

Abstract
Rapid expression of critical stress response factors is a key survival strategy for diseased or stressed cells. During cell stress, translation is inhibited, and a pre-existing pool of cytoplasmic mRNA–protein complexes reversibly assembles into cytoplasmic stress granules (SGs). Gle1 is a conserved modulator of RNA-dependent DEAD-box proteins required for mRNA export, translation, and stress responses. Proper Gle1 function is critical as reflected by some human diseases such as developmental and neurodegenerative disorders and some cancers linked to gle1 mutations. However, the mechanism by which Gle1 controls SG formation is incompletely understood. Here, we show that human Gle1 is regulated by phosphorylation during heat shock stress. In HeLa cells, stress-induced Gle1 hyperphosphorylation was dynamic, primarily in the cytoplasmic pool, and followed changes in translation factors. MS analysis identified 14 phosphorylation sites in the Gle1A isoform, six of which clustered in an intrinsically disordered, low-complexity N-terminal region flanking the coil-coiled self-association domain. Of note, two mitogen-activated protein kinases (MAPKs), extracellular signal–regulated kinase (ERK) and c-Jun N-terminal kinase (JNK), phosphorylated the Gle1A N-terminal domain, priming it for phosphorylation by glycogen synthase kinase 3 (GSK3). A phosphomimetic gle1A6D variant (in which six putative Ser/Thr phosphorylation sites were substituted with Asp) perturbed self-association and inhibited DEAD-box helicase 3 (X-linked) (DDX3) ATPase activity. Expression of alanine-substituted, phosphodeficient GFP-gle1A6A promoted SG assembly, whereas GFP-gle1A6D enhanced SG disassembly. We propose that MAPKs and GSK3 phosphorylate Gle1A and thereby coordinate SG dynamics by altering DDX3 function.
Keywords: stress granule, phosphorylation, mitogen-activated protein kinase (MAPK), post-translational modification (PTM), translation regulation, glycogen synthase kinase 3 (GSK-3), neurodegeneration, stress response, DDX3, DEAD-box proteins, Gle1
Introduction
A key survival pathway for cells impacted by disease states and environmental stresses is the rapid expression of critical stress response factors. To achieve this, nuclear export of nonessential transcripts is blocked, whereas temporally induced mRNA transcripts in protein complexes (mRNPs)3 are selectively exported through nuclear pore complexes (NPCs) and then translated. Concurrently, general translation is inhibited, and a pre-existing pool of cytoplasmic mRNPs reversibly assembles into cytoplasmic stress granule (SG) complexes of RNA processing machinery, translation factors, and signaling proteins (1). In addition to the changes in gene expression profiles, the dynamics of mRNP occupancy in SGs affect rates of mRNA decay, global translational activity, and cell signaling cascades (2–5). Increasing evidence suggests the involvement of altered SG assembly and disassembly in the pathogenesis of neurodegenerative diseases; however, the underlying means by which changes in SG dynamics impact diseases is not well understood (6–9).
Several recent reports propose mechanisms for controlling the highly dynamic process of SG assembly and disassembly. SG components have liquid droplet properties and assemble through phase transitions in response to changing temperature, pH, or ionic strength (7, 10, 11). For some components, post-translational modifications (PTMs) mediate their SG assembly (12, 13). For example, phosphorylation of the SG component G3BP by casein kinase 2 promotes dissociation of G3BP from SGs and triggers SG disassembly (14). Notably, aberrant activity of mitogen-activated kinases (MAPKs) is linked to amyotrophic lateral sclerosis (ALS), a neurodegenerative disease characterized by SG accumulation (15). Finally, SG dynamics are altered by the actions of RNA-dependent DEAD-box proteins (Dbps in yeast/DDXs in humans) and their cofactors, possibly through remodeling of RNA–protein complexes or unwinding RNA–RNA duplexes (16).
The essential and highly conserved Dbp/DDX modulator Gle1 plays key roles in nuclear mRNA export, translation initiation and termination, and SG dynamics (17–24). In each of these roles, Gle1 binds to and modulates the ATPase activity of distinct Dbps/DDXs bound to RNA. In humans, the GLE1 gene is alternatively spliced to generate at least two isoforms, Gle1A and Gle1B (25). Both human isoforms share high sequence similarity and common functional motifs: an amino (N)-terminal region that interacts with the human NPC component Nup155; a coiled-coil region that is involved in Gle1 self-association; a carboxyl (C)-terminal domain that interacts with DDX19B, DDX3, and inositol hexakisphosphate (IP6); and a shuttling domain that mediates its translocation between the nucleus and cytoplasm (21, 25–27). Gle1B exhibits pancellular localization with pronounced steady-state enrichment at the NPCs that is partially dependent on a unique 39-amino acid C-terminal extension that mediates Nup42 binding (25, 26). Our work further shows that Nup42 interaction and IP6 are independently required to activate Dbp5/DDX19B for proper mRNA export (26). In contrast, Gle1A lacks the Nup42-binding domain, and it is not functional in mRNA export at NPCs (17). Instead, Gle1A localizes predominantly in the cytoplasm (25) where it interacts with DDX3 to modulate SG dynamics and translational repression in response to stress (17, 28). Thus, Gle1A and Gle1B reside in distinct subcellular pools and perform nonoverlapping functions. These specificities provide human Gle1A and Gle1B with the capacity to regulate multiple steps of gene expression simultaneously, a critical aspect of the cellular stress response.
Several mutations in GLE1 are linked with human diseases, including ALS, multiple forms of arthrogryposis multiplex congenita, a broad array of developmental defects, and cancer (29–33). Our prior studies of human gle1 variants linked to ALS and the arthrogryposis multiplex congenita disease lethal congenital contracture syndrome 1 (LCCS1) suggest that proper subcellular localization and the separable functions of each Gle1 isoform are central to both mechanisms of pathogenesis (17, 21, 28). Consistent with this premise is the observation that Gle1 subcellular localization is disrupted in mouse models of Huntington's disease, which exhibit nuclear localization of Gle1 in the brain cortex (34). Thus, controlled Gle1 subcellular localization and segregation of isoform-specific functions are critical for normal cell physiology.
Based on Gle1's intracellular dynamics and roles in stress responses, we speculated that mechanisms might exist to control Gle1 function in response to stress or disease. Here, we show that, under stress conditions, human Gle1 is hyperphosphorylated by MAPK and GSK3 in an N-terminal, low-complexity region. The basally phosphorylated pool of Gle1A promotes SG assembly and stimulates DDX3 activity, whereas Gle1A hyperphosphorylation promotes SG disassembly, inhibits DDX3 activity, and is disrupted in its capacity to oligomerize in vitro. We propose that the Gle1A hyperphosphorylation cycle effectively alters Gle1A self-association and DDX3 functions. These insights allow a further understanding of how perturbing Gle1 might contribute to different disease states and be a key connection between reported neurodegeneration and SG dysfunction.
Results
Gle1 is phosphorylated during stress
We previously found a critical role for Gle1A in the SG response (17). However, the molecular events directing Gle1A activity during stress were not defined. To test whether PTMs might be involved, HeLa cell lysates from cells grown under stress conditions were analyzed by SDS-PAGE and anti-Gle1 immunoblotting. As shown in Fig. 1A, Gle1 in either heat shock– or arsenite-stressed cell lysates exhibited reduced electrophoretic mobility (lanes 4 and 7) compared with that of the untreated sample (lane 1). Similar shifts in mobility were detected for Gle1 in RPE-1 and HEK-293 cells following heat stress (data not shown). To test whether this slower migration was due to phosphorylation, cell lysates from untreated control, heat-shocked, or sodium arsenite–treated cells were incubated with λ-phosphatase (λ-PPase) in the presence or absence of phosphatase inhibitors. The λ-PPase treatment resulted in collapse of the slower migrating Gle1 band in lysates from stress-treated cells (Fig. 1A, lanes 5 and 8), whereas no change was observed in the presence of both λ-PPase and phosphatase inhibitors (Fig. 1A, lanes 6 and 9). Notably, a minor fraction of basally phosphorylated Gle1 was reproducibly observed in control cell lysates, whereas lysates from stress-treated cells contained a significantly greater proportion of phosphorylated Gle1 (Fig. 1A, compare lane 1 with lanes 4 and 7). To examine Gle1 phosphorylation by an alternative method, we utilized Phos-tag gel electrophoresis followed by immunoblot analysis (35). Three distinct bands for Gle1 were observed in heat-shocked cell lysates (Fig. 1B, lane 2), indicating varying degrees of Gle1 phosphorylation. Interestingly, treatment with the broad-spectrum kinase inhibitor K252a prior to heat shock abolished the altered Gle1 mobility (Fig. S1A and Fig. 1B, lane 3). Thus, upon stress treatment, phosphorylation of endogenous Gle1 was enhanced.
Figure 1.

Cytoplasmic Gle1 is dynamically phosphorylated in response to stress. A and B, Gle1 is phosphorylated during stress. A, HeLa cells were either left untreated, exposed to heat shock at 45 °C, or treated with 0.5 mm sodium arsenite for 60 min. Cell lysates were prepared and incubated with either phosphatase buffer alone, λ-PPase, or λ-PPase and phosphatase inhibitors together for 30 min. Samples were resolved by SDS-PAGE and immunoblotted with anti-Gle1 antibodies. B, HeLa cells were either left untreated (UT) or preincubated with the broad-spectrum kinase inhibitor K252a (1 μm) for 60 min at 37 °C followed by heat shock at 45 °C for 60 min. Cell lysates were resolved on a Phos-tag SDS-PAGE gel and immunoblotted with anti-Gle1 antibodies. C and D, Gle1 phosphorylation follows MAPK activation (C) and phosphorylation (p) of translation factors (D). HeLa cells subjected to heat shock at 45 °C for 60 min followed by a recovery phase at 37 °C were harvested at the indicated time points. Lysates were analyzed by immunoblotting using the indicated antibodies. E, phosphorylated Gle1 localizes to the cytoplasm. Following heat shock at 45 °C for 60 min, nuclear (N) and cytosolic (C) HeLa cell fractions were prepared, resolved by SDS-PAGE, and immunoblotted with anti-Gle1, anti-lamin B1, and anti-GAPDH antibodies. T, total cell lysate.
To test whether Gle1 phosphorylation occurred independently in the C-terminal (residues 400–698) and/or N-terminal (residues 1–400) regions of the protein, plasmids expressing GFP-GLE11–400 or GFP-GLE1400–698 were transfected into HeLa cells. Of note (as shown in Fig. S1 and data not shown), heat shock treatment consistently reduced the steady-state level of all tested, exogenously expressed, GFP-tagged Gle1 proteins through a mechanism that remains to be determined. Analysis of respective cell lysates showed that only the GFP-Gle11–400 protein exhibited two distinct electrophoretic mobilities on a Phos-tag gel with an increased proportion of the slower migrating band present following stress (Fig. S1B, lanes 2 and 3). As observed for endogenous Gle1, K252a pretreatment decreased the amount of phosphorylated Gle1 present (Fig. S1B, compare lane 3 with lane 4). Because in silico phosphorylation analysis predicted a clustering of potential Gle1 phosphorylation sites within the first N-terminal 120 amino acids (data not shown), we also examined GFP-Gle1120–698 protein and found no electrophoretic shift in response to stress (Fig. S1B, lanes 10 and 11). Taken together, we concluded that residues in the N-terminal region of Gle1 were phosphorylated under these stress conditions.
The Gle1 phosphorylation state correlates with preceding cycles of MAPK and translation factor phosphorylation
To investigate the temporal dynamics of Gle1 phosphorylation during stress and recovery, a time course analysis of heat shock–induced phosphorylation was conducted. Cell lysates were collected at time 0 and every 30 min throughout the 45 °C heat stress and subsequent 37 °C recovery phase. A dramatic increase in Gle1 phosphorylation was observed from the 60-min time point during heat stress to the 120-min time point in the recovery phase (Fig. 1, C and D, lanes 1–4). This level of Gle1 phosphorylation persisted in the recovery phase for approximately 3 h (240 min experimental time point; lane 5) after which it slowly diminished and was undetectable 9 h post-stress treatment (lane 8).
As both yeast Gle1 and human Gle1A function to regulate translation (17, 19, 20), the time course of Gle1 phosphorylation was further assessed by evaluating the concomitant phosphorylation state of various translation regulators (Fig. 1D). During stress, MAPKs and mechanistic targets of rapamycin (mTORs) are activated to repress global translation initiation and stimulate translation of stress response proteins (36). Changes in eIF2α and 4EBP1 phosphorylation states are well characterized and effectively halt translation initiation by, respectively, blocking formation of the ternary complex and eIF4F complex (37–39). As cells recover from stress, the phosphorylation states of these proteins are reversed, and global translation resumes. Immunoblotting was used to assess kinase activation (Fig. 1C) and to analyze translation factor phosphorylation (Fig. 1D) in cell lysates from the heat shock time course. As shown in Fig. 1C, the stress-induced phosphorylation cycles of ERK and JNK slightly preceded Gle1 phosphorylation and dephosphorylation (Fig. 1C). Strong phosphorylation of JNK was detected within 30 min (Fig. 1C, lane 2) of heat stress, whereas, consistent with other reports and quite reproducibly (40, 41), a rapid early burst of ERK phosphorylation was observed at 30 min of stress treatment (lane 2) followed by a later peak of phosphorylation at 120 min (lane 4). JNK and ERK phosphorylation was reduced by the 240-min time point (lane 5) and continued to decrease throughout the monitored recovery period. Consistent with their roles in triggering the onset of translational reprogramming (42), the cycles of eIF2α phosphorylation and 4EBP dephosphorylation also preceded the timing of Gle1 phosphorylation changes (Fig. 1D). Overall, stress-induced Gle1 phosphorylation correlated with a response to MAPK activation and translation repression cues, and Gle1 dephosphorylation correlated with MAPK deactivation and translation activation.
Based on the link to translation factor changes, we reasoned that cytoplasmic Gle1 might be the predominant target for phosphorylation compared with the nuclear pool. To test this, nuclear and cytosolic fractions were prepared from untreated and heat-shocked HeLa cells, and Gle1 phosphorylation was analyzed by immunoblotting (Fig. 1E). Lamin B1 and GAPDH were controls for nuclear and cytosolic fractions, respectively. The slower migrating, phosphorylated Gle1 was observed in the cytosolic fraction for both untreated and stressed cells (Fig. 1E, lanes 2 and 5). In contrast, Gle1 detected in the nuclear fraction migrated at the molecular mass of nonphosphorylated Gle1 (Fig. 1E, lanes 3 and 6). Thus, in both untreated and heat shock–stressed cells, phosphorylated Gle1 was predominantly cytoplasmic. The presence of phosphorylated Gle1 in the cytoplasm may reflect the targeting of only cytoplasmic Gle1 by kinases or, alternatively, rapid export of Gle1 out of the nucleus following phosphorylation.
Gle1A is phosphorylated at periodically repeating serine/threonine residues in the intrinsically disordered N-terminal region
Studies suggest that protein phosphorylation often occurs within intrinsically disordered regions (43, 44). Based on our analysis (Fig. 2A) and as recently reported (45), the first 120 residues of the Gle1 N-terminal region are predicted to comprise an intrinsically disordered, low-complexity domain. Together with the domain mapping analysis in Fig. S1B, we speculated that the Gle1 residues modified by stress-induced phosphorylation reside in this predicted low-complexity domain. Given that we observed stress-induced phosphorylation specifically in the cytoplasmic fraction of endogenous Gle1 (Fig. 1E) and the predominantly cytoplasmic isoform Gle1A, but not Gle1B, is specifically required during stress to modulate translation (17), we selected Gle1A for a proteomics approach to identify the phosphorylated amino acid residues. HeLa cells ectopically expressing GFP-GLE1A were either left untreated or treated with 0.5 mm sodium arsenite for 60 min. GFP-Gle1A protein was then isolated by immunoprecipitation, excised from an SDS-PAGE gel (Fig. S2), trypsin-digested, and processed for MS. In total, MS/MS analysis identified 14 Gle1A phosphorylation sites in samples subjected to stress. The locations of modified residues are shown in the domain map of Fig. 2B. Phosphorylation of five residues (Ser88, Ser93, Ser99, Thr427, and Ser465) was also detected in Gle1A isolated from untreated cells, consistent with the observed basal Gle1 phosphorylation (Fig. 2B, denoted in gray). The majority of stress-induced phosphorylation sites concentrated in two small clusters adjacent to either the N- or C-terminal boundary of the coiled-coil domain. With regard to the hyperphosphorylated N-terminal region (Ser88, Ser92, Ser93, Ser99, and Thr102), we validated the modifications at residues Ser88, Ser99, and Thr102 by manual inspection of the MS/MS scans (Fig. S3). Although the MS/MS scans obtained for the Ser92 and Ser93 sites were less conclusive, evidence for phosphorylation of all five residues in the N-terminal cluster as well as for Ser96 is also reported in published large-scale data sets from high-throughput phosphoproteomic screens (46–48).
Figure 2.

Gle1 is hyperphosphorylated in a low-complexity domain in response to stress. A, the N-terminal region of Gle1A is predicted to be a highly disordered, low-complexity domain. Disorder potential was predicted using the IUPred web server. Scores above 0.5 indicate a high degree of predicted highly disordered. B, schematic representation of phosphorylated Gle1A residues identified by MS. The diagram depicts the functional and structural domains of Gle1A (adapted from Folkmann et al. (21)) and location of phosphorylated residues therein. Gle1A residues shown in gray were phosphorylated in both untreated and stress-treated samples. Residues detected only in stress-treated samples are shown in black. Phosphorylation of Ser41 was detected only in untreated samples. The asterisk indicates the position of the PFQ insertion generated by the gle1-Finmajor mutation. C, a periodicity is evident in the pattern of phosphorylation events across the N-terminal cluster of phosphorylated amino acids. Dotted arrows across two consecutive SXXXS(P) motifs in green show the in silico prediction of GSK3 priming by MAPK phosphorylation. Purple residues illustrate the PXXP polyproline motif of amino acids 97–103. D, modification of the Ser88–Thr102 cluster alters Gle1A electrophoretic mobility. HeLa cells transfected with plasmids expressing GFP-GLE1A, GFP-gle1A6A, or GFP-gle1A6D were either untreated or exposed to 0.5 mm sodium arsenite for 60 min. Cell lysates were resolved by Phos-tag SDS-PAGE and immunoblotted with anti-GFP antibodies. E, phosphorylation alters the b-isox precipitation of Gle1. Nuclear (N) and cytosolic (C) fractions prepared from HeLa cells exposed to 45 °C heat shock for 60 min were incubated with 100 μm b-isox, pelleted by centrifugation, and analyzed by immunoblotting with anti-Gle1 and anti-actin antibodies. Sup, supernatant.
The Ser88/Ser92/Ser93/Ser96/Ser99/Thr102 cluster of phosphorylated Gle1A residues (hereafter termed the Ser88–Thr102 cluster) lies within the N-terminal 120-amino acid region of Gle1A responsible for the phosphatase-sensitive mobility shift according to our domain mapping analysis (Fig. S1B). Although our MS data indicated that three of these residues (Ser88, Ser93, and Ser99) were also phosphorylated in Gle1A isolated from untreated cells, a quantitative analysis of individual phosphorylation levels was not possible because the multiply phosphorylated peptides eluted together in our LC-MS experiments. Therefore, we could not definitively exclude increased phosphorylation at these residues as a possible contributor to this change in mass. Conversely, the C-terminal region of Gle1 did not demonstrate a mobility shift under stress conditions (Fig. S1B), suggesting that the degree of stress-induced modification at the seven identified C-terminal phosphorylation sites is less significant. These data therefore singled out the N-terminal Ser88–Thr102 cluster as a putative region of hyperphosphorylation during stress.
Remarkably, a striking periodicity was evident in the potential pattern of phosphorylation events across this N-terminal cluster, including modifications occurring at every third residue from Ser93 to Thr102 and within two sequential SXXXS(P) motifs (where S(P) represents phosphorylation of serine) (Fig. 2C). Two polyproline II helix peptide motifs (PXXP), which when phosphorylated could markedly alter Gle1 structure and interactions, are also present in this sequence adjacent to Ser96 and containing Ser99 and Thr102 (PXXP; Fig. 2C) (49–51). PhosphoNet in silico kinase prediction analysis across this sequence suggested that Gle1 residues Ser88, Ser99, and Thr102 are potential targets for phosphorylation by ERK, and that Ser93 and Ser96 are targeted by JNK. Furthermore, when primed by Ser96 phosphorylation, GSK3 was predicted to sequentially phosphorylate Ser92 and Ser88 (Fig. 2C). Taken together, the Gle1 phosphorylation domain mapping and in silico analysis strongly indicated that Ser88–Thr102 is phosphorylated en masse following the stress-induced activation of MAPK/ERK1/2 and Akt/GSK3 cascades with potentially important functional repercussions.
Given the preponderance of data implicating the entire Ser88–Thr102 cluster as a likely target of massive stress-induced MAPK and GSK3 phosphorylation, we investigated the impact that global modification of these residues has on Gle1 function. To test whether modifications in the Ser88–Thr102 cluster were responsible for the mobility shift of Gle1 from cells grown under stress conditions, mutations in the coding sequences were generated that result in nonphosphorylatable alanines (gle1A6A) or phosphomimetic aspartic acids (gle1A6D) in place of the six putative serine or threonine phosphorylation sites. Plasmids expressing GFP-GLE1A, GFP-gle1A6A, or GFP-gle1A6D were transfected into HeLa cells, and lysates from untreated or arsenite-treated samples were analyzed by Phos-tag electrophoresis and anti-GFP immunoblotting. Arsenite stress treatment resulted in reduced electrophoretic mobility of GFP-Gle1A compared with untreated protein (Fig. 2D, compare lane 4 with lane 1), whereas no mobility shift was observed for GFP-gle1A6A (lanes 2 and 5). GFP-gle1A6D exhibited slower mobility irrespective of treatment (Fig. 2E, lanes 3 and 6) as would be predicted for a phosphomimetic form. Thus, modification of residues in the Ser88–Thr102 cluster was required to produce the gel mobility shift in response to stress.
Phosphorylation of Gle1 alters its biochemical properties
Others have reported that phosphorylation events within low-complexity domains alter the respective protein's polymerization characteristics and function (43, 44, 52, 53). Because low-complexity domains can mediate the type of phase transitions that potentially drive SG dynamics (11), phosphorylation of SG components may serve to regulate that process. Others also have demonstrated that the isoxazole (b-isox) compound specifically interacts with highly disordered, low-complexity domains and results in protein precipitation. Indeed, many SG components precipitate with b-isox (52, 54), and a recent study shows that phosphorylation of Cdc19's low-complexity domain abolishes its precipitation by b-isox (53, 55). Thus, we established assays to measure the propensity for Gle1 precipitation by b-isox as a test of the effect of phosphorylation on its biochemical properties. The nuclear and cytoplasmic fractions isolated from heat-shocked HeLa cells were treated with b-isox, and the resulting pelleted and soluble portions were tested for Gle1 precipitation by immunoblotting (Fig. 2E). Consistent with our data in Fig. 1E, phosphorylated Gle1 was in the total cytoplasmic fraction (Fig. 2E, lane 5). In the presence of b-isox, the phosphodeficient Gle1 in the nuclear fraction was distributed between the soluble and pelleted fractions (Fig. 2E, lanes 1 and 3). However, phosphorylated Gle1 was only detected in the supernatant following b-isox incubation (Fig. 2E, lane 4). The same distribution was observed in untreated cells with basally phosphorylated Gle1 remaining soluble when incubated with b-isox (data not shown). These data indicated that the N-terminal low-complexity domain of phosphodeficient Gle1 is highly disordered and prone to aggregation like that of many SG components. Conversely, Gle1 hyperphosphorylation appeared to change the protein's biochemical properties, reducing its b-isox precipitation propensity. Given the high degree of disorder inherently surrounding many of the Gle1 phosphorylation sites, it is likely that the altered behavior arises from charge-based disruption of intramolecular bonds or oligomerization interactions. This effect was previously described for FUS, which is also phosphorylated in a low-complexity domain. Modification of FUS does not alter the disordered structure of the domain, but rather the increased negative charge introduced by multiple phosphorylation events (or the phosphomimetics) decreases intramolecular and intermolecular interactions and thereby disrupts FUS aggregation (56).
MAPKs prime Gle1 for phosphorylation by GSK3
ERK and JNK belong to the MAPK family and play critical roles in stress responses (57, 58). Translation inhibitors such as cycloheximide and anisomycin activate MAPKs (59) and prevent polysome disassembly, trapping the ribosomes in different states (60) and blocking SG formation to varying degrees as shown by anti-G3BP immunofluorescence (Fig. S4A). In agreement with the prediction of MAPK sequence motifs in the Ser88–Thr102 cluster, treatment of HeLa cells with either cycloheximide or anisomycin (in the absence or presence of stress) dramatically enhanced the phosphorylation of Gle1 (Fig. S4B). Thus, increased Gle1 phosphorylation was potentially not dependent on SG assembly but might be linked to the regulation of translation.
To test whether MAPKs are required for Gle1 phosphorylation, specific kinase inhibitors of the three major MAPK subgroups (ERK1/2, JNK, and p38) were tested for inhibition of Gle1 phosphorylation in HeLa cells exposed to heat shock. The resulting cell lysates were analyzed by anti-Gle1 immunoblotting. Addition of p38 inhibitor had no impact upon Gle1 phosphorylation (Fig. 3A, lanes 3 and 7). Similarly, inhibition of either JNK or ERK alone did not alter the relative degree of Gle1 phosphorylation (Fig. 3A, lanes 4, 5, and 6) compared to control with no inhibitors (Fig. 3A, lane 2). However, in the presence of inhibitors for both JNK and ERK, a faster migrating Gle1 band indicative of reduced phosphorylation was observed (Fig. 3A, lane 8). The intermediate mobility of this band might indicate less than 100% inhibition of ERK and JNK or represent the actions of another kinase on Gle1. This latter possibility was supported by the greater efficacy of the MEK1/2-targeted ERK inhibitor U0126 than the direct ERK inhibitor FR180204 at preventing Gle1 phosphorylation when combined with JNK inhibitor (Fig. 3A, lanes 8 and 9, respectively). In sum, these results suggested that actions of MAPKs, including the ERK and JNK, are involved in Gle1 phosphorylation.
Figure 3.

Phosphorylation events by the MAPKs ERK and JNK prime GSK3 to phosphorylate Gle1. A and B, ERK and JNK phosphorylate Gle1 in the N-terminal Ser88–Thr102 cluster. A, HeLa cells were untreated (UT) or heat-shocked at 45 °C for 60 min in the presence of the indicated kinase inhibitors (inh) (p38 (SB203580), ERK (FR180204), MEK-ERK (U0126), and/or JNK (JNK-IN-8)), lysed, and immunoblotted with the indicated antibodies. The asterisk indicates a nonspecific band detected by anti-phospho (p)-JNK antibody. B, bacterially expressed and purified recombinant Gle11–360 or gle11–360 6A proteins were incubated with recombinant active ERK and/or JNK in the presence of radioactive [γ-32P]ATP. Reactions were terminated by addition of Laemmli sample denaturation buffer and resolved by SDS-PAGE, and radioisotope incorporation was measured by autoradiography (Autorad). Coomassie stain demonstrates equivalent protein loading. C–E, phosphorylation of Gle1 by ERK and JNK primes GSK3 to phosphorylate Gle1. C, schematic diagram describes the sequential in vitro kinase assay. D and E, GSK3 phosphorylation occurs in the N terminus of Gle1 within the Ser88–Thr102 cluster of phosphorylation sites. D, purified Gle11–360 was preincubated with ERK and JNK followed by the addition of active recombinant GSK3 in the presence of [γ-32P]ATP. ERK and JNK activity was inhibited during GSK3 incubation as indicated, and samples were analyzed for phosphorylation as in B. E, sequential in vitro kinase reactions were performed as in D for purified Gle11–360 or gle11–360 6A.
To further investigate whether ERK and JNK can directly phosphorylate Gle1, recombinant purified N-terminal Gle11–360 or gle11–360 6A was incubated with or without purified active ERK and/or JNK in the presence of [γ-32P]ATP. In the absence of exogenous substrate, autophosphorylation of JNK was observed (Fig. 3B, lane 2). Incubation of Gle11–360 with ERK and/or JNK resulted in phosphorylation of the N-terminal Gle1 protein (Fig. 3B, lanes 4, 5, and 6). Phosphorylation of gle11–360 6A was minimal compared with WT (Fig. 3B; lanes 8, 9, and 10), and phosphorylation of the C-terminal Gle1360–698 protein was also negligible (Fig. S5). Together, these data demonstrated that ERK and JNK specifically phosphorylate Gle1 and that phosphorylation requires the N-terminal Ser88–Thr102 cluster of residues.
Based on the sequence motifs surrounding the Ser88–Thr102 cluster, we also tested whether Gle1 is phosphorylated by GSK3. GSK3 typically targets substrates primed by prior phosphorylation events at serine or threonine residues located four residues C-terminal to the GSK3 phosphorylation site (61). We reasoned that Ser92 might be a GSK3 target site that is primed by ERK or JNK phosphorylation at Ser96 (Fig. 2C). To determine whether the MAPKs prime Gle1 for GSK3 phosphorylation, we performed sequential in vitro kinase assays. Purified Gle11–360 was preincubated with ERK and JNK followed by the addition of active recombinant GSK3 in the presence of [γ-32P]ATP (Fig. 3C, schematic). A minor band corresponding to GSK3 autophosphorylation was detected in the absence of any Gle1 protein (Fig. 3D, lane 15). As expected, incubation with GSK3 alone resulted in minimal phosphorylation of Gle11–360 (Fig. 3D, lane 4). However, when preincubated with ERK or JNK, robust phosphorylation of Gle11–360 by GSK3 was detected (Fig. 3D, lanes 5-7). To investigate whether this phosphorylation signal arises solely from primed GSK3 activity and not from residual MAPK activity, ERK and/or JNK specific inhibitors were added to the reactions after the respective MAPK preincubation step. In the absence of GSK3, addition of the MAPK inhibitors after pretreatment, but prior to incubation with [γ-32P]ATP, completely abolished the incorporation of radiolabeled 32P into Gle1 (Fig. 3D, lanes 8–10). In contrast, if GSK3 was included after addition of the MAPK inhibitors, dramatic Gle1 phosphorylation was observed (Fig. 3, D, lanes 12–14, and E, lanes 3–5). Consistent with its lack of phosphorylation by ERK and JNK, GSK3 also did not phosphorylate gle11–360 6A (Fig. 3E, lanes 8–10). Together, these results strongly implicated a kinase cascade of ERK/JNK priming events followed by GSK3 phosphorylation in regulating Gle1A function during stress.
The Gle1 phosphorylation cycle directs its role in SG assembly and disassembly
Given that Gle1A is required for proper SG assembly (17) and stress-induced Gle1A phosphorylation occurred concurrently with changes in translation factors (Fig. 1D), we speculated that Gle1A phosphorylation might play a role during SG assembly and/or disassembly. In ectopic expression experiments, both GFP-gle1A6A and GFP-gle1A6D localized to SGs upon heat shock (Fig. S6). To assay for SG perturbations resulting from specific Gle1 phosphorylation states, we performed knockdown/addback experiments using our previously established methods (17). Non-targeting (CTRL) or GLE1 siRNA–treated HeLa cells expressing either GFP, GFP-GLE1A, GFP-gle1A6A, or GFP-gle1A6D were heat-shocked for 60 min and processed for anti-G3BP immunofluorescence analysis (Fig. 4A). In CTRL siRNA–treated cells after heat shock for 60 min (Fig. 4A), the expression of phosphomimetic GFP-gle1A6D behaved similarly to that of GFP alone with no significant difference in the number of SGs per cell. In contrast, GFP-GLE1A and GFP-gle1A6A expression resulted in substantially fewer SGs per cell in the heat-shocked, CTRL siRNA–treated samples (Fig. 4A). Thus, as we previously reported for GFP-GLE1A (17, 28), overexpression of GFP-gle1A6A also altered SG morphology. Consistent with our previous reports, the number of SGs also significantly increased in GLE1 siRNA–treated cells expressing GFP compared with CTRL siRNA–treated cells expressing GFP (p value <0.0001), and this phenotype was rescued by expressing either GFP-GLE1A, GFP-gle1A6A, or GFP-gle1A6D (Fig. 4A). Interestingly, in the heat-shocked GLE1 siRNA–treated cells, GFP-gle1A6A expression resulted in greater rescue of the SG defect than GFP-GLE1A or GFP-gle1A6D (Fig. 4A). This analysis indicated that Gle1A-mediated SG assembly is not dependent on phosphorylation of the N-terminal Ser88–Thr102 cluster.
Figure 4.

Gle1A phosphorylation state alters SG assembly and disassembly. A, gle1A6A promotes SG assembly. CTRL or GLE1 siRNA–treated HeLa cells transfected with GFP, GFP-GLE1A, GFP-gle1A6A, or GFP-gle1A6D and heat-shocked at 45 °C for 60 min were processed for anti-G3BP immunofluorescence and analyzed for the number of SGs present. B, Gle1A phosphorylation promotes disassembly of SG granules. CTRL or GLE1 siRNA–treated cells transfected with GFP, GFP-GLE1A, GFP-gle1A6A, or GFP-gle1A6D were heat-shocked at 45 °C for 60 min and processed for anti-G3BP immunofluorescence before and after recovery at 37 °C for 60 min, and the number of cells without SGs postrecovery was quantified. Analysis was performed using a two-tailed unpaired t test, and error bars represent S.E.
Because Gle1 dephosphorylation coincided with the release of translation factors from inhibitory phosphorylation states (Fig. 1D), we speculated that Gle1 dephosphorylation might be required for SG disassembly. As a measure of SG disassembly, the number of cells with no anti-G3BP–labeled SGs was quantified for each knockdown/addback condition wherein, after heat shock for 60 min, duplicate cell samples were allowed to recover at 37 °C for 60 min (Fig. 4B). Compared with GFP alone, an increase in the number of cells without SGs would indicate enhanced SG disassembly during the 60-min recovery period. As reported previously (28), GFP-GLE1A expression, in CTRL and GLE1 siRNA–treated cells, resulted in a greater percentage of cells with no SGs compared with GFP alone (Fig. 4B). Thus, GFP-Gle1A promoted SG disassembly. Expression of GFP-gle1A6D in CTRL and GLE1 siRNA–treated cells resulted in similarly high levels of cells lacking SGs within 60 min of recovery from heat shock as with GFP-GLE1A expression. This suggested that GFP-gle1A6D mediates SG disassembly to the same extent as WT GFP-Gle1A within a 60-min recovery phase. However, with GFP-gle1A6A expression when endogenous Gle1 is depleted (GLE1 siRNA cells), fewer cells were without SGs compared with GFP-GLE1A or GFP-gle1A6D (Fig. 4B). Thus, GFP-gle1A6A alone was not as effective as WT or GFP-gle1A6D in mediating SG disassembly during the 60-min recovery period. Collectively, the impact of Gle1 on SG dynamics was distinctly phosphodependent. These results suggested that, during stress, an unphosphorylated or basally phosphorylated pool of Gle1A promotes SG assembly. Then, during recovery from stress, increasing Gle1A hyperphosphorylation in the Ser88–Thr102 cluster facilitates SG disassembly.
Phosphorylation of the Ser88–Thr102 cluster in Gle1A inhibits DDX3 ATPase activity
Our prior studies in human cells linked the role of Gle1A function in SG dynamics to the regulation of DDX3 (17). DDX3 modulates translation through interaction with the preinitiation complex and is required for proper SG assembly (60). Because Gle1A modification at the Ser88–Thr102 cluster promoted SG disassembly (Fig. 4B), we tested for the impact of Gle1A phosphorylation on DDX3 ATPase activity. In vitro ATPase assays were conducted with bacterially expressed, purified recombinant DDX3 and Gle1A, gle16A, or gle1A6D at various molar ratios. As expected, DDX3 exhibited RNA-dependent ATPase activity (Fig. 5, bar 3) (62), which was further stimulated by addition of Gle1A (Fig. 5, bars 7 and 8). Addition of gle1A6A also stimulated DDX3 activity to a level equivalent to WT Gle1A (Fig. 5, bars 9 and 10). However, addition of gle1A6D inhibited DDX3 ATPase activity in a dose-dependent manner (Fig. 5, bars 11–13). IP6 did not appear to play a role in Gle1 stimulation of DDX3 as its addition to the assay had no effect (Fig. 5, bars 8, 10, and 14). We concluded that the phosphorylation state of Gle1 alters its ability to stimulate DDX3. From these data, we further speculated that unphosphorylated or basally phosphorylated Gle1 promotes SG assembly by activating DDX3. Likewise, once the stress is removed, inhibition of DDX3 activity by hyperphosphorylated Gle1 would reverse the process and support SG disassembly.
Figure 5.
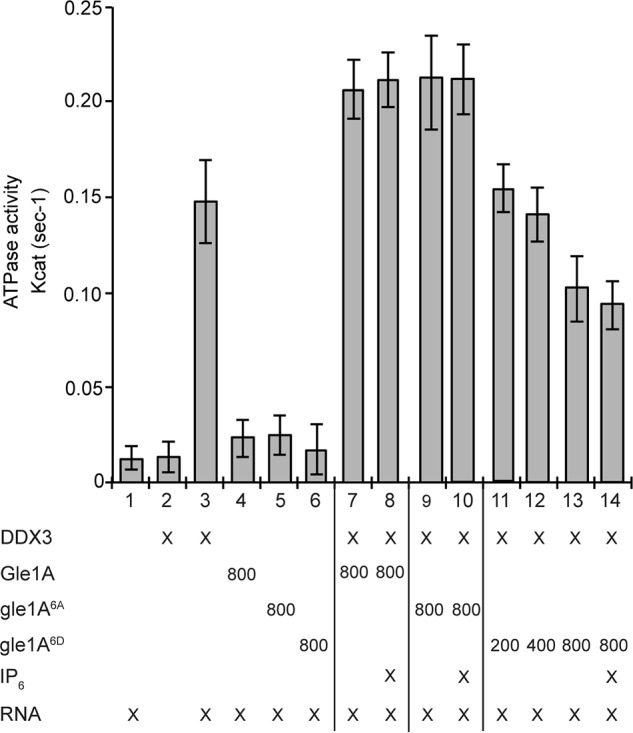
The phosphomimetic gle1A6D inhibits DDX3 RNA-dependent ATPase activity. Colorimetric NADH-coupled ATPase assays were performed with bacterially expressed and purified recombinant DDX3 in the presence of 2.5 mm ATP, 2.5 mm MgCl2, and 0.5 μm RNA-DNA hairpin oligonucleotide. Modulation of DDX3 ATPase activity was tested for bacterially expressed and purified Gle1A (800 nm), gle1A6A (800 nm), or gle1A6D (200, 400, and 800 nm) with or without 1 μm IP6. Error bars represent S.E.
Gle1A is a stable component of SGs
The SG assembly factors typically designated as nucleators, such as G3BP and TIA-1, are largely mobile within SGs and exhibit rapid exchange rates of 2–3 s (63, 64). In contrast, RNA-binding proteins such as YB1 and HuR (63) and the Fas-activated serine/threonine phosphoprotein (FAST) (64) are stable SG components. Although Gle1A function and phosphorylation state modulate several aspects of the stress response, including SG assembly, disassembly, and DDX3 activity, it was unclear whether Gle1A is transiently localized at SGs like a nucleator or whether it is a stable SG component. To address this question, the exchange rate of Gle1A between SGs and the cytoplasm was examined under heat shock conditions. HeLa cells ectopically expressing GFP-GLE1A and mCHERRY-G3BP were subjected to heat shock at 45 °C for 45 min, and then fluorescence recovery after photobleaching (FRAP) was measured only for SGs that had colocalized mCherry-G3BP and GFP-Gle1A. Large GFP-Gle1 aggregates devoid of mCherry-G3BP were excluded from analysis. SGs were photobleached using a 561 nm laser for mCherry-G3BP FRAP (Fig. 6A, top panel) or using a 488 nm laser for GFP-Gle1A FRAP (Fig. 6A, bottom panel). Consistent with published studies (63, 64), mCherry-G3BP moved rapidly in and out of SGs (mobile fraction of 63.8%) and exhibited a fast half-time recovery of 4.4 s. In contrast, GFP-Gle1A was relatively immobile in stress granules (mobile fraction of 6.1%). No significant difference in mobility was observed for the phosphodeficient GFP-Gle1A6A (mobile fraction of 7.8%) and phosphomimetic GFP-Gle1A6D (mobile fraction of 10.2%) compared with GFP-Gle1A (Fig. S7). These results suggested that GFP-Gle1 is a stable component of SGs.
Figure 6.
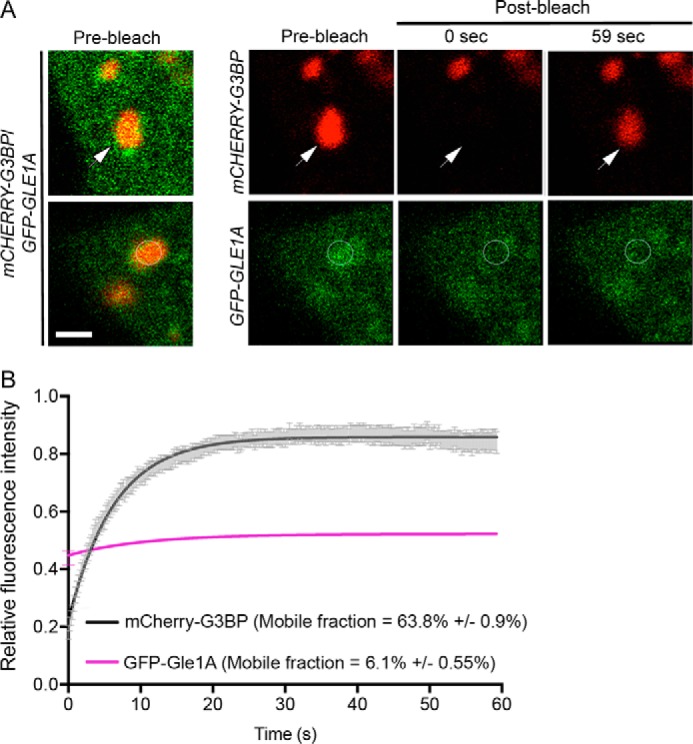
GFP-Gle1A is a stable component of SGs. A, FRAP analysis of G3BP (top panel) and Gle1A (bottom panel) was conducted in heat-shocked HeLa cells cotransfected with mCHERRY-G3BP and GFP-GLE1A. Heat shock was conducted at 45 °C for 45 min prior to FRAP assay at 45 °C. SGs containing both proteins (merged prebleach images, example indicated by arrowhead) were bleached using the argon 561 nm laser (top panel) or the argon 488 nm laser (bottom panel) and then imaged for recovery up to 59 s. Images are representative of three independent experiments. B, graph illustrating the fluorescence intensity measured in bleached ROIs over time. Data from at least 25 cells per condition were analyzed to determine mobility with error calculated as S.D. Scale bar, 5 μm.
Gle1 self-association is required for SG assembly and perturbed in vitro with gle1A6D
The full N-terminal domain of Gle1 from residues 1 to 360, containing both the intrinsically disordered, low-complexity region and a coiled-coil domain, self-associates in vitro to form higher-order disk-like oligomer structures (21). Because the two major clusters of phosphorylated Gle1 residues map to regions flanking the coiled-coil domain, we investigated whether phosphorylation altered the disk-like, oligomerized Gle1 structures. High-molecular-mass complexes of purified, recombinant Gle11–360, gle11–360 6A, and gle11–360 6D were isolated and analyzed by negative-stain EM. As expected, Gle11–360 formed organized disk-like structures (Fig. 7A). The gle11–360 6A protein also formed higher-order symmetrical disks that were indistinguishable from Gle11–360 (Fig. 7B). However, gle11–360 6D assembled into less ordered, more elliptical disks (Fig. 7C). Quantification of the disk circularity, as shown by the outline of disk shape and major/minor axis cross-hairs depicted in Fig. 7, A–C, insets, documented that gle11–360 6D disks were more elongated compared with Gle11–360 or gle11–360 6A disks (Fig. 7D). Thus, the phosphomimetic variant slightly altered Gle1 self-association in vitro, further suggesting that Gle1 hyperphosphorylation might perturb Gle1 self-association in a cellular context.
Figure 7.

Gle1A self-association is perturbed in vitro by modification of Ser88–Thr102 and is required for proper SG response. A–C, phosphorylation of Gle1A perturbs Gle1A association. Representative EM images for bacterially expressed and purified recombinant Gle1A1–360 (A), gle1A1–360 6A (B), and gle1A1–360 6D (C) proteins are shown. Insets depict disk outline (dashed lines) with major and minor axes indicated (solid lines). Scale bar, 50 nm. D, circularity was quantified from at least 171 disks per protein and analyzed by unpaired t test. Major axis/minor axis represents the ratio of longest to shortest diameter of the elliptical as shown by insets in A–C. A ratio of 1 indicates a circle. Error bars represent S.D. E and F, GFP-gle1A-Finmajor exhibits reduced capacity for rescuing SG defects. E, CTRL or GLE1 siRNA–treated cells were transfected with GFP, GFP-GLE1A, or GFP-gle1A-Finmajor plasmids, heat-shocked for 60 min at 45 °C, and processed for immunofluorescence using anti-G3BP antibodies. Scale bar, 10 μm. F, quantification from three independent experiments, performed in duplicate, is shown for the number of SGs in CTRL or GLE1 siRNA–treated cells expressing the indicated plasmids. Data were quantified for at least 65 cells per condition and analyzed using a two-tailed, unpaired t test. Error bars represent S.E.
To evaluate whether proper self-association of Gle1 plays a role in SG assembly, we also examined the LCCS1-causative gle1-Finmajor mutation that perturbs Gle1 self-association and leads to a defect in mRNA export (21). The gle1-Finmajor mutation results in a PFQ insertion in the coiled-coil domain of Gle1 (31), and in vitro analysis shows gle1-Finmajor disk-like structures are deformed (21), although in a different manner than gle11–360 6D disks. GFP-gle1A-Finmajor expression in heat-shocked GLE1 siRNA cells rescued the SG defects, however not as robustly as compared with GFP-GLE1A (Fig. 7, E and F). The extent of SG rescue by GFP-gle1A-Finmajor expression was similar to that observed for GFP-gle1A6D (Fig. 4B). Thus, proper self-association was required for effective SG assembly. Moreover, the N-terminal domain of Gle1 was specifically implicated in regulation of SG biology, as either the PFQ insertion in the coiled-coil domain or phosphomimetic alterations in the intrinsically disordered, low-complexity region impacted SG dynamics.
Discussion
Although phosphorylation is a highly temporal and rapidly reversible PTM found on many SG regulatory proteins, it has not been fully resolved how such stress-induced phosphorylation events might control SG dynamics. Here, we demonstrate that human Gle1 is hyperphosphorylated in response to cellular stress, with this phosphorylation persisting until modifications that alter translation factors are reversed. As represented in the cartoon model (Fig. 8), we show that Gle1A residues in the N-terminal Ser88–Thr102 cluster are substrates for sequential phosphorylation by the MAPKs ERK and JNK and by GSK3. Furthermore, the effect of mutations that change the coding sequence to that for alanine or aspartic acid at these residues suggests that the phosphorylation status of the Ser88–Thr102 cluster impacts SG assembly/disassembly dynamics as well as DDX3 activity. In the early stages of stress response, we propose that Gle1A in its basally modified state self-associates via its N-terminal domain and promotes SG assembly. We further posit that the accumulation of phosphorylation events in the Ser88–Thr102 cluster over the course of the stress response changes the low-complexity character of Gle1A's N terminus, altering Gle1A self-association and thereby promoting SG disassembly. As such, the stress-induced phosphorylation of Gle1A and subsequent dephosphorylation during recovery provide a critical layer of control over the gene expression pathway and cell survival. It remains to be determined whether each of the six phosphosites in the Ser88–Thr102 cluster or specific combinations thereof are necessary for these functional changes. This will be a focus of our future studies.
Figure 8.
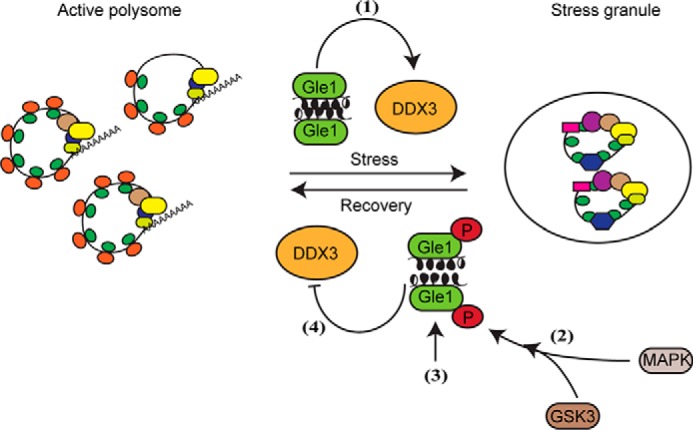
Model of MAPK and GSK3 phosphoregulation of Gle1A. During initiation of the stress response, Gle1A in its basally modified, self-associated state activates DDX3 functions and promotes SG formation (1). As eIF2α is phosphorylated and the integrated stress response proceeds, the activation of MAPK cascades stimulate ERK1/2 and JNK to phosphorylate a cluster of N-terminal Gle1A residues, which then primes GSK3 to phosphorylate additional residues in the Gle1A cluster (2). Accumulation of the multiple phosphate groups over the course of the stress response alters the low-complexity character of the N terminus and perturbs Gle1 self-association (3). In this state, hyperphosphorylated Gle1A inhibits DDX3 activity and thereby promotes SG disassembly as translation resumes (4).
We found that a sequential cascade of stress-induced MAPK and GSK3 activity underlies Gle1 phosphorylation. During stress conditions, multiple signaling pathways are triggered to mediate a multifaceted stress response. MAPKs and GSK3 are implicated in proper SG function and disease. For instance, aberrant MAPK p38 activity induces a gain of function for the ALS-linked SG component FUS (65), and, conversely, inhibition of MAPK signaling improves survival of SOD1 E100G mutant motor neurons derived from ALS patient induced pluripotent stem cells (66). Likewise, tight regulation of GSK3 activity appears to underlie proper SG responses, given that GSK3 inhibition dramatically reduces stress granule formation (12, 67), and aberrant activation of GSK3 is associated with ALS (68). Moreover, several reports show reductions in ALS phenotypes using GSK3 inhibitors, positioning it as a potential therapeutic target (69, 70). Given our prior studies of ALS-linked gle1 mutations that alter Gle1 function (28, 30), phosphoregulation of Gle1 by both MAPK and GSK3 phosphorylation offers insights into new mechanisms by which stress responses in ALS cells might be modulated.
A model for how Gle1 phosphorylation regulates SG biology is based on several pieces of evidence. We previously reported that Gle1A regulates SGs by modulating DDX3's role in translation repression. In brief, heat shock stress of Gle1-depleted HeLa cells results in defective assembly of mature SGs and loss of translation inhibition, both of which are rescued by DDX3 overexpression (17). Here, we show mutagenesis data indicating that Gle1A phosphorylation states modulate the ATPase activity of DDX3 and regulate SG assembly and disassembly. The phosphodeficient gle1A6A stimulates DDX3 ATPase activity and rescues SG assembly defects in Gle1-depleted cells, arguing that Gle1A stimulates SG assembly in the absence of stress-induced hyperphosphorylation. In contrast, the phosphomimetic gle1A6D inhibits DDX3 ATPase activity and promotes SG disassembly. This suggests that Gle1A drives DDX3 ATPase activity to stimulate SG assembly. Although previous studies have argued that DDX3 does not require its ATPase activity for translational repression (62), acting instead by displacement of eIF4E in the cap-binding complex (71, 72), interpretation of this prior work is complicated by experimental conditions based on overexpression. Furthermore, another study recently reported conflicting data, showing that a medulloblastoma-linked ATPase-dead variant of DDX3 is unable to repress translation following arsenite stress (73). Taken together with our findings, it is plausible that Gle1A regulation of DDX3 catalytic activity does play a role in translation inhibition and SG assembly, perhaps via modulation of RNA structure and proteins associated with translation machinery or by mRNP remodeling in SGs (models are reviewed in Ref. 74). Similarly, eIF4B and eIF4G modulate the ATPase activity of eIF4A, which is required for SG assembly (75, 76). Moreover, this mode of regulating ribonucleoprotein bodies appears to be conserved, since recent work in budding yeast showed that the Not1 cofactor stimulates the ATPase activity of Dhh1, which controls P body assembly and disassembly (77). Our finding suggesting that Gle1A hyperphosphorylation alters DDX3 activity and SG dynamics adds an additional layer of complexity to this model. Future studies will be necessary to provide a better understanding of how Gle1A modulates DDX3 functions in vivo during SG assembly and translation inhibition.
If SG assembly is driven in large part by phase separation as proposed in current models (78), then Gle1A phosphorylation may directly alter this physical property and thereby diminish SG formation. There are numerous examples of SG components that, when phosphorylated, have altered SG assembly properties (10, 11, 36, 54, 79). For example, G3BP is subject to phosphorylation in its intrinsically disordered region, which abrogates its function as an SG nucleator (13). Likewise, the ALS-linked SG nucleator FUS is hyperphosphorylated in its low-complexity domain by DNA-dependent protein kinase following DNA damage, which reduces its phase separation properties and aggregation (56). Both examples mirror the effects of phosphorylation on Gle1A, wherein hyperphosphorylation of its intrinsically disordered N-terminal domain changes its function in SG assembly compared with the nonhyperphosphorylated form. Our data suggest that this does not result from a loss of Gle1A recruitment to SGs when it is hyperphosphorylated because both GFP-gle1A6A and GFP-gle1A6D localize to stress granules (Fig. S6). Indeed, recruitment to SGs does not appear to be a factor in dictating the effect of Gle1 on SGs, as Gle1B also localizes to SGs but is incapable of supporting SG dynamics (17).
Although the mechanism by which Gle1A's phosphorylation state dictates DDX3 activity is not resolved, several pieces of data point to Gle1 self-association as a potential switch. We know that Gle1 self-associates via its N-terminal 360 amino acids and that disruption of Gle1B self-association by the LCCS1-linked gle1-Finmajor mutation (annotated in Fig. 2) impedes its ability to stimulate DDX19B-mediated mRNA export (21). It remains to be determined whether Gle1 self-association is a common determinant of its ability to stimulate other Dbps/DDXs. However, our analysis of the phosphodeficient gle1A6A and phosphomimetic gle1A6D suggests that phosphorylation may dictate the oligomerization state of Gle1A and thereby modulate its regulation of DDX3 and SG dynamics. Phosphorylation often alters protein function and interactions by changing the character of intrinsically disordered, low-complexity domains (51, 80, 81), and the Ser88–Thr102 cluster lies within a low-complexity domain of Gle1. Intriguingly, multimerization is required for the RNA helicase activity of Ded1, the yeast homologue of human DDX3 (82). It follows that Gle1A oligomerization might also be required to stimulate a multimeric form of DDX3 during the stress response.
Altered DDX3 activation may be only one of the ways that a phosphosensitive Gle1 oligomerization state could modulate function. Alternatively, but not mutually exclusive, Gle1 oligomers might serve as scaffolds for higher-order organization of SGs by promoting multivalent interactions. In a similar way, the assembly of membraneless organelles such as germ granules and paraspeckles is initiated by self-association of coiled-coil domains in PGL1/3 and Drosophila behavior/human splicing proteins, respectively (83, 84). Recent work also suggests that SGs are made up of dynamic shells surrounding stable core substructures, which are thought to assemble through stable interactions between low-complexity domains in the RNA-binding proteins that decorate stalled mRNPs (16). The FRAP data presented here show that Gle1A is a stable component of SGs. Moreover, SG maturation is promoted by WT Gle1A or gle16A overexpression (both of which are capable of uniform oligomerization in vitro) and not by gle16D (Figs. 4A and 7, A–D). Thus, it is possible that a scaffold of nonhyperphosphorylated Gle1A oligomers supports SG core formation through recruitment of SG nucleators and that hyperphosphorylation dismantles this Gle1A scaffold as part of SG disassembly. However, despite its disparate ability to oligomerize in vitro, gle16D localization is similarly immobile compared with WT Gle1A or gle16A. Additional studies are required to understand the role of Gle1A oligomerization in SG biology.
It is also intriguing that the PFQ insertion in the Gle1 coiled-coil domain from the LCCS1 disease-linked gle1-Finmajor allele impacts Gle1 self-association and SG dynamics in a manner comparable with that caused by aspartic acid substitutions that mimic stress-induced hyperphosphorylation in the intrinsically disordered, low-complexity region (gle1A6D) (21) (Fig. 7, E and F). This suggests that the pathologies underlying LCCS1 may be due to both altered mRNA export (21) and perturbed SG dynamics. As such, multiple coincident defects in the gene expression pathway and in recovery from stress responses might contribute to the disease pathology.
Although Gle1A is localized to the cytoplasm at steady state, it shuttles in and out of the nucleus. It is unclear whether Gle1 is hyperphosphorylated in the nucleus or in the cytoplasm, but our data demonstrate that it is localized in the cytoplasm when phosphorylated. Gle1 phosphorylation is not dependent upon formation of SGs because translation inhibitors that block SG assembly (Fig. S4A) but activate MAPKs are still able to stimulate Gle1 hyperphosphorylation (Fig. S4B). This suggests that Gle1 hyperphosphorylation might also occur under conditions that do not stimulate SG assembly, and therefore may regulate other functions of Gle1. Furthermore, the longer persistence of Gle1 hyperphosphorylation through the period when eIF2α and 4EBP phosphorylation states have begun to reverse (Fig. 1D) suggests that the Gle1 phosphorylation state may act as a homeostatic signal.
Gle1 serves as a critical lynchpin in multiple steps of gene expression from mRNA export to translation (17). In this light, the advantage of having multiple constraints on Gle1 function is clear from the expression of functionally distinct isoforms to their discrete subcellular compartmentalization and targeting for specific interaction partners. Dynamic phosphorylation of Gle1A adds another essential layer to this complex array of regulatory mechanisms. This also reveals further potential key connections between SG and Gle1 dysfunction in neurodegenerative diseases, including ALS. Future studies will focus on combining structural and mechanistic analyses to further define how Gle1 hyperphosphorylation regulates individual steps in gene expression.
Experimental procedures
Cell culture, treatments, and reagents
HeLa cells were cultured in complete DMEM (Gibco) supplemented with 10% fetal bovine serum (Atlanta Biologicals, Flowery Branch, GA) at 37 °C incubator in 5% CO2. For heat shock stress, cells were incubated at 45 °C in the absence of CO2. To induce oxidative stress, cells were treated with 0.5 mm sodium arsenite for 60 min at 37 °C. Kinase inhibitors were obtained from the following sources: broad-spectrum kinase inhibitor K252a (Tocris, Minneapolis, MN), JNK inhibitor JNK-IN-8 (Calbiochem, Darmstadt, Germany), ERK inhibitor UO126 (Cell Signaling Technology, Danvers, MA) and MEK-ERK inhibitor FR 180204 (Tocris). AllStars negative control siRNA and GLE1 siRNAs were supplied by Qiagen (Germantown, MD).
Plasmids and transfections
Plasmids expressing GFP and siRNA-resistant GFP-GLE1A (pSW3909) were described previously (17). Plasmids expressing siRNA-resistant GFP-gle1A6A (pSW4360) and phosphomimetic GFP-gle1A6D (pSW4361) were generated by modification of pSW3909 using a QuikChange Multi Site-Directed Mutagenesis kit (Agilent Genomics, Santa Clara, CA). GFP-GLE1120–698, GFP-GLE11–400, and GFP-GLE1400–698 plasmids were constructed by PCR using cloned Pfu DNA polymerase (Agilent Genomics). GFP-gle1A-Finmajor plasmid (pSW4453) was constructed by restriction digestion of pSW3945 (21) and pSW3909 and ligation of products. Bacterial expression plasmids encoding His-MBP-Gle11–360 (pSW4392), His-MBP-gle11–360 6A (pSW4396), and His-MBP-gle11–360 6D (pSW4397) were generated by subcloning pSW3909, pSW4360, and pSW4361, respectively, into the bacterial expression vector pLM302 using Gibson Assembly (New England Biolabs, Ipswich, MA). To generate the bacterial expression plasmid encoding MBP-Gle1A-His, GLE1A was PCR-amplified from pSW3909 and subcloned into pAT107 by Gibson Assembly. The DDX3-HIS expression vector was made by subcloning DDX3 from pSW4105 (17) into the bacterial expression vector pET30A. siRNA and plasmid transfections were performed as described (17).
Immunoblotting, immunofluorescence, and SG quantification
Immunoblotting and indirect immunofluorescence were performed as described (17). Briefly, HeLa cells were plated on 1.5-mm round coverslips in a 24-well plate (Fisher Scientific) for immunofluorescence. Following transfection and 60-min heat shock treatment at 45 °C, cells were fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton X-100. Coverslips were blocked with 10% FBS in PBS prior to sequential incubations with anti-G3BP antibody (1:300; BD Transduction Laboratories) and Alexa Fluor–conjugated secondary antibodies (Life Technologies). Imaging was performed using a 63× 1.4 numerical aperture oil-immersion objective on a confocal microscope (Leica TCS SP5), and measurements of SG number and SG recovery were performed as described (17). Briefly, postimage processing was done using the ImageJ plug-in 3D object counter to quantify the number of SGs per cell. The 3D objects counter measurement parameters were set to “surface,” and the minimum size filter was set to 3. For SG recovery, the number of cells with SGs was determined as 100 × (Number of cells with G3BP-positive foci)/(Total number of cells). Prism6 (GraphPad, La Jolla, CA) was used to graph data from at least three independent experiments as mean ± S.E. and perform unpaired, two-tailed Student's t tests of statistical significance. For immunoblotting, the following antibodies were used: anti-Gle1 (ASW48.2; 1:1000) (17); anti-GFP (Life Technologies, catalogue number A6455; 1:1000); anti-actin (Sigma-Aldrich, catalogue number A5441; 1:5000); anti-lamin (Abcam, Cambridge, MA, catalogue number ab16048; 1:1000); and anti-phospho-eIF2α (Ser51, catalogue number 3398; 1:1000); anti-ERK1/2 (catalogue number 9102; 1:1000); anti-phospho ERK1/2 (Thr202/Tyr204; catalogue number 4370; 1:1000); anti-JNK (catalogue number 9252; 1:1000); anti-phospho JNK (Thr183/Tyr185; catalogue number 4668; 1:1000); anti-phospho 4EBP1 (Thr37/46; catalogue number 2855; 1:1000); and anti-GAPDH (14C10; catalogue number 2118; 1:1000) (all from Cell Signaling Technology). Phos-tag acrylamide was purchased from Wako Chemicals (Richmond, VA).
Fluorescence recovery after photobleaching
HeLa cells expressing mCHERRY-G3BP and GFP-GLE1A were heat-shocked at 45 °C for 45 min prior to FRAP assay and subsequently imaged for 45 min. A Zeiss LSM710 confocal microscope was prewarmed to 45 °C with 5% CO2; set to 1024 × 1024 frame size, eight-bit pixel depth, optical zoom of 3, and 1 airy unit; and used with a 63× objective. The region of interest (ROI) for photobleaching was defined using mCherry-G3BP as a marker of SGs. Three prebleach images were acquired, and then three bleaching iterations of the ROI were performed at a scan speed of 2 using a 561 nm laser (Fig. 6) or a 488 nm laser (Fig. S7), each at 100% intensity. Postbleach images were acquired at 200-ms intervals. Localization of mCherry-G3BP (Fig. 6) or GFP-Gle1A (Fig. S7) was used to confirm SG locations in postbleach images. Analyses of FRAP recovery were performed as described previously (21) using the following equation,
| (Eq. 1) |
where Fbkg is background fluorescence, F(t)ROI is intensity of bleaching ROI at each time point, F(t)cell is intensity of the entire cell at each time point, F(i)cell is initial whole-cell intensity, and F(i)ROI is initial bleaching ROI intensity. Nonregression analysis was used to determine the recovery kinetics and fitted to the following one-phase association equation using GraphPad Prism software,
| (Eq. 2) |
where Y0 is the Y value when X (time) is zero, Plateau is the Y value at infinite times, and K is the rate constant. Span is the difference between Y0 and Plateau and represents the mobile fraction, which was multiplied by 100 and expressed as a percentage of total fluorescence intensity. An unpaired two-tailed t test was used to test for statistical differences on half-life and rate constant.
Phos-tag gel electrophoresis
HeLa cells were lysed with radioimmunoprecipitation assay buffer (Sigma-Aldrich) supplemented with cOmplete EDTA-free protease inhibitor mixture (Roche Applied Science), 50 mm NaF, and 100 μm Zn2+. A 5.5% Phos-tag acrylamide gel was prepared according to the manufacturer's instructions, and lysates were resolved by electrophoresis at 25 mA for 5–6 h at room temperature. The gel was washed two times with Tris-glycine transfer buffer containing 2 mm EDTA, once with transfer buffer alone, and then wet transferred to nitrocellulose membrane overnight at 25 V at 4 °C. The membrane was blocked with 5% milk blocking buffer and processed for immunoblotting with the indicated antibodies.
λ-Phosphatase assay
Following the indicated treatments, equivalent cell numbers were lysed in a buffer containing 50 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1.5 mm MgCl2, 1% NP-40, and protease inhibitor mixture. Equal volumes of cell lysate were incubated at 30 °C for 30 min with phosphatase buffer alone (10× λ PPase buffer and 10× MnCl2) or λ-phosphatase enzyme (500 units; New England Biolabs) in the presence or absence of 50 mm NaF and 1× PhosSTOP (Roche Applied Science). Reactions were terminated by addition of 6× Laemmli sample denaturation buffer, and samples were analyzed by immunoblotting.
Subcellular fractionation
Following the indicated treatments, HeLa cells were trypsinized and collected by centrifugation at 100 × g for 3 min. Nuclear and cytosolic fractions were prepared using the NE-PER kit (Thermo Fisher) following the manufacturer's instructions. In brief, cell pellets were lysed in ice-cold CER I buffer on ice for 10 min. CER II buffer was added, and lysates were vortexed and further incubated for 1 min on ice. Samples were centrifuged at 16,000 × g for 7 min after which supernatants were transferred to new tubes and labeled as cytoplasmic fractions. The pellets were washed once in CER I buffer and then resuspended in NER solution to lyse the nuclei. Samples were centrifuged for 10 min at ∼16,000 × g, and supernatants were labeled as nuclear fractions.
B-isox precipitation of proteins
Nuclear and cytosolic fractions were incubated with 100 μm b-isox (Sigma-Aldrich) for 1 h at 4 °C with shaking. Following incubation, reactions were centrifuged at 16,000 × g for 15 min at 4 °C. Pellets were washed twice with 20 mm Tris-HCl (pH 7.5), 150 mm NaCl, 5 mm MgCl2, 20 mm β-mercaptoethanol, 0.5% NP-40, and protease inhibitors. Supernatant and pellets were resuspended in Laemmli sample denaturation buffer, heated at 95 °C for 5 min, and analyzed by immunoblotting.
In vitro kinase assay
For in vitro kinase assays with MAPKs, recombinant bacterially produced proteins were incubated with either 100 units of ERK enzyme (New England Biolabs) or 100 ng of JNK enzyme (Thermo Scientific, Waltham, MA) for 45 min at 30 °C in 30-μl reactions with the supplied kinase reaction buffer (New England Biolabs) supplemented with 100 μm ATP and 1 μCi of [γ-32P]ATP. Reactions were stopped by addition of Laemmli sample denaturation buffer, and then samples were heated at 95 °C for 5 min and resolved by 7.5% SDS-gel electrophoresis. Gels were dried and analyzed by autoradiography. 32P incorporation was detected and quantified with a Typhoon FLA7000 biomolecular imager (GE Healthcare). For in vitro kinase assays with GSK3, the indicated proteins were incubated with ERK and/or JNK in 30-μl reactions with kinase buffer supplemented with 100 μm ATP for 60 min at 30 °C. GSK-3 (125 units; New England Biolabs) and 1 μCi of [γ-32P]ATP were then added to the reactions in the presence or absence of ERK and or JNK inhibitors, and all reaction volumes were adjusted to 40 μl. After incubation for 45 min at 30 °C, reactions were terminated by addition of Laemmli sample denaturation buffer. Samples were heated at 95 °C for 5 min, resolved by SDS-PAGE, and analyzed by autoradiography.
Immunoprecipitation
24 h post-transfection, HeLa cells were either left untreated or treated with 0.5 mm sodium arsenite for 60 min at 37 °C. Cells were then lysed in 500 μl of lysis buffer containing 20 mm Tris-HCl (pH 7.5), 150 mm NaCl, 2 mm MgCl2, 1 mm EDTA, 0.5% NP-40, and protease and phosphatase inhibitors. Lysates were spun at 16,000 × g for 10 min at 4 °C. Agarose beads conjugated to anti-GFP VHH single domain antibody (GFP-TRAP, ChromoTek, Hauppauge, NY) were added to the supernatant, and the slurry was incubated with shaking for 2 h at 4 °C. Bound complexes were pelleted at 2500 × g for 1 min, washed five times with lysis buffer, and eluted with 2× Laemmli sample denaturation buffer. Proteins were resolved by 7.5% SDS-gel electrophoresis, and the gel was stained with colloidal blue. Protein bands corresponding to GFP-Gle1A were excised from the gel and processed for MS.
Mass spectrometry
Acrylamide gel samples containing GFP-Gle1A were cut into 1-mm3 pieces and treated with 45 mm DTT, and available Cys residues were carbamidomethylated with 100 mm iodoacetamide. The samples were destained with 50% MeCN in 25 mm ammonium bicarbonate, and proteins were digested with trypsin (10 ng/μl) in 25 mm ammonium bicarbonate overnight at 37 °C. Peptides were extracted by gel dehydration (60% MeCN, 0.1% TFA), the extract was dried by SpeedVac centrifugation, and peptides were reconstituted in 0.1% formic acid. The peptide solutions were then loaded onto a capillary reverse-phase analytical column (360-μm outer diameter × 100-μm inner diameter) using an Eksigent NanoLC HPLC and autosampler. The analytical column was packed with C18 reverse-phase resin (Jupiter, 3-μm beads, 300 Å, Phenomenex, Torrance, CA) directly into a laser-pulled emitter tip. Mobile-phase solvents consisted of 0.1% formic acid, 99.9% water (solvent A) and 0.1% formic acid, 99.9% acetonitrile (solvent B). A 90-min gradient was performed, and eluting peptides were mass-analyzed on an LTQ Orbitrap Velos mass spectrometer (Thermo Scientific) equipped with a nanoelectrospray ionization source. The instrument was operated using a data-dependent method with dynamic exclusion enabled. Full-scan (m/z 300–2000) spectra were acquired with the Orbitrap, and the top 16 most abundant ions in each MS scan were selected for fragmentation via collision-induced dissociation in the LTQ. An isolation width of 2 m/z, activation time of 10 ms, and 35% normalized collision energy were used to generate MS2 spectra. For additional LC-MS/MS analysis, the instrument was operated using a combination method of data-dependent (top 12) and targeted scan events. Targeted scans were acquired for m/z 985.43 and m/z 657.29, which correspond to phosphorylated Gle1 peptide SPDASSAFSPASPATPNGTK. For identification of peptides, tandem mass spectra were searched with Sequest (Thermo Scientific) against a human subset database created from the UniProtKB protein database. Variable modifications of +57.0214 on Cys (carbamidomethylation); +15.9949 on Met (oxidation); and +79.9663 on Ser, Thr, and Tyr (phosphorylation) were included for database searching. Search results were assembled using Scaffold 4.3.2 (Proteome Software, Portland, OR).
Protein purification
Plasmids were transformed into Escherichia coli Rosetta cells and cultured in Terrific Broth under kanamycin and chloramphenicol selection. Expression was induced with 0.2 mm isopropyl 1-thio-β-d-galactopyranoside at an A600 of 0.8 for 18 h at 18 °C. Bacteria were lysed by sonication in 20 mm HEPES (pH 7.5), 400 mm NaCl, 0.5 mm TCEP, and 20% glycerol supplemented with cOmplete EDTA-free protease inhibitor mixture and 2 mm phenylmethylsulfonyl fluoride. MBP-Gle1A1–360-His was purified by nickel-immobilized metal affinity chromatography (IMAC). His-PPS was added to the pooled peak elution fractions to cleave the His-MBP tag, and fractions were dialyzed overnight to remove imidazole. Gle1A1–360 was then separated from the tag and His-PPS by nickel-IMAC (Thermo Scientific) and applied to an S200 column equilibrated with buffer containing 20 mm HEPES (pH 7.5), 150 mm NaCl, 20% glycerol, and 0.5 mm TCEP for final purification. His-MBP–tagged full-length Gle1A proteins were purified by amylose resin chromatography according to the manufacturer's recommendations followed by nickel-IMAC. His-tagged DDX3 was purified by nickel-IMAC and dialyzed overnight in buffer containing 20 mm HEPES (pH 7.5), 50 mm NaCl, 0.5 mm TCEP, 2 mm MgCl2, and 20% glycerol. The protein was further purified by heparin chromatography and eluted with a salt gradient from 50 to 1000 mm NaCl.
Electron microscopy
Uranyl formate–stained samples were prepared as described (85). Samples were imaged on an FEI Morgagni electron microscope operated at an acceleration voltage of 100 kV. Images were recorded at a magnification of 28,000× and collected using a 1000 × 1000 charge-coupled device camera (ATM). Particle shape was quantified by taking the ratio of the longest axis (major axis) and the axis perpendicular to the major axis (minor axis).
ATPase assay
Colorimetric enzyme-coupled ATPase rate assays were performed as described (86). Briefly, reaction mixtures (100 μl) containing 40 mm Tris-HCl (pH 8.0); 50 mm NaCl; 0.5 mm MgCl2; 1 mm DTT; 20 units of SUPERase In RNase inhibitor; 0.01% (v/v) Nonidet P-40; 6 mm phosphoenolpyruvate; 1.2 mm NADH; 2.5 mm ATP; 2.1 units of pyruvate kinase/lactate dehydrogenase; 0.5 μm RNA–DNA hairpin oligo; 200 nm DDX3; 1 μm IP6 (as indicated); and 200, 400, or 800 nm Gle1A. A340 was monitored every 40 s for 40 min at 37 °C in a BioTek Synergy HT microplate reader.
Author contributions
A. and S. R. W. conceptualization; A., A. C. M., and M. S. data curation; A., A. C. M., and M. S. formal analysis; A., A. C. M., and M. S. investigation; A., A. C. M., and M. S. methodology; A. writing-original draft; A. C. M., M. S., T. R. D., and S. R. W. writing-review and editing; T. R. D. and S. R. W. supervision; T. R. D. and S. R. W. project administration; S. R. W. funding acquisition.
Supplementary Material
Acknowledgments
We thank members of the Wente laboratory for technical assistance, discussions, and reading the manuscript. We acknowledge Kristie Rose, Ph.D., and the staff of the Proteomics Core in the Vanderbilt Mass Spectrometry Research Center for assistance with analysis of our samples. The LTQ Orbitrap Velos mass spectrometer used in these studies was purchased with funds from National Institutes of Health S10 Grant RR027714 awarded to the Vanderbilt Proteomics Shared Resource. Microscopy experiments were performed in part through the use of the Vanderbilt Cell Imaging Shared Resource, supported by National Institutes of Health Grants CA68485, DK20593, DK58404, DK59637, and EY08126.
This work was supported by National Institutes of Health Grant 5R37GM051219 (to S. R. W.) and the Vanderbilt International Scholar Program. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S7.
- mRNP
- messenger ribonucleoprotein
- NPC
- nuclear pore complex
- SG
- stress granule
- MAPK
- mitogen-activated protein kinase
- GSK3
- glycogen synthase kinase 3
- ERK
- extracellular signal-regulated kinase
- JNK
- c-Jun N-terminal kinase
- DDX
- DEAD-box helicase (X-linked)
- IP6
- inositol hexakisphosphate
- LCCS1
- lethal congenital contracture syndrome 1
- λ-PPase
- λ-phosphatase
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- b-isox
- biotin-isoxazole
- FUS
- fused in sarcoma
- CTRL
- control
- FRAP
- fluorescence recovery after photobleaching
- 4EBP
- eIF4E-binding protein
- ROI
- region of interest
- NP-40
- Nonidet P-40
- CER
- cytoplasmic extraction reagent
- NER
- nuclear extraction reagent
- TCEP
- tris(2-carboxyethyl)phosphine
- IMAC
- immobilized metal affinity chromatography
- MBP
- maltose-binding protein
- PTM
- post-translational modification
- PPS
- human rhinovirus 3C protease.
References
- 1. Sheinberger J., and Shav-Tal Y. (2017) mRNPs meet stress granules. FEBS Lett. 591, 2534–2542 10.1002/1873-3468.12765 [DOI] [PubMed] [Google Scholar]
- 2. Anderson P., and Kedersha N. (2008) Stress granules: the Tao of RNA triage. Trends Biochem. Sci. 33, 141–150 10.1016/j.tibs.2007.12.003 [DOI] [PubMed] [Google Scholar]
- 3. Buchan J. R., and Parker R. (2009) Eukaryotic stress granules: the ins and out of translation. Mol. Cell 36, 932–941 10.1016/j.molcel.2009.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Protter D. S. W., and Parker R. (2016) Principles and properties of stress granules. Trends Cell Biol. 26, 668–679 10.1016/j.tcb.2016.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thomas M. G., Loschi M., Desbats M. A., and Boccaccio G. L. (2011) RNA granules: the good, the bad and the ugly. Cell. Signal. 23, 324–334 10.1016/j.cellsig.2010.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Buchan J. R., Kolaitis R.-M., Taylor J. P., and Parker R. (2013) Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 153, 1461–1474 10.1016/j.cell.2013.05.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Patel A., Lee H. O., Jawerth L., Maharana S., Jahnel M., Hein M. Y., Stoynov S., Mahamid J., Saha S., Franzmann T. M., Pozniakovski A., Poser I., Maghelli N., Royer L. A., Weigert M., et al. (2015) A Liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162, 1066–1077 10.1016/j.cell.2015.07.047 [DOI] [PubMed] [Google Scholar]
- 8. Ramaswami M., Taylor J. P., and Parker R. (2013) Altered “ribostasis”: RNA-protein granule formation or persistence in the development of degenerative disorders. Cell 154, 727–736 10.1016/j.cell.2013.07.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wolozin B. (2012) Regulated protein aggregation: stress granules and neurodegeneration. Mol. Neurodegener. 7, 56–56 10.1186/1750-1326-7-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lin Y., Protter D. S., Rosen M. K., and Parker R. (2015) Formation and maturation of phase-separated liquid droplets by RNA-binding proteins. Mol. Cell 60, 208–219 10.1016/j.molcel.2015.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Molliex A., Temirov J., Lee J., Coughlin M., Kanagaraj A. P., Kim H. J., Mittag T., and Taylor J. P. (2015) Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163, 123–133 10.1016/j.cell.2015.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Arimoto K., Fukuda H., Imajoh-Ohmi S., Saito H., and Takekawa M. (2008) Formation of stress granules inhibits apoptosis by suppressing stress-responsive MAPK pathways. Nat. Cell Biol. 10, 1324–1332 10.1038/ncb1791 [DOI] [PubMed] [Google Scholar]
- 13. Tourrière H., Chebli K., Zekri L., Courselaud B., Blanchard J. M., Bertrand E., and Tazi J. (2003) The RasGAP-associated endoribonuclease G3BP assembles stress granules. J. Cell Biol. 160, 823–831 10.1083/jcb.200212128 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14. Reineke L. C., Tsai W.-C., Jain A., Kaelber J. T., Jung S. Y., and Lloyd R. E. (2017) Casein kinase 2 is linked to stress granule dynamics through phosphorylation of the stress granule nucleating protein G3BP1. Mol. Cell. Biol. 37, e00596-16 10.1128/MCB.00596-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim E. K., and Choi E.-J. (2010) Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 1802, 396–405 10.1016/j.bbadis.2009.12.009 [DOI] [PubMed] [Google Scholar]
- 16. Jain S., Wheeler J. R., Walters R. W., Agrawal A., Barsic A., and Parker R. (2016) ATPase-modulated stress granules contain a diverse proteome and substructure. Cell 164, 487–498 10.1016/j.cell.2015.12.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Aditi, Folkmann A. W., and Wente S. R. (2015) Cytoplasmic hGle1A regulates stress granules by modulation of translation. Mol. Biol. Cell 26, 1476–1490 10.1091/mbc.E14-11-1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Alcázar-Román A. R., Tran E. J., Guo S., and Wente S. R. (2006) Inositol hexakisphosphate and Gle1 activate the DEAD-box protein Dbp5 for nuclear mRNA export. Nat. Cell Biol. 8, 711–716 10.1038/ncb1427 [DOI] [PubMed] [Google Scholar]
- 19. Bolger T. A., and Wente S. R. (2011) Gle1 is a multifunctional DEAD-box protein regulator that modulates Ded1 in translation initiation. J. Biol. Chem. 286, 39750–39759 10.1074/jbc.M111.299321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bolger T. A., Folkmann A. W., Tran E. J., and Wente S. R. (2008) The mRNA export factor Gle1 and inositol hexakisphosphate regulate distinct stages of translation. Cell 134, 624–633 10.1016/j.cell.2008.06.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Folkmann A. W., Collier S. E., Zhan X., Aditi, Ohi M. D., and Wente S. R. (2013) Gle1 functions during mRNA export in an oligomeric complex that is altered in human disease. Cell 155, 582–593 10.1016/j.cell.2013.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jao L.-E., Akef A., and Wente S. R. (2017) A role for Gle1, a regulator of DEAD-box RNA helicases, at centrosomes and basal bodies. Mol. Biol. Cell 28, 120–127 10.1091/mbc.e16-09-0675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Montpetit B., Thomsen N. D., Helmke K. J., Seeliger M. A., Berger J. M., and Weis K. (2011) A conserved mechanism of DEAD-box ATPase activation by nucleoporins and InsP6 in mRNA export. Nature 472, 238–242 10.1038/nature09862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Weirich C. S., Erzberger J. P., Flick J. S., Berger J. M., Thorner J., and Weis K. (2006) Activation of the DExD/H-box protein Dbp5 by the nuclear-pore protein Gle1 and its coactivator InsP6 is required for mRNA export. Nat. Cell Biol. 8, 668–676 10.1038/ncb1424 [DOI] [PubMed] [Google Scholar]
- 25. Kendirgi F., Barry D. M., Griffis E. R., Powers M. A., and Wente S. R. (2003) An essential role for hGle1 nucleocytoplasmic shuttling in mRNA export. J. Cell Biol. 160, 1029–1040 10.1083/jcb.200211081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Adams R. L., Mason A. C., Glass L., Aditi, and Wente S. R. (2017) Nup42 and IP6 coordinate Gle1 stimulation of Dbp5/DDX19B for mRNA export in yeast and human cells. Traffic 18, 776–790 10.1111/tra.12526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rayala H. J., Kendirgi F., Barry D. M., Majerus P. W., and Wente S. R. (2004) The mRNA export factor human Gle1 interacts with the nuclear pore complex protein Nup155. Mol. Cell. Proteomics 3, 145–155 10.1074/mcp.M300106-MCP200 [DOI] [PubMed] [Google Scholar]
- 28. Aditi, Glass L., Dawson T. R., and Wente S. R. (2016) An amyotrophic lateral sclerosis-linked mutation in GLE1 alters the cellular pool of human Gle1 functional isoforms. Adv. Biol. Regul. 62, 25–36 10.1016/j.jbior.2015.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Al-Qattan M. M., Shamseldin H. E., and Alkuraya F. S. (2012) Familial dorsalization of the skin of the proximal palm and the instep of the sole of the foot. Gene 500, 216–219 10.1016/j.gene.2012.03.009 [DOI] [PubMed] [Google Scholar]
- 30. Kaneb H. M., Folkmann A. W., Belzil V. V., Jao L.-E., Leblond C. S., Girard S. L., Daoud H., Noreau A., Rochefort D., Hince P., Szuto A., Levert A., Vidal S., André-Guimont C., Camu W., et al. (2015) Deleterious mutations in the essential mRNA metabolism factor, hGle1, in amyotrophic lateral sclerosis. Hum. Mol. Genet. 24, 1363–1373 10.1093/hmg/ddu545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nousiainen H. O., Kestilä M., Pakkasjärvi N., Honkala H., Kuure S., Tallila J., Vuopala K., Ignatius J., Herva R., and Peltonen L. (2008) Mutations in mRNA export mediator GLE1 result in a fetal motoneuron disease. Nat. Genet. 40, 155–157 10.1038/ng.2007.65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pont M. J., van der Lee D. I., van der Meijden E. D., van Bergen C. A., Kester M. G., Honders M. W., Vermaat M., Eefting M., Marijt E. W., Kielbasa S. M., Hoen P. A., Falkenburg J. H., and Griffioen M. (2016) Integrated whole genome and transcriptome analysis identified a therapeutic minor histocompatibility antigen in a splice variant of ITGB2. Clin. Cancer Res. 22, 4185–4196 10.1158/1078-0432.CCR-15-2307 [DOI] [PubMed] [Google Scholar]
- 33. Tzschach A., Grasshoff U., Schäferhoff K., Bonin M., Dufke A., Wolff M., Haas-Lude K., Bevot A., and Riess O. (2012) Interstitial 9q34.11-q34.13 deletion in a patient with severe intellectual disability, hydrocephalus, and cleft lip/palate. Am. J. Med. Genet. A 158A, 1709–1712 10.1002/ajmg.a.35398 [DOI] [PubMed] [Google Scholar]
- 34. Gasset-Rosa F., Chillon-Marinas C., Goginashvili A., Atwal R. S., Artates J. W., Tabet R., Wheeler V. C., Bang A. G., Cleveland D. W., and Lagier-Tourenne C. (2017) Polyglutamine-expanded huntingtin exacerbates age-related disruption of nuclear integrity and nucleocytoplasmic transport. Neuron 94, 48–57.e4 10.1016/j.neuron.2017.03.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kinoshita E., Kinoshita-Kikuta E., Takiyama K., and Koike T. (2006) Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol. Cell. Proteomics 5, 749–757 10.1074/mcp.T500024-MCP200 [DOI] [PubMed] [Google Scholar]
- 36. Panas M. D., Ivanov P., and Anderson P. (2016) Mechanistic insights into mammalian stress granule dynamics. J. Cell Biol. 215, 313–323 10.1083/jcb.201609081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gebauer F., and Hentze M. W. (2004) Molecular mechanisms of translational control. Nat. Rev. Mol. Cell Biol. 5, 827–835 10.1038/nrm1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sonenberg N., and Hinnebusch A. G. (2009) Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136, 731–745 10.1016/j.cell.2009.01.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Spriggs K. A., Bushell M., and Willis A. E. (2010) Translational regulation of gene expression during conditions of cell stress. Mol. Cell 40, 228–237 10.1016/j.molcel.2010.09.028 [DOI] [PubMed] [Google Scholar]
- 40. Mebratu Y., and Tesfaigzi Y. (2009) How ERK1/2 activation controls cell proliferation and cell death: is subcellular localization the answer? Cell Cycle 8, 1168–1175 10.4161/cc.8.8.8147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stasyk T., Schiefermeier N., Skvortsov S., Zwierzina H., Peränen J., Bonn G. K., and Huber L. A. (2007) Identification of endosomal epidermal growth factor receptor signaling targets by functional organelle proteomics. Mol. Cell. Proteomics. 6, 908–922 10.1074/mcp.M600463-MCP200 [DOI] [PubMed] [Google Scholar]
- 42. Jackson R. J., Hellen C. U., and Pestova T. V. (2010) The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 11, 113–127 10.1038/nrm2838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Iakoucheva L. M., Radivojac P., Brown C. J., O'Connor T. R., Sikes J. G., Obradovic Z., and Dunker A. K. (2004) The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 32, 1037–1049 10.1093/nar/gkh253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wright P. E., and Dyson H. J. (2015) Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 16, 18–29 10.1038/nrm3920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Uversky V. N. (2017) The roles of intrinsic disorder-based liquid-liquid phase transitions in the “Dr. Jekyll-Mr. Hyde” behavior of proteins involved in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Autophagy 13, 2115–2162 10.1080/15548627.2017.1384889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Klammer M., Kaminski M., Zedler A., Oppermann F., Blencke S., Marx S., Müller S., Tebbe A., Godl K., and Schaab C. (2012) Phosphosignature predicts dasatinib response in non-small cell lung cancer. Mol. Cell. Proteomics 11, 651–668 10.1074/mcp.M111.016410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sharma K., D'Souza R. C., Tyanova S., Schaab C., Wiśniewski J. R., Cox J., and Mann M. (2014) Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep. 8, 1583–1594 10.1016/j.celrep.2014.07.036 [DOI] [PubMed] [Google Scholar]
- 48. Zhou H., Di Palma S., Preisinger C., Peng M., Polat A. N., Heck A. J., and Mohammed S. (2013) Toward a comprehensive characterization of a human cancer cell phosphoproteome. J. Proteome Res. 12, 260–271 10.1021/pr300630k [DOI] [PubMed] [Google Scholar]
- 49. Adzhubei A. A., and Sternberg M. J. (1993) Left-handed polyproline II helices commonly occur in globular proteins. J. Mol. Biol. 229, 472–493 10.1006/jmbi.1993.1047 [DOI] [PubMed] [Google Scholar]
- 50. Ren S., Uversky V. N., Chen Z., Dunker A. K., and Obradovic Z. (2008) Short linear motifs recognized by SH2, SH3 and Ser/Thr kinase domains are conserved in disordered protein regions. BMC Genomics 9, Suppl. 2, S26 10.1186/1471-2164-9-S2-S26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tyanova S., Cox J., Olsen J., Mann M., and Frishman D. (2013) Phosphorylation variation during the cell cycle scales with structural propensities of proteins. PLoS Comput. Biol. 9, e1002842 10.1371/journal.pcbi.1002842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Han T. W., Kato M., Xie S., Wu L. C., Mirzaei H., Pei J., Chen M., Xie Y., Allen J., Xiao G., and McKnight S. L. (2012) Cell-free formation of RNA granules: bound RNAs identify features and components of cellular assemblies. Cell 149, 768–779 10.1016/j.cell.2012.04.016 [DOI] [PubMed] [Google Scholar]
- 53. Kwon I., Kato M., Xiang S., Wu L., Theodoropoulos P., Mirzaei H., Han T., Xie S., Corden J. L., and McKnight S. L. (2013) Phosphorylation-regulated binding of RNA polymerase II to fibrous polymers of low-complexity domains. Cell 155, 1049–1060 10.1016/j.cell.2013.10.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kato M., Han T. W., Xie S., Shi K., Du X., Wu L. C., Mirzaei H., Goldsmith E. J., Longgood J., Pei J., Grishin N. V., Frantz D. E., Schneider J. W., Chen S., Li L., et al. (2012) Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell 149, 753–767 10.1016/j.cell.2012.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Saad S., Cereghetti G., Feng Y., Picotti P., Peter M., and Dechant R. (2017) Reversible protein aggregation is a protective mechanism to ensure cell cycle restart after stress. Nat. Cell Biol. 19, 1202–1213 10.1038/ncb3600 [DOI] [PubMed] [Google Scholar]
- 56. Monahan Z., Ryan V. H., Janke A. M., Burke K. A., Rhoads S. N., Zerze G. H., O'Meally R., Dignon G. L., Conicella A. E., Zheng W., Best R. B., Cole R. N., Mittal J., Shewmaker F., and Fawzi N. L. (2017) Phosphorylation of the FUS low-complexity domain disrupts phase separation, aggregation, and toxicity. EMBO J. 36, 2951–2967 10.15252/embj.201696394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Morrison D. K. (2012) MAP kinase pathways. Cold Spring Harb. Perspect. Biol. 4, a011254 10.1101/cshperspect.a011254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhang W., and Liu H. T. (2002) MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 12, 9–18 10.1038/sj.cr.7290105 [DOI] [PubMed] [Google Scholar]
- 59. Zinck R., Cahill M. A., Kracht M., Sachsenmaier C., Hipskind R. A., and Nordheim A. (1995) Protein synthesis inhibitors reveal differential regulation of mitogen-activated protein kinase and stress-activated protein kinase pathways that converge on Elk-1. Mol. Cell. Biol. 15, 4930–4938 10.1128/MCB.15.9.4930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ingolia N. T. (2016) Ribosome footprint profiling of translation throughout the genome. Cell 165, 22–33 10.1016/j.cell.2016.02.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cohen P., and Frame S. (2001) The renaissance of GSK3. Nat. Rev. Mol. Cell Biol. 2, 769–776 10.1038/35096075 [DOI] [PubMed] [Google Scholar]
- 62. Shih J. W., Wang W. T., Tsai T. Y., Kuo C. Y., Li H. K., and Wu Lee Y. H. (2012) Critical roles of RNA helicase DDX3 and its interactions with eIF4E/PABP1 in stress granule assembly and stress response. Biochem. J. 441, 119–129 10.1042/BJ20110739 [DOI] [PubMed] [Google Scholar]
- 63. Bley N., Lederer M., Pfalz B., Reinke C., Fuchs T., Glaß M., Möller B., and Hüttelmaier S. (2015) Stress granules are dispensable for mRNA stabilization during cellular stress. Nucleic Acids Res. 43, e26 10.1093/nar/gku1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kedersha N., Stoecklin G., Ayodele M., Yacono P., Lykke-Andersen J., Fritzler M. J., Scheuner D., Kaufman R. J., Golan D. E., and Anderson P. (2005) Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J. Cell Biol. 169, 871–884 10.1083/jcb.200502088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sama R. R., Fallini C., Gatto R., McKeon J. E., Song Y., Rotunno M. S., Penaranda S., Abdurakhmanov I., Landers J. E., Morfini G., Brady S. T., and Bosco D. A. (2017) ALS-linked FUS exerts a gain of toxic function involving aberrant p38 MAPK activation. Sci. Rep. 7, 115 10.1038/s41598-017-00091-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bhinge A., Namboori S. C., Zhang X., VanDongen A. M. J., and Stanton L. W. (2017) Genetic correction of SOD1 mutant iPSCs reveals ERK and JNK activated AP1 as a driver of neurodegeneration in amyotrophic lateral sclerosis. Stem Cell Reports 8, 856–869 10.1016/j.stemcr.2017.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Moujalled D., James J. L., Parker S. J., Lidgerwood G. E., Duncan C., Meyerowitz J., Nonaka T., Hasegawa M., Kanninen K. M., Grubman A., Liddell J. R., Crouch P. J., and White A. R. (2013) Kinase inhibitor screening identifies cyclin-dependent kinases and glycogen synthase kinase 3 as potential modulators of TDP-43 cytosolic accumulation during cell Stress. PLoS One 8, e67433 10.1371/journal.pone.0067433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ambegaokar S. S., and Jackson G. R. (2011) Functional genomic screen and network analysis reveal novel modifiers of tauopathy dissociated from tau phosphorylation. Hum. Mol. Genet. 20, 4947–4977 10.1093/hmg/ddr432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. de Munck E., Palomo V., Muñoz-Sáez E., Perez D. I., Gómez-Miguel B., Solas M. T., Gil C., Martínez A., and Arahuetes R. M. (2016) Small GSK-3 inhibitor shows efficacy in a motor neuron disease murine model modulating autophagy. PLoS One 11, e0162723 10.1371/journal.pone.0162723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yang Y. M., Gupta S. K., Kim K. J., Powers B. E., Cerqueira A., Wainger B. J., Ngo H. D., Rosowski K. A., Schein P. A., Ackeifi C. A., Arvanites A. C., Davidow L. S., Woolf C. J., and Rubin L. L. (2013) A small molecule screen in stem-cell-derived motor neurons identifies a kinase inhibitor as a candidate therapeutic for ALS. Cell Stem Cell 12, 713–726 10.1016/j.stem.2013.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Shih J.-W., Tsai T.-Y., Chao C.-H., and Wu Lee Y.-H. (2008) Candidate tumor suppressor DDX3 RNA helicase specifically represses cap-dependent translation by acting as an eIF4E inhibitory protein. Oncogene 27, 700–714 10.1038/sj.onc.1210687 [DOI] [PubMed] [Google Scholar]
- 72. Soto-Rifo R., Rubilar P. S., and Ohlmann T. (2013) The DEAD-box helicase DDX3 substitutes for the cap-binding protein eIF4E to promote compartmentalized translation initiation of the HIV-1 genomic RNA. Nucleic Acids Res. 41, 6286–6299 10.1093/nar/gkt306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Oh S., Flynn R. A., Floor S. N., Purzner J., Martin L., Do B. T., Schubert S., Vaka D., Morrissy S., Li Y., Kool M., Hovestadt V., Jones D. T., Northcott P. A., Risch T., et al. (2016) Medulloblastoma-associated DDX3 variant selectively alters the translational response to stress. Oncotarget 7, 28169–28182 10.18632/oncotarget.8612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Soto-Rifo R., and Ohlmann T. (2013) The role of the DEAD-box RNA helicase DDX3 in mRNA metabolism. Wiley Interdiscip. Rev. RNA 4, 369–385 10.1002/wrna.1165 [DOI] [PubMed] [Google Scholar]
- 75. Andreou A. Z., and Klostermeier D. (2014) eIF4B and eIF4G jointly stimulate eIF4A ATPase and unwinding activities by modulation of the eIF4A conformational cycle. J. Mol. Biol. 426, 51–61 10.1016/j.jmb.2013.09.027 [DOI] [PubMed] [Google Scholar]
- 76. Mazroui R., Sukarieh R., Bordeleau M.-E., Kaufman R. J., Northcote P., Tanaka J., Gallouzi I., and Pelletier J. (2006) Inhibition of ribosome recruitment induces stress granule formation independently of eukaryotic initiation factor 2α phosphorylation. Mol. Biol. Cell. 17, 4212–4219 10.1091/mbc.e06-04-0318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Mugler C. F., Hondele M., Heinrich S., Sachdev R., Vallotton P., Koek A. Y., Chan L. Y., and Weis K. (2016) ATPase activity of the DEAD-box protein Dhh1 controls processing body formation. eLife 5, e18746 10.7554/eLife.18746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kedersha N., Ivanov P., and Anderson P. (2013) Stress granules and cell signaling: more than just a passing phase? Trends Biochem. Sci. 38, 494–506 10.1016/j.tibs.2013.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Aguzzi A., and Altmeyer M. (2016) Phase separation: linking cellular compartmentalization to disease. Trends Cell Biol. 26, 547–558 10.1016/j.tcb.2016.03.004 [DOI] [PubMed] [Google Scholar]
- 80. Nishi H., Hashimoto K., and Panchenko A. R. (2011) Phosphorylation in protein-protein binding: effect on stability and function. Structure 19, 1807–1815 10.1016/j.str.2011.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Xin F., and Radivojac P. (2012) Post-translational modifications induce significant yet not extreme changes to protein structure. Bioinformatics 28, 2905–2913 10.1093/bioinformatics/bts541 [DOI] [PubMed] [Google Scholar]
- 82. Putnam A. A., Gao Z., Liu F., Jia H., Yang Q., and Jankowsky E. (2015) Division of labor in an oligomer of the DEAD-Box RNA helicase Ded1p. Mol. Cell 59, 541–552 10.1016/j.molcel.2015.06.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hanazawa M., Yonetani M., and Sugimoto A. (2011) PGL proteins self associate and bind RNPs to mediate germ granule assembly in C. elegans. J. Cell Biol. 192, 929–937 10.1083/jcb.201010106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Knott G. J., Bond C. S., and Fox A. H. (2016) The DBHS proteins SFPQ, NONO and PSPC1: a multipurpose molecular scaffold. Nucleic Acids Res. 44, 3989–4004 10.1093/nar/gkw271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ohi M., Li Y., Cheng Y., and Walz T. (2004) Negative staining and image classification—powerful tools in modern electron microscopy. Biol. Proced. Online 6, 23–34 10.1251/bpo70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kornberg A., and Pricer W. E. Jr. (1951) Enzymatic phosphorylation of adenosine and 2,6-diaminopurine riboside. J. Biol. Chem. 193, 481–495 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.