

Abstract
NADH–quinone oxidoreductase (respiratory complex I) couples NADH-to-quinone electron transfer to the translocation of protons across the membrane. Even though the architecture of the quinone-access channel in the enzyme has been modeled by X-ray crystallography and cryo-EM, conflicting findings raise the question whether the models fully reflect physiologically relevant states present throughout the catalytic cycle. To gain further insights into the structural features of the binding pocket for quinone/inhibitor, we performed chemical biology experiments using bovine heart sub-mitochondrial particles. We synthesized ubiquinones that are oversized (SF–UQs) or lipid-like (PC–UQs) and are highly unlikely to enter and transit the predicted narrow channel. We found that SF–UQs and PC–UQs can be catalytically reduced by complex I, albeit only at moderate or low rates. Moreover, quinone-site inhibitors completely blocked the catalytic reduction and the membrane potential formation coupled to this reduction. Photoaffinity-labeling experiments revealed that amiloride-type inhibitors bind to the interfacial domain of multiple core subunits (49 kDa, ND1, and PSST) and the 39-kDa supernumerary subunit, although the latter does not make up the channel cavity in the current models. The binding of amilorides to the multiple target subunits was remarkably suppressed by other quinone-site inhibitors and SF–UQs. Taken together, the present results are difficult to reconcile with the current channel models. On the basis of comprehensive interpretations of the present results and of previous findings, we discuss the physiological relevance of these models.
Keywords: bioenergetics, mitochondria, Complex I, ubiquinone, enzyme inhibitor, chemical biology, photoaffinity labeling, amilorides, NADH–quinone oxidoreductase
Introduction
Proton-translocating NADH–quinone oxidoreductase (respiratory complex I) couples electron transfer from NADH to the quinone with the translocation of protons across the membrane. The electrochemical proton gradient produced by the enzyme drives energy-consuming reactions, such as ATP synthesis and substrate transport (1–3). Complex I3 is the largest of the respiratory chain enzymes and one of the major sources of superoxide production in mammalian mitochondria (4). The X-ray crystallographic structures of complex I from Thermus thermophilus (5) and Yarrowia lipolytica (6) were modeled at resolutions of 3.3 and 3.6 Å, respectively. The entire structures of mammalian complex I, including all 45 subunits (31 of which are the supernumerary subunits), from bovine (Bos taurus) (7, 8), ovine (Ovis aries) (9), porcine (Sus scrofa domesticus) (10), and mouse (Mus musculus) (11) hearts and human (Homo sapiens) HEK293F cells (12) were modeled by single-particle cryo-electron microscopy (cryo-EM). One of the striking findings obtained with T. thermophilus complex I (5) is the identification of a long and narrow channel, which extends from the membrane interior to the Fe-S cluster N2 (∼30 Å long) and is a completely enclosed tunnel with only a narrow entry point (∼3 × 5 Å diameter) for quinone/inhibitors; however, this has not yet been confirmed experimentally. Moreover, it was revealed that the link continues over the membrane domain as the central axis of potentially ionized or protonated residues (5), which may play critical roles in the transmission of conformational charges initially caused by the quinone reduction and in proton translocation across the membrane. Similar structural models were reported for yeast and mammalian complex I (6–12). These developments in structural works have led to the consensus that the quinone reduction deep in the predicted quinone-access channel plays a key role in the energy conversion processes; however, the mechanism responsible for the processes remains largely elusive.
The unique structure of the quinone-access channel was first modeled in T. thermophilus complex I (5). Because the so-called quinone-site inhibitors are considered to bind to the channel interior (5, 6, 13), we hereafter refer to this channel as the “quinone/inhibitor-access channel.” The narrow entry point in the membrane interior was framed by TMH1, TMH6, and amphipathic α-helix1 from the Nqo8 subunit (ND1 in the bovine enzyme) and TMH1 from the Nqo7 subunit (ND3). The channel is sufficiently long to accommodate ubiquinones (UQs) having seven to nine isoprenyl tails. Different laboratories reported similar architectures for the channel in yeast (6), bovine (7), ovine (9), and mouse (11) complex I; however, the channels were considerably shorter in yeast and ovine enzymes than in bacterial and bovine enzymes because the inner part of the channel around some functionally critical amino acid residues (e.g. His-59 and Tyr-108 in the 49-kDa subunit) was closed by the β1–β2 loop of the 49-kDa subunit. From this, the yeast and ovine enzymes were supposed to be in the deactive state. Hirst and co-workers (8, 11) recently reported that the structural changes accompanying deactivation may be common to the bovine and mouse enzymes. Considering the unusually long substrate-binding channel, definitions of how UQs of varying isoprenyl chain length (UQ1–UQ10) enter and transit the channel to be reduced, thereby eliciting the same proton-pumping stoichiometry, remain elusive (13, 14).
The findings of chemical biology studies previously conducted in our laboratory (15–18) via different techniques using bovine heart SMPs are difficult to be reconciled with the quinone/inhibitor-access channel models (5–11), as summarized under the “Discussion.” Therefore, our studies raise the question of whether the channel models fully reflect physiologically relevant states present throughout the catalytic cycle. In this context, it is important to note that the channel in the static state was postulated to undergo structural rearrangement to allow UQs to move into and out of the channel because the planar quinone head-ring is wider (∼6 Å across) than the diameter of the entry point (5, 11). We herein performed experiments from different two angles. First, we examined whether complex I catalyzes the reduction of oversized or lipid-like UQs (SF–UQs and PC–UQs, respectively, Fig. 1), which are highly unlikely to enter and transit the predicted channel (∼30 Å long) due to extensive physical restrictions. Second, because the photoreactive amiloride PRA1 (Fig. 2) was shown to label a supernumerary subunit (not a core subunit) (17), the binding positions of a series of amiloride-type inhibitors were further investigated by a photoaffinity labeling technique. The reasons why we selected these two subjects are as follows.
Figure 1.

Structures of SF–UQs and PC–UQs synthesized in this study. Other reagents mentioned in the text are also shown. As an index of the hydrophobicities of nonionized forms of SF–UQs, their retention times (minutes) in the HPLC analysis are listed in parentheses (see “Experimental procedures”). The average retention times of UQ3 and UQ4 were 14.0 and 17.8 min, respectively. Side views of the ubiquinone head-ring, 2,6-di-tert-butylphenol, and 2,6-di(TMS-1,3-butadiynyl)phenol are shown in a space-filling model (oxygen atoms are colored in red). The lengths of the models indicate the distances between the corresponding two (C)–H atoms.
Figure 2.

Structures of photoreactive amilorides synthesized in this study. PRA1 and PRA2 used in our previous work (17) are also shown. A photolabile azido group attached to each compound is indicated by a gray circle. The concentration in parentheses is the average IC50 value, which is the molar concentration (nanomolar) needed to reduce the control NADH oxidase activity in bovine heart SMPs (30 μg of proteins/ml) by 50%. As a reference, the IC50 value of bullatacin was 1.1 (± 0.09) nm under the same experimental conditions.
According to the current channel models (5–9), complex I only catalyzes the reduction of UQs that enter and transit the narrow channel to the reaction site near the Fe-S cluster N2 located at the “top” of the channel. Given this convincing experiment to verify the physiological relevance of the models may prove that the enzyme is indeed unable to catalyze the reduction of UQs, which are incapable of reaching the reaction site for some physical reason. Therefore, to prevent the quinone head-ring from reaching the reaction site by physical factors, we synthesized two types of UQs: oversized UQs that have a bulky “block” in the terminus of the short side chain (SF–UQs, Fig. 1) and a hybrid of phosphatidylcholine (PC) and UQ (PC–UQs, Fig. 1), in which the UQ head-ring is attached to the terminus of the sn-2 acyl chain. Detailed design concepts for these UQs are described at the beginning of “Results.”
Concerning the second subject, amiloride-type inhibitors were once considered to bind to any or all Na+/H+-antiporter–like subunits of complex I (i.e. the ND2, ND4, and ND5 subunits) (19, 20). However, we previously demonstrated using a photoaffinity-labeling technique that amilorides do not bind to any of the antiporter-like subunits; they specifically bind to the interface of the hydrophilic and membrane arms (17). To be more specific, the photoreactive amilorides PRA1 and PRA2 (Fig. 2), which are very similar in structure, but possess a photolabile azido (−N=N+=N−) group at the opposite ends of their molecules, are labeled the B14.5a supernumerary and 49-kDa core subunits, respectively. Because quinone-site inhibitors are considered to enter the channel irrespective of different chemical frameworks (5, 6, 13), PRA1 labeling the supernumerary subunit is inconsistent with the channel models because the channel cavity is formed solely by the core subunits (49 kDa, PSST, and ND1). Because the entire structural model of mammalian complex I was unavailable when we published our previous study (17), further intensive studies to characterize the unique action manner of amilorides are needed. On addressing this task, it should be mentioned that advances were achieved in the syntheses of amilorides after the publication of our previous study (17). We succeeded in producing ∼10-fold more potent amilorides (amide-type) than PRA1 and PRA2 (guanidine-type) and establishing a synthetic procedure to incorporate radioactive 125I into amilorides as a detection tag. These advances together have ensured that the photoaffinity labeling experiments are conducted with the lowest possible concentrations of photoreactive amilorides (<10 nm), which are <1/100 of the concentrations of PRA1 and PRA2 used previously. This advantage must minimize the probability of nonspecific labeling, which is a primary cause of false-positive results.
The first set of experiments conducted in this study revealed that oversized SF–UQs and lipid-like PC–UQs can accept electrons from the physiological reaction site of UQ. The second set of experiments showed that amilorides bind at the interfacial domain of multiple core subunits (49 kDa, ND1, and PSST) and the 39-kDa supernumerary subunit, although 39 kDa does not make up the channel cavity in the current models. These results are difficult to explain by the channel models. On the basis of the comprehensive interpretations of the present results and previous findings (15–18), we discussed the physiological relevance of the channel models.
Results
Molecular design of SF–UQs and PC–UQs
We synthesized SF–UQs and PC–UQs by the procedures described in Schemes S1–S3. The molecular design concepts of these UQs are as follows.
The 2,6-disubstituted phenol moiety, which was attached to a terminus of the side chain of SF–UQs as a block, is bulky (Fig. 1); the diameters of 2,6-di-tert-butylphenol (∼9 Å across) and 2,6-di(TMS-1,3-butadiynyl)phenol (∼18 Å) are much wider than that of the channel of bovine complex I (the entry point is ∼3 × 5 Å, see Ref. 7). Therefore, even if the quinone head-ring enters the channel, the head-ring may be unable to reach the reaction site because of crucial steric hindrance at the entrance and/or somewhere along the narrow channel.
The 2,6-di-tert-butylphenol moiety imitated the structure of SF6847 (Fig. 1), which is one of the most efficient protonophore-type uncouplers (21, 22); therefore, this structural unit itself may be inherently free from restricted movement in the lipid bilayer environment. Because the substituted phenol moiety of SF–UQ1–SF–UQ3 and SF–UQ6 is weakly acidic (pKa ≒ ∼8.0) due to the presence of strong electron-withdrawing substituents at the para position (i.e. −C N and −COOR) (21), ∼25% of SF–UQs exists as anionic form in the reaction buffer (pH 7.4), although a negative charge is delocalized over the long-electron–conjugated system. We anticipated that this electronic nature may improve the solubility of hydrophobic SF–UQs in the buffer and, in turn, partition into SMPs. In contrast, because the pKa values of the phenol moiety of SF–UQ4 and SF–UQ5 are higher than 9 due to the lack of the electron-withdrawing substituent, almost all of them exist as nonionized forms under the experimental conditions used. As an index of the hydrophobicities of the nonionized forms of SF–UQs, their retention times in HPLC analysis using a reverse-phase column were listed in Fig. 1.
N and −COOR) (21), ∼25% of SF–UQs exists as anionic form in the reaction buffer (pH 7.4), although a negative charge is delocalized over the long-electron–conjugated system. We anticipated that this electronic nature may improve the solubility of hydrophobic SF–UQs in the buffer and, in turn, partition into SMPs. In contrast, because the pKa values of the phenol moiety of SF–UQ4 and SF–UQ5 are higher than 9 due to the lack of the electron-withdrawing substituent, almost all of them exist as nonionized forms under the experimental conditions used. As an index of the hydrophobicities of the nonionized forms of SF–UQs, their retention times in HPLC analysis using a reverse-phase column were listed in Fig. 1.
Phospholipid-based fluorescent molecular probes, which partition into the membrane environment from external aqueous medium, have been used in biophysical studies on the mitochondrial membrane (23, 24). PC–UQs are hybrid compounds of UQ and PC, which has an oleoyl group at the sn-1 glycerol position. Because of structural similarities, PC–UQs are anticipated to behave like PC in the SMP membrane. Electron densities for 10–20 tightly-bound phospholipids (PC, phosphatidylethanolamine, and cardiolipin) were identified between the core membrane subunits in ovine, porcine, and mouse heart mitochondrial complex I (9–11). There was no bound phospholipid along the quinone/inhibitor-access channel in the models. Given the lipid-like nature, it is hard to image that whole PC–UQ molecules directly enter the channel via the narrow entry point. Collectively, because of the extensive physical restrictions incorporated in SF–UQs and PC–UQs, the likelihood that their quinone head-ring reaches the reaction site located deep in the channel is quite low.
Electron transfer activities of SF–UQs
The electron transfer activities of SF–UQs were determined in the NADH:SF–UQ oxidoreductase assay using bovine heart SMPs by monitoring NADH oxidation in the presence of complexes III and IV inhibitors (antimycin A and KCN, respectively). This reaction was initiated by the addition of NADH (100 μm) to the reaction medium (2.5 ml) containing SMPs at a final protein concentration of 90 μg/ml.
SF–UQ1 worked as an electron acceptor from complex I (Fig. 3A), and its electron transfer activity varied in a concentration-dependent manner (Fig. 3B). The electron transfer activity of UQ3 measured under the same experimental conditions was shown in Fig. 3, A and B, as a reference. The electron transfer activity of SF–UQ1 was considerably lower than that of UQ2, but superior to that of UQ4, which has an isoprenyl tail with a moderate length. A potential reason for the apparently low electron–transfer efficiencies of UQ3 and UQ4 may be their low solubilities in water, thereby preventing monomeric dispersion in the assay medium and, consequently, efficient partitioning into the lipid membrane phase in SMPs (25, 26). In support of this, UQ4 was shown to work as an efficient electron acceptor that is superior to UQ2 in bovine complex I–reconstituted proteoliposomes, in which a definite concentration of UQ4 was initially incorporated as one of the membrane components (13). Thus, the low solubility of SF–UQ1 in water may be one of the reasons for its poor electron transfer activity.
Figure 3.

Electron transfer activities of SF–UQs and PC–UQs. A, measurement of NADH:SF–UQ1 oxidoreductase activity in SMPs. Panel a, NADH oxidation in the NADH:SF–UQ1 oxidoreductase assay was monitored in the absence (black trace) and presence (gray trace) of bullatacin (100 nm). The reaction was initiated by the addition of NADH (a final concentration of 100 μm). The arrow indicates the addition of bullatacin (100 nm). Panel b, NADH oxidation in the NADH:UQ3 oxidoreductase assay was monitored in the absence (black trace) and presence (gray trace) of bullatacin (100 nm). A final SMP protein concentration was 90 μg/ml. Data are representative of three independent experiments. B, concentration dependence of NADH:SF–UQ1 oxidoreductase activity. As a reference, NADH:UQ3 oxidoreductase activity was also shown. Squares and circles represent SF–UQ1 and UQ3, respectively. Filled and empty symbols represent the absence and presence of bullatacin (100 nm), respectively. Final NADH and SMP protein concentrations were 100 μm and 90 μg/ml, respectively. Values show means ± S.E. (n = 3). C, summary of the electron transfer activities of SF–UQs and PC–UQs. The rates of NADH oxidation in the NADH:quinone oxidoreductase assay in the absence (black) and presence (gray) of bullatacin (100 nm) are listed. The concentrations of SF–UQs and PC–UQs were set to 50 and 20 μm, respectively. As a reference, the electron transfer activities of UQ2, UQ3, and UQ4 (50 μm each) are listed. Graphed values are means ± S.E. (n = 3–5).
The electron transfer activity of SF–UQ1 was almost completely blocked by various quinone-site inhibitors, such as rotenone, fenpyroximate, and bullatacin, irrespective of the timing of the addition of inhibitors (before or during the catalytic reaction), as shown in Fig. 3, A and B, taking bullatacin (100 nm) as an example. This result indicates that SF–UQ1 accepts electrons predominantly from the quinone-binding pocket in complex I (i.e. the physiological site), and not from the FMN site located in the hydrophilic 51-kDa subunit. Note that residual NADH oxidation rates in the presence of excess quinone-site inhibitors were similar among UQ2–UQ4 and all synthetic UQs examined below.
Because the electron transfer activities of all SF–UQs were saturated in a range of 30–50 μm, the activities measured at 50 μm were summarized in Fig. 3C. The electron transfer activity of SF–UQ3 was weaker than those of SF–UQ1 and SF–UQ2 possibly due to its lower solubility in reaction buffer. The activities of SF–UQ2 and SF–UQ3 were almost completely blocked by bullatacin (100 nm), indicating that SF–UQ2 and SF–UQ3 accept electrons from the quinone-binding pocket.
The electron transfer activity of SF–UQ4 was slightly weaker than those of SF–UQ1 and SF–UQ2 (Fig. 3C). The activity of SF–UQ5 was superior to that of SF–UQ4 and close to that of UQ3 (Fig. 3C). This result is attributable to the solubility of less hydrophobic SF–UQ5 (HPLC retention time 11.7 min) in buffer being greater than that of SF–UQ4 (14.0 min). The electron transfer activities of SF–UQ4 and SF–UQ5 were also significantly blocked by bullatacin.
Surprisingly, the short-chain SF–UQ6 possessing an extremely bulky block (∼18 Å) functioned as an electron acceptor, similar to UQ4 (Fig. 3C). Its electron transfer activity was almost completely blocked by various quinone-site inhibitors, as observed for other SF–UQs. Because the hydrophobicity of SF–UQ6 (HPLC retention time 18.3 min) is fairly greater than that of SF–UQ4 (14.0 min), the lower pKa value of SF–UQ6 (∼8.0) due to the presence of the electron-withdrawing substituents may improve, even only slightly, its solubility in buffer and, in turn, the partitioning into SMPs.
Electron transfer activities of PC–UQs
The electron transfer activities of PC–UQs were also assessed as described above. Because the activities of PC–UQs were saturated in a range of 10–20 μm, the activities measured at 20 μm were summarized in Fig. 3C. PC–UQ1–PC–UQ3 worked as electron acceptors from complex I (Fig. 3C), although their electron transfer activities were lower than that of UQ3. The poor electron transfer activities of PC–UQ1–PC–UQ3 may also be related to their low solubilities in reaction buffer. In support of this, the order of apparent activities (PC–UQ1 > PC–UQ2 > PC–UQ3) correlates with their hydrophobicities. In comparison of PC–UQ4 and PC–UQ3, which have an identical total number of carbon atoms in the sn-2 acyl chain, the electron transfer activity of PC–UQ4 was slightly superior to that of PC–UQ3, suggesting that the natural isoprenyl structure is somewhat favorable for the electron transfer activity. The electron transfer activities of all PC–UQs were remarkably blocked by different quinone-site inhibitors (Fig. 3C, bullatacin in this case), suggesting that their reduction takes place at the quinone-binding pocket in complex I.
Membrane potential formation coupled with the reduction of SF–UQs and PC–UQs
The catalytic reduction of SF–UQs and PC–UQs was almost completely blocked by different quinone-site inhibitors, which strongly suggests that their reduction is coupled to proton translocation across the membrane domain. To verify this, we investigated whether NADH:PC–UQ (or SF–UQ) oxidoreduction generates a membrane potential across the SMP membrane by monitoring changes in the absorbance of oxonol VI in the presence of nigericin (a K+/H+ exchanger). As a control, we confirmed that the reduction of UQ2 forms the membrane potential and is completely dissipated by uncoupler SF6847 (Fig. 4, trace a (black)). UQ2 did not generate a membrane potential in the presence of quinone-site inhibitors (trace a (gray), bullatacin in this case).
Figure 4.

Membrane potential formation coupled with the reduction of PC–UQs or SF–UQs. The membrane potential generated by NADH:PC–UQ (or SF–UQ) oxidoreduction was monitored by following changes in the absorbance of oxonol VI. The SMP protein concentration was set to 90 μg/ml. The arrows indicate the addition of NADH (100 μm) and SF6847 (0.2 μm). Traces are as follows: a, UQ2 (50 μm); b, PC–UQ4 (50 μm); c, SF–UQ1 (50 μm); d, SF–UQ5 (50 μm). The black and gray traces represent the absorbance of oxonol VI in the absence and presence of bullatacin (20 nm), respectively. Data are representative of at least three independent experiments.
PC–UQ4 (20 μm) generated a membrane potential at a concentration range exhibiting the electron transfer activity (Fig. 4, trace b (black)). The transient formation of membrane potential may be due to the limited concentrations of PC–UQ4 partitioned into SMPs, as mentioned above. The membrane potential formation was completely blocked in the presence of bullatacin (trace b (gray)). These results indicate that the catalytic reduction of PC–UQ4 is coupled to the proton translocation. PC–UQ1–PC–UQ3 also generated a membrane potential at a concentration range exhibiting the electron transfer activity. The membrane potential formation coupled with their reduction was completely blocked by quinone-site inhibitors (data not shown).
It is important to note that because SF–UQ1–SF–UQ3 and SF–UQ6 inherently function as protonophores due to their SF6847-mimic structures, the membrane potential that may be formed by coupling with their reduction was not determined, as shown with SF–UQ1 as an example (Fig. 4, trace c). However, the membrane potential generated by SF–UQ4 and SF–UQ5 reduction could be monitored (Fig. 4, trace d for SF–UQ5) because their protonophoric abilities are significantly weaker than those of other SF–UQs due to their large pKa values (>9) (we note that the optimal pKa of acidic uncouplers for protonophoric activity at pH 7.4 is in the range of 7.5–8.0, see Refs. 21, 27).
Nevertheless, to confirm the uncoupling effect of SF–UQs in SMPs, we investigated whether they dissipate the membrane potential formed by ATP hydrolysis by ATP synthase. The potent uncoupler SF6847, used as a control, completely dissipated the membrane potential (Fig. 5, trace a). All SF–UQs, except for SF–UQ4 and SF–UQ5, completely dissipated the membrane potential at 50 μm, which was the concentration used in the electron transfer assay above (Fig. 3C), as shown in Fig. 5 (trace b) taking SF–UQ1 as an example. Their protonophoric efficiencies varied depending on the spacer's length. The longer the spacer, the weaker the protonophoric efficiency became, as observed in the comparison between SF–UQ1 and SF–UQ3 (Fig. 5, traces c and d, respectively); SF–UQ1, but not SF–UQ3, completely dissipated the membrane potential, even at 5.0 μm.
Figure 5.
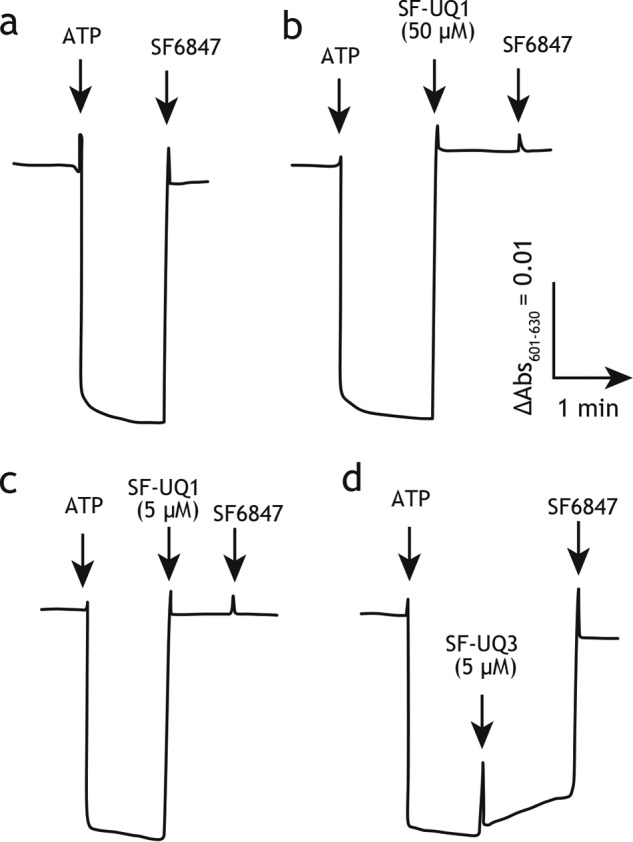
Effects of SF–UQs on the membrane potential formed by ATP hydrolysis. The membrane potential generated by ATP hydrolysis by ATPase was monitored by following changes in the absorbance of oxonol VI. The SMP protein concentration was set to 90 μg/ml. The arrows indicate the addition of ATP (4.0 mm) and SF6847 (0.1 μm). Traces are as follows: a, SF6847 (0.1 μm); b, SF–UQ1 (50 μm); c, SF–UQ1 (5.0 μm); d, SF–UQ3 (5.0 μm). Data are representative of at least three independent experiments.
Superoxide production involved in the reduction of PC–UQs and SF–UQs
We measured superoxide production during the catalytic turnover of NADH: SF–UQ (or PC–UQ) oxidoreduction in SMPs. We preliminarily confirmed that the superoxide-dependent oxidation of epinephrine in this assay is fully sensitive to superoxide dismutase. The extent of superoxide production involved in the reduction of SF–UQs (50 μm each), except SF–UQ6, and PC–UQs (20 μm each) was generally comparable with that induced by UQ3 or UQ4 reduction (Fig. 6). The amount of superoxide produced in the NADH:SF–UQ6 oxidoreduction assay was slightly higher than with other SF–UQs presumably because its ubisemiquinone intermediate would be insufficiently stabilized due to marked steric hindrance in the side chain moiety.
Figure 6.

Superoxide production from complex I in SMPs. The rates of superoxide production during the NADH:quinone oxidoreductase reaction in the absence (gray) and presence (black) of bullatacin (100 nm) are listed. The SMP protein concentration was set to 90 μg/ml. Graphed values are means ± S.E. (n = 3).
As observed for ordinary short-chain UQs (28), superoxide production induced by SF–UQs and PC–UQs was moderately or slightly enhanced in the presence of quinone-site inhibitor bullatacin; however, the extent of enhancement was significantly lower than that observed for UQ2. Short-chain UQs (such as UQ1 and UQ2) in the reaction buffer are thought to serve as redox intermediates between the FMN site and molecular oxygen (29, 30); however, the function of short-chain UQs remains elusive because the relative rates of superoxide production do not correlate in a simple manner with their hydrophobicities (30).
Syntheses of photoreactive amilorides
Based on the results of structure–activity relationship studies on amilorides (31, 32), we newly synthesized four photoreactive 125I-labeled amilorides (Fig. 2 and Schemes S4 and S5). The synthetic procedures for these amilorides are described in the supporting information. We named the derivatives possessing a photolabile azido group in the toxophoric pyrazinoyl ring ([125I]PRA4 and [125I]PRA6) and in the side chain moiety ([125I]PRA3 and [125I]PRA5) as series A and B, respectively. [125I]PRA3 and [125I]PRA4 have the prototypical pyrazinoyl guanidine skeleton, which is considered to be a critical structural unit as the inhibitors of various antiporters. [125I]PRA5 and [125I]PRA6 are amide-type amiloride derivatives that elicit more potent inhibitory activities than guanidine-type derivatives with bovine complex I (32). The inhibitory potencies determined in terms of IC50 values using their cold derivatives are listed in the parentheses in Fig. 2. The potencies of PRA5 (19 ± 3 nm) and PRA6 (IC50 = 30 ± 7 nm) were ∼1000-fold more potent than those of commercially available amilorides such as 5-(N-ethyl-N-isopropyl)amiloride and benzamil, but weaker than traditional quinone-site inhibitors such as rotenone and piericidin A; their IC50 values are at one- to two-digit nanomolar levels.
Photoaffinity labeling of complex I by series A amilorides
To identify the binding position of the toxophoric pyrazinoyl ring of amilorides, bovine SMPs (4.0 mg of proteins/ml) were incubated with series A derivatives ([125I]PRA4 or [125I]PRA6, 10 nm each), irradiated with a UV lamp on ice, and the radiolabeled complex I was isolated by BN-PAGE, followed by resolution on Laemmli- or Schägger-type SDS gels. As shown in Fig. 7A, complex I labeled by [125I]PRA4 provided a single radioactive band at ∼50 kDa on a 12.5% Laemmli-type SDS gel, which corresponds to the 49-kDa core subunit (confirmed by MS, Table S1). The [125I]PRA4-labeled complex I was also resolved by doubled SDS-PAGE using 10 and 16% Schägger-type SDS gels (Fig. 7C) (33). Strong radioactivity was observed at the position corresponding to the 49-kDa subunit. This result is consistent with our previous finding showing that PRA2 (Fig. 2), possessing a terminal alkyne group instead of radioactive 125I in the side chain moiety, binds to the 49-kDa subunit (17).
Figure 7.

Photoaffinity labeling of bovine SMPs by photoreactive 125I-amilorides. SMPs (4.0 mg of proteins/ml) were cross-linked by a photoreactive amiloride ([125I]PRA3, [125I]PRA4, [125I]PRA5, or [125I]PRA6, 10 nm each), followed by the purification of complex I by BN-PAGE and electroelution. A, isolated complex I was resolved on a 10% Schägger-type SDS gel (10% T, 3% C, containing 6.0 m urea), and the gel was subjected to CBB staining and autoradiography. An asterisk indicates an ADP/ATP carrier (AAC). B, complex I, which was separated on the 10% Schägger-type SDS gel, was further resolved on a second dimension 16% Schägger-type SDS gel (16% T, 3% C, doubled SDS-PAGE), followed by silver staining. C, SDS gel was subjected to autoradiography. Proteins equivalent to ∼200 μg of SMPs were loaded onto each well. The target subunits (49-kDa, ND1, PSST, and 39-kDa subunits) were identified by MS (shown by red arrowheads, see Table S1). All data are representative of at least three independent experiments.
We note that irrespective of different chemical frameworks, complex I inhibitors having a photolabile phenylazido unit concomitantly bind to the ADP/ATP carrier (the most abundant protein in the inner mitochondrial membrane) and 3-hydroxybutyrate dehydrogenase for a previously unknown reason (34, 35), although the labeling yields to these proteins vary depending on the experiments. The band at ∼30 kDa marked with star represents the two proteins (Fig. 7A). These proteins are also indicated by AAC in Fig. 7C.
The labeling of complex I by [125I]PRA6 gave two radioactive bands at ∼50 and ∼20 kDa on 12.5% Laemmli-type SDS gel with relative intensities of ∼1:1 (Fig. 7A). They were identified as the 49-kDa and ND1 subunits by MS, respectively (Table S1). In support of this, radioactivities migrated to the silver-stained protein spots corresponding to the 49-kDa and ND1 subunits (Fig. 7B), which migrated on or above a diagonal of hydrophilic proteins, respectively (Fig. 7C). Thus, photoaffinity labeling using series A amilorides indicated that the toxophoric pyrazinoyl moiety binds to the 49-kDa or interface of the 49-kDa and ND1 core subunits.
Photoaffinity labeling of bovine SMP by series B amilorides
Series B amilorides were subjected to the photoaffinity-labeling experiments, as conducted above. When complex I in SMPs was labeled by [125I]PRA3, a strong radioactivity migrated to the protein band at ∼40 kDa on a 12.5% Laemmli-type SDS gel (Fig. 7A). The [125I]PRA3–labeled complex I was also resolved by doubled SDS-PAGE (Fig. 7C). The tryptic digests of this protein contained the sequence corresponding to the 39-kDa supernumerary subunit (Table S1), which is positioned adjacent to the 49-kDa, PSST, and ND1 core subunits (7–11).
However, labeling by [125I]PRA5 afforded two radioactive bands at ∼50 and ∼15 kDa with almost equivalent relative intensities, including the 49-kDa subunit (Fig. 7A). When the [125I]PRA5-labeled complex I was resolved on a doubled SDS gel, radioactivity incorporated into the ∼15-kDa protein migrated to the spot corresponding to the PSST core subunit (Fig. 7C), which was confirmed by MS and Western blotting (Table S1 and Fig. S1, respectively). The results obtained using series B amilorides indicated that the flexible side chains of amilorides bind to the interfacial domain formed by the 49-kDa and PSST core subunits and the 39-kDa supernumerary subunit, although the 39-kDa subunit does not form the quinone-access channel in the current models (7–11).
It should be noted that judging from the radioactivity incorporated into the 39-kDa subunit, the labeling yield of [125I]PRA3 was significantly lower (∼1/5) than that of [125I]PRA4 as well as the amide-type amilorides ([125I]PRA5 and [125I]PRA6). Because the labeling yield is affected by the reactivity of the local protein microenvironment interacting with the photolabile azido group, the lower yield is not simply due to the lower binding affinity of [125I]PRA3 than those of other amilorides.
Analysis of the 49-kDa subunit labeled by [125I]PRA4, [125I]PRA5, and [125I]PRA6
The 49-kDa core subunit was specifically labeled by three amiloride derivatives, [125I]PRA4, [125I]PRA5, and [125I]PRA6. This subunit was also the target protein of other amilorides (PRA2 (17) and AAT (32)). To roughly localize the site labeled by [125I]PRA4, the [125I]PRA4-labeled 49-kDa subunit was excised from a Laemmli-type SDS gel and partially digested with V8-protease in the gel (35, 37). Partial digestion reproducibly gave several radioactive fragments on a 15% Tris-EDTA mapping gel, which converged on a major ∼10-kDa fragment (Fig. 8A). The N-terminal sequence of the corresponding CBB-stained spot was identified as H2N-25TAHWK29 by Edman degradation, suggesting that [125I]PRA4 binds to the region of Thr-25–Glu-115 (10.5 kDa, Fig. 8C).
Figure 8.

Localization of the site labeled by [125I]PRA4, [125I]PRA5, and [125I]PRA6 in the 49-kDa subunit. A, partial V8-protease digestion (Cleveland mapping) of the 49-kDa subunit labeled by [125I]PRA4, [125I]PRA5, or [125I]PRA6. SMPs (4.0 mg of proteins/ml) were cross-linked by [125I]PRA4, [125I]PRA5, or [125I]PRA6 (10 nm each), followed by the isolation of complex I by BN-PAGE and resolution of complex I subunits by 12.5% Laemmli-type SDS-PAGE. The CBB-stained gel piece of the 49-kDa subunit was digested with V8-protease in a 15% Tris-EDTA mapping gel as described under “Experimental procedures.” The gel was stained by CBB and subjected to autoradiography. The N-terminal sequences of the CBB-stained spots (V8–25 kDa, V8–12 kDa, and V8–10 kDa, denoted by arrows) were determined by Edman degradation. Proteins equivalent to ∼100 μg of SMPs were loaded onto each well. B, 49-kDa subunit labeled by [125I]PRA4 was exhaustively digested with V8-protease or Lys-C, as described under “Experimental procedures.” The digests were analyzed on a 16% Schägger-type SDS gel (16% T, 6% C, containing 6.0 m urea). Proteins equivalent to ∼50 μg of SMPs were loaded onto each well. C, schematic presentation of the partial digestion of the 49-kDa subunit by V8-protease. The predicted cleavage sites are denoted by arrowheads and marked with their residue numbers in the matured sequences of the bovine 49-kDa subunit (SwissProt entry P17694). D, schematic presentation of the exhaustive digestion of the 49-kDa subunit by V8-protease or Lys-C in the region covering “V8–10 kDa” (Thr-25–Glu-115). All data are representative of three independent experiments.
When the [125I]PRA4-labeled 49-kDa subunit was exhaustively digested with V8-protease, the digests converged on a radioactive band with an apparent molecular mass of ∼6 kDa (Fig. 8B) on a 16.5% Schägger-type SDS gel. Considering the results of partial digestion and theoretical cleavage sites by V8-protease, the ∼6-kDa band is predicted to be the peptide Thr-25–Glu-67 with a calculated mass of 4.8 kDa. The [125I]PRA4-labeled 49-kDa subunit was then digested with lysylendopeptidase (Lys-C) to provide dominant radioactive bands at ∼5 kDa. Careful examination of the theoretical cleavage patterns for these proteases strongly suggests the assignment of the Lys-C digest to the peptide Asp-41–Lys-75 (3.8 kDa), which is consistent with the results of partial/exhaustive V8 digestion. Therefore, the position labeled by [125I]PRA4 in the 49-kDa subunit is within the N-terminal region Asp-41–Glu-67 (Fig. 8D).
The digestion of the [125I]PRA5- and [125I]PRA6-labeled 49-kDa subunit by V8-protease and Lys-C afforded the same fragmentation patterns as those of the [125I]PRA4-labeled 49-kDa subunit (Fig. 8A), although the recovery of radioactivities varied among the amilorides used. Therefore, we concluded that the position labeled by [125I]PRA5 and [125I]PRA6 is also within the N-terminal region Asp-41–Glu-67.
Analysis of the ND1 subunit labeled by [125I]PRA6
[125I]PRA6 concomitantly labeled the ND1 and 49-kDa core subunits. The [125I]PRA6-labeled ND1 subunit was isolated from an SDS gel, digested with Lys-C or endoprotease Asp-N (Asp-N), followed by the resolution of digests on a 16.5% Schägger-type SDS gel. The Lys-C and Asp-N digestion gave single radioactive bands with apparent molecular masses of ∼15 and ∼8 kDa, respectively, on the Tricine gel (Fig. 9A). On the basis of the theoretical cleavage sites by the proteases, these digests appeared to be the peptides Tyr-127–Lys-262 (15.2 kDa) and Asp-199–Tyr-282 (9.7 kDa), respectively. These results suggest that the position labeled by [125I]PRA6 in the ND1 subunit is within the region of Asp-199–Lys-262 (Fig. 9B), which includes the matrix-side third loop connecting TMH5–6. It is important to note that this loop is adjacent to the N-terminal domain (Asp-41–Glu-67) of the 49-kDa subunit (7–9), which was identified as the concomitant binding position of [125I]PRA6, as described above. Therefore, the binding position of [125I]PRA6 in the ND1 subunit may be in the third loop.
Figure 9.
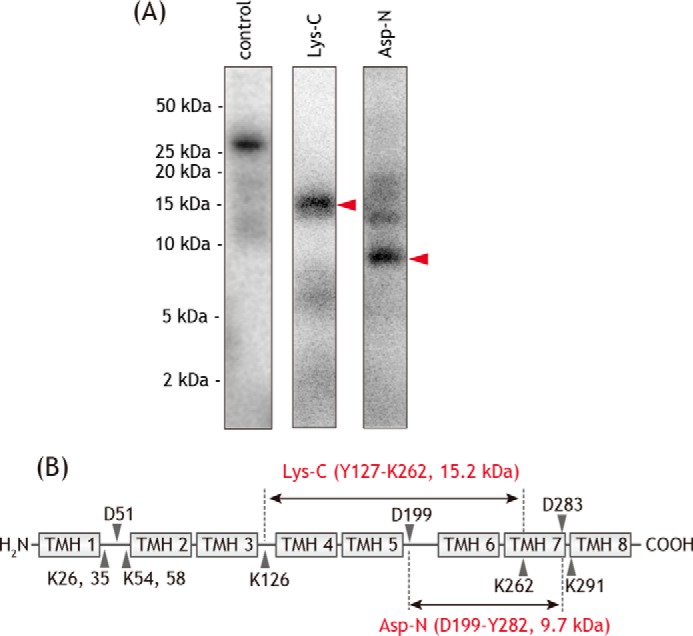
Localization of the site labeled by [125I]PRA6 in the ND1 subunit. A, ND1 subunit labeled by [125I]PRA6 was exhaustively digested with Lys-C or Asp-N. The digests were resolved on a 16% Schägger-type SDS gel (16% T, 6% C, containing 6.0 m urea), followed by autoradiography. Proteins equivalent to ∼50 μg of SMPs were loaded onto each well. B, schematic representation of the exhaustive digestion of the ND1 subunit with Lys-C or Asp-N. The TMHs were assigned according to the structures of bovine (7) and ovine (9) complexes I. Predicted cleavage sites are denoted by arrowheads and marked with their residue numbers in the matured sequences of the bovine ND1 subunit (SwissProt entry P03887). We note that the patterns of digestion were identical to those labeled by photoreactive quinazoline [125I]AzQ (37). All data are representative of three independent experiments.
Analysis of the PSST subunit labeled by [125I]PRA5
[125I]PRA5 concomitantly labeled the PSST and 49-kDa core subunits. The [125I]PRA5-labeled PSST subunit was isolated from the SDS gel and digested with Lys-C or Asp-N. The digests were resolved on a 16.5% Schägger-type SDS gel to provide predominant radioactive bands at ∼9 and ∼8 kDa, respectively (Fig. 10A). Because the Asp-N digest contained the tryptic digest 33LDDLINWAR41 (m/z 1115.6), this digest is predicted to be the peptide Pro-1–Tyr-67 (7.2 kDa, Fig. 10B) containing two uncleavable aspartic acids (34DD35). Based on the results of Asp-N digestion and theoretical cleavable sites by Lys-C, the Lys-C digest must be the peptide Leu-33–Lys-95 (7.1 kDa, Fig. 10B). Together, the region labeled by [125I]PRA5 in the PSST subunit was assigned to the peptide Leu-33–Tyr-67 (Fig. 10B). This region overlaps with the region labeled by the photoreactive fenpyroximate ([125I]APF), although the labeled regions in the 49-kDa subunit differ between [125I]PRA5 (Asp-41–Glu-67) and [125I]APF (Asp-160–Arg-174) (35).
Figure 10.

Localization of the site labeled by [125I]PRA5 in the PSST subunit. A, PSST subunit labeled by [125I]PRA5 was exhaustively digested with Lys-C or Asp-N, and the digests were resolved on a 16% Schägger-type SDS gel (16% T, 6% C, containing 6.0 m urea), followed by autoradiography. Proteins equivalent to ∼50 μg of SMPs were loaded onto each well. B, schematic representation of the exhaustive digestion of the PSST subunit with Lys-C or Asp-N. The peptide 33LDDLINWAR41 containing two uncleavable aspartic acids (34DD35) are colored in red. The predicted cleavage sites are denoted by arrows and marked with their residue numbers in the matured sequences of the bovine PSST subunit (SwissProt entry P42026). We note that the patterns of digestion are identical to those labeled by photoreactive fenpyroximate [125I]APF (35). All data are representative of three independent experiments.
Analysis of the 39-kDa subunit labeled by [125I]PRA3
To roughly localize the position labeled by [125I]PRA3 in the 39-kDa subunit, the [125I]PRA3-labeled subunit was subjected to in-gel partial digestion with V8-protease according to the same procedures used for the 49-kDa subunit above. Partial digestion gave a radioactive fragment with an apparent molecular mass of ∼17 kDa on a 15% Tris-EDTA gel (Fig. 11A), the tryptic digests of which were identified by MS as 178FLNYFANIR186 (m/z 1157.6), 269LFEISPFEPWTTR281 (m/z 1622.9), and 293ILPHLPGLEDLGVEATPLELK313 (m/z 2254.5). Based on the internal sequence and apparent molecular mass, the fragment appeared to be the region Asp-176–Glu-316 (16.2 kDa, Fig. 11C). Moreover, the [125I]PRA3-labeled 39-kDa subunit was exhaustively digested with Lys-C, and the digests were resolved on a 16.5% Schägger-type SDS gel, providing a major radioactive band at ∼7 kDa (Fig. 11B). Given the results for V8-partial digestion, the ∼7-kDa fragment must be the peptide Thr-227–Lys-283 with a calculated mass of 6.8 kDa (Fig. 11C). Whereas the 39-kDa supernumerary subunit is mostly hydrophilic, this labeled region contains hydrophobic segments (38), as discussed later.
Figure 11.

Localization of the site labeled by [125I]PRA3 in the 39-kDa subunit. A, complex I labeled by [125I]PRA3 was separated on a 12.5% Laemmli-type SDS gel. The CBB-stained gel piece of the 39-kDa subunit was partially digested with V8-protease in a 15% Tris-EDTA mapping gel, followed by CBB staining and autoradiography. Proteins equivalent to ∼100 μg of SMPs were loaded onto each well. The CBB-stained spot (V8–17 kDa), which corresponds to the radioactive V8–17 kDa band, was assigned to the peptide Asp-176–Glu-316 by MS. B, 39-kDa subunit labeled by [125I]PRA3 was exhaustively digested with Lys-C, and the digests were resolved on a 16% Schägger-type SDS gel (16% T, 6% C, containing 6.0 m urea), followed by autoradiography. Proteins equivalent to ∼50 μg of SMPs were loaded onto each well. C, schematic representation of the partial and exhaustive digestion of the 39-kDa subunit with V8-protease and Lys-C, respectively. The predicted cleavage sites are denoted by arrows and marked with their residue numbers in the matured sequences of the bovine 39-kDa subunit (SwissProt entry P34943). All data are representative of three independent experiments.
Competition between photoreactive amilorides and different quinone-site inhibitors
Bovine SMPs (2.0 mg of proteins/ml) were cross-linked by a photoreactive amiloride ([125I]PRA3, [125I]PRA4, [125I]PRA5, or [125I]PRA6, 5.0 nm each) in the presence of excess quinone-site inhibitors (amiloride-5m (32), quinazoline, fenpyroximate, bullatacin, rotenone, and piericidin A), including nonradioactive amiloride derivatives (PRA3, PRA4, PRA5, and PRA6), followed by the isolation of complex I by BN-PAGE and the resolution of complex I subunits by SDS-PAGE. The specific binding of the four 125I-amilorides to complex I was remarkably suppressed in the presence of different quinone-site inhibitors (5.0 μm each) (Fig. 12A). Concerning [125I]PRA5 (labeled PSST and 49 kDa) and [125I]PRA6 (labeled ND1 and 49 kDa), the suppressive effects on the labeling of the PSST and ND1 subunits, respectively, were shown in Fig. 12A; the extent of the suppression of the labeling against the 49-kDa subunit was similar. Nonradioactive amilorides (50 μm each) markedly suppressed not only the binding of corresponding 125I-amilorides but also that of structurally different 125I-amilorides (Fig. 12B). To make the competition clearer, a 10-fold higher concentration of nonradioactive amilorides (50 μm) than ordinary inhibitors (5.0 μm) was used because the inhibitory potencies of the former, in terms of the IC50 values, are 10–100-fold weaker than those of the latter. The results of the competition tests indicate that the binding positions of the amilorides overlap with or are close to those of various quinone-site inhibitors. We note that it is impractical in these experiments to discriminate between direct displacement by competitors and indirect displacement due to structural changes in the enzyme induced by the binding of competitors.
Figure 12.
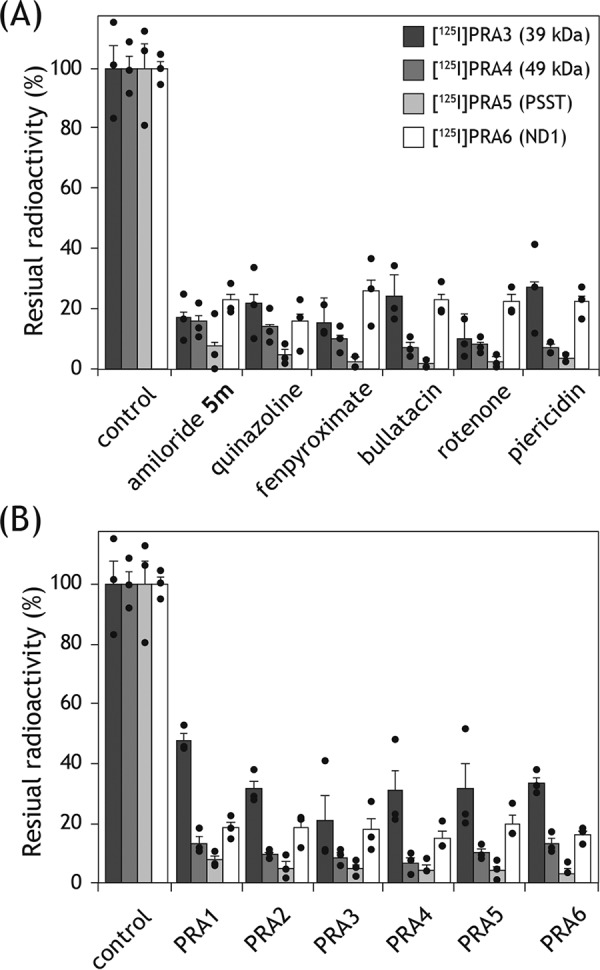
Competition test between photoreactive 125I-amilorides and other inhibitors. A, SMPs (2.0 mg of proteins/ml) were cross-linked by [125I]PRA3, [125I]PRA4, [125I]PRA5, or [125I]PRA6 (5.0 nm each) in the presence of excess quinone-site inhibitors (amiloride-5m (32), quinazoline, fenpyroximate, bullatacin, rotenone, and piericidin A), followed by the isolation of complex I by BN-PAGE and resolution of complex I subunits by SDS-PAGE. The residual radioactivities in the target subunits were quantified. B, competition test was conducted in the presence of excess nonradioactive amiloride derivatives (PRA1–PRA6) under the same experimental conditions. Graphed values are means ± S.E. (n = 3).
Competition between photoreactive amilorides and UQs
We previously showed that UQ2 moderately suppresses the labeling of complex I by PRA1 and PRA2 (17). Therefore, we examined whether oversized UQs (SF–UQ2 and SF–UQ6) suppress the labeling by [125I]PRA3, [125I]PRA4, [125I]PRA5, or [125I]PRA6. As a reference, SMPs (2.0 mg of proteins/ml) were subjected to photoaffinity labeling by each of the four 125I-amilorides (5.0 nm) in the absence or presence of excess UQ2 (50 μm) according to the same procedures described above. UQ2 significantly suppressed the labeling of target subunit(s) by individual 125I-amilorides (Fig. 13A). SF–UQ2 also suppressed the labeling; the extent of suppression was comparable with that by UQ2 (Fig. 13A). These results strongly suggest that the binding positions of amilorides overlap (at least partially) with those of UQs.
Figure 13.
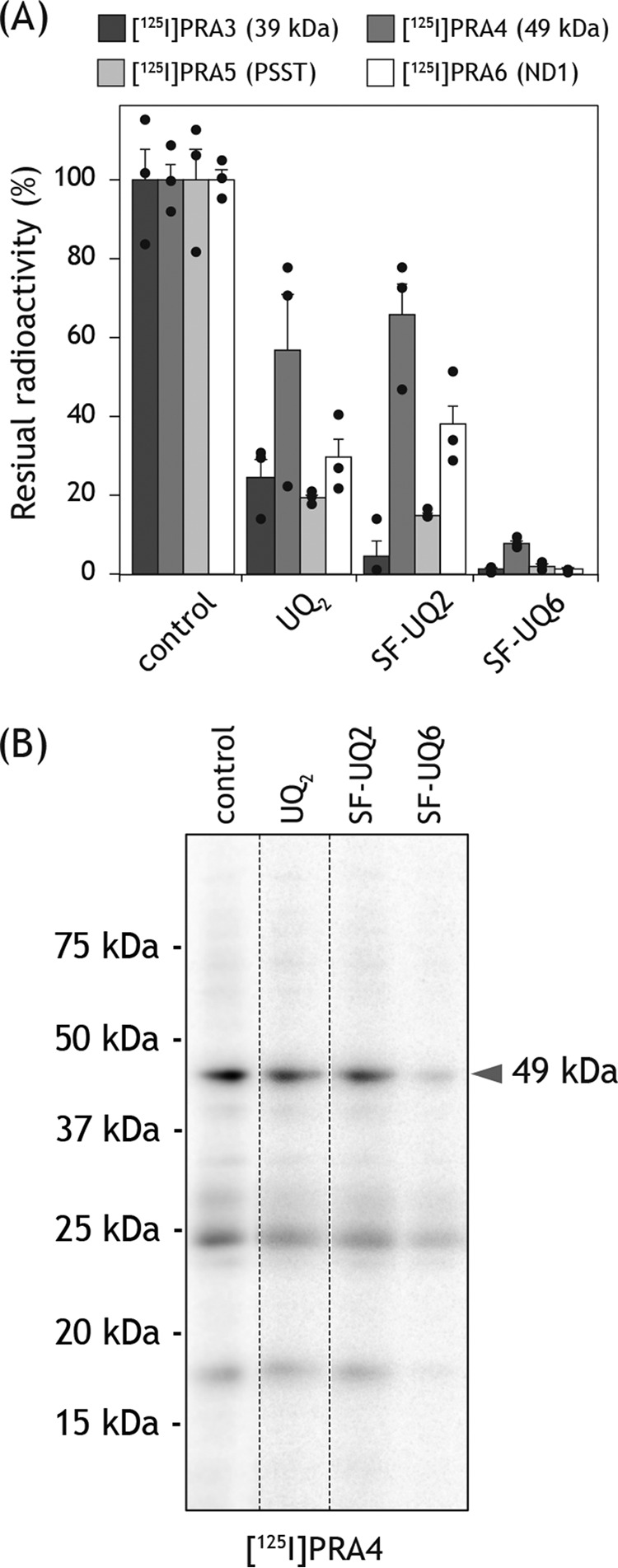
Competition test between photoreactive 125I-amilorides and quinones. A, SMPs (2.0 mg of proteins/ml) were cross-linked by [125I]PRA3, [125I]PRA4, [125I]PRA5, or [125I]PRA6 (5.0 nm each) in the presence of excess quinone (UQ2, SF–UQ2, and SF–UQ6), as conducted in Fig. 12. The residual radioactivities in the target subunits were quantified. Graphed values are means ± S.E. (n = 3). B, gel image of the SDS-PAGE analysis used for the competition test between [125I]PRA4 and quinones.
Although SF–UQ6 markedly suppressed the labeling of target subunit(s) by 125I-amilorides (Fig. 13A), the nonspecific binding of 125I-amilorides to multiple nontarget subunits was also distinctly decreased, which was detected from the background radioactivities in SDS-polyacrylamide gels, as shown in Fig. 13B with [125I]PRA4 as an example. Given the extremely hydrophobic property of SF–UQ6, the latter was presumably because this quinone nonspecifically bound to nontarget subunits due to hydrophobic interaction and prevented the nonspecific binding of 125I-amilorides to the nontarget subunits. Therefore, we cannot exclude the possibility that the suppression of the 125I-amiloride labeling of target subunits by SF–UQ6 was caused by the specific and nonspecific effects.
Discussion
Respiratory complex I has long been a “black box”; however, the recent rapid advances in structural biology studies (5–12) along with computational simulations (39–41), which were conducted based on the crystal structure of the T. thermophilus enzyme (5), provided invaluable information on the structure, functions, and assembly of the enzyme. Careful inspection of inconsistencies as well as consistencies between the biochemical/biophysical data obtained to date and the structural works may contribute to deepening our understanding of the enzyme.
Catalytic reduction of SF–UQs and PC–UQs in complex I
Based on the results obtained in this study, we conclude that the quinone head-rings of SF–UQ4, SF–UQ5, and PC–UQs indeed access the physiological reaction site of intact complex I as follows. First, the reduction of these UQs was catalyzed with the formation of membrane potential. Second, the electron transfer and membrane potential formation were almost completely blocked by various quinone-site inhibitors. Third, impediment of the reduction of these UQs by quinone-site inhibitors enhanced superoxide production. These features meet the criteria of “physiological” quinone substrates for complex I, as observed for ordinary short-chain UQs such as UQ1 and UQ2. Although the membrane potential formation by SF–UQ1–SF–UQ3 and SF–UQ6 in SMPs was not confirmed due to their uncoupling effect, they may access the reaction site because their electron transfer activities were completely blocked by quinone-site inhibitors, and the impediment of their reduction enhanced superoxide production. Of particular interest is that even SF–UQ6, which has a bulky and rigid block, did not completely lose its electron-accepting ability.
The electron transfer efficiencies of SF–UQs and PC–UQs were significantly lower than that of UQ2 in our assay system. Low efficiencies per se do not necessarily dispute the occurrence of catalytic reduction at the quinone reaction site because (i) the apparent electron transfer efficiency of hydrophobic UQ4 is also low in SMPs, and (ii) the efficiencies of UQ2 and decyl-UQ (an alkyl derivative of UQ2) markedly vary even with slight structural modifications in the side chain as well as the quinone head-ring (42, 43). Therefore, the result that their head-rings reached the reaction site is difficult to reconcile with the channel models.
To elucidate how UQs bind and transit in the channel, Fedor et al. (13) performed computational simulations of UQ-bound complex I using UQ1, UQ2, UQ4, UQ6, UQ8, and UQ10. This simulation study provided interesting insights that may explain, in part, the kinetics of complex I catalysis with UQs of various isoprene chain lengths; however, some hypothetical ideas were needed to account for discontinuous variations in kinetic parameters (e.g. pseudo second-order rate constant (kcat/Km) and binding affinity (Kd = koff/kon)) between the short (UQ1 and UQ2 (or UQ4)) and long (UQ6 (or UQ4)–UQ10) analogs. It should be reminded that because this simulation study was conducted on the premise of the predicted channel structure (7), it did not necessarily verify whether the structure in question is physiologically relevant.
Irrespective of the physiological relevance of the channel models, the reduction of the quinone head-ring takes place nearby the Fe-S cluster N2 (a direct electron donor to UQs), which is ∼20 Å away from the membrane surface (5–11). Therefore, the head-rings of SF–UQs and PC–UQs need to reach this position to be reduced. Although we cannot exclude the possibility that SF–UQs and PC–UQs approached the reaction site via different routes, it is unclear how they achieved this from the lipid–bilayer membrane. If they enter and transit the currently predicted channel, the channel cavity must undergo remarkable structural rearrangements, which may be more drastic than those currently supposed for the active/deactive transition (6–11). If the currently predicted entrance is not the entrance for SF–UQs and PC–UQs, where is their entry point? Regarding PC–UQs, we cannot exclude the possibility that they wedge into the channel interior from crevices, if any, on the channel wall. To address this question, comprehensive interpretations of the results obtained in the photoaffinity labeling experiments using different types of quinone-site inhibitors may be helpful. Therefore, by summarizing the photoaffinity labeling results, the potential area will be discussed in the last section.
Binding positions of amilorides in complex I
We conducted the photoaffinity labeling using a series of amilorides with intact complex I. The results obtained with series A amilorides strongly suggest that the toxophoric pyrazinoyl group of amilorides binds to the interface of the 49-kDa and ND1 subunits. This labeled region is identical to that labeled by the quinazoline-type inhibitor [125I]AzQ (34, 37). The labeling patterns of series B amilorides were more complicated presumably because the binding positions of the flexible side chains, to which a photolabile azido group attached, vary depending on different pyrazinoyl and side-chain structures. The regions labeled by the four 125I-amilorides are summarized in the structural model of ovine complex I (Fig. 14). We here used the ovine enzyme (9) because the matrix-side third loop (Thr-201–Ala-217) connecting the TMH5–6 of the ND1 subunit, which is included in the [125I]PRA6-labeled region, is disordered in the deactive state of the bovine enzyme (7, 8). Concerning the [125I]PRA6-labeled region in the ND1 subunit, only this loop was colored (yellow) in Fig. 14 for simplification. The regions labeled by the four 125I-amilorides are in contact or close to one another. Although the labeled regions in the 49-kDa (Asp-41–Glu-67) and 39-kDa (Thr-227–Lys-283) subunits seem to be somewhat distant, we cannot rule out the possibility that the position of the hydrophobic segment of the 39-kDa subunit (red circle in Fig. 14B) is closer to the membrane domain side in intact complex I than the position modeled by cryo-EM (7–9), as discussed later.
Figure 14.

Regions labeled by photoreactive 125I-amilorides. A, 49-kDa, PSST, ND1, and 39-kDa subunits in ovine complex I (PDB entry 5LNK (9)) are colored in pink, blue, orange, and green, respectively. B, close up of the labeled regions. The labeled regions are shown in spheres: Val-44–Glu-67 in the 49-kDa subunit (labeled by [125I]PRA4, [125I]PRA5, and [125I]PRA6); Leu-33–Tyr-67 in the PSST subunit (labeled by [125I]PRA5); Thr-201–Ala-217 in the ND1 subunit (labeled by [125I]PRA6); and Thr-227–Pro-252 and Pro-277–Lys-283 in the 39-kDa subunit (labeled by [125I]PRA3). The quinone/inhibitor-access channel predicted in ovine complex I was generated using MOLE and shown in black. The red circle marks the hydrophobic segment in the 39-kDa subunit (see the text). In ovine complex I (9), Asp-41–Leu-43 and Phe-253–Glu-276 in the 49- and 39-kDa subunits, respectively, were unmodeled.
Series A amilorides efficiently suppressed the labeling by series B, and vice versa (Fig. 12B). Given the very similar structures, these results are reasonable. The labeling of multiple subunits (including the 39-kDa subunit) by both series of amilorides was significantly suppressed by other quinone-site inhibitors (Fig. 12A), indicating that the binding positions of amilorides and other inhibitors overlap (at least partially) or are close to one another. The labeling was also suppressed by short-chain UQ2, SF–UQ2, and SF–UQ6 (Fig. 13). Collectively, these results suggest that some of the labeled region (Thr-227–Lys-283) in the 39-kDa supernumerary subunit also forms a part of the binding pocket for quinone/inhibitor, presumably the entry domain. However, this is inconsistent with the channel models, even after consideration of different structural integrity of the C-terminal domain of the 39-kDa subunit (including Thr-227–Lys-283) in the deactive state between the bovine (8) and ovine (9) enzymes.
Structural works showed that the 39-kDa supernumerary subunit flanks the quinone module in the hydrophilic arm and is adjacent to the core subunits forming the quinone/inhibitor-access channel (PSST, 49 kDa, and ND1) (6–10). Because the 39-kDa subunit is essential for stabilizing the junction between the membrane and matrix arms of complex I (44), it is conceivable that this subunit has some specific yet heretofore unknown structural/functional roles for the mitochondrial enzyme. The 39-kDa subunit is predominantly hydrophilic but contains hydrophobic segments in the C-terminal domain, which could be anchored into the membrane part via hydrophobic transmembrane α-helices (38). Because the [125I]PRA3-labeled region (Thr-227–Lys-283) in the 39-kDa subunit contains the α-helices, we speculate that the position of some of this labeled region is close to the membrane domain side in intact complex I in SMPs. In support of this, Babot et al. (45) showed that the 39-kDa subunit is cross-linked to the ND3 subunit (at the loop connecting TMH1–2) by a heterobifunctional cross-linker SPDP (6.8 Å length) in the deactive form of the enzyme in SMPs. This speculation may explain why amiloride analogs labeled the 49- and 39-kDa subunits.
In a previous photoaffinity-labeling study (17), we concluded that PRA1 and PRA2 (Fig. 2) label the B14.5a supernumerary and 49-kDa core subunits, respectively. Referring to the entire structural models (7–9), the B14.5a subunit runs over the hydrophilic domain surface behind the entry point of the quinone-accessing channel. Therefore, we cannot rule out the possibility that the assignment of this subunit as the PRA1-labeled subunit may be a false-positive result due to the high concentration of PRA1 (10 μm) used. In this study, we used the lowest possible concentrations of 125I-amilorides (5∼10 nm) to minimize the probability of nonspecific labeling.
Physiological relevance of the quinone/inhibitor-access channel models
We previously reported three types of chemical biology studies, which are also difficult to explain by the channel models: (i) reactivity of the chemical tag incorporated into Asp-160 in the 49-kDa subunit (49-kDa Asp-160) against externally added bulky reagents (15, 18); (ii) different binding positions of various types of quinone-site inhibitors revealed by photoaffinity labeling (17, 34–37, 46); and (iii) the existence of “decoupling UQs,” the catalytic reduction of which is not coupled with proton translocation (16, 47). We will briefly summarize them below and discuss the physiological relevance of the channel models based on comprehensive interpretations of the present results and previous findings.
First, we investigated reactivities of the chemical tags attached to 49-kDa Asp-160 via ligand-directed tosyl (LDT) chemistry (Fig. S2) using intact complex I in bovine SMPs (15, 18). We found that externally added bulky TAMRA-DIBO and BODIPY-tetrazine directly react with the azido group (Asp-160(COO)-(CH2)3-N3) and cyclopropene group (Asp-160(COO)-(CH2)3-cyclopropene) attached to 49-kDa Asp-160, respectively. Because TAMRA-DIBO (∼14 × 20 Å) and BODIPY-tetrazine (∼9 × 20 Å) are both markedly bulkier than the diameters of the channel entrance (∼3 × 5 Å) and body, it is impractical to consider that whole reagents entered and transited the channel to react with the modified 49-kDa Asp-160.
Second, we performed the photoaffinity-labeling studies using different types of quinone-site inhibitors (fenpyroximate (35), quinazoline (34, 37), and acetogenin (46)) with bovine SMPs. The regions labeled by the inhibitors are illustrated in the structural model of ovine complex I (Fig. S3). The binding positions of these inhibitors do not necessarily face the channel interior (Fig. S3, black). For example, fenpyroximate ([125I]APF and [125I]AIF (3-azido-5-iodobenzyl fenpyroximate) binds at the interface between the PSST (Ser-43–Arg-66) and 49-kDa (Asp-160–Arg-174) subunits in a manner such that the toxophoric pyrazole ring and side chain of the inhibitor orient toward the PSST and 49-kDa subunits, respectively (Fig. S3A) (35). This localization was corroborated by a recent genetic study (48), which demonstrated that a mutation (H92R) in the PSST homolog in phytophagous mite (Tetranychus urticae), corresponding to His-61 in bovine PSST, is associated with significant resistance to fenpyroximate. The labeled area faces a part of the “top” of the channel, whereas the presumed location of fenpyroximate is not inside the channel. Quinazoline ([125I]AzQ) binds to the interface between the N-terminal region (Asp-41–Arg-63) of the 49-kDa subunit and the matrix-side third loop connecting TMH5–6 of the ND1 subunit (Fig. S3B) (34, 37). This area is near the channel but does not form the channel cavity. Acetogenin ([125I]DANA (diazinylated natural acetogenin)) binds to the ND1 subunit in a manner such that the bis-tetrahydrofuran ring and γ-lactone are directed toward the loop connecting TMH5–6 and the region spanning TMH4–5 (Val-144–Glu-192), respectively (Fig. S3C) (46). This area forms a part of the channel, but acetogenin is positioned outside the channel. Collectively, the results of photoaffinity labeling experiments, including the present results for amilorides, strongly suggest that the binding positions of different inhibitors are distributed around the predicted channel.
Third, we produced decoupling UQs (e.g. QT in Fig. 1), the catalytic reduction of which by complex I in bovine SMPs is decoupled from proton translocation (16, 47). QT, which was originally synthesized as the reagent applicable to LDT chemistry, efficiently reacted with the 49-kDa Asp-160. It is conceivable that decoupling UQs accept electrons from the Fe-S cluster N2, but at somewhat different positions from that of ordinary short-chain UQs, such as UQ2, because of their unique side-chain structures. Accordingly, the catalytic reduction of decoupling UQs may be unable to induce the predicted structural changes inside the quinone-binding pocket, which is essential for transmitting the redox energy released in the electron transfer reaction to the membrane domain for driving proton translocation (39, 41). However, the channel models exclude the possibility that quinones possessing various chemical frameworks bind to the narrow reaction site in different positions or manners (5–9).
With the present results and previous findings taken together, it is likely that the binding pocket for quinone/inhibitor is considerably “open” to allow a wide range of specific ligands access to the deep inside. The potential entrance of this open space may be the area at which the regions labeled by different photoreactive inhibitors are in contact or close to one other; these regions are the N-terminal Asp-41–Glu-67 of 49 kDa (labeled by quinazoline, [125I]PRA4, [125I]PRA5, and [125I]PRA6); Leu-33–Tyr-67 of PSST (labeled by fenpyroximate and [125I]PRA5); the loop connecting TMH5–6 of ND1 (labeled by quinazoline, bullatacin, and [125I]PRA6); and the C-terminal Thr-227–Lys-283 of 39 kDa (labeled by [125I]PRA3). These labeled regions are represented by red spheres in the deactive state of ovine complex I (Fig. 15), which actually correspond to the regions labeled by the amiloride derivatives (Fig. 14). Because the photoreactive inhibitors examined are all competitive with each other, we tentatively propose a potential common entry area of the open space (yellow circle in Fig. 15). Overall conformational flexibility of this area was emphasized in the mammalian enzymes to explain the active/deactive transition because some parts of this area are disordered in the deactive state (e.g. the loop connecting TMH5–6 of ND1 and a part of the C-terminal domain of 39 kDa) (7–9, 11). Although this area does not include the entry point of the channel (Fig. 15, black) predicted in the current models, the possibility that enzyme particles, in which the area is open, were discarded during image processing and/or 3D classification may not be ruled out. Altogether, a variety of ligands, including quinones, would bind to or enter through this area, but in a somewhat different manner reflecting their diverse chemical structures.
Figure 15.

Summary of binding sites of different quinone-site inhibitors. Ovine complex I was used (PDB entry 5LNK (9)). A, N-terminal region (Val-44–Glu-67) of 49 kDa (labeled by quinazoline (34, 37), [125I]PRA4, [125I]PRA5, and [125I]PRA6), Leu-33–Tyr-67 of PSST (labeled by fenpyroximate (35) and [125I]PRA5), the loop connecting TMH5–6 of ND1 (labeled by quinazoline (34), bullatacin (46), and [125I]PRA6), and the C-terminal domain (Thr-227–Pro-252 and Pro-277–Lys-283) of 39 kDa (labeled by [125I]PRA3) are represented in red spheres. B, close up of the binding sites. The colors of remaining parts of the 49-kDa, PSST, ND1, and 39-kDa subunits are identical to those used in Fig. 14. The yellow circle marks the putative entry area to the open binding pocket for quinone/inhibitor. The quinone/inhibitor-access channel predicted in the current models is shown in black. Asp-41–Leu-43 and Phe-253–Glu-276 in the 49- and 39-kDa subunits, respectively, were unmodeled in ovine complex I (9).
The structures of the quinone/inhibitor-access channel in complex I purified from different biological sources are somewhat different. Because the differences were discussed in detail in previous studies (6–11) and partly in this study, here we do not particularly mention them. Variations in complex I structures would arise not only from putative different enzyme states (the active or deactive state) and/or their medium resolution but also from some differences in the enzyme preparations, as pointed out by Sazanov and co-workers (49). As is sometimes the case with membrane-bound proteins, structural data are inconsistent with the data obtained in organello by biochemical/biophysical approaches, making comparisons difficult (e.g. voltage-dependent anion channel 1 (50, 51), ADP/ATP carrier (52, 53), and Na+-pumping NADH–quinone oxidoreductase (54, 55)). In conclusion, it remains debatable whether the current quinone/inhibitor-access channel models fully reflect the physiologically relevant states present throughout the catalytic cycle, including the proposed structural changes accompanying the active/deactive transition. Various structural models set under different experimental conditions (for example, UQ-bound and/or different inhibitors-bound states) with higher resolution are needed to solve the current contradictions.
Experimental procedures
Materials
Ubiquinone-1 (UQ1)–ubiquinone-3 (UQ3) were kind gifts from Eisai (Tokyo, Japan). Ubiquinone-4 (UQ4) was purchased from Sigma. Protein standards (Precision Plus Protein Standards, Hercules, CA) for SDS-PAGE were purchased from Bio-Rad. [125I]NaI was purchased from PerkinElmer Life Sciences. Other reagents were all of analytical grade.
Synthesis of SF–UQs, PC–UQs, and photoreactive 125I-amilorides
The synthetic procedures for SF–UQs, PC–UQs, and photoreactive 125I-amilorides are described in supporting information. All compounds synthesized were characterized by 1H and 13C nuclear magnetic resonance (NMR) spectroscopy and MS.
Analysis of synthetic short-chain UQs by reverse-phase HPLC
The hydrophobicities of short-chain UQs were assessed by retention times in reverse-phase column chromatography. HPLC analysis of short-chain UQs was conducted with a Shimadzu LC-20AD HPLC system (Shimadzu, Kyoto, Japan) equipped with a triple quadrupole mass spectrometer LC-MS 8040 (Shimadzu). UQs were separated on a reverse-phase column (Luna 5 μm phenyl–hexyl, 2.0 × 150 mm, Phenomenex, Torrance, CA). The mobile phase is composed of methanol and 0.1% aqueous formic acid delivered at a flow rate of 0.2 ml/min, and a linear gradient of methanol formed from 80 to 100% over 20 min. Elution profiles were monitored by UV absorbance at 280 nm and total ion chromatogram.
Preparation of bovine heart SMPs and measurement of complex I activity
Mitochondria were isolated from bovine heart. SMPs were prepared by the method of Matsuno-Yagi and Hatefi (56) and stored in buffer containing 250 mm sucrose and 10 mm Tris-HCl (pH 7.4) at −80 °C until used. NADH oxidase activity in SMPs was measured spectrometrically with a Shimadzu UV-3000 instrument (340 nm, ϵ = 6.2 mm−1 cm−1) at 30 °C. The reaction medium (2.5 ml) contained 0.25 m sucrose, 1.0 mm MgCl2, and 50 mm phosphate buffer (pH 7.4). The final mitochondrial protein concentration was 30 μg of proteins/ml. The reaction was initiated by adding 50 μm NADH after the equilibration of SMP with an inhibitor for 4 min. The IC50 values of inhibitors were calculated by Prism (version 4, GraphPad, La Jolla, CA) using sigmoid dose-response curve fitting.
NADH–quinone oxidoreductase activity was also measured under the same experimental conditions, except that the reaction medium (2.5 ml) contained 0.80 μm antimycin A and 4.0 mm KCN (16). The final protein concentration was 90 μg of proteins/ml. The reaction was initiated by adding 100 μm NADH after the equilibration of SMP with a synthetic quinone for 4 min.
Measurement of membrane potential formation
Membrane potential formation coupled with NADH–quinone oxidoreduction was measured by following changes in the absorbance of oxonol VI (601–630 nm) with a Shimadzu UV-3000 instrument in the dual-wavelength mode in reaction medium (2.5 ml) containing 0.25 m sucrose, 1.0 mm MgCl2, 0.8 μm antimycin A, 4.0 mm KCN, 2.5 mm oligomycin, 0.10 μm nigericin, 1.0 μm oxonol VI, and 50 mm phosphate buffer (pH 7.4) at 30 °C (16, 32). The final mitochondrial protein concentration was set to 90 μg of proteins/ml. The reaction was initiated by adding 100 μm NADH after the equilibration of SMP with a short-chain UQ for 4 min. A protonophoric uncoupler SF6847 was used at a final concentration of 0.10 μm to confirm the complete dissipation of the membrane potential. ATP-driven membrane potential formation was also measured under the same experimental conditions, but without oligomycin. The reaction was started by adding 4.0 mm ATP.
Measurement of superoxide production
Superoxide production from complex I was determined by the superoxide-dependent oxidation of epinephrine to adrenochrome with a Shimadzu UV-3000 instrument (485–575 nm, ϵ = 2.96 mm−1 cm−1) in the dual-wavelength mode at 30 °C (16). The reaction medium (2.5 ml) contained 0.25 m sucrose, 1.0 mm epinephrine, 1.0 mm EDTA, 1.0 μm catalase, 0.80 μm antimycin A, 1.0 μm MOA-stilbene, and 10 mm Tris-HCl (pH 7.4). The final mitochondrial protein concentration was set to 90 μg/ml. The reaction was initiated by adding 100 μm NADH after the equilibration of SMP with a quinone analog for 4 min. Superoxide dismutase (bovine liver, Sigma) was used at a concentration of 60 units/ml to evaluate the specificity of the assay.
Photoaffinity labeling of bovine SMPs by photoreactive 125I-amilorides
Bovine SMPs (2.0–4.0 mg of proteins/ml) were incubated with a photoreactive 125I-amiloride ([125I]PRA3, [125I]PRA4, [125I]PRA5, or [125I]PRA6, 5–10 nm) in buffer containing 0.25 m sucrose, 1.0 mm MgCl2, and 50 mm phosphate buffer (pH 7.4) at room temperature for 10 min, followed by the treatment with 50 μm NADH for a further 5 min. The mixture was irradiated with a long wavelength UV lamp (Black-lay model B-100A, UVP, Upland, CA) on ice for 10 min at a distance of 10 cm from the light source (35). When the competition test was conducted, the mixture was incubated with other complex I inhibitors or UQ analogs for 10 min at room temperature prior to the treatment with the photoreactive 125I-amiloride.
Electrophoresis
To isolate complex I, SMPs labeled by a photoreactive 125I-amiloride were solubilized in sample buffer containing 50 mm Bistris-HCl (pH 7.2), 50 mm NaCl, 10% (w/v) glycerol, 1% (w/v) n-dodecyl d-maltoside, and 0.001% (w/v) Ponceau S on ice for 1 h, and the samples were separated by BN-PAGE (57) using a 4–16% precast gel system (Life Technologies, Inc.) according to the manufacturer's protocol. The isolated complex I was further resolved on a Laemmli-type 12.5% SDS gel (58) or Schägger-type 10% SDS gel containing 6.0 m urea (33).
Doubled SDS-PAGE was conducted as described previously (17, 33). In brief, the labeled complex I was separated on a first-dimension 10% Schägger-type gel (10% T, 3% C, containing 6.0 m urea). The gel slice was then acidified with 100 mm Tris-HCl (pH 2.0) for 30 min, followed by second-dimension separation on a 16% Schägger-type gel (16% T, 3% C). Typically, complex I equivalent to 200 μg of SMPs was separated on a mini-size gel (80 × 90 mm, 1 mm). The resolved proteins were visualized by CBB or mass spectrometry (MS)-compatible silver staining (Wako Silver Stain MS kit, Wako Pure Chemicals, Osaka, Japan). The migration patterns of the labeled proteins were visualized with the FLA-5100 bioimaging analyzer (Fuji Film) or Typhoon-FLA 9500 bioimaging analyzer (GE Healthcare, Buckinghamshire, UK). The radioactivity of each band was quantified using MultiGauge (Fuji Film) or ImageQuant (GE Healthcare).
Proteomic analysis
Regarding the partial digestion (Cleveland mapping (32)) of labeled subunits, CBB-stained bands of the target subunits were digested with V8-protease (Roche Applied Science, Penzberg, Germany) in a 15% Tris-EDTA mapping gel according to the previously described procedures (17, 18). Partial digests were identified by Edman degradation or MS.
For the exhaustive digestion of complex I subunits, the target subunits were recovered from the SDS gel by electroelution or direct diffusion in 10 mm Tris-HCl buffer (pH 8.0) containing 0.025% (w/v) SDS. The isolated subunits were digested with lysyl endopeptidase (Wako Pure Chemicals, Osaka, Japan), endoprotease Asp-N (Roche Applied Science), or V8-protease in 50 mm Tris HCl-buffer containing 0.1% SDS, 50 mm NaPi buffer containing 0.01% SDS, or 50 mm NaHCO3 containing 0.01% SDS, respectively. The digests were resolved on a Schägger-type SDS gel.
Regarding the identification of proteins (peptide mass fingerprinting), proteins were digested “in-gel” with trypsin (Promega, Fitchburg, WI) according to previous procedures (17). A mass spectrometric analysis was conducted using Bruker Autoflex III Smartbeam (MALDI-TOF MS, Bruker Daltonics, Billerica, MA) or AXIMA-Performance (MALDI-TOF MS, Shimadzu). N-terminal amino acid residues were examined with a Procise 494 HT protein sequencing system (Applied Life Sciences, Foster City, CA) at the APRO Life Science Institute, Inc. (Tokushima, Japan).
The synthetic procedures for SF–UQs, PC–UQs, and photoreactive 125I-amilorides are shown in Schemes S1–S5, detection of the PSST subunit on the doubled SDS gel is shown in Fig. S1; schematic diagram of the pinpoint chemical modification of the 49-kDa Asp-160 is shown in Fig. S2; summary of photoaffinity labeling studies using different quinone-site inhibitors is shown in Fig. S3; and the proteins identified by MALDI-TOF MS are described in the Table S1.
Author contributions
S. U., H. K., M. M., and H. M. data curation; S. U., H. K., M. M., and H. M. formal analysis; S. U., H. K., M. M., and H. M. investigation; S. U., M. M., and H. M. methodology; S. U., H. K., M. M., and H. M. writing-original draft; M. M. and H. M. funding acquisition; H. M. supervision; H. M. project administration.
Supplementary Material
Acknowledgments
We thank Drs. Kentaro Ifuku and Fumihiko Sato (Division of Integrated Life Science, Graduate School of Biostudies, Kyoto University) for allowing us access to their MALDI-TOF MS (Bruker Autoflex III Smartbeam), as well as for their helpful advice on the experiments. The experiments involving radioisotope techniques were performed at the Radioisotope Research Center, Kyoto University.
This work was supported by JSPS KAKENHI Grants JP26292060 and JP18H02147 (to H. M.), Grant JP18K05458 (to M. M.), and by The Uehara Memorial Foundation (to M. M.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S3, Table S1, Schemes S1–S5, and supporting Refs. 1–11
- complex I
- proton-translocating NADH–quinone oxidoreductase
- Asp-N
- endoprotease Asp-N
- BN-PAGE
- blue native-polyacrylamide gel electrophoresis
- Lys-C
- lysyl endopeptidase
- PC
- phosphatidylcholine
- TMH
- transmembrane helix
- SMP
- submitochondrial particle
- UQn
- ubiquinone-n
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- PDB
- Protein Data Bank
- CBB
- Coomassie Brilliant Blue
- APF
- 4-azidophenyl fenpyroximate
- AzQ
- azido-quinazoline
- TAMRA-DIBO
- 6-carboxy-N,N,N′,N′-tetramethylrhodamine-dibenzocyclootyne
- TMS
- trimethylsilyl.
References
- 1. Hirst J. (2013) Mitochondrial complex I. Annu. Rev. Biochem. 82, 551–575 10.1146/annurev-biochem-070511-103700 [DOI] [PubMed] [Google Scholar]
- 2. Sazanov L. A. (2015) A giant molecular proton pump: structure and mechanism of respiratory complex I. Nat. Mol. Cell Biol. 16, 375–388 10.1038/nrm3997 [DOI] [PubMed] [Google Scholar]
- 3. Wirth C., Brandt U., Hunte C., and Zickermann V. (2016) Structure and function of mitochondrial complex I. Biochim. Biophys. Acta 1857, 902–914 10.1016/j.bbabio.2016.02.013 [DOI] [PubMed] [Google Scholar]
- 4. Wong H.-S., Dighe P. A., Mezera V., Monternier P.-A., and Brand M. D. (2017) Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J. Biol. Chem. 292, 16804–16809 10.1074/jbc.R117.789271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baradaran R., Berrisford J. M., Minhas G. S., and Sazanov L. A. (2013) Crystal structure of the entire respiratory complex I. Nature 494, 443–448 10.1038/nature11871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zickermann V., Wirth C., Nasiri H., Siegmund K., Schwalbe H., Hunte C., and Brandt U. (2015) Mechanistic insight from the crystal structure of mitochondrial complex I. Science 347, 44–49 10.1126/science.1259859 [DOI] [PubMed] [Google Scholar]
- 7. Zhu J., Vinothkumar K. R., and Hirst J. (2016) Structure of mammalian respiratory complex I. Nature 536, 354–358 10.1038/nature19095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Blaza J. N., Vinothkumar K. R., and Hirst J. (2018) Structure of the deactive state of mammalian respiratory complex I. Structure 26, 312–319.e9 10.1016/j.str.2017.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fiedorczuk K., Letts J. A., Degliesposti G., Kaszuba K., Skehel M., and Sazanov L. A. (2016) Atomic structure of the entire mammalian mitochondrial complex I. Nature 538, 406–410 10.1038/nature19794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wu M., Gu J., Guo R., Huang Y., and Yang M. (2016) Structure of mammalian respiratory supercomplex I1III2IV1. Cell 167, 1598–1609.e10 10.1016/j.cell.2016.11.012 [DOI] [PubMed] [Google Scholar]
- 11. Agip A. A., Blaza J. N., Bridges H. R., Viscomi C., Rawson S., Muench S. P., and Hirst J. (2018) Cryo-EM structures of complex I from mouse heart mitochondria in two biochemically defined states. Nat. Struct. Mol. Biol. 25, 548–556 10.1038/s41594-018-0073-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guo R., Zong S., Wu M., Gu J., and Yang M. (2017) Architecture of human mitochondrial respiratory megacomplex I2III2IV2. Cell 170, 1247–1257.e12 10.1016/j.cell.2017.07.050 [DOI] [PubMed] [Google Scholar]
- 13. Fedor J. G., Jones A. J. Y., Di Luca A., Kaila V. R. I., and Hirst J. (2017) Correlating kinetic and structural data on ubiquinone binding and reduction by respiratory complex I. Proc. Natl. Acad. Sci. U.S.A. 114, 12737–12742 10.1073/pnas.1714074114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Haapanen O., and Sharma V. (2018) A modeling and simulation perspective on the mechanism and function of respiratory complex I. Biochim. Biophys. Acta Bioenerg. 1859, 510–523 10.1016/j.bbabio.2018.04.001 [DOI] [PubMed] [Google Scholar]
- 15. Masuya T., Murai M., Morisaka H., and Miyoshi H. (2014) Pinpoint chemical modification of Asp-160 in the 49 kDa subunit of bovine mitochondrial complex I via a combination of ligand-directed tosyl chemistry and click chemistry. Biochemistry 53, 7816–7823 10.1021/bi501342w [DOI] [PubMed] [Google Scholar]
- 16. Okuda K., Murai M., Aburaya S., Aoki W., and Miyoshi H. (2016) Reduction of synthetic ubiquinone QT catalyzed by bovine mitochondrial complex I is decoupled from proton translocation. Biochemistry 55, 470–481 10.1021/acs.biochem.5b01090 [DOI] [PubMed] [Google Scholar]
- 17. Murai M., Murakami S., Ito T., and Miyoshi H. (2015) Amilorides bind to the quinone binding pocket of bovine mitochondrial complex I. Biochemistry 54, 2739–2746 10.1021/acs.biochem.5b00187 [DOI] [PubMed] [Google Scholar]
- 18. Masuya T., Murai M., Ito T., Aburaya S., Aoki W., and Miyoshi H. (2017) Pinpoint chemical modification of the quinone-access channel of mitochondrial complex I via a two-step conjugation reaction. Biochemistry 56, 4279–4287 10.1021/acs.biochem.7b00612 [DOI] [PubMed] [Google Scholar]
- 19. Nakamaru-Ogiso E., Seo B. B., Yagi T., and Matsuno-Yagi A. (2003) Amiloride inhibition of the proton-translocating NADH–quinone oxidoreductase of mammals and bacteria. FEBS Lett. 549, 43–46 10.1016/S0014-5793(03)00766-X [DOI] [PubMed] [Google Scholar]
- 20. Stolpe S., and Friedrich T. (2004) The Escherichia coli NADH-ubiquinone oxidoreductase (complex I) is a primary proton pump but may be capable of secondary sodium antiport. J. Biol. Chem. 279, 18377–18383 10.1074/jbc.M311242200 [DOI] [PubMed] [Google Scholar]
- 21. Miyoshi H., Nishioka T., and Fujita T. (1987) Quantitative relationship between protonophoric and uncoupling activities of analogs of SF6847 (2,6-di-tert-butyl-4-(2,2-dicyanovinyl)phenol). Biochim. Biophys. Acta 891, 293–299 10.1016/0005-2728(87)90224-6 [DOI] [PubMed] [Google Scholar]
- 22. Miyoshi H., Tsujishita H., Tokutake N., and Fujita T. (1990) Quantitative analysis of uncoupling activity of substituted phenols with physicochemical substituent and molecular parameters. Biochim. Biophys. Acta 1016, 99–106 10.1016/0005-2728(90)90011-R [DOI] [PubMed] [Google Scholar]
- 23. Chazotte B., and Hackenbrock C. R. (1988) The multi-collisional, obstructed, long-range diffusional nature of mitochondrial electron transport. J. Biol. Chem. 263, 14359–14367 [PubMed] [Google Scholar]
- 24. Chazotte B., and Hackenbrock C. R. (1989) Lateral diffusion as a rate-limiting step in ubiquinone-mediated mitochondrial electron transport. J. Biol. Chem. 264, 4978–4985 [PubMed] [Google Scholar]
- 25. Estornell E., Fato R., Pallotti F., and Lenaz G. (1993) Assay conditions for the mitochondrial NADH:coenzyme Q oxidoreductase. FEBS Lett. 332, 127–131 10.1016/0014-5793(93)80498-J [DOI] [PubMed] [Google Scholar]
- 26. Fato R., Estornell E., Di Bernardo S., Pallotti F., Parenti Castelli G., and Lenaz G. (1996) Steady-state kinetics of the reduction of coenzyme Q analogs by complex I (NADH:ubiquinone oxidoreductase) in bovine heart mitochondria and submitochondrial particles. Biochemistry 35, 2705–2716 10.1021/bi9516034 [DOI] [PubMed] [Google Scholar]
- 27. Benz R., and McLaughlin S. (1983) The molecular mechanism of action of the proton ionophore FCCP (carbonylcyanide p-trifluoromethoxyphenylhydrazone). Biophys. J. 41, 381–398 10.1016/S0006-3495(83)84449-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lambert A. J., and Brand M. D. (2004) Inhibitors of the quinone-binding site allow rapid superoxide production from mitochondrial NADH-ubiquinone oxidoreductase (complex I). J. Biol. Chem. 279, 39414–39420 10.1074/jbc.M406576200 [DOI] [PubMed] [Google Scholar]
- 29. Galkin A., and Brandt U. (2005) Superoxide radical formation by pure complex I (NADH:ubiquinone oxidoreductase) from Yarrowia lipolytica. J. Biol. Chem. 280, 30129–30135 10.1074/jbc.M504709200 [DOI] [PubMed] [Google Scholar]
- 30. King M. S., Sharpley M. S., and Hirst J. (2009) Reduction of hydrophilic ubiquinones by the flavin in mitochondrial NADH:ubiquinone oxidoreductase (complex I) and production of reactive oxygen species. Biochemistry 48, 2053–2062 10.1021/bi802282h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Murai M., Habu S., Murakami S., Ito T., and Miyoshi H. (2015) Production of new amilorides as potent inhibitors of mitochondrial respiratory complex I. Biosci. Biotechnol. Biochem. 79, 1061–1066 10.1080/09168451.2015.1010479 [DOI] [PubMed] [Google Scholar]
- 32. Ito T., Murai M., Morisaka H., and Miyoshi H. (2015) Identification of the binding position of amilorides in the quinone binding pocket of mitochondrial complex I. Biochemistry 54, 3677–3686 10.1021/acs.biochem.5b00385 [DOI] [PubMed] [Google Scholar]
- 33. Rais I., Karas M., and Schägger H. (2004) Two-dimensional electrophoresis for the isolation of integral membrane proteins and mass spectrometric identification. Proteomics 4, 2567–2571 10.1002/pmic.200400829 [DOI] [PubMed] [Google Scholar]
- 34. Murai M., Sekiguchi K., Nishioka T., and Miyoshi H. (2009) Characterization of the inhibitor binding site in mitochondrial NADH-ubiquinone oxidoreductase by photoaffinity labeling using a quinazoline-type inhibitor. Biochemistry 48, 688–698 10.1021/bi8019977 [DOI] [PubMed] [Google Scholar]
- 35. Shiraishi Y., Murai M., Sakiyama N., Ifuku K., and Miyoshi H. (2012) Fenpyroximate binds to the interface between PSST and 49 kDa subunits in mitochondrial NADH-ubiquinone oxidoreductase. Biochemistry 51, 1953–1963 10.1021/bi300047h [DOI] [PubMed] [Google Scholar]
- 36. Cleveland D. W., Fischer S. G., Kirschner M. W., and Laemmli U. K. (1977) Peptide mapping by limited proteolysis in sodium dodecyl sulfate and analysis by gel electrophoresis. J. Biol. Chem. 252, 1102–1106 [PubMed] [Google Scholar]
- 37. Murai M., Mashimo Y., Hirst J., and Miyoshi H. (2011) Exploring interactions between the 49 kDa and ND1 subunits in mitochondrial NADH-ubiquinone oxidoreductase (complex I) by photoaffinity labeling. Biochemistry 50, 6901–6908 10.1021/bi200883c [DOI] [PubMed] [Google Scholar]
- 38. Fearnley I. M., Finel M., Skehel J. M., and Walker J. E. (1991) NADH:ubiquinone oxidoreductase from bovine heart mitochondria. cDNA sequences of the import precursors of the nuclear-encoded 39 kDa and 42 kDa subunits. Biochem. J. 278, 821–829 10.1042/bj2780821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sharma V., Belevich G., Gamiz-Hernandez A. P., Róg T., Vattulainen I., Verkhovskaya M. L., Wikström M., Hummer G., and Kaila V. R. (2015) Redox-induced activation of the proton pump in the respiratory complex I. Proc. Natl. Acad. Sci. U.S.A. 112, 11571–11576 10.1073/pnas.1503761112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Di Luca A., Gamiz-Hernandez A. P., and Kaila V. R. I. (2017) Symmetry-related proton transfer pathways in respiratory complex I. Proc. Natl. Acad. Sci. U.S.A. 114, E6314–E6321 10.1073/pnas.1706278114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gamiz-Hernandez A. P., Jussupow A., Johansson M. P., and Kaila V. R. (2017) Terminal electron-proton transfer dynamics in the quinone reduction of respiratory complex I. J. Am. Chem. Soc. 139, 16282–16288 10.1021/jacs.7b08486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ohshima M., Miyoshi H., Sakamoto K., Takegami K., Iwata J., Kuwabara K., Iwamura H., and Yagi T. (1998) Characterization of the ubiquinone reduction site of mitochondrial complex I using bulky synthetic ubiquinones. Biochemistry 37, 6436–6445 10.1021/bi9800202 [DOI] [PubMed] [Google Scholar]
- 43. Sakamoto K., Miyoshi H., Ohshima M., Kuwabara K., Kano K., Akagi T., Mogi T., and Iwamura H. (1998) Role of isoprenyl tail of ubiquinone in reaction with respiratory enzymes: studies with bovine heart mitochondrial complex I and Escherichia coli bo-type ubiquinol oxidase. Biochemistry 37, 15106–15113 10.1021/bi981193u [DOI] [PubMed] [Google Scholar]
- 44. Stroud D. A., Formosa L. E., Wijeyeratne X. W., Nguyen T. N., and Ryan M. T. (2013) Gene knockout using transcription activator-like effector nucleases (TALENs) reveals that human NDUFA9 protein is essential for stabilizing the junction between membrane and matrix arms of complex I. J. Biol. Chem. 288, 1685–1690 10.1074/jbc.C112.436766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Babot M., Labarbuta P., Birch A., Kee S., Fuszard M., Botting C. H., Wittig I., Heide H., and Galkin A. (2014) ND3, ND1 and 39 kDa subunits are more exposed in the de-active form of bovine mitochondrial complex I. Biochim. Biophys. Acta 1837, 929–939 10.1016/j.bbabio.2014.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nakanishi S., Abe M., Yamamoto S., Murai M., and Miyoshi H. (2011) Bis-THF motif of acetogenin binds to the third matrix-side loop of ND1 subunit in mitochondrial NADH-ubiquinone oxidoreductase. Biochim. Biophys. Acta 1807, 1170–1176 10.1016/j.bbabio.2011.05.012 [DOI] [PubMed] [Google Scholar]
- 47. Masuya T., Okuda K., Murai M., and Miyoshi H. (2016) Characterization of the reaction of decoupling ubiquinone with bovine mitochondrial respiratory complex I. Biosci. Biotechnol. Biochem. 80, 1464–1469 10.1080/09168451.2016.1179095 [DOI] [PubMed] [Google Scholar]
- 48. Bajda S., Dermauw W., Panteleri R., Sugimoto N., Douris V., Tirry L., Osakabe M., Vontas J., and Van Leeuwen T. (2017) A mutation in the PSST homologue of complex I (NADH:ubiquinone oxidoreductase) from Tetranychus urticae is associated with resistance to METI acaricides. Insect Biochem. Mol. Biol. 80, 79–90 10.1016/j.ibmb.2016.11.010 [DOI] [PubMed] [Google Scholar]
- 49. Letts J. A., Degliesposti G., Fiedorczuk K., Skehel M., and Sazanov L. (2016) Purification of ovine respiratory complex I results in a high active and stable preparation. J. Biol. Chem. 291, 24657–24675 10.1074/jbc.M116.735142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Colombini M. (2009) The published 3D structure of the VDAC channel: native or not? Trends Biochem. Sci. 34, 382–389 10.1016/j.tibs.2009.05.001 [DOI] [PubMed] [Google Scholar]
- 51. Hiller S., Abramson J., Mannella C., Wagner G., and Zeth K. (2010) The 3D structures of VDAC represent a native conformation. Trends Biochem. Sci. 35, 514–521 10.1016/j.tibs.2010.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rey M., Forest E., and Pelosi L. (2012) Exploring the conformational dynamics of the bovine ADP/ATP carrier in mitochondria. Biochemistry 51, 9727–9735 10.1021/bi300759x [DOI] [PubMed] [Google Scholar]
- 53. Crichton P. G., Lee Y., Ruprecht J. J., Cerson E., Thangaratnarajah C., King M. S., and Kunji E. R. (2015) Trends in thermostability provide information on the nature of substrate, inhibitor, and lipid interactions with mitochondrial carriers. J. Biol. Chem. 290, 8206–8217 10.1074/jbc.M114.616607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Steuber J., Vohl G., Casutt M. S., Vorburger T., Diederichs K., and Fritz G. (2014) Structure of the V. cholerae Na+-pumping NADH:quinone oxidoreductase. Nature 516, 62–67 10.1038/nature14003 [DOI] [PubMed] [Google Scholar]
- 55. Ito T., Murai M., Ninokura S., Kitazumi Y., Mezic K. G., Cress B. F., Koffas M. A. G., Morgan J. E., Barquera B., and Miyoshi H. (2017) Identification of the binding sites for ubiquinone and inhibitors in the Na+-pumping NADH-ubiquinone oxidoreductase of Vibrio cholerae by photoaffinity labeling. J. Biol. Chem. 292, 7727–7742 10.1074/jbc.M117.781393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Matsuno-Yagi A., and Hatefi Y. (1985) Studies on the mechanism of oxidative phosphorylation. Catalytic site cooperativity in ATP synthesis. J. Biol. Chem. 260, 14424–14427 [PubMed] [Google Scholar]
- 57. Wittig I., Braun H. P., and Schägger H. (2006) Blue native PAGE. Nat. Protoc. 1, 418–428 10.1038/nprot.2006.62 [DOI] [PubMed] [Google Scholar]
- 58. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 10.1038/227680a0 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.