehyde-3-phosphate dehydrogenase (clone 6C5) from Meridian (Memphis, TN)l antitrypsin (AB1823) from Chemicon International (Temecula, CA)l and anti-E-cadherin (clone 36, catalog no. 20820) from Transduction Laboratories (San Diego, CA). Protease activity was measured by adding the following substrates: for cathepsin C AMC-Gly-Phe-CHN2 from MP Biomedicals (Eschwege, Germany); for neutrophil elastase methyl-O-succinyl-Ala-Ala-Pro-Val-AMC from EMD Chemicals (Gibbstown, NJ); for cathepsin G Suc-AAPF-pNA and proteinase 3 MeOSuc-AAPV-pNA from Enzo Life Sciences (Farmingdale, NY); for trypsin R110-(CBZ-Ile-Pro-Arg)2 from Invitrogen; and for chymotrypsin Suc-Ala-Ala-Pro-Phe-AMC from Bachem (Bubendorf, Switzerland).
Induction of acute pancreatitis in mice
All animal experiments were performed according to state institutional guidelines and animal facility protocols after prior approval by the institutional animal care committee (Landesamt für Landwirtschaft, Lebensmittelsicherheit und Fischerei, Mecklenburg-Vorpommern). WT C67BL/6 mice were obtained from Charles River Laboratories (Sulzfeld, Germany). CTSC−/− mice were kindly provided by Dr. Christine Pham (Washington University School of Medicine, St. Louis, MO) (25). A mild form of pancreatitis was induced by hourly intraperitoneal injections of caerulein (50 μg/kg/body weight) for up to 8 h (32). Severe acute pancreatitis was induced by partial ligation of the pancreatic duct as previously established (23). Tissue was harvested immediately after sacrificing the animals. Pancreas and lung samples were frozen in liquid nitrogen and stored at −80 °C for later analysis. For histology, tissue was fixed in 4.5% formalin for paraffin embedding or embedded in TissueTec® O.C.T.TM compound (Sakura Finetek, Alphen aan den Rijn, The Netherlands) for cryo sections. Serum samples were collected and stored at −20 °C.
Isolation of acinar cells
Acinar cells were prepared by collagenase digestion as previously described (35). Cells were maintained and stimulated in Dulbecco's modified Eagle's medium containing 10 mm HEPES and 2% BSA with 1 μm CCK. Protease activity was monitored for up to 30 min after CCK stimulation (36). Untreated cells served as controls. In vivo measurement of protease activation in living acinar cells was performed in cell medium (pH 7.4) containing 24.5 mm HEPES, 96 mm NaCl, 11.5 mm glucose, 6 mm KCl, 1 mm MgCl2·6H2O, 0.5 mm CaCl2·2H2O, 2.5 mm NaH2PO4·H2O, 5 mm sodium fumarate, 5 mm sodium glutamate, 5 mm sodium pyruvate, 1% BSA, and DMEM as described previously (37). Trypsin activity was measured using 10 μm R110-IPR and CTSB using 20 μm AMC-Arg2 as substrate. Necrosis was determined by measurement of propidium iodide incorporation in acinar cells (38). For analysis of E-cadherin cleavage immunoblots from isolated acinar cells were generated, which were incubated with CCK, NE (2 μg/ml), CTSG (5 milliunits/ml), or PR3 (2 μg/ml). All measurements were done in triplicate.
Isolation of neutrophils
Leukocytes were isolated from murine spleens of CTSC−/− and WT mice. The resulting cell suspension was passed over a 70-μm sterile nylon filter (BD Falcon, Amsterdam, The Netherlands) to remove cell aggregates and debris (39). After lysis of erythrocytes with 155 mm NH4Cl, 10 mm KHCO3, and 0.1 mm EDTA, the cells were counted and transferred to sterile PBS. Neutrophils were isolated from total leukocytes using MACS® cell separation kit according to the manufacturer's instructions (Miltenyi Biotech, Bergisch-Gladbach, Germany) by using Ly6g as a specific antibody.
Cell migration
Real-time cell analysis of migration was performed using the xCELLigence DP device (Roche Diagnostics) according to the manufacturer's instructions. Briefly, cells were added to the upper chamber of a two-chamber device separated by a porous membrane to allow migration directly through the pores to the bottom side of the membrane. This leads to an increase in electrical impedance and is displayed as a dimensionless parameter termed cell index. The cell index represents the capacity of cell migration, and the slope of the curve is related to the migration of cells. 50,000 cells/well, suspended in culture medium without FCS, were then seeded in the upper chamber. The lower wells were filled with or without 3% FCS. The cell indices were measured with the real-time cell analysis software (version 1.2, Roche Diagnostics).
Assessment of acinar cell contact dissociation
For biovolume experiments, the biovolume ratio was determined using the CASY I cell analyzing system as described previously (33, 40). Briefly, acini were isolated by collagenase digestion and incubated with neutrophil elastase (2 μg/ml), cathepsin G (5 milliunits/ml), proteinase-3 (2 μg/ml), or buffer alone. Measurements under different experimental conditions are shown for single cells (11–23 μm) and intact acini (24–80 μm). The results are expressed as percentages of control incubations with buffer alone. The biovolume percentages are representative of four experiments in each group. The data show means (S.D.) for triplicate measurements.
Biochemical assays
Serum lipase and amylase activities were measured by photometric assays (Roche–Hitachi) as kinetic over 30 min at 37 °C. Purified enzymes served as controls (Sigma). Protease activity was determined from either whole pancreatic tissue, acinar cell, or neutrophil homogenates. Trypsin, chymotrypsin, neutrophil elastase, and proteinase 3 activity were measured at 37 °C in a buffer containing 100 mm Tris, 5 mm CaCl2, pH 8.0, by adding 10 μm of R110-Ile-Pro-Arg, Suc-Ala-Ala-Pro-Phe-AMC, R110-Ala4, and MeOSuc-AAPV-pNA substrates. Because neutrophil elastase also cleaves the PR3 substrate, the samples were pretreated with 50 μg of NE inhibitor (elastase inhibitor II, Calbiochem, CA) to eliminate NE activity. Cathepsin G activity was measured in a buffer 100 mm HEPES, pH 7.5, using the substrate Suc-AAPF-pNA. Cathepsin C activity was measured in 50 mm tri-sodium citrate-dihydrate at pH 6.0 containing 2 mm DTT and 10 μm Gly-Phe-CHN2 substrate. Trypsinogen content was measured as trypsin activity after preincubation with enterokinase (20 milliunits) over a period of 30 min. Protease activity was corrected for protein content using the MicroBCA kit (Pierce).
MPO activity measurement was performed as previously described (41). Briefly pancreatic tissue was homogenized on ice in 20 mm potassium phosphate buffer (pH 7.4) and centrifuged at 20,000 × g at 4 °C for 10 min. The pellet was resuspended in 50 mm potassium phosphate buffer (pH 6.0) containing 0.5% cetyltrimethylammonium bromide. The suspension was frozen and thawed in cycles, sonicated twice, and centrifuged at 20,000 × g at 4 °C for 10 min. MPO activity was assayed in 50 mm potassium phosphate buffer (pH 6.0) containing 0.53 mm O-dianisidine and 0.15 mm H2O2. The initial increase in absorbance was measured at room temperature with a Spectramax Spectrophotometer (Molecular Devices, Sunnyvale, CA). The results were expressed in units of MPO activity on the basis of 1 unit being able to oxidize 1 μmol H2O2/min/mg pancreatic protein (35).
Isolated neutrophils were homogenized, lysed, and centrifuged. Supernatant was harvested, and protein content was determined using the MicroBCA kit (Pierce). For different experimental settings active enzyme for CTSB (10 μm), trypsin (10 μm), or CTSC (10 μm) was added and incubated for 30 min at 37 °C, and the activity of neutrophil elastase was determined by fluorometry.
For in vitro proteolytic cleavage of trypsinogen either enterokinase (10 milliunits) or cathepsin C enzyme (10 milliunits) was added to 40 μg/ml bovine trypsinogen. Aliquots were removed at 0, 1, and 3 h. Reaction was stopped by immediately adding loading buffer containing 10% β-mercaptoethanol, boiling at 96 °C for 5 min, and subsequently freezing to −20 °C.
Histology and immunofluorescence
Paraffin sections were used for hematoxylin and eosin staining. For assessment of damage, a modified score adapted from Niederau et al. was used (42). The scoring was based on the extent of necrosis, vacuolization of acinar cells, and invasion of inflammatory cells into the pancreas. A minimum of five visual fields from each animal was investigated. Immunofluorescence staining of F4/80 was performed in paraffin sections. For antigen retrieval, a target retrieval solution from DAKO (Carpinteria, CA) was used. Antibody was used in a dilution of 1:50 in 20% fetal calf serum (FCS) from PAN Biotech (Aidenbach, Germany). Immunofluorescence stainings of Ly6g, CD68, CTSC, and co-immunostainings of CTSC and CTSD were performed in cryo-embedded tissues. Therefore 1–2-μm-thick slides were fixed in acetone at −20 °C for 30 min. 20% FCS was used for blocking. Primary antibodies were used in a dilution of 1:20 to 1:100 in 20% FCS, and incubation was performed over night at 4 °C.
Scoring of infiltrating cells were performed by counting numbers of cells per visual field. A minimum of 10 visual fields for each animal were quantified. Secondary antibody incubation in cryo-embedded, and paraffin sections were performed for 1 h at room temperature. The nuclei were stained by DAPI, and the slides were mounted with DACO mounting medium (Agilent Technologies Inc., Santa Clara, CA) for immunofluorescence.
Western blotting
The samples were homogenized and lysed in buffer containing 25 mm HEPES, 75 mm NaCl, 0.5% Triton X-100, 5% glycerin, 1 mm EDTA in the presence of 1 mm phenylmethylsulfonyl fluoride, 5 mm Na4P2O7, 10 mm NaF, and 1 μg/ml aprotinin. Protein concentrations were determined by MicroBCA kit (Pierce). Equal amounts of proteins were loaded on polyacrylamide gel and transferred onto nitrocellulose membrane for immunoblots as described previously (21, 37).
Statistical analysis
All data are expressed as means ± S.E. from at least five animals per experiment in each group. Statistical analyses were performed by SigmaPlot (Systat Software Inc., Erkrath, Germany) and SigmaStat (Systat Software Inc.) using the unpaired two-tailored Student's t test or analysis of variances for samples without normality. Differences were considered significant for p < 0.05.
Author contributions
D. S. J., J. A., M. S., F. U. W., and A.A.A. data curation; D. S. J., J. A., and M. S. formal analysis; D. S. J., J. A., and B. K. validation; D. S. J., J. A., and A. A. A. investigation; D. S. J., B. K., and A. A. A. methodology; D. S. J., J. A., B. K., M. S., F. U. W., J. M., M. M. L., and A. A. A. writing-review and editing; M. S., F. U. W., M. M. L., and A. A. A. supervision; F. U. W., J. M., M. M. L., and A. A. A. project administration; J. M., M. M. L., and A. A. A. conceptualization; A. A. A. writing-original draft.
Acknowledgments
We thank Christine T. N. Pham (Washington University School of Medicine, St. Louis, MO) for providing CTSC−/− mice; Kathrin Gladrow and Norina Loth for technical assistance; and Walter Halangk and Thomas Wartmann (both of the University of Magdeburg) for helpful criticism and discussions.
This work was supported by Deutsche Forschungsgemeinschaft Grant AG 203/2-1 (to A. A. A.) with additional support from Forschungsgemeinschaft Grant GRK1947(A3) and PePPP (Protein misfolding, ER-stress and protein degradation) Project Grant ESF/14-BM-A55-0045/16 funded by the European Union and the state of Mecklenburg–Western Pomerania. The authors declare that they have no conflicts of interest with the contents of this article.
- CTSB
- cathepsin B
- CTSC
- cathepsin C
- CTSD
- cathepsin D
- CTSG
- cathepsin G
- CTSL
- cathepsin L
- AMC
- 7-amino-4-methylcoumarin
- CCK
- cholecystokinin
- MPO
- myeloperoxidase
- NE
- neutrophil elastase
- PR3
- proteinase 3
- FCS
- fetal calf serum.
References
- 1. Pandol S. J., Saluja A. K., Imrie C. W., and Banks P. A. (2007) Acute pancreatitis: bench to the bedside. Gastroenterology 132, 1127–1151 10.1053/j.gastro.2007.01.055 [DOI] [PubMed] [Google Scholar]
- 2. Hofbauer B., Saluja A. K., Lerch M. M., Bhagat L., Bhatia M., Lee H. S., Frossard J. L., Adler G., and Steer M. L. (1998) Intra-acinar cell activation of trypsinogen during caerulein-induced pancreatitis in rats. Am. J. Physiol. 275, G352–G362 [DOI] [PubMed] [Google Scholar]
- 3. Mayerle J., Dummer A., Sendler M., Malla S. R., van den Brandt C., Teller S., Aghdassi A., Nitsche C., and Lerch M. M. (2012) Differential roles of inflammatory cells in pancreatitis. J. Gastroenterol. Hepatol. 27, 47–51 10.1111/j.1440-1746.2011.07011.x [DOI] [PubMed] [Google Scholar]
- 4. Halangk W., Lerch M. M., Brandt-Nedelev B., Roth W., Ruthenbuerger M., Reinheckel T., Domschke W., Lippert H., Peters C., and Deussing J. (2000) Role of cathepsin B in intracellular trypsinogen activation and the onset of acute pancreatitis. J. Clin. Invest. 106, 773–781 10.1172/JCI9411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Saluja A. K., Donovan E. A., Yamanaka K., Yamaguchi Y., Hofbauer B., and Steer M. L. (1997) Cerulein-induced in vitro activation of trypsinogen in rat pancreatic acini is mediated by cathepsin B. Gastroenterology 113, 304–310 10.1016/S0016-5085(97)70108-2 [DOI] [PubMed] [Google Scholar]
- 6. Wartmann T., Mayerle J., Kähne T., Sahin-Tóth M., Ruthenbürger M., Matthias R., Kruse A., Reinheckel T., Peters C., Weiss F. U., Sendler M., Lippert H., Schulz H. U., Aghdassi A., Dummer A., et al. (2010) Cathepsin L inactivates human trypsinogen, whereas cathepsin L-deletion reduces the severity of pancreatitis in mice. Gastroenterology 138, 726–737 10.1053/j.gastro.2009.10.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Aghdassi A. A., John D. S., Sendler M., Weiss F. U., Reinheckel T., Mayerle J., and Lerch M. M. (2018) Cathepsin D regulates cathepsin B activation and disease severity predominantly in inflammatory cells during experimental pancreatitis. J. Biol. Chem. 293, 1018–1029 10.1074/jbc.M117.814772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rao N. V., Rao G. V., and Hoidal J. R. (1997) Human dipeptidyl-peptidase I: gene characterization, localization, and expression. J. Biol. Chem. 272, 10260–10265 10.1074/jbc.272.15.10260 [DOI] [PubMed] [Google Scholar]
- 9. Turk D., Janjić V., Stern I., Podobnik M., Lamba D., Dahl S. W., Lauritzen C., Pedersen J., Turk V., and Turk B. (2001) Structure of human dipeptidyl peptidase I (cathepsin C): exclusion domain added to an endopeptidase framework creates the machine for activation of granular serine proteases. EMBO J. 20, 6570–6582 10.1093/emboj/20.23.6570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Turk B., Dolenc I., and Turk V. (1998) Dipeptidly-peptidase I. In Handbook of Proteolytic Enzymes (Barrett A. J., Rawlings N. D., and Woessner J. F. Jr., eds) pp. 631–634, Academic Press, London, UK [Google Scholar]
- 11. Planta R. J., Gorter J., and Gruber M. (1964) The catalytic properties of cathepsin C. Biochim. Biophys. Acta 89, 511–519 [DOI] [PubMed] [Google Scholar]
- 12. McGuire M. J., Lipsky P. E., and Thiele D. L. (1992) Purification and characterization of dipeptidyl peptidase I from human spleen. Arch. Biochem. Biophys. 295, 280–288 10.1016/0003-9861(92)90519-3 [DOI] [PubMed] [Google Scholar]
- 13. Thiele D. L., and Lipsky P. E. (1990) The action of leucyl-leucine methyl ester on cytotoxic lymphocytes requires uptake by a novel dipeptide-specific facilitated transport system and dipeptidyl peptidase I-mediated conversion to membranolytic products. J. Exp. Med. 172, 183–194 10.1084/jem.172.1.183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Korkmaz B., Lesner A., Letast S., Mahdi Y. K., Jourdan M. L., Dallet-Choisy S., Marchand-Adam S., Kellenberger C., Viaud-Massuard M. C., Jenne D. E., and Gauthier F. (2013) Neutrophil proteinase 3 and dipeptidyl peptidase I (cathepsin C) as pharmacological targets in granulomatosis with polyangiitis (Wegener granulomatosis). Semin. Immunopathol. 35, 411–421 10.1007/s00281-013-0362-z [DOI] [PubMed] [Google Scholar]
- 15. Dahl S. W., Halkier T., Lauritzen C., Dolenc I., Pedersen J., Turk V., and Turk B. (2001) Human recombinant pro-dipeptidyl peptidase I (cathepsin C) can be activated by cathepsins L and S but not by autocatalytic processing. Biochemistry 40, 1671–1678 10.1021/bi001693z [DOI] [PubMed] [Google Scholar]
- 16. Rowan A. D., Mason P., Mach L., and Mort J. S. (1992) Rat procathepsin B. Proteolytic processing to the mature form in vitro. J. Biol. Chem. 267, 15993–15999 [PubMed] [Google Scholar]
- 17. Chiari H. (1896) About autodigestion of the human pancreas. Zeitschrift für Heilkunde 17, 69–96 [Google Scholar]
- 18. Lerch M. M., Saluja A. K., Dawra R., Ramaraò P., Saluja M., and Steer M. L. (1992) Acute necrotizing pancreatitis in the opossum: earliest morphological changes involve acinar cells. Gastroenterology 103, 205–213 10.1016/0016-5085(92)91114-J [DOI] [PubMed] [Google Scholar]
- 19. Gorelick F. S., and Lerch M. M. (2017) Do animal models of acute pancreatitis reproduce human disease? Cell Mol. Gastroenterol. Hepatol. 4, 251–262 10.1016/j.jcmgh.2017.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schütte K., and Malfertheiner P. (2008) Markers for predicting severity and progression of acute pancreatitis. Best Pract. Res. Clin. Gastroenterol. 22, 75–90 10.1016/j.bpg.2007.10.013 [DOI] [PubMed] [Google Scholar]
- 21. Sendler M., Dummer A., Weiss F. U., Krüger B., Wartmann T., Scharffetter-Kochanek K., van Rooijen N., Malla S. R., Aghdassi A., Halangk W., Lerch M. M., and Mayerle J. (2013) Tumour necrosis factor α secretion induces protease activation and acinar cell necrosis in acute experimental pancreatitis in mice. Gut 62, 430–439 10.1136/gutjnl-2011-300771 [DOI] [PubMed] [Google Scholar]
- 22. Gukovskaya A. S., Vaquero E., Zaninovic V., Gorelick F. S., Lusis A. J., Brennan M. L., Holland S., and Pandol S. J. (2002) Neutrophils and NADPH oxidase mediate intrapancreatic trypsin activation in murine experimental acute pancreatitis. Gastroenterology 122, 974–984 10.1053/gast.2002.32409 [DOI] [PubMed] [Google Scholar]
- 23. Sendler M., Weiss F. U., Golchert J., Homuth G., van den Brandt C., Mahajan U. M., Partecke L. I., Döring P., Gukovsky I., Gukovskaya A. S., Wagh P. R., Lerch M. M., and Mayerle J. (2018) Cathepsin B-mediated activation of trypsinogen in endocytosing macrophages increases severity of pancreatitis in mice. Gastroenterology 154, 704–718.e10 10.1053/j.gastro.2017.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brown G. R., McGuire M. J., and Thiele D. L. (1993) Dipeptidyl peptidase I is enriched in granules of in vitro- and in vivo-activated cytotoxic T lymphocytes. J. Immunol. 150, 4733–4742 [PubMed] [Google Scholar]
- 25. Pham C. T., and Ley T. J. (1999) Dipeptidyl peptidase I is required for the processing and activation of granzymes A and B in vivo. Proc. Natl. Acad. Sci. U.S.A. 96, 8627–8632 10.1073/pnas.96.15.8627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sendler M., Beyer G., Mahajan U. M., Kauschke V., Maertin S., Schurmann C., Homuth G., Völker U., Völzke H., Halangk W., Wartmann T., Weiss F. U., Hegyi P., Lerch M. M., and Mayerle J. (2015) Complement component 5 mediates development of fibrosis, via activation of stellate cells, in 2 mouse models of chronic pancreatitis. Gastroenterology 149, 765–776 10.1053/j.gastro.2015.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schreiber A., Pham C. T., Hu Y., Schneider W., Luft F. C., and Kettritz R. (2012) Neutrophil serine proteases promote IL-1β generation and injury in necrotizing crescentic glomerulonephritis. J. Am. Soc. Nephrol. 23, 470–482 10.1681/ASN.2010080892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Adkison A. M., Raptis S. Z., Kelley D. G., and Pham C. T. (2002) Dipeptidyl peptidase I activates neutrophil-derived serine proteases and regulates the development of acute experimental arthritis. J. Clin. Invest. 109, 363–371 10.1172/JCI0213462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hu Y., and Pham C. T. (2005) Dipeptidyl peptidase I regulates the development of collagen-induced arthritis. Arthritis Rheum. 52, 2553–2558 10.1002/art.21192 [DOI] [PubMed] [Google Scholar]
- 30. Jenne D. E., and Tschopp J. (1988) Granzymes, a family of serine proteases released from granules of cytolytic T lymphocytes upon T cell receptor stimulation. Immunol. Rev. 103, 53–71 10.1111/j.1600-065X.1988.tb00749.x [DOI] [PubMed] [Google Scholar]
- 31. Caputo A., Garner R. S., Winkler U., Hudig D., and Bleackley R. C. (1993) Activation of recombinant murine cytotoxic cell proteinase-1 requires deletion of an amino-terminal dipeptide. J. Biol. Chem. 268, 17672–17675 [PubMed] [Google Scholar]
- 32. Lerch M. M., Lutz M. P., Weidenbach H., Müller-Pillasch F., Gress T. M., Leser J., and Adler G. (1997) Dissociation and reassembly of adherens junctions during experimental acute pancreatitis. Gastroenterology 113, 1355–1366 10.1053/gast.1997.v113.pm9322531 [DOI] [PubMed] [Google Scholar]
- 33. Mayerle J., Schnekenburger J., Krüger B., Kellermann J., Ruthenbürger M., Weiss F. U., Nalli A., Domschke W., and Lerch M. M. (2005) Extracellular cleavage of E-cadherin by leukocyte elastase during acute experimental pancreatitis in rats. Gastroenterology 129, 1251–1267 10.1053/j.gastro.2005.08.002 [DOI] [PubMed] [Google Scholar]
- 34. Hermant B., Bibert S., Concord E., Dublet B., Weidenhaupt M., Vernet T., and Gulino-Debrac D. (2003) Identification of proteases involved in the proteolysis of vascular endothelium cadherin during neutrophil transmigration. J. Biol. Chem. 278, 14002–14012 10.1074/jbc.M300351200 [DOI] [PubMed] [Google Scholar]
- 35. Halangk W., Krüger B., Ruthenbürger M., Stürzebecher J., Albrecht E., Lippert H., and Lerch M. M. (2002) Trypsin activity is not involved in premature, intrapancreatic trypsinogen activation. Am. J. Physiol. Gastrointest. Liver Physiol. 282, G367–G374 10.1152/ajpgi.00315.2001 [DOI] [PubMed] [Google Scholar]
- 36. Lerch M. M., Saluja A. K., Rünzi M., Dawra R., and Steer M. L. (1995) Luminal endocytosis and intracellular targeting by acinar cells during early biliary pancreatitis in the opossum. J. Clin. Invest. 95, 2222–2231 10.1172/JCI117912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sendler M., Maertin S., John D., Persike M., Weiss F. U., Krüger B., Wartmann T., Wagh P., Halangk W., Schaschke N., Mayerle J., and Lerch M. M. (2016) Cathepsin B activity initiates apoptosis via digestive protease activation in pancreatic acinar cells and experimental pancreatitis. J. Biol. Chem. 291, 14717–14731 10.1074/jbc.M116.718999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mooren F. C., Turi S., Gunzel D., Schlue W. R., Domschke W., Singh J., and Lerch M. M. (2001) Calcium-magnesium interactions in pancreatic acinar cells. FASEB J. 15, 659–672 10.1096/fj.00-0172com [DOI] [PubMed] [Google Scholar]
- 39. Essin K., Salanova B., Kettritz R., Sausbier M., Luft F. C., Kraus D., Bohn E., Autenrieth I. B., Peschel A., Ruth P., and Gollasch M. (2007) Large-conductance calcium-activated potassium channel activity is absent in human and mouse neutrophils and is not required for innate immunity. Am. J. Physiol. Cell Physiol. 293, C45–C54 10.1152/ajpcell.00450.2006 [DOI] [PubMed] [Google Scholar]
- 40. Schnekenburger J., Mayerle J., Krüger B., Buchwalow I., Weiss F. U., Albrecht E., Samoilova V. E., Domschke W., and Lerch M. M. (2005) Protein tyrosine phosphatase κ and SHP-1 are involved in the regulation of cell–cell contacts at adherens junctions in the exocrine pancreas. Gut. 54, 1445–1455 10.1136/gut.2004.063164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zenker M., Mayerle J., Lerch M. M., Tagariello A., Zerres K., Durie P. R., Beier M., Hülskamp G., Guzman C., Rehder H., Beemer F. A., Hamel B., Vanlieferinghen P., Gershoni-Baruch R., Vieira M. W., et al. (2005) Deficiency of UBR1, a ubiquitin ligase of the N-end rule pathway, causes pancreatic dysfunction, malformations and mental retardation (Johanson-Blizzard syndrome). Nat. Genet. 37, 1345–1350 10.1038/ng1681 [DOI] [PubMed] [Google Scholar]
- 42. Niederau C., Ferrell L. D., and Grendell J. H. (1985) Caerulein-induced acute necrotizing pancreatitis in mice: protective effects of proglumide, benzotript, and secretin. Gastroenterology 88, 1192–1204 10.1016/S0016-5085(85)80079-2 [DOI] [PubMed] [Google Scholar]